
Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress

 , , ,
, , , 
Abstract

1. Introduction
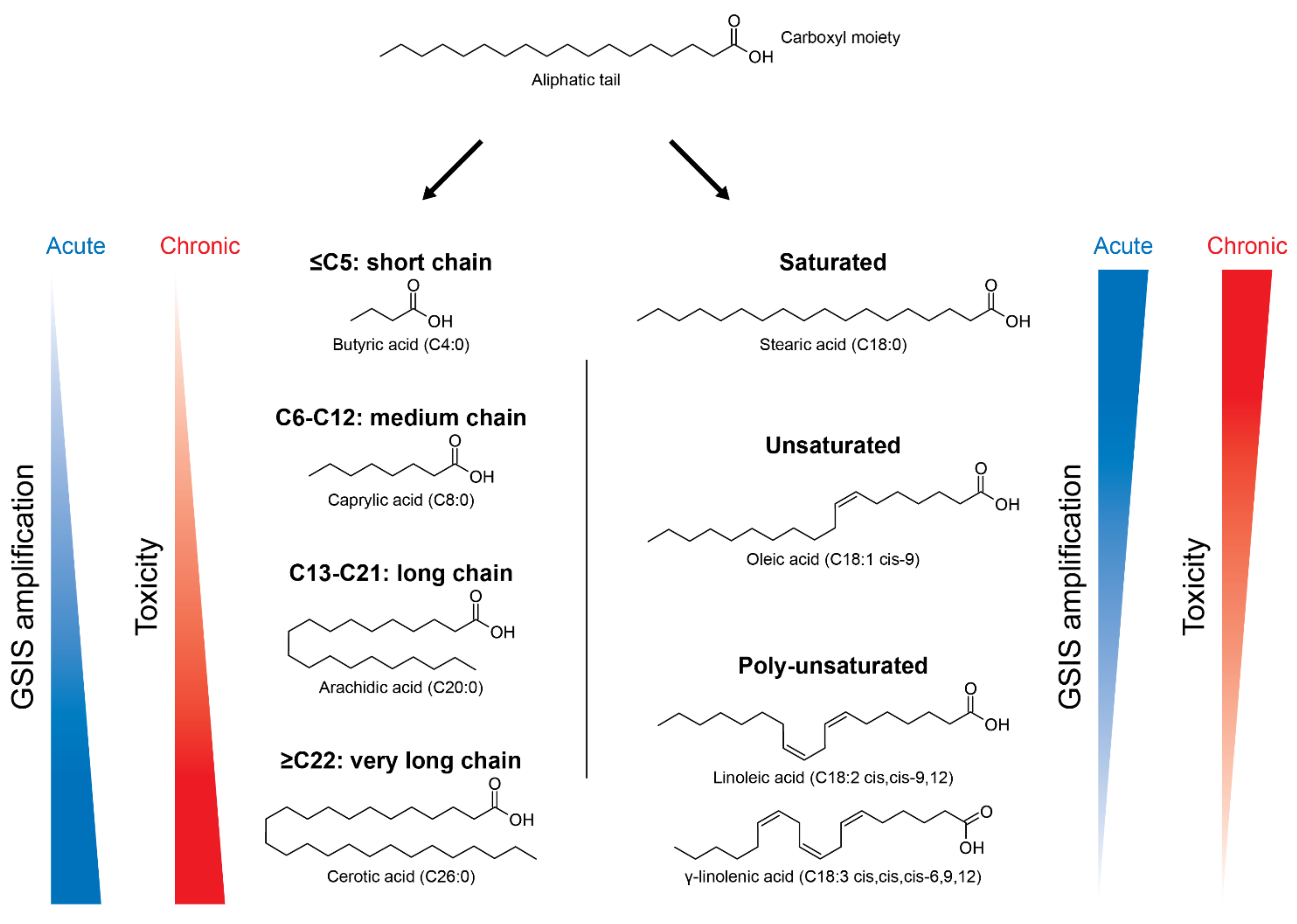
2. Fatty Acids and the Obesity-Related Hyperglycemia in T2D
3. Acute Versus Chronic Effects of FAs in Pancreatic β-Cells
3.1. FAs-Increased Glucose-Stimulated Insulin Secretion (GSIS)
3.2. FAs-Induced Lipotoxicity in β-Cells
4. Oxidative Stress in Lipotoxicity
4.1. Reactive Oxygen Species (ROS): Duality of Effects
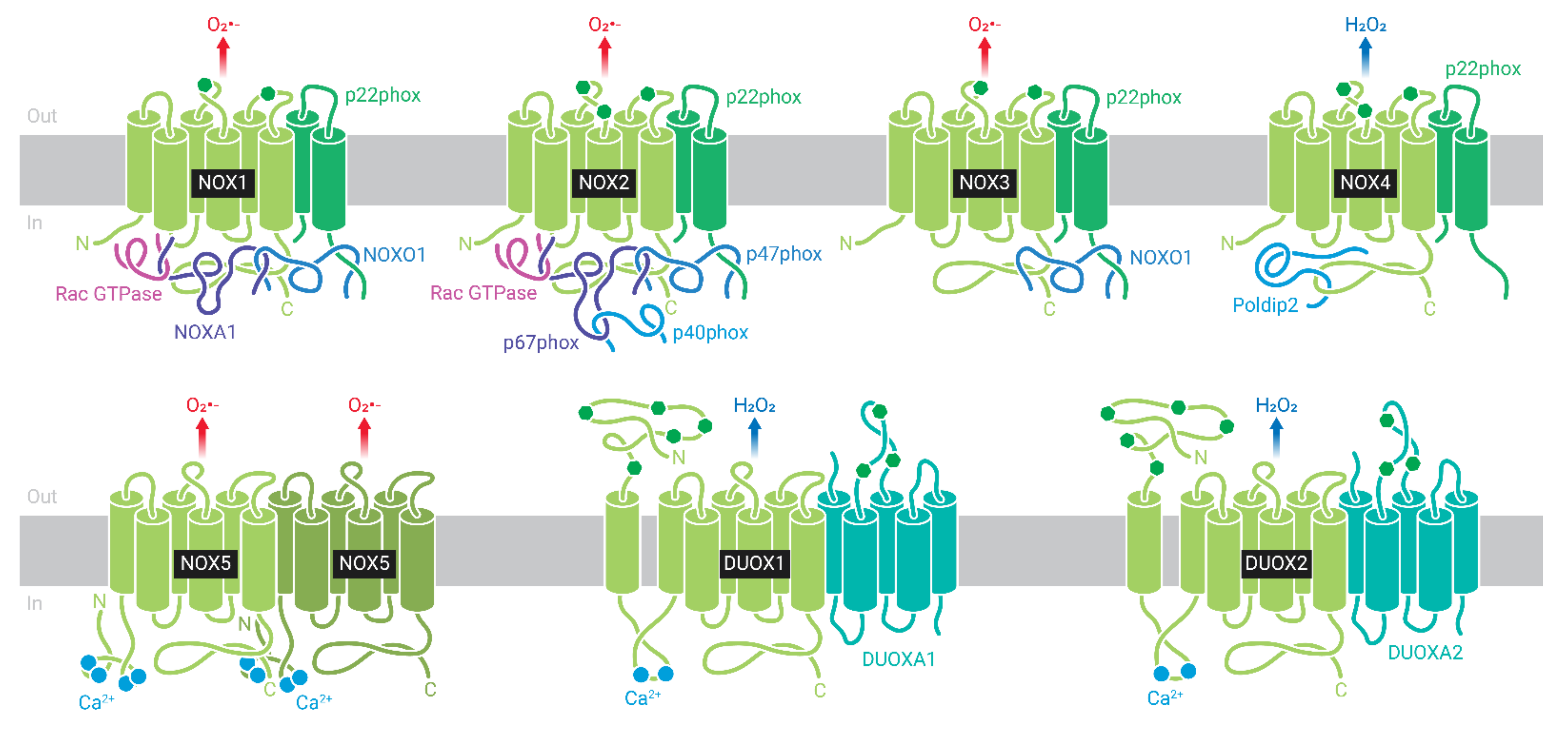
4.2. NOX in Lipotoxicity
5. ER Stress in Lipotoxicity
6. NOX and ER Stress
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef]
- Grarup, N.; Sandholt, C.H.; Hansen, T.; Pedersen, O. Genetic susceptibility to type 2 diabetes and obesity: From genome-wide association studies to rare variants and beyond. Diabetologia 2014, 57, 1528–1541. [Google Scholar] [CrossRef]
- Meigs, J.B.; Cupples, L.A.; Wilson, P.W. Parental transmission of type 2 diabetes: The Framingham Offspring Study. Diabetes 2000, 49, 2201–2207. [Google Scholar] [CrossRef]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53 (Suppl. 3), S16–S21. [Google Scholar] [CrossRef]
- Cali, A.M.; Man, C.D.; Cobelli, C.; Dziura, J.; Seyal, A.; Shaw, M.; Allen, K.; Chen, S.; Caprio, S. Primary defects in beta-cell function further exacerbated by worsening of insulin resistance mark the development of impaired glucose tolerance in obese adolescents. Diabetes Care 2009, 32, 456–461. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef]
- Burdge, G.C.; Calder, P.C. Introduction to fatty acids and lipids. World Rev. Nutr. Diet. 2015, 112, 1–16. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Kamp, F. How are free fatty acids transported in membranes? Is it by proteins or by free diffusion through the lipids? Diabetes 1999, 48, 2255–2269. [Google Scholar] [CrossRef]
