
Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches




Abstract
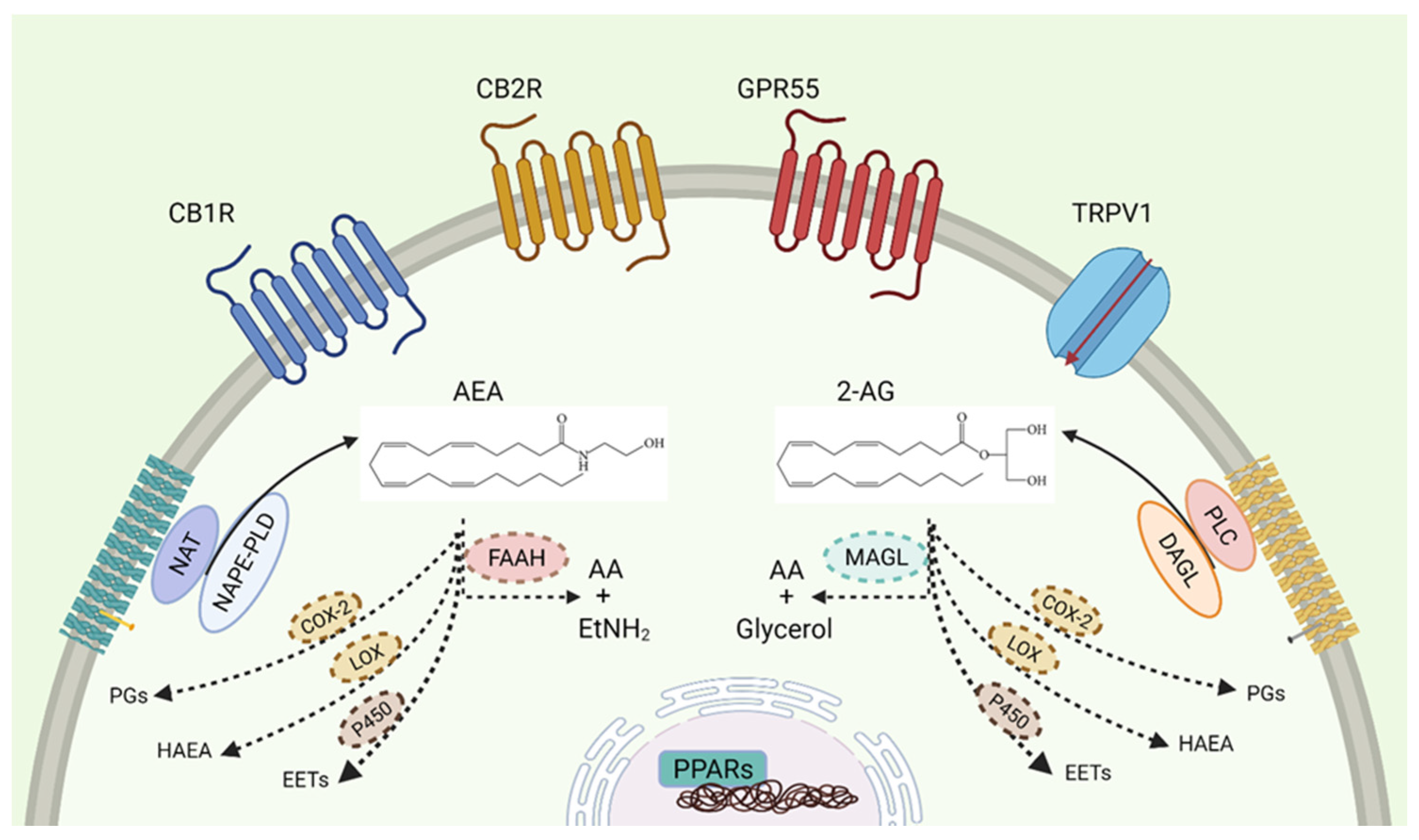
1. The Endocannabinoid System
1.1. Receptors
1.2. Cannabinoid Receptor Agonists
1.3. Other Agonists
1.4. Cannabinoid Receptor Antagonists/Inverse Agonists
1.5. Cannabinoid Enzymatic Dysregulations in Cancer
1.6. MAGL Inhibitors
1.7. FAAH Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COMPOUND | TUMOUR | ACTION | REF. |
|---|---|---|---|
| ∆9-THC (classical) | Gastro-intestinal cancer | Induction of apoptosis through CB1R-mediated inhibition of RAS-MAPK/ERK and PI3K-Akt survival signalling cascades | [164] |
| HCC | Anti-proliferative action associated with accumulation of ceramide, ER-stress and PPARγ activity Autophagy-mediated cell death in combination with JWH-015 | [69,165] | |
| Lung cancer | Inhibition of tumour cell growth (reduction in 3H thymidine and 14C-uridine uptake) Inhibition of EGF-induced proliferation/migration and invasion, reduction in EGF-induced phosphorylation of ERK1/2, ERK1/2 and Akt | [166,167] | |
| Breast cancer | Disruption of HER2-CB2R heteromers leading to HER2-proteasome degradation Induction of cell cycle arrest through Cdc2 downregulation, leading to apoptosis Reduction in 17β-oestradiol-induced proliferation | [90,168,169] | |
| Prostate cancer | Induction of apoptosis independent from CBRs | [170] | |
| Pancreatic tumour | Induction of apoptosis through de novo synthesis of ceramide and consequent upregulation of ER stress related genes p8, ATF-4 and TRB3 | [171] | |
| Brain cancer | Inhibition of cell proliferation, induction of cycle arrest, ROS production and apoptosis, given alone or in combination with CBD Autophagy-mediated cancer cell death Inhibition of MMP-2 expression and cell invasion in cultured glioma cells via ceramide accumulation and activation of p8 stress protein Increase in radiosensivity in combination with CBD | [132,172,173,174,175] | |
| Endometrial cancer | Inhibition of migration through down regulation of MMP-9 | [176] | |
| Leukaemia | Induction of apoptosis via MAPK pathway Reversion of multidrug resistance together with CBD Sensitisation to cytotoxic effects of chemotherapy | [177,178,179] | |
| Melanoma | Induction of cell cycle arrest through Akt inhibition, activation of autophagy-mediated apoptosis | [180,181] | |
| WIN 55,212-2 (aminoalkyndole) | Gastro-intestinal cancer | Inhibition of cell proliferation and induction of apoptosis. Inhibition of Akt, downregulation of MMP-2 and VEGF-A Inhibition of cell migration/invasion and EMT markers through COX2 downregulation | [182,183,184] |
| Prostate cancer | Inhibition of cell growth, induction of apoptosis, decrease in AR, PSA, PCNA and VEGF in LNCaP Prevention of neuroendocrine differentiation of LNCaP by inhibition of PI3K/Akt/mTOR axis and stimulation of AMPK | [185,186] | |
| Renal carcinoma | Inhibition of proliferation and cell viability. Induction of G0/G1 cell cycle arrest, apoptosis and reduced proliferation into 3D spheres | [187] | |
| Osteosarcoma | Inhibition on cell migration with reduction in MMP-2 and MMP-9 | [188] | |
| Lung and testicular cancer | Induction of apoptosis | [189] | |
| AEA (eicosanoid) | Gastro-intestinal cancer | Induction of G0/G1 cell cycle arrest and apoptosis Reduction in cell proliferation through activation of Wnt5a non-canonical pathway Inhibition of cell proliferation induced by FAS-death receptor translocation in lipid rafts, mediated by GPR55 activation | [99,190,191] |
| Lung cancer | Reduction in tumour cell spreading, mimicking the anti-invasive action of FAAH inhibitors (same effect given by 2-AG, OEA, PEA) | [163] | |
| Breast cancer | Inhibition of cell proliferation through downregulation of adenylate cyclase and activation of MAPK, exerting downregulation on prolactin and tyrosine kinase levels | [192,193,194] | |
| Prostate cancer | Reduction in EGF-induced cell proliferation, induction of apoptosis and necrosis through EGFR downregulation Induction of apoptosis mediated by activation of ERK and inhibition of AKT signalling pathways (same effect given by 2-AG and Met-F-AEA) | [195,196] | |
| Non-melanoma skin cancer | Induction of apoptosis mediated by oxidative stress and CBR-independent signalling | [197] | |
| Lymphoma | Reduction of tumour cell viability | [198] | |
| R(+)-Methanandamide (stable AEA analogue) | Prostate cancer | Inhibition of cell growth in prostate cells (PC-3) | [98] |
| Cervical cancer | Activation of apoptosis mediated by COX-2 and subsequent prostaglandins synthesis via PPARγ | [97] | |
| Gastro-intestinal cancer | Induction of G0/G1 cell cycle arrest and necrosis | [99] | |
| Met-F-AEA (stable AEA analogue) | Breast cancer | Induction of cell cycle arrest correlated with Chk1 activation, Cdc25A degradation and downregulation of Cdk2 activity Inhibition of adhesion and migration, interfering with the RhoA/ROCK signalling pathway and FAK phosphorylation | [100,101,102] |
| Melanoma | Inhibition of cell growth | [106] | |
| Lung cancer | Induction of G0/G1 cell cycle arrest leading to apoptosis (in combination with UR597) | [103] | |
| Gastro-intestinal cancer | Increase in AEA availability, induction of oestrogen receptor β expression, decrease in proliferation rate due to CB1 up-regulation through the transcriptional activation of CNR1 promoter (CRC) | [104] | |
| Thyroid cancer | Induction of apoptosis via p53 and p21 | [105] | |
| PEA | Brain cancer | Induction of cell death | [52] |
| Melanoma | Reduction of melanoma cell survival in combination with URB597 | [108] | |
| Breast cancer | Increase in cytotoxic effect of AEA | [109] | |
| ACEA | Gastro-intestinal cancer | Activation of apoptosis through TNF-α–mediated ceramide de novo synthesis | [110] |
| HCC | Reduction of cell viability, invasion and MMP-2/MMP-9 expression | [111] | |
| Breast cancer | Inhibition of invasion in breast cancer stem cells | [112] | |
| Pancreatic cancer | Induction of ROS-mediated autophagy via activation of AMPK, inhibition of energetic metabolism. Decrease in GAPDH and PMK2 expression. Increase the anticancer potential of gemcitabine | [113] | |
| JWH-015 | Prostate cancer | Inhibition of cell growth and apoptosis induction via de novo synthesis of ceramide. Signalling pathways include JNK activation and Akt inhibition. | [98] |
| Breast cancer | Reduction of tumour growth, chemotaxis and wound healing. (block of the chemokine receptor CXCR4 signalling) Inhibition of EGFR activation in ERα breast cancer cells | [115,199] | |
| Lung cancer | Attenuation of growth factor-directed in vitro chemotaxis and chemo-invasion. Reduction in focal adhesion complex. Inhibition of Akt phosphorylation and reduction in MMP-9 expression and activity | [25] | |
| JWH-133 | Brain cancer | Inhibition of glioma cell viability | [118] |
| Breast cancer | Decrease in cell proliferation, induction of apoptosis, inhibition of cell migration | [119] | |
| Melanoma | Decrease in trans-endothelial migration in vitro | [117] |
| COMPOUND | TUMOUR | ACTION | REF. |
|---|---|---|---|
| SR141716 (CB1R selective antagonist) | Gastro-intestinal cancer | Induction of G2/M cell cycle arrest and mitotic catastrophe Synergic effect in combination with oxaliplatin, blocking cell proliferation Impact in chemoresistance and cancer stemness, retain of architecture and heterogeneity of human healthy organoids in ex vivo cultures through inhibition of Wnt/β-catenin canonical pathway | [126,129,200,201] |
| Brain cancer | Induction of cell proliferation arrest, caspase-dependent apoptosis and upregulation of the NKG2D ligand MICA/B | [128] | |
| Breast cancer | Inhibition of cell proliferation via CB1R-interaction with lipid rafts | [127] | |
| CBD (antagonist, inverse agonist and negative allosteric modulator at CB1R/partial agonist at CB2R) | Gastro-intestinal cancer | Induction of G0/G1 cell cycle arrest through downregulation of CDK2-cyclin E. Activation of mitochondrial-dependent apoptosis pathway by increasing ROS production Reduction of cell migration Protection of DNA from oxidative damage, increase in endocannabinoid levels, reduction in proliferation through CB1R, TRPV1 and PPARγ involvement. Reduction of invasion and cell migration Induction of apoptosis through excessive ROS production, ER stress and Noxa activation | [130,131,139,151] |
| Lung cancer | Induction of PPARγ dependent apoptosis through increased levels of COX2-dependent prostaglandins Reduction in cell migration accompanied with decreased PAI-1 Induction of ICAM-1 in cancer cells leading to lymphokine-activated killer (LAK) cell-mediated cytotoxicity Upregulation of ICAM-1 and TIMP-1 levels, decreasing cell migration via CBRs, TRPV1 and p42/44 MAPK | [135,140,141,202] | |
| Breast cancer | Induction of a crosstalk between apoptosis and autophagy in mediating cancer cell death Inhibition of cell proliferation, induction of apoptosis, ER stress (MDA-MB-231). Induction of cell cycle arrest at G1/S phase (MCF-7) via CBRs or TRPV1 receptors Induction of apoptosis through downregulation of mTOR, cyclin D1 and upregulation of PPARγ (T47-D, MDA-MB-231) Inhibition of EGF-induced cell proliferation, colony formation, migration and invasion. Downregulation in cytokine production Reduction of proliferation and invasion through Id-1 downregulation Increase uptake of DOXO and induction of apoptosis, via activation of TRPV2 (TNBC) | [133,136,137,138,142,145] | |
| Prostate cancer | Cytotoxic effects and downregulation of CB1R, CB2R, VEGF, PSA, IL-6, IL-8 in LNCaP. Reduction of spheroid formation in LNCaP stem cells Cytotoxic activity, cell cycle arrest, apoptosis induction. Induction of apoptosis in LNCaP partially due to TRPM8 antagonism and accompanied by downregulation of AR, p53, elevated ROS. Synergistic anti-proliferative effects with docetaxel and/or bicalutamide in DU-145 and/or LNCaP cells | [134,203] | |
| Brain cancer | Inhibition of cell proliferation, modulation of cell cycle, increase in ROS levels and apoptosis when given in combination with ∆9-THC Increase in ROS production derived from upregulation of HSP super family genes. Decrease in cytotoxic effects through HSP upregulation. HSP inhibitors in combination with CBD lead to increased cytotoxicity respect to CBD alone Inhibition of cell invasion through Id-1 downregulation Inhibition of cell proliferation and invasiveness through downregulation in proteins specifically involved in growth, invasion and angiogenesis, downregulation of ERK, Akt, and HIF-1α Inhibition of cell proliferation, induction of apoptosis and chemosensitivity to TMZ, BCNU, and DOXO through TRPV2 activation | [132,143,144,146,147] | |
| AM251 (CB1R inverse agonist/GPR55 agonist) | Pancreatic cancer | Induction of apoptosis via receptor-independent mechanisms | [204] |
| Gastro-intestinal cancer | Reversion of the Met-F-AEA anti-proliferative effect | [104] | |
| Breast cancer | Reversion of the effect of ACEA on the decrease in the invasive potential of breast cancer stem cells | [112] | |
| Renal cell carcinoma | Decrease in proliferation, induction of apoptosis by upregulating Bax and decreasing Bcl-2. Inhibition of cell migration | [148] | |
| 6-iodopravadoline (AM-630) (CB2R inverse agonist) | Renal cell carcinoma | Inhibition of cell proliferation, induction of cell cycle arrest in G2/M phase, anti-migratory effects | [149] |
| CID16020046 (selective GPR55 antagonist) | Gastro-intestinal cancer | Decrease in migration and adhesion to endothelial cell | [151] |
| Inhibition of cell proliferation and ERK1/2 phosphorylation | [150] | ||
| Breast cancer | Decrease filopodia formation and migration | [72] | |
| Reduction in chemoresistance through downregulation of MDR (e.g., BCRP) | [152] |
2. The Tumour Microenvironment
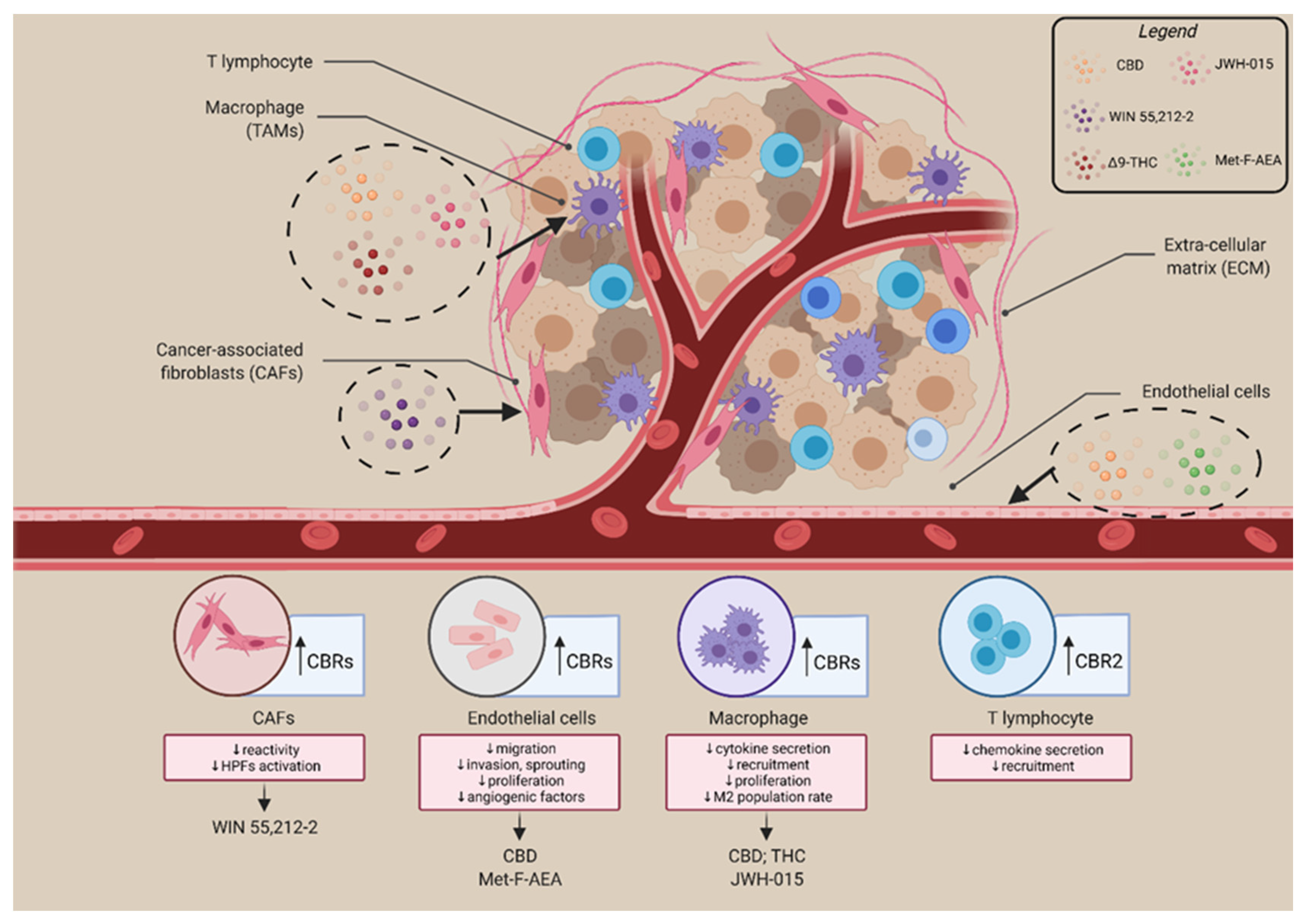
3. The Involvement of the Endocannabinoid System in the Tumour Microenvironment
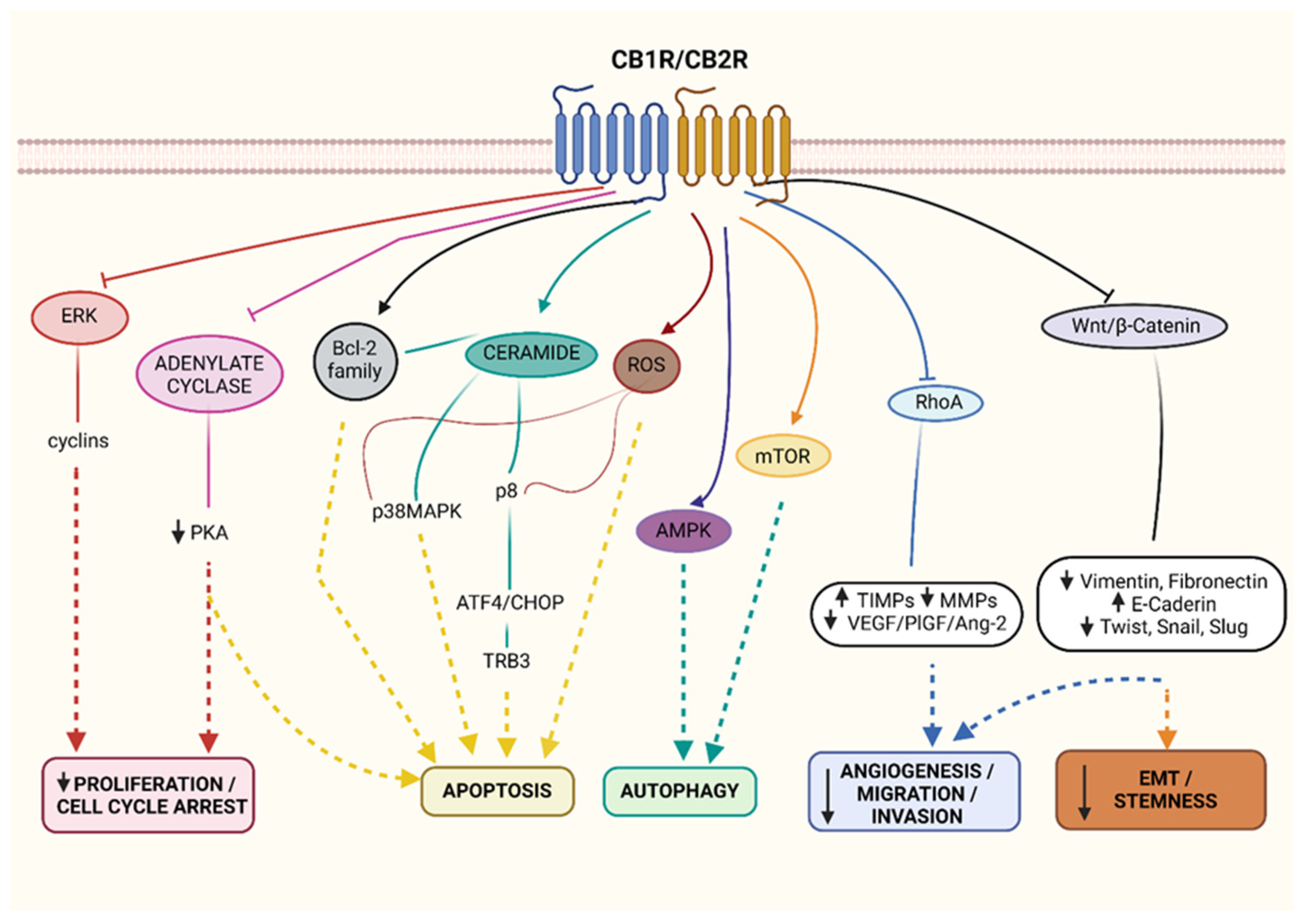
4. Cannabinoid-Based Antineoplastic Treatment—Preclinical Studies
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmid, P.C.; Schwartz, K.D.; Smith, C.N.; Krebsbach, R.J.; Berdyshev, E.V.; Schmid, H.H. A sensitive endocannabinoid assay. The simultaneous analysis of N-acylethanolamines and 2-monoacylglycerols. Chem. Phys. Lipids 2000, 104, 185–191. [Google Scholar] [CrossRef]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.-C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef]
- Cadas, H.; Gaillet, S.; Beltramo, M.; Venance, L.; Piomelli, D. Biosynthesis of an Endogenous Cannabinoid Precursor in Neurons and its Control by Calcium and cAMP. J. Neurosci. 1996, 16, 3934–3942. [Google Scholar] [CrossRef]
- Stella, N.; Schweitzer, P.J.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef]
- Bisogno, T.; Berrendero, F.; Ambrosino, G.; Cebeira, M.; Ramos, J.; Fernandez-Ruiz, J.; Di Marzo, V. Brain Regional Distribution of Endocannabinoids: Implications for Their Biosynthesis and Biological Function. Biochem. Biophys. Res. Commun. 1999, 256, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.-J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The endocannabinoid system: An overview. Front. Behav. Neurosci. 2012, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid Oxygenation by Cyclooxygenases, Lipoxygenases, and Cytochromes P450: Cross-Talk between the Eicosanoid and Endocannabinoid Signaling Pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef] [PubMed]
- Valdeolivas, S.; Pazos, M.R.; Bisogno, T.; Piscitelli, F.; Iannotti, F.A.; Allarà, M.; Sagredo, O.; Di Marzo, V.; Fernández-Ruiz, J. The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: A potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis. 2013, 4, e862. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Ni, J.; Ling, K.-H.J.; Acheampong, A.; Tang-Liu, D.D.-S.; Burk, R.; Cravatt, B.F.; Woodward, D. Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J. Lipid Res. 2004, 45, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Hillard, C.J. The Endocannabinoid Signaling System in the CNS. Int. Rev. Neurobiol. 2015, 125, 1–47. [Google Scholar] [CrossRef]
- Almogi-Hazan, O.; Or, R. Cannabis, the Endocannabinoid System and Immunity—the Journey from the Bedside to the Bench and Back. Int. J. Mol. Sci. 2020, 21, 4448. [Google Scholar] [CrossRef]
- Silvestri, C.; Di Marzo, V. The Endocannabinoid System in Energy Homeostasis and the Etiopathology of Metabolic Disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Maurya, N.; Velmurugan, B.K. Therapeutic applications of cannabinoids. Chem. Interact. 2018, 293, 77–88. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Martín-Sabroso, C.; Torres-Suárez, A.I. Insights into the effects of the endocannabinoid system in cancer: A review. Br. J. Pharmacol. 2018, 175, 2566–2580. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Sadek, B.; Goyal, S.N.; Sinha, S.; Kamal, M.A.; Ojha, S. Small Molecules from Nature Targeting G-Protein Coupled Cannabinoid Receptors: Potential Leads for Drug Discovery and Development. Evid.-Based Complement. Altern. Med. 2015, 2015, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, E.; Kathmann, M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001, 22, 565–572. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Strangman, N.M.; Patrick, S.L.; Hohmann, A.G.; Tsou, K.; Walker, J. Evidence for a role of endogenous cannabinoids in the modulation of acute and tonic pain sensitivity. Brain Res. 1998, 813, 323–328. [Google Scholar] [CrossRef]
- Matias, I.; Di Marzo, V. Endocannabinoids and the control of energy balance. Trends Endocrinol. Metab. 2007, 18, 27–37. [Google Scholar] [CrossRef]
- Bonz, A.; Laser, M.; Küllmer, S.; Kniesch, S.; Babin-Ebell, J.; Popp, V.; Ertl, G.; Wagner, J.A. Cannabinoids Acting on CB1 Receptors Decrease Contractile Performance in Human Atrial Muscle. J. Cardiovasc. Pharmacol. 2003, 41, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, M.T.; Oddi, S.; Latini, L.; Pasquariello, N.; Florenzano, F.; Bernardi, G.; Molinari, M.; Maccarrone, M. Selective CB2 Receptor Agonism Protects Central Neurons from Remote Axotomy-Induced Apoptosis through the PI3K/Akt Pathway. J. Neurosci. 2009, 29, 4564–4570. [Google Scholar] [CrossRef] [PubMed]