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef]
- Jay, A.G.; Simard, J.R.; Huang, N.; Hamilton, J.A. SSO and other putative inhibitors of FA transport across membranes by CD36 disrupt intracellular metabolism, but do not affect FA translocation. J. Lipid. Res. 2020, 61, 790–807. [Google Scholar] [CrossRef]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef]
- Bays, H.; Mandarino, L.; DeFronzo, R.A. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: Peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab. 2004, 89, 463–478. [Google Scholar] [CrossRef]
- Bays, H.E.; González-Campoy, J.M.; Bray, G.A.; Kitabchi, A.E.; Bergman, D.A.; Schorr, A.B.; Rodbard, H.W.; Henry, R.R. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert. Rev. Cardiovasc. Ther. 2008, 6, 343–368. [Google Scholar] [CrossRef]
- Haber, E.P.; Ximenes, H.M.; Procópio, J.; Carvalho, C.R.; Curi, R.; Carpinelli, A.R. Pleiotropic effects of fatty acids on pancreatic beta-cells. J. Cell Physiol. 2003, 194, 1–12. [Google Scholar] [CrossRef]
- Haber, E.P.; Procópio, J.; Carvalho, C.R.; Carpinelli, A.R.; Newsholme, P.; Curi, R. New insights into fatty acid modulation of pancreatic beta-cell function. Int. Rev. Cytol. 2006, 248, 1–41. [Google Scholar] [CrossRef]
- Bermudez, B.; Ortega-Gomez, A.; Varela, L.M.; Villar, J.; Abia, R.; Muriana, F.J.; Lopez, S. Clustering effects on postprandial insulin secretion and sensitivity in response to meals with different fatty acid compositions. Food Funct. 2014, 5, 1374–1380. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef]