- Pyszniak, M.; Tabarkiewicz, J.; Łuszczki, J.J. Endocannabinoid system as a regulator of tumor cell malignancy—biological pathways and clinical significance. OncoTargets Ther. 2016, 9, 4323–4336. [Google Scholar] [CrossRef]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid Receptors, CB1 and CB2, as Novel Targets for Inhibition of Non–Small Cell Lung Cancer Growth and Metastasis. Cancer Prev. Res. 2010, 4, 65–75. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The Endocannabinoid System: A Target for Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Falasca, M.; Ferro, R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Biol. Regul. 2016, 60, 88–93. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.; Schröder, R.; Kargl, J.K.; Platzer, W.; Martini, L.; Arthur, S.; Penman, J.; Whistler, J.L.; Kostenis, E.; et al. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br. J. Pharmacol. 2010, 160, 604–614. [Google Scholar] [CrossRef] [PubMed]
- AlSuleimani, Y.M.; Hiley, C.R. The GPR55 agonist lysophosphatidylinositol relaxes rat mesenteric resistance artery and induces Ca2+ release in rat mesenteric artery endothelial cells. Br. J. Pharmacol. 2015, 172, 3043–3057. [Google Scholar] [CrossRef]
- Whyte, L.S.; Ryberg, E.; Sims, N.A.; Ridge, S.A.; Mackie, K.; Greasley, P.J.; Ross, R.A.; Rogers, M.J. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc Natl Acad Sci USA 2009, 106, 16511–16516. [Google Scholar] [CrossRef]
- Wu, C.S.; Chen, H.; Sun, H.; Zhu, J.; Jew, C.P.; Wager-Miller, J.; Straiker, A.; Spencer, C.; Bradshaw, H.; Mackie, K.; et al. GPR55, a G-Protein Coupled Receptor for Lysophosphatidylinositol, Plays a Role in Motor Coordination. PLoS ONE 2013, 8, e60314. [Google Scholar] [CrossRef] [PubMed]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 2008, 139, 225–236. [Google Scholar] [CrossRef]
- Guerrero-Alba, R.; Barragan-Iglesias, P.; González-Hernández, A.; Valdez-Moráles, E.E.; Granados-Soto, V.; Condés-Lara, M.; Rodríguez, M.G.; Marichal-Cancino, B.A. Some Prospective Alternatives for Treating Pain: The Endocannabinoid System and Its Putative Receptors GPR18 and GPR55. Front. Pharmacol. 2019, 9, 1496. [Google Scholar] [CrossRef]
- Tudurí, E.; Imbernon, M.; Bautista, R.J.H.; Tojo, M.; Fernø, J.; Diéguez, C.; Nogueiras, R. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017, 58, R191–R202. [Google Scholar] [CrossRef]
- Ford, L.A.; Roelofs, A.J.; Anavi-Goffer, S.; Mowat, L.; Simpson, D.G.; Irving, A.J.; Rogers, M.J.; Rajnicek, A.M.; Ross, R.A. A role for L-α-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br. J. Pharmacol. 2010, 160, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Andradas, C.; Caffarel, M.M.; Pérez-Gómez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2011, 30, 245–252. [Google Scholar] [CrossRef]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2010, 30, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, E.; Andradas, C.; Flores, J.M.; Quintanilla, M.; Paramio, J.M.; Guzmán, M.; Sanchez, C. The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas. Oncogene 2012, 32, 2534–2542. [Google Scholar] [CrossRef]
- Li, L.; Chen, C.; Chiang, C.; Xiao, T.; Chen, Y.; Zhao, Y.; Zheng, D. The Impact of TRPV1 on Cancer Pathogenesis and Therapy: A Systematic Review. Int. J. Biol. Sci. 2021, 17, 2034–2049. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.J.M.; Ciotu, C.I.; Szallasi, A. The Mysteries of Capsaicin-Sensitive Afferents. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Helliwell, R.J.; McLatchie, L.M.; Clarke, M.; Winter, J.; Bevan, S.; McIntyre, P. Capsaicin sensitivity is associated with the expression of the vanilloid (capsaicin) receptor (VR1) mRNA in adult rat sensory ganglia. Neurosci. Lett. 1998, 250, 177–180. [Google Scholar] [CrossRef]
- Kark, T.; Bagi, Z.; Lizanecz, E.; Pásztor, E.T.; Erdei, N.; Czikora, A.; Papp, Z.; Edes, I.; Porszasz, R.; Toth, A. Tissue-Specific Regulation of Microvascular Diameter: Opposite Functional Roles of Neuronal and Smooth Muscle Located Vanilloid Receptor-1. Mol. Pharmacol. 2008, 73, 1405–1412. [Google Scholar] [CrossRef]
- Cavanaugh, D.J.; Chesler, A.T.; Jackson, A.C.; Sigal, Y.M.; Yamanaka, H.; Grant, R.; O’Donnell, D.; Nicoll, R.A.; Shah, N.M.; Julius, D.; et al. Trpv1 Reporter Mice Reveal Highly Restricted Brain Distribution and Functional Expression in Arteriolar Smooth Muscle Cells. J. Neurosci. 2011, 31, 5067–5077. [Google Scholar] [CrossRef] [PubMed]
- Birder, L.; Nakamura, Y.; Kiss, S.; Nealen, M.; Barrick, S.; Kanai, A.; Wang, E.; Ruiz, G.; De Groat, W.; Apodaca, G.; et al. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat. Neurosci. 2002, 5, 856–860. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The Cloned Capsaicin Receptor Integrates Multiple Pain-Producing Stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Hartel, M.; di Mola, F.F.; Selvaggi, F.; Mascetta, G.; Wente, M.N.; Felix, K.; Giese, N.A.; Hinz, U.; di Sebastiano, P.; Büchler, M.W.; et al. Vanilloids in pancreatic cancer: Potential for chemotherapy and pain management. Gut 2006, 55, 519–528. [Google Scholar] [CrossRef]
- Marincsák, R.; Tóth, B.I.; Czifra, G.; Márton, I.; Redl, P.; Tar, I.; Tóth, L.; Kovács, L.; Bíró, T. Increased expression of TRPV1 in squamous cell carcinoma of the human tongue. Oral Dis. 2009, 15, 328–335. [Google Scholar] [CrossRef]
- Weber, L.V.; Al-Refae, K.; Wölk, G.; Bonatz, G.; Altmüller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer Targets Ther. 2016, 8, 243–252. [Google Scholar] [CrossRef]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.J.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232–1238. [Google Scholar] [CrossRef]
- Stienstra, R.; Duval, C.; Muller, M.; Kersten, S. PPARs, Obesity, and Inflammation. PPAR Res. 2006, 2007, 095974. [Google Scholar] [CrossRef]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of mRNAs of Peroxisome Proliferator-Activated Receptors and Liver X Receptor- in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Xue, J.; Avery, J.; Wu, J.; Lind, S.E.; Ding, W.-Q. Activation of Peroxisome Proliferator-activated Receptor α (PPARα) Suppresses Hypoxia-inducible Factor-1α (HIF-1α) Signaling in Cancer Cells. J. Biol. Chem. 2012, 287, 35161–35169. [Google Scholar] [CrossRef]
- Shigeto, T.; Yokoyama, Y.; Xin, B.; Mizunuma, H. Peroxisome proliferator-activated receptor α and γ ligands inhibit the growth of human ovarian cancer. Oncol. Rep. 2007, 18, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Elbegdorj, O.; Westkaemper, R.B.; Zhang, Y. A homology modeling study toward the understanding of three-dimensional structure and putative pharmacological profile of the G-protein coupled receptor GPR55. J. Mol. Graph. Model. 2013, 39, 50–60. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.; Kargl, J.; Andradas, C.; Brown, A.; Irving, A.; Sanchez, C.; Waldhoer, M. Minireview: Recent Developments in the Physiology and Pathology of the Lysophosphatidylinositol-Sensitive Receptor GPR55. Mol. Endocrinol. 2011, 25, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.S.; Shamsizadeh, A.; Azhdari-Zarmehri, H.; Roohbakhsh, A. Orphan G protein-coupled receptors: The role in CNS disorders. Biomed. Pharmacother. 2018, 98, 222–232. [Google Scholar] [CrossRef]
- Pecze, L.; Blum, W.; Schwaller, B. Mechanism of capsaicin receptor TRPV1-mediated toxicity in pain-sensing neurons focusing on the effects of Na+/Ca2+ fluxes and the Ca2+-binding protein calretinin. Biochim. Biophys. Acta Bioenerg. 2013, 1833, 1680–1691. [Google Scholar] [CrossRef]
- Romanovsky, A.A.; Almeida, M.C.; Garami, A.; Steiner, A.; Norman, M.H.; Morrison, S.F.; Nakamura, K.; Burmeister, J.J.; Nucci, T.B. The Transient Receptor Potential Vanilloid-1 Channel in Thermoregulation: A Thermosensor It Is Not. Pharmacol. Rev. 2009, 61, 228–261. [Google Scholar] [CrossRef]
- Cortright, D.N.; Krause, J.E.; Broom, D.C. TRP channels and pain. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Adcock, J.J. TRPV1 receptors in sensitisation of cough and pain reflexes. Pulm. Pharmacol. Ther. 2009, 22, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoids and the gastrointestinal tract. Gut 2001, 48, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Benz, A.H.; Renné, C.; Maronde, E.; Koch, M.; Grabiec, U.; Kallendrusch, S.; Rengstl, B.; Newrzela, S.; Hartmann, S.; Hansmann, M.-L.; et al. Expression and Functional Relevance of Cannabinoid Receptor 1 in Hodgkin Lymphoma. PLoS ONE 2013, 8, e81675. [Google Scholar] [CrossRef]
- Perez-Gomez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; García-Taboada, E.; Villa-Morales, M.C.; Moreno, E.; Hamann, S.; Martin-Villar, E.; Flores, J.M.; et al. Role of Cannabinoid Receptor CB2 in HER2 Pro-oncogenic Signaling in Breast Cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Teixeira, N.A.; Correia-Da-Silva, G. Cannabinoids as Modulators of Cell Death: Clinical Applications and Future Directions. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 63–88. [Google Scholar] [CrossRef]
- Javid, F.A.; Phillips, R.M.; Afshinjavid, S.; Verde, R.; Ligresti, A. Cannabinoid pharmacology in cancer research: A new hope for cancer patients? Eur. J. Pharmacol. 2016, 775, 1–14. [Google Scholar] [CrossRef]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzmán, M.; Velasco, G.; Díaz-Laviada, I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: Role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef]
- Andradas, C.; Benito, S.B.; Castillo-Lluva, S.; Pilla, P.D.; Alarcia, R.D.; Garcia, A.J.; García-Taboada, E.; Hernando-Llorente, R.; Soriano, J.; Hamann, S.; et al. Activation of the orphan receptor GPR55 by lysophosphatidylinositol promotes metastasis in triple-negative breast cancer. Oncotarget 2016, 7, 47565–47575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-L.; Guo, X.; Song, Y.-P.; Zhu, C.-Y.; Zou, W. The LPI/GPR55 axis enhances human breast cancer cell migration via HBXIP and p-MLC signaling. Acta Pharmacol. Sin. 2017, 39, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biol. Regul. 2015, 57, 217–227. [Google Scholar] [CrossRef]
- Oka, S.; Kimura, S.; Toshida, T.; Ota, R.; Yamashita, A.; Sugiura, T. Lysophosphatidylinositol induces rapid phosphorylation of p38 mitogen-activated protein kinase and activating transcription factor 2 in HEK293 cells expressing GPR55 and IM-9 lymphoblastoid cells. J. Biochem. 2010, 147, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Czifra, G.; Varga, A.; Nyeste, K.; Marincsák, R.; Tóth, B.I.; Kovács, I.; Kovács, L.; Bíró, T. Increased expressions of cannabinoid receptor-1 and transient receptor potential vanilloid-1 in human prostate carcinoma. J. Cancer Res. Clin. Oncol. 2008, 135, 507–514. [Google Scholar] [CrossRef]
- Zhai, K.; Liskova, A.; Kubatka, P.; Büsselberg, D. Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells. Int. J. Mol. Sci. 2020, 21, 4177. [Google Scholar] [CrossRef]
- Hu, F.; Sun, W.W.; Zhao, X.T.; Cui, Z.J.; Yang, W.X. TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem. Biophys. Res. Commun. 2008, 369, 989–993. [Google Scholar] [CrossRef]
- Liu, T.; Wang, G.; Tao, H.; Yang, Z.; Wang, Y.; Meng, Z.; Cao, R.; Xiao, Y.; Wang, X.; Zhou, J. Capsaicin mediates caspases activation and induces apoptosis through P38 and JNK MAPK pathways in human renal carcinoma. BMC Cancer 2016, 16, 790. [Google Scholar] [CrossRef]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Giangaspero, F.; Santoni, G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef]
- Ip, S.-W.; Lan, S.-H.; Lu, H.-F.; Huang, A.-C.; Yang, J.-S.; Lin, J.-P.; Huang, H.-Y.; Lien, J.-C.; Ho, C.-C.; Chiu, C.-F.; et al. Capsaicin mediates apoptosis in human nasopharyngeal carcinoma NPC-TW 039 cells through mitochondrial depolarization and endoplasmic reticulum stress. Hum. Exp. Toxicol. 2011, 31, 539–549. [Google Scholar] [CrossRef]
- Katsuda, K.; Kataoka, M.; Uno, F.; Murakami, T.; Kondo, T.; Roth, J.A.; Tanaka, N.; Fujiwara, T. Activation of caspase-3 and cleavage of Rb are associated with p16-mediated apoptosis in human non-small cell lung cancer cells. Oncogene 2002, 21, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.M.; Colvin, E.S.; Chen, Y.-C.; Geiss, S.L.; Eller, L.E.; Fueger, P.T. Upregulation of p21 activates the intrinsic apoptotic pathway in β-cells. Am. J. Physiol. Metab. 2013, 304, E1281–E1290. [Google Scholar] [CrossRef][Green Version]
- Cheng, E.H.-Y.; Kirsch, D.G.; Clem, R.J.; Ravi, R.; Kastan, M.B.; Bedi, A.; Ueno, K.; Hardwick, J.M. Conversion of Bcl-2 to a Bax-like Death Effector by Caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef]
- Gil, Y.-G.; Kang, M.-K. Capsaicin induces apoptosis and terminal differentiation in human glioma A172 cells. Life Sci. 2008, 82, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, C.; Fruchart, J.-C.; Staels, B. PPARα and PPARγ dual agonists for the treatment of type 2 diabetes and the metabolic syndrome. Curr. Opin. Pharmacol. 2006, 6, 606–614. [Google Scholar] [CrossRef]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Perez-Gomez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB2-GPR55 Receptor Heteromers Modulates Cancer Cell Signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef]
- Scarlett, K.A.; White, E.S.Z.; Coke, C.J.; Carter, J.R.; Bryant, L.K.; Hinton, C.V. Agonist-induced CXCR4 and CB2 Heterodimerization Inhibits Gα13/RhoA-mediated Migration. Mol. Cancer Res. 2018, 16, 728–739. [Google Scholar] [CrossRef]
- Blasco-Benito, S.; Moreno, E.; Seijo-Vila, M.; Tundidor, I.; Andradas, C.; Caffarel, M.M.; Caro-Villalobos, M.; Urigüen, L.; Diez-Alarcia, R.; Moreno-Bueno, G.; et al. Therapeutic targeting of HER2–CB2R heteromers in HER2-positive breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Nikan, M.; Nabavi, S.M.; Manayi, A. Ligands for cannabinoid receptors, promising anticancer agents. Life Sci. 2016, 146, 124–130. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147, S163–S171. [Google Scholar] [CrossRef]
- Soethoudt, M.; Grether, U.; Fingerle, J.; Grim, T.W.; Fezza, F.; De Petrocellis, L.; Ullmer, C.; Rothenhäusler, B.; Perret, C.; Van Gils, N.; et al. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 2017, 8, 13958. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Gavva, N.R.; Klionsky, L.; Qu, Y.; Shi, L.; Tamir, R.; Edenson, S.; Zhang, T.J.; Viswanadhan, V.N.; Tóth, A.; Pearce, L.V.; et al. Molecular Determinants of Vanilloid Sensitivity in TRPV1. J. Biol. Chem. 2004, 279, 20283–20295. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Di Marzo, V. An introduction to the endocannabinoid system: From the early to the latest concepts. Best Pr. Res. Clin. Endocrinol. Metab. 2009, 23, 1–15. [Google Scholar] [CrossRef]
- Eichele, K.; Ramer, R.; Hinz, B. R(+)-Methanandamide-Induced Apoptosis of Human Cervical Carcinoma Cells Involves A Cyclooxygenase-2-Dependent Pathway. Pharm. Res. 2008, 26, 346–355. [Google Scholar] [CrossRef]
- Olea-Herrero, N.; Vara, D.; Malagarie-Cazenave, S.; Diazlaviada, I. Inhibition of human tumour prostate PC-3 cell growth by cannabinoids R(+)-Methanandamide and JWH-015: Involvement of CB2. Br. J. Cancer 2009, 101, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; García-Hernández, V.M.; Ruiz-Garcia, E.; Meneses-García, A.; Herrera-Gomez, A.; Aguilar-Ponce, J.L.; Montes-Servín, E.; Prospero-García, O.; Del Angel, S.A. Comparing the effects of endogenous and synthetic cannabinoid receptor agonists on survival of gastric cancer cells. Life Sci. 2016, 165, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Pisanti, S.; Malfitano, A.M.; Bifulco, M. The anandamide analog, Met-F-AEA, controls human breast cancer cell migration via the RHOA/RHO kinase signaling pathway. Endocr.-Related Cancer 2008, 15, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, A.M.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Pisanti, S.; Crescenzi, E.; Bifulco, M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006, 580, 6076–6082. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014, 5, 2475–2486. [Google Scholar] [CrossRef]
- Proto, M.C.; Gazzerro, P.; Di Croce, L.; Santoro, A.; Malfitano, A.M.; Pisanti, S.; Laezza, C.; Bifulco, M. Interaction of endocannabinoid system and steroid Hormones in the control of colon cancer cell growth. J. Cell. Physiol. 2011, 227, 250–258. [Google Scholar] [CrossRef]