- Kunau, W.H.; Dommes, V.; Schulz, H. beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: A century of continued progress. Prog. Lipid. Res. 1995, 34, 267–342. [Google Scholar] [CrossRef]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Itoh, Y.; Hinuma, S. GPR40, a free fatty acid receptor on pancreatic beta cells, regulates insulin secretion. Hepatol. Res. 2005, 33, 171–173. [Google Scholar] [CrossRef]
- Gravena, C.; Mathias, P.C.; Ashcroft, S.J. Acute effects of fatty acids on insulin secretion from rat and human islets of Langerhans. J. Endocrinol. 2002, 173, 73–80. [Google Scholar] [CrossRef][Green Version]
- Carpinelli, A.R.; Picinato, M.C.; Stevanato, E.; Oliveira, H.R.; Curi, R. Insulin secretion induced by palmitate--a process fully dependent on glucose concentration. Diabetes Metab. 2002, 28 Pt 2, 3S37-44. [Google Scholar]
- Deeney, J.T.; Gromada, J.; Høy, M.; Olsen, H.L.; Rhodes, C.J.; Prentki, M.; Berggren, P.O.; Corkey, B.E. Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI beta-cells). J. Biol. Chem. 2000, 275, 9363–9368. [Google Scholar] [CrossRef]
- Stein, D.T.; Esser, V.; Stevenson, B.E.; Lane, K.E.; Whiteside, J.H.; Daniels, M.B.; Chen, S.; McGarry, J.D. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J. Clin. investig. 1996, 97, 2728–2735. [Google Scholar] [CrossRef]
- Stein, D.T.; Stevenson, B.E.; Chester, M.W.; Basit, M.; Daniels, M.B.; Turley, S.D.; McGarry, J.D. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J. Clin. Investig. 1997, 100, 398–403. [Google Scholar] [CrossRef]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Shapiro, H.; Shachar, S.; Sekler, I.; Hershfinkel, M.; Walker, M.D. Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem. Biophys Res. Commun. 2005, 