- Cozzolino, R.; Calì, G.; Bifulco, M.; Laccetti, P. A metabolically stable analogue of anandamide, Met-F-AEA, inhibits human thyroid carcinoma cell lines by activation of apoptosis. Investig. New Drugs 2009, 28, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, I.; Bánki, B.; Márk, Á.; Varga, N.; Tóvári, J.; Ladányi, A.; Rásó, E.; Tímár, J. Revisiting CB1 Receptor as Drug Target in Human Melanoma. Pathol. Oncol. Res. 2012, 18, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2016, 174, 1349–1365. [Google Scholar] [CrossRef]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Bisogno, T.; Ligresti, A.; Bifulco, M.; Melck, D.; Di Marzo, V. Effect on cancer cell proliferation of palmitoylethanolamide, a fatty acid amide interacting with both the cannabinoid and vanilloid signalling systems. Fundam. Clin. Pharmacol. 2002, 16, 297–302. [Google Scholar] [CrossRef]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid Receptor Activation Induces Apoptosis through Tumor Necrosis Factor α–Mediated Ceramide De novo Synthesis in Colon Cancer Cells. Clin. Cancer Res. 2008, 14, 7691–7700. [Google Scholar] [CrossRef]
- Pourkhalili, N.; Ghahremani, M.H.; Farsandaj, N.; Tavajohi, S.; Majdzadeh, M.; Parsa, M.; Lavasani, N.J.; Ostad, S.N. Evaluation of anti-invasion effect of cannabinoids on human hepatocarcinoma cells. Toxicol. Mech. Methods 2012, 23, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, F.; Ostad, S.N.; Aliebrahimi, S.; Daman, Z. Anti-invasion effects of cannabinoids agonist and antagonist on human breast cancer stem cells. Iran. J. Pharm. Res. IJPR 2017, 16, 1479–1486. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29552056 (accessed on 2 August 2021). [PubMed]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Pozza, E.D.; Scupoli, M.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef]
- Nasser, M.W.; Qamri, Z.; Deol, Y.S.; Smith, D.; Shilo, K.; Zou, X.; Ganju, R.K. Crosstalk between Chemokine Receptor CXCR4 and Cannabinoid Receptor CB2 in Modulating Breast Cancer Growth and Invasion. PLoS ONE 2011, 6, e23901. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Ahirwar, D.; Ravi, J.; Nasser, M.W.; Ganju, R.K. Novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR and IGF-I/IGF-IR pathways in breast cancer. Oncotarget 2016, 8, 29668–29678. [Google Scholar] [CrossRef]
- Haskó, J.; Fazakas, C.; Molnár, J.; Nyúl-Tóth, Á.; Herman, H.; Hermenean, A.; Wilhelm, I.; Persidsky, Y.; Krizbai, I.A. CB2 Receptor Activation Inhibits Melanoma Cell Transmigration through the Blood-Brain Barrier. Int. J. Mol. Sci. 2014, 15, 8063–8074. [Google Scholar] [CrossRef]
- Sanchez, C.; de Ceballos, M.L.; Gomez del Pulgar, T.; Rueda, D.; Corbacho, C.; Velasco, G.; Galve-Roperh, I.; Huffman, J.W.; y Cajal, S.R.; Guzmán, M. Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res. 2001, 61, 5784–5798. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11479216 (accessed on 7 June 2020).
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3129. [Google Scholar] [CrossRef]
- McPartland, J.M.; Duncan, M.; Di Marzo, V.; Pertwee, R. Are cannabidiol and Δ9-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol. 2015, 172, 737–753. [Google Scholar] [CrossRef]
- Ożarowski, M.; Karpiński, T.; Zielińska, A.; Souto, E.; Wielgus, K. Cannabidiol in Neurological and Neoplastic Diseases: Latest Developments on the Molecular Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 4294. [Google Scholar] [CrossRef]
- Sharir, H.; Abood, M.E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther. 2010, 126, 301–313. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Jones, B.; Korchev, Y.; Bloom, S.R.; Pacchetti, B.; Anand, P.; Sodergren, M.H. CBD Effects on TRPV1 Signaling Pathways in Cultured DRG Neurons. J. Pain Res. 2020, 13, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharmacol. 2007, 152, 984–986. [Google Scholar] [CrossRef]
- Santoro, A.; Pisanti, S.; Grimaldi, C.; Izzo, A.; Borrelli, F.; Proto, M.C.; Malfitano, A.M.; Gazzerro, P.; Laezza, C.; Bifulco, M. Rimonabant inhibits human colon cancer cell growth and reduces the formation of precancerous lesions in the mouse colon. Int. J. Cancer 2009, 125, 996–1003. [Google Scholar] [CrossRef]
- Sarnataro, D.; Pisanti, S.; Santoro, A.; Gazzerro, P.; Malfitano, A.M.; Laezza, C.; Bifulco, M. The Cannabinoid CB1 Receptor Antagonist Rimonabant (SR141716) Inhibits Human Breast Cancer Cell Proliferation through a Lipid Raft-Mediated Mechanism. Mol. Pharmacol. 2006, 70, 1298–1306. [Google Scholar] [CrossRef]
- Ciaglia, E.; Torelli, G.; Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Malfitano, A.M.; Fiore, D.; Zottola, A.C.P.; Proto, M.C.; et al. Cannabinoid receptor CB1 regulates STAT3 activity and its expression dictates the responsiveness to SR141716 treatment in human glioma patients’ cells. Oncotarget 2015, 6, 15464–15481. [Google Scholar] [CrossRef]
- Fiore, D.; Ramesh, P.; Proto, M.C.; Piscopo, C.; Franceschelli, S.; Anzelmo, S.; Medema, J.P.; Bifulco, M.; Gazzerro, P. Rimonabant Kills Colon Cancer Stem Cells without Inducing Toxicity in Normal Colon Organoids. Front. Pharmacol. 2018, 8, 949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qin, Y.; Pan, Z.; Li, M.; Liu, X.; Chen, X.; Qu, G.; Zhou, L.; Xu, M.; Zheng, Q.; et al. Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells. Biomolecules 2019, 9, 302. [Google Scholar] [CrossRef]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Marcu, J.P.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Horowitz, M.P.; Lee, J.; Pakdel, A.; Allison, J.; Limbad, C.; Moore, D.H.; et al. Cannabidiol Enhances the Inhibitory Effects of Δ9-Tetrahydrocannabinol on Human Glioblastoma Cell Proliferation and Survival. Mol. Cancer Ther. 2010, 9, 180–189. [Google Scholar] [CrossRef]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor Activity of Plant Cannabinoids with Emphasis on the Effect of Cannabidiol on Human Breast Carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growthin vitroandin vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2012, 168, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Heinemann, K.; Merkord, J.; Rohde, H.; Salamon, A.; Linnebacher, M.; Hinz, B. COX-2 and PPAR-γ Confer Cannabidiol-Induced Apoptosis of Human Lung Cancer Cells. Mol. Cancer Ther. 2012, 12, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.S.; Marie, M.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol Induces Programmed Cell Death in Breast Cancer Cells by Coordinating the Cross-talk between Apoptosis and Autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.-Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef]
- Aviello, G.; Romano, B.; Borrelli, F.; Capasso, R.; Gallo, L.; Piscitelli, F.; Di Marzo, V.; Izzo, A.A. Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J. Mol. Med. 2012, 90, 925–934. [Google Scholar] [CrossRef]
- Ramer, R.; Rohde, A.; Merkord, J.; Rohde, H.; Hinz, B. Decrease of Plasminogen Activator Inhibitor-1 May Contribute to the Anti-Invasive Action of Cannabidiol on Human Lung Cancer Cells. Pharm. Res. 2010, 27, 2162–2174. [Google Scholar] [CrossRef]
- Ramer, R.; Bublitz, K.; Freimuth, N.; Merkord, J.; Rohde, H.; Haustein, M.; Borchert, P.; Schmuhl, E.; Linnebacher, M.; Hinz, B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J. 2011, 26, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Massi, P.; Cinquina, V.; Valenti, M.; Bolognini, D.; Gariboldi, M.; Monti, E.; Rubino, T.; Parolaro, D. Cannabidiol, a Non-Psychoactive Cannabinoid Compound, Inhibits Proliferation and Invasion in U87-MG and T98G Glioma Cells through a Multitarget Effect. PLoS ONE 2013, 8, e76918. [Google Scholar] [CrossRef]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 Is a Key Transcriptional Regulator of Glioblastoma Aggressiveness and a Novel Therapeutic Target. Cancer Res. 2012, 73, 1559–1569. [Google Scholar] [CrossRef]
- Elbaz, M.; Ahirwar, D.; Xiaoli, Z.; Zhou, X.; Lustberg, M.; Nasser, M.W.; Shilo, K.; Ganju, R.K. TRPV2 is a novel biomarker and therapeutic target in triple negative breast cancer. Oncotarget 2016, 9, 33459–33470. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2012, 34, 48–57. [Google Scholar] [CrossRef]
- Scott, K.; Dennis, J.L.; Dalgleish, A.G.; Liu, W.M. Inhibiting Heat Shock Proteins Can Potentiate the Cytotoxic Effect of Cannabidiol in Human Glioma Cells. Anticancer. Res. 2015, 35, 5827–5837. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26504004 (accessed on 23 October 2020).
- Wang, J.; Xu, Y.; Zou, Y.; Zhu, L.; Dong, B.; Huang, J.; Chen, Y.; Xue, W.; Huang, Y.; Kong, W.; et al. Overexpression of cannabinoid receptor 1 promotes renal cell carcinoma progression. Tumor Biol. 2016, 37, 16237–16247. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Zhu, L.; Zou, Y.; Kong, W.; Dong, B.; Huang, J.; Chen, Y.; Xue, W.; Huang, Y.; et al. Cannabinoid receptor 2 as a novel target for promotion of renal cell carcinoma prognosis and progression. J. Cancer Res. Clin. Oncol. 2017, 144, 39–52. [Google Scholar] [CrossRef]
- Hasenoehrl, C.; Feuersinger, D.; Sturm, E.M.; Bärnthaler, T.; Heitzer, E.; Graf, R.; Grill, M.; Pichler, M.; Beck, S.; Butcher, L.; et al. G protein-coupled receptor GPR55 promotes colorectal cancer and has opposing effects to cannabinoid receptor 1. Int. J. Cancer 2018, 142, 121–132. [Google Scholar] [CrossRef]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stancic, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br. J. Pharmacol. 2015, 173, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.S.; Bernier, M.; Wainer, I.W. Selective GPR55 antagonism reduces chemoresistance in cancer cells. Pharmacol. Res. 2016, 111, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Ayakannu, T.; Taylor, A.H.; Bari, M.; Mastrangelo, N.; Maccarrone, M.; Konje, J.C. Expression and Function of the Endocannabinoid Modulating Enzymes Fatty Acid Amide Hydrolase and N-Acylphosphatidylethanolamine-Specific Phospholipase D in Endometrial Carcinoma. Front. Oncol. 2019, 9, 1363. [Google Scholar] [CrossRef] [PubMed]
- Endsley, M.P.; Thill, R.; Choudhry, I.; Williams, C.L.; Kajdacsy-Balla, A.; Campbell, W.B.; Nithipatikom, K. Expression and function of fatty acid amide hydrolase in prostate cancer. Int. J. Cancer 2008, 123, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-κB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 1–13. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef]
- Ma, M.; Bai, J.; Ling, Y.; Chang, W.; Xie, G.; Li, R.; Wang, G.; Tao, K. Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2850–2856. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol Lipase: A Novel Potential Therapeutic Target and Prognostic Indicator for Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef]
- Marino, S.; de Ridder, D.; Bishop, R.T.; Renema, N.; Ponzetti, M.; Sophocleous, A.; Capulli, M.; Aljeffery, A.; Carrasco, G.; Gens, M.D.; et al. Paradoxical effects of JZL184, an inhibitor of monoacylglycerol lipase, on bone remodelling in healthy and cancer-bearing mice. EBioMedicine 2019, 44, 452–466. [Google Scholar] [CrossRef]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Orrego-González, E.; Londoño-Tobón, L.; Ardila-González, J.; Polania-Tovar, D.; Valencia-Cárdenas, A.; Meerbeke, A.V.-V. Cannabinoid Effects on Experimental Colorectal Cancer Models Reduce Aberrant Crypt Foci (ACF) and Tumor Volume: A Systematic Review. Evid.-Based Complement. Altern. Med. 2020, 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Ramer, R.; Dithmer, S.; Ivanov, I.; Merkord, J.; Hinz, B. Fatty acid amide hydrolase inhibitors confer anti-invasive and antimetastatic effects on lung cancer cells. Oncotarget 2016, 7, 15047–15064. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Patsos, H.A.; Williams, A.C.; Paraskeva, C. The cannabinoid δ9-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int. J. Cancer 2007, 121, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Morell, C.M.; Rodríguez-Henche, N.; Díaz-Laviada, I. Involvement of PPARγ in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis. 2013, 4, e618. [Google Scholar] [CrossRef]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic Activity of Cannabinoids2. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef]
- Preet, A.; Ganju, R.K.; E Groopman, J. Δ9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene 2007, 27, 339–346. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sanchez, C. Δ9-Tetrahydrocannabinol Inhibits Cell Cycle Progression in Human Breast Cancer Cells through Cdc2 Regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef]
- Von Bueren, A.O.; Schlumpf, M.; Lichtensteiger, W. Delta(9)-tetrahydrocannabinol inhibits 17beta-estradiol-induced proliferation and fails to activate androgen and estrogen receptors in MCF7 human breast cancer cells. Anticancer. Res. 2008, 28. Available online: http://www.ncbi.nlm.nih.gov/pubmed/18383828 (accessed on 20 September 2021).
- Ruiz, L.; Miguel, A.; Díaz-Laviada, I. Δ9-Tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. 1999, 458, 400–404. [Google Scholar] [CrossRef]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L.; et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef]
- Galanti, G.; Fisher, T.; Kventsel, I.; Shoham, J.; Gallily, R.; Mechoulam, R.; Lavie, G.; Amariglio, N.; Rechavi, G.; Toren, A. Δ9-Tetrahydrocannabinol inhibits cell cycle progression by downregulation of E2F1 in human glioblastoma multiforme cells. Acta Oncol. 2008, 47, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Tiedra, S.; Fabrias, G.; Dávila, D.; Salanueva, J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Salazar, M.; Carracedo, A.; Lorente, M.; Egia, A.; González-Feria, L.; Haro, A.; Velasco, G.; Guzmán, M. Cannabinoids Inhibit Glioma Cell Invasion by Down-regulating Matrix Metalloproteinase-2 Expression. Cancer Res. 2008, 68, 1945–1952. [Google Scholar] [CrossRef]
- Scott, K.; Dalgleish, A.G.; Liu, W.M. The Combination of Cannabidiol and Δ9-Tetrahydrocannabinol Enhances the Anticancer Effects of Radiation in an Orthotopic Murine Glioma Model. Mol. Cancer Ther. 2014, 13, 2955–2967. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, W.; Shen, K.; Shen, W. ∆9-tetrahydrocannabinol inhibits epithelial-mesenchymal transition and metastasis by targeting matrix metalloproteinase-9 in endometrial cancer. Oncol. Lett. 2018, 15, 8527–8535. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Poele, R.T.; Shamash, J.; Chaplin, T.; Propper, D.; Joel, S.; Oliver, T.; Liu, W.M. Cannabis-induced cytotoxicity in leukemic cell lines: The role of the cannabinoid receptors and the MAPK pathway. Blood 2005, 105, 1214–1221. [Google Scholar] [CrossRef]
- Holland, M.; Panetta, J.; Hoskins, J.; Bebawy, M.; Roufogalis, B.; Allen, J.; Arnold, J. The effects of cannabinoids on P-glycoprotein transport and expression in multidrug resistant cells. Biochem. Pharmacol. 2006, 71, 1146–1154. [Google Scholar] [CrossRef]
- Scott, K.A.; Dalgleish, A.G.; Liu, W.M. Anticancer effects of phytocannabinoids used with chemotherapy in leukaemia cells can be improved by altering the sequence of their administration. Int. J. Oncol. 2017, 51, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Anagnostou, M.E.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting Cannabinoid-Induced Cytotoxic Autophagy to Drive Melanoma Cell Death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.-S.; Park, H.; Cho, Y.K.; Lee, I.S.; Kim, S.W.; Choi, M.-G.; Chung, I.-S.; Han, K.-H.; Park, J.M. Effect of a synthetic cannabinoid agonist on the proliferation and invasion of gastric cancer cells. J. Cell. Biochem. 2010, 110, 321–332. [Google Scholar] [CrossRef]
- Xian, X.-S.; Park, H.; Choi, M.-G.; Park, J.M. Cannabinoid Receptor Agonist as an Alternative Drug in 5-Fluorouracil-Resistant Gastric Cancer Cells. Anticancer Res. 2013, 33, 2541–2547. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23749906 (accessed on 5 July 2020).
- Xian, X.; Huang, L.; Zhang, B.; Wu, C.; Cui, J.; Wang, Z. WIN 55,212-2 Inhibits the Epithelial Mesenchymal Transition of Gastric Cancer Cells via COX-2 Signals. Cell. Physiol. Biochem. 2016, 39, 2149–2157. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Mukhtar, H. Cannabinoid Receptor as a Novel Target for the Treatment of Prostate Cancer. Cancer Res. 2005, 65, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Bort, A.; Vara-Ciruelos, D.; Ramos-Torres, A.; Rodríguez-Henche, N.; Díaz-Laviada, I. The cannabinoid WIN 55,212-2 prevents neuroendocrine differentiation of LNCaP prostate cancer cells. Prostate Cancer Prostatic Dis. 2016, 19, 248–257. [Google Scholar] [CrossRef]
- Khan, M.I.; Sobocińska, A.A.; Brodaczewska, K.K.; Zielniok, K.; Gajewska, M.; Kieda, C.; Czarnecka, A.M.; Szczylik, C. Involvement of the CB2 cannabinoid receptor in cell growth inhibition and G0/G1 cell cycle arrest via the cannabinoid agonist WIN 55,212–2 in renal cell carcinoma. BMC Cancer 2018, 18, 583. [Google Scholar] [CrossRef]
- Notaro, A.; Emanuele, S.; Geraci, F.; D’Anneo, A.; Lauricella, M.; Calvaruso, G.; Giuliano, M. WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner. Int. J. Mol. Sci. 2019, 20, 5235. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Radtke, A.; Decker, J.; Koch, M.; Belge, G. The Synthetic Cannabinoid WIN 55,212-2 Elicits Death in Human Cancer Cell Lines. Anticancer. Res. 2017, 37, 6341–6345. [Google Scholar] [CrossRef]
- DeMorrow, S.; Francis, H.; Gaudio, E.; Venter, J.; Franchitto, A.; Kopriva, S.; Onori, P.; Mancinelli, R.; Frampton, G.; Coufal, M.; et al. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the noncanonical Wnt signaling pathway. Am. J. Physiol. Liver Physiol. 2008, 295, G1150–G1158. [Google Scholar] [CrossRef]
- Huang, L.; Ramirez, J.C.; Frampton, G.A.; Golden, L.E.; Quinn, M.A.; Pae, H.Y.; Horvat, D.; Liang, L.-J.; DeMorrow, S. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab. Investig. 2011, 91, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Melck, D.; Rueda, D.; Galve-Roperh, I.; De Petrocellis, L.; Guzmán, M.; Di Marzo, V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999, 463, 235–240. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef]
- Melck, D.; De Petrocellis, L.; Orlando, P.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. Suppression of Nerve Growth Factor Trk Receptors and Prolactin Receptors by Endocannabinoids Leads to Inhibition of Human Breast and Prostate Cancer Cell Proliferation. Endocrinology 2000, 141, 118–126. [Google Scholar] [CrossRef]
- Mimeault, M.; Pommery, N.; Wattez, N.; Bailly, C.; Hénichart, J.-P. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: Implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate 2003, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Serradell, O.; Poblete, C.E.; Sanchez, C.; Castellón, E.A.; Gallegos, I.; Huidobro, C.; Llanos, M.N.; Contreras, H.R. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol. Rep. 2015, 33, 1599–1608. [Google Scholar] [CrossRef]
- Soliman, E.; Van Dross, R. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: Receptor-independent endocannabinoid signaling. Mol. Carcinog. 2015, 55, 1807–1821. [Google Scholar] [CrossRef]
- Flygare, J.; Gustafsson, K.; Kimby, E.; Christensson, B.; Sander, B. Cannabinoid receptor ligands mediate growth inhibition and cell death in mantle cell lymphoma. FEBS Lett. 2005, 579, 6885–6889. [Google Scholar] [CrossRef] [PubMed]
- Vanderah, T.W.; Hanlon, K.E.; Lozano-Ondoua, A.N.; Umaretiya, P.J.; Symons-Liguori, A.M.; Chandramouli, A.; Moy, J.K.; Kwass, W.K.; Mantyh, P.W.; Nelson, M.A. Modulation of breast cancer cell viability by a cannabinoid receptor 2 agonist, JWH-015, is calcium dependent. Breast Cancer Targets Ther. 2016, 8, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Gazzerro, P.; Malfitano, A.M.; Proto, M.C.; Santoro, A.; Pisanti, S.; Caruso, M.G.; Notarnicola, M.; Messa, C.; Laezza, C.; Misso, G.; et al. Synergistic inhibition of human colon cancer cell growth by the cannabinoid CB1 receptor antagonist rimonabant and oxaliplatin. Oncol. Rep. 2010, 23, 171–175. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19956878 (accessed on 1 December 2020).
- Proto, M.C.; Fiore, D.; Piscopo, C.; Franceschelli, S.; Bizzarro, V.; Laezza, C.; Lauro, G.; Feoli, A.; Tosco, A.; Bifulco, G.; et al. Inhibition of Wnt/β-Catenin pathway and Histone acetyltransferase activity by Rimonabant: A therapeutic target for colon cancer. Sci. Rep. 2017, 7, 11678. [Google Scholar] [CrossRef] [PubMed]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem. Pharmacol. 2014, 92, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Hudson, J.B.; Adomat, H.; Guns, E.; Cox, M.E. In Vitro Anticancer Activity of Plant-Derived Cannabidiol on Prostate Cancer Cell Lines. Pharmacol. Pharm. 2014, 05, 806–820. [Google Scholar] [CrossRef]
- Fogli, S.; Nieri, P.; Chicca, A.; Adinolfi, B.; Mariotti, V.; Iacopetti, P.; Breschi, M.C.; Pellegrini, S. Cannabinoid derivatives induce cell death in pancreatic MIA PaCa-2 cells via a receptor-independent mechanism. FEBS Lett. 2006, 580, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Truffi, M.; Sorrentino, L.; Corsi, F. Fibroblasts in the Tumor Microenvironment. Neuropilin 2020, 1234, 15–29. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment—New Findings and Future Perspectives. Front. Cell Dev. Biol. 2020, 8, 776. [Google Scholar] [CrossRef]
- Bernard, J.J.; Wellberg, E.A. The Tumor Promotional Role of Adipocytes in the Breast Cancer Microenvironment and Macroenvironment. Am. J. Pathol. 2021, 191, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Kong, X.; Qiu, X.; Huang, C.; Wong, P.-P. The Emerging Roles of Pericytes in Modulating Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 676342. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Deville, S.S.; Cordes, N. The Extracellular, Cellular, and Nuclear Stiffness, a Trinity in the Cancer Resistome—A Review. Front. Oncol. 2019, 9, 1376. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Giaccia, E.L.L.A.J. The ever-expanding role of HIF in tumour and stromal biology. Nature 2016, 18, 356–365. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef]
- Pasquier, J.; Ghiabi, P.; Chouchane, L.; Razzouk, K.; Rafii, S.; Rafii, A. Angiocrine endothelium: From physiology to cancer. J. Transl. Med. 2020, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Guerrouahen, B.S.; Pasquier, J.; Satheesh, N.J.; Suhre, K.; Rafii, A. Nesting of colon and ovarian cancer cells in the endothelial niche is associated with alterations in glycan and lipid metabolism. Sci. Rep. 2017, 7, 39999. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, E.; Wu, N.C.; Cadavid, J.L.; McGuigan, A.P. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br. J. Cancer 2020, 122, 931–942. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal Activation of Prostate Cancer Cells and Cancer-Associated Fibroblasts Stimulates Epithelial-Mesenchymal Transition and Cancer Stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.; Marín-Jiménez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and Functions of CAFs Secretome during Cancer Progression and Therapy. Front. Cell Dev. Biol. 2021, 8, 621070. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-De-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2013, 33, 2423–2431. [Google Scholar] [CrossRef]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Harrell, C.R.; Markovic, B.S.; Fellabaum, C.; Arsenijevic, A.; Djonov, V.; Volarevic, V. Molecular mechanisms underlying therapeutic potential of pericytes. J. Biomed. Sci. 2018, 25, 21. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Barik, S.; Banerjee, S.; Ghosh, T.; Mallick, A.; Majumdar, S.B.; Goswami, K.K.; Bhuniya, A.; Banerjee, S.; Baral, R.; et al. Tumor-Derived Vascular Pericytes Anergize Th Cells. J. Immunol. 2013, 191, 971–981. [Google Scholar] [CrossRef]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Attané, C.; Muller, C. Drilling for Oil: Tumor-Surrounding Adipocytes Fueling Cancer. Trends Cancer 2020, 6, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Braile, M.; Marcella, S.; Marone, G.; Galdiero, M.; Varricchi, G.; Loffredo, S. The Interplay between the Immune and the Endocannabinoid Systems in Cancer. Cells 2021, 10, 1282. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; Pochard, P.; Orlando, P.; Salzet, M.; Pestel, J.; Di Marzo, V. Presence and regulation of the endocannabinoid system in human dendritic cells. JBIC J. Biol. Inorg. Chem. 2002, 269, 3771–3778. [Google Scholar] [CrossRef] [PubMed]
- López, A.J.S.; Román-Vega, L.; Tojeiro, E.R.; Giuffrida, A.; García-Merino, A. Regulation of cannabinoid receptor gene expression and endocannabinoid levels in lymphocyte subsets by interferon-β: A longitudinal study in multiple sclerosis patients. Clin. Exp. Immunol. 2014, 179, 119–127. [Google Scholar] [CrossRef]
- Castaneda, J.T.; Harui, A.; Roth, M.D. Regulation of Cell Surface CB2 Receptor during Human B Cell Activation and Differentiation. J. Neuroimmune Pharmacol. 2017, 12, 544–554. [Google Scholar] [CrossRef]
- Sugamura, K.; Sugiyama, S.; Nozaki, T.; Matsuzawa, Y.; Izumiya, Y.; Miyata, K.; Nakayama, M.; Kaikita, K.; Obata, T.; Takeya, M.; et al. Activated Endocannabinoid System in Coronary Artery Disease and Antiinflammatory Effects of Cannabinoid 1 Receptor Blockade on Macrophages. Circulation 2009, 119, 28–36. [Google Scholar] [CrossRef]
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid CB1and CB2receptors in immune cells. Acta Physiol. 2015, 214, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Dittel, B.N. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol. Res. 2011, 51, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ren, Y.; Dai, Z.-J.; Wu, C.-J.; Ji, Y.-H.; Xu, J. IL-6, IL-8 and TNF-α levels correlate with disease stage in breast cancer patients. Adv. Clin. Exp. Med. 2017, 26, 421–426. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2020, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Kienzl, M.; Kargl, J.; Schicho, R. The Immune Endocannabinoid System of the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 8929. [Google Scholar] [CrossRef]
- Staiano, R.I.; Loffredo, S.; Borriello, F.; Iannotti, F.; Piscitelli, F.; Orlando, P.; Secondo, A.; Granata, F.; Lepore, M.T.; Fiorelli, A.; et al. Human lung-resident macrophages express CB1 and CB2 receptors whose activation inhibits the release of angiogenic and lymphangiogenic factors. J. Leukoc. Biol. 2015, 99, 531–540. [Google Scholar] [CrossRef]
- Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T.; Gaffal, E. Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci. 2015, 138, 35–40. [Google Scholar] [CrossRef]
- Gruber, T.; Robatel, S.; Kremenovic, M.; Bäriswyl, L.; Gertsch, J.; Schenk, M. Cannabinoid Receptor Type-2 in B Cells Is Associated with Tumor Immunity in Melanoma. Cancers 2021, 13, 1934. [Google Scholar] [CrossRef]
- Ravi, J.; Elbaz, M.; Wani, N.A.; Nasser, M.W.; Ganju, R.K. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol. Carcinog. 2016, 55, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Gaffal, E.; Kemter, A.M.; Scheu, S.; Dantas, R.L.; Vogt, J.; Baune, B.; Tüting, T.; Zimmer, A.; Alferink, J. Cannabinoid Receptor 2 Modulates Maturation of Dendritic Cells and Their Capacity to Induce Hapten-Induced Contact Hypersensitivity. Int. J. Mol. Sci. 2020, 21, 475. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Yang, L.; Wang, B.; Cui, L.; Li, C.; Zhuo, Y.; Zhang, L.; Zhang, S.; Zhang, Q.; Wang, X. The role of 2-arachidonoylglycerol in the regulation of the tumor-immune microenvironment in murine models of pancreatic cancer. Biomed. Pharmacother. 2019, 115, 108952. [Google Scholar] [CrossRef]
- Suk, K.-T.; Mederacke, I.; Gwak, G.-Y.; Cho, S.W.; Adeyemi, A.; Friedman, R.; Schwabe, R.F. Opposite roles of cannabinoid receptors 1 and 2 in hepatocarcinogenesis. Gut 2016, 65, 1721–1732. [Google Scholar] [CrossRef]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef]
- Yin, J.; Kim, S.S.; Choi, E.; Oh, Y.T.; Lin, W.; Kim, T.-H.; Sa, J.K.; Hong, J.H.; Park, S.H.; Kwon, H.J.; et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat. Commun. 2020, 11, 2978. [Google Scholar] [CrossRef]
- Ghosh, S.; Preet, A.; Groopman, J.; Ganju, R. Cannabinoid receptor CB2 modulates the CXCL12/CXCR4-mediated chemotaxis of T lymphocytes. Mol. Immunol. 2006, 43, 2169–2179. [Google Scholar] [CrossRef]
- Taylor, L.; Christou, I.; Kapellos, T.S.; Buchan, A.; Brodermann, M.H.; Gianella-Borradori, M.; Russell, A.; Iqbal, A.J.; Greaves, D.R. Primary Macrophage Chemotaxis Induced by Cannabinoid Receptor 2 Agonists Occurs Independently of the CB2 Receptor. Sci. Rep. 2015, 5, 10682. [Google Scholar] [CrossRef]
- Montecucco, F.; Burger, F.; Mach, F.; Steffens, S. CB2 cannabinoid receptor agonist JWH-015 modulates human monocyte migration through defined intracellular signaling pathways. Am. J. Physiol. Circ. Physiol. 2008, 294, H1145–H1155. [Google Scholar] [CrossRef]
- Raborn, E.S.; Marciano-Cabral, F.; Buckley, N.E.; Martin, B.R.; Cabral, G.A. The Cannabinoid Delta-9-tetrahydrocannabinol Mediates Inhibition of Macrophage Chemotaxis to RANTES/CCL5: Linkage to the CB2 Receptor. J. Neuroimmune Pharmacol. 2007, 3, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Steffens, S.; Veillard, N.R.; Arnaud, C.; Pelli, G.; Burger, F.; Staub, C.; Zimmer, A.; Frossard, J.-L. Fran Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature 2005, 434, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Pietrovito, L.; Iozzo, M.; Bacci, M.; Giannoni, E.; Chiarugi, P. Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression. Int. J. Mol. Sci. 2020, 21, 787. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Rybałtowska-Kawałko, P.; Skrzydlewska, E. Rutin as a Mediator of Lipid Metabolism and Cellular Signaling Pathways Interactions in Fibroblasts Altered by UVA and UVB Radiation. Oxidative Med. Cell. Longev. 2017, 2017, 4721352. [Google Scholar] [CrossRef]
- Bobrov, M.Y.; Bezuglov, V.V.; Khaspekov, L.G.; Illarioshkin, S.N.; Novosadova, E.V.; Grivennikov, I.A. Expression of Type I Cannabinoid Receptors at Different Stages of Neuronal Differentiation of Human Fibroblasts. Bull. Exp. Biol. Med. 2017, 163, 272–275. [Google Scholar] [CrossRef]
- Solinas, M.; Massi, P.; Cantelmo, A.; Cattaneo, M.; Cammarota, R.; Bartolini, D.; Cinquina, V.; Valenti, M.; Vicentini, L.; Noonan, D.; et al. Cannabidiol inhibits angiogenesis by multiple mechanisms. Br. J. Pharmacol. 2012, 167, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Picardi, P.; Ciaglia, E.; Proto, M.; Pisanti, S. Anandamide inhibits breast tumor-induced angiogenesis. Transl. Med. UniSa 2014, 10, 8–12. [Google Scholar]
- Pisanti, S.; Borselli, C.; Oliviero, O.; Laezza, C.; Gazzerro, P.; Bifulco, M. Antiangiogenic activity of the endocannabinoid anandamide: Correlation to its tumor-suppressor efficacy. J. Cell. Physiol. 2006, 211, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.-K.; Chou, P.-H.; Ng, S.-K.; Lin, W.-Y.; Wei, T.-T. Cannabinoids orchestrate cross-talk between cancer cells and endothelial cells in colorectal cancer. Cancer Gene Ther. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Zhou, X.; Cheng, J.; Yu, J.; Wu, J.; Jiang, C. Cannabinoids Regulate the Diameter of Pericyte-Containing Retinal Capillaries in Rats. Cell. Physiol. Biochem. 2017, 43, 2088–2101. [Google Scholar] [CrossRef]
- Bellocchio, L.; Cervino, C.; Vicennati, V.; Pasquali, R.; Pagotto, U. Cannabinoid Type 1 Receptor: Another Arrow in the Adipocytes’ Bow. J. Neuroendocr. 2008, 20, 130–138. [Google Scholar] [CrossRef]
- Gasperi, V.; Fezza, F.; Pasquariello, N.; Bari, M.; Oddi, S.; Agrò, A.F.; Maccarrone, M. Endocannabinoids in adipocytes during differentiation and their role in glucose uptake. Experientia 2006, 64, 219–229. [Google Scholar] [CrossRef]
- Mazier, W.; Saucisse, N.; Gatta-Cherifi, B.; Cota, D. The Endocannabinoid System: Pivotal Orchestrator of Obesity and Metabolic Disease. Trends Endocrinol. Metab. 2015, 26, 524–537. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Yang, C.; et al. Tumour-originated exosomal miR-155 triggers cancer-associated cachexia to promote tumour progression. Mol. Cancer 2018, 17, 155. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Duarte, M.J.; Blázquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sanchez, C.; Velasco, G.; González-Feria, L. A pilot clinical study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Bali, C. Cannabis Extract Treatment for Terminal Acute Lymphoblastic Leukemia with a Philadelphia Chromosome Mutation. Case Rep. Oncol. 2013, 6, 585–592. [Google Scholar] [CrossRef]
- Foroughi, M.; Hendson, G.; Sargent, M.A.; Steinbok, P. Spontaneous regression of septum pellucidum/forniceal pilocytic astrocytomas—possible role of Cannabis inhalation. Child’s Nerv. Syst. 2011, 27, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S.C. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br. J. Cancer 2021, 124, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzmán, M.; Velasco, G. A Combined Preclinical Therapy of Cannabinoids and Temozolomide against Glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Pagano, E.; Romano, B.; Panzera, S.; Maiello, F.; Coppola, D.; De Petrocellis, L.; Buono, L.; Orlando, P.; Izzo, A.A. Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-derived non-psychotropic cannabinoid. Carcinogenesis 2014, 35, 2787–2797. [Google Scholar] [CrossRef]
- Chen, L.; Chen, H.; Li, Y.; Li, L.; Qiu, Y.; Ren, J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol. Rep. 2015, 34, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Haybaeck, J.; Stančić, A.; Andersen, L.; Marsche, G.; Heinemann, A.; Schicho, R. O-1602, an atypical cannabinoid, inhibits tumor growth in colitis-associated colon cancer through multiple mechanisms. J. Mol. Med. 2012, 91, 449–458. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2018, 176, 1384–1394. [Google Scholar] [CrossRef]
- Blázquez, C.; González-Feria, L.; Álvarez, L.; Haro, A.; Casanova, M.L.; Guzmán, M. Cannabinoids Inhibit the Vascular Endothelial Growth Factor Pathway in Gliomas. Cancer Res. 2004, 64, 5617–5623. [Google Scholar] [CrossRef]
- Kogan, N.M.; Blázquez, C.; Álvarez, L.; Gallily, R.; Schlesinger, M.; Guzmán, M.; Mechoulam, R. A Cannabinoid Quinone Inhibits Angiogenesis by Targeting Vascular Endothelial Cells. Mol. Pharmacol. 2006, 70, 51–59. [Google Scholar] [CrossRef]
- Honarmand, M.; Namazi, F.; Mohammadi, A.; Nazifi, S. Can cannabidiol inhibit angiogenesis in colon cancer? Comp. Haematol. Int. 2018, 28, 165–172. [Google Scholar] [CrossRef]
- Ladin, D.A.; Soliman, E.; Griffin, L.; Van Dross, R. Preclinical and Clinical Assessment of Cannabinoids as Anti-Cancer Agents. Front. Pharmacol. 2016, 7, 361. [Google Scholar] [CrossRef]
- Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef] [PubMed]
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2018, 180, 346–356. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Ceruti, S.; Colombo, A.; Abbracchio, M.P.; Parolaro, D. Antitumor Effects of Cannabidiol, a Nonpsychoactive Cannabinoid, on Human Glioma Cell Lines. J. Pharmacol. Exp. Ther. 2003, 308, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulam, R.; Alvarez, L.; Guzmán, M.; Galve-Roperh, I. Cannabinoids Induce Glioma Stem-like Cell Differentiation and Inhibit Gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.D.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [PubMed]
- Kogan, N.M.; Schlesinger, M.; Peters, M.; Marincheva, G.; Beeri, R.; Mechoulam, R. A Cannabinoid Anticancer Quinone, HU-331, Is More Potent and Less Cardiotoxic than Doxorubicin: A Comparative in Vivo Study. J. Pharmacol. Exp. Ther. 2007, 322, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Andradas, C.; Perez-Gomez, E.; Guzmán, M.; Sanchez, C. Cannabinoids: A new hope for breast cancer therapy? Cancer Treat. Rev. 2012, 38, 911–918. [Google Scholar] [CrossRef] [PubMed]




| RECEPTORS | LOCALIZATION | FUNCTION | TUMOUR | REF. |
|---|---|---|---|---|
| CB1R | Central nervous system Peripheral tissues (e.g., liver, heart, skeletal muscle, adipose tissue, gastro-intestinal tract). | Neurotransmitters release Role in memory Motor coordination Emotional processes | Ovarian tumour Digestive tract Hodgkin lymphoma Prostate cancer | [18,19,20,21,22,24,25,26] |
| CB2R | Lymphoid organs Immune cells Nervous system | Anti-inflammatory Immunosuppressive | Breast cancer Pancreatic tumour Thyroid cancer Prostate cancer | [23,25,26,27] |
| GPR55 | Brain Spleen Bones Adipose tissue Langerhans islets | Vascular tone Bone turnover Motor coordination Inflammatory pain Neurological disorders Metabolic/immune dysregulation | Glioma Melanoma Breast cancer Pancreatic tumour | [28,29,30,31,32,33,34,35,36,37,38,39,40] |
| TRPV1 | Dorsal root neurons Trigeminal Arteriolar smooth muscle cells Bladder urothelium | Thermoregulation Involved in cough Bladder hyperactivity | Brain tumour Pancreatic tumour Breast cancer Prostate cancer Squamous cell carcinoma | [41,42,43,44,45,46,47,48,49,50,51,52] |
| PPARα | Liver Heart Muscles | Involved in fatty acid catabolism Inflammatory processes | Colon cancer Ovarian tumour Breast cancer Prostate cancer | [53,54,55,56] |
| PPARγ (γ1, γ2, γ3) | γ1: ubiquitous γ2: adipose tissue γ3: macrophages | Adipocyte formation Insulin sensitivity Inflammation |
| EFFECTS | TUMOUR TYPES and MEDIATORS | REF. |
|---|---|---|
| Tumour growth | Glioma (JWH-133; THC; CBD) Breast cancer (JWH-133; THC; WIN 55,212-2; JZL184) Prostate cancer (CBD; JWH-015; WIN 55,212-2; JZL184) CRC (CBD; JWH-015; URB597; URB-602; HU-331; O-1602) Melanoma (THC; CBD; WIN 55,212-2; URB597) HCC (JZL184) | [119,134,157,159,160,181,185,248,278,279,280,281] |
| Angiogenesis | Glioma (JWH-133) CRC (CBD; URB-602; CBG; HU-331) Melanoma (THC; CBD; WIN 55,212-2) Breast cancer (WIN 55,212-2) | [98,104,108,118,119,131,161,180,181,248,279,282,283,284,285] |
| MMPs expr. | Brain cancer (JWH-133; THC) | [174,286] |
| Apoptosis | CRC (CBD) Prostate cancer (CBD) Melanoma (THC; CBD) | [131,134,181,248] |
| Metastatic incidence | CRC (CBD) Breast cancer (THC; CBD; WIN 55,212-2; JZL184) Melanoma (THC; CBD; WIN 55,212-2; ACEA) Prostate cancer (WIN 55,212-2; JZL184) Lung cancer (URB597) HCC (JZL184) | [106,119,131,157,159,160,163,180,287,288] |
| Survival | Glioma (CBD; JZL184) Breast cancer (CBD) Melanoma (THC) | [101,255,287,288,289,290,291] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iozzo, M.; Sgrignani, G.; Comito, G.; Chiarugi, P.; Giannoni, E. Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches. Cells 2021, 10, 3396. https://doi.org/10.3390/cells10123396
Iozzo M, Sgrignani G, Comito G, Chiarugi P, Giannoni E. Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches. Cells. 2021; 10(12):3396. https://doi.org/10.3390/cells10123396
Chicago/Turabian StyleIozzo, Marta, Giovanna Sgrignani, Giuseppina Comito, Paola Chiarugi, and Elisa Giannoni. 2021. "Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches" Cells 10, no. 12: 3396. https://doi.org/10.3390/cells10123396
APA StyleIozzo, M., Sgrignani, G., Comito, G., Chiarugi, P., & Giannoni, E. (2021). Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches. Cells, 10(12), 3396. https://doi.org/10.3390/cells10123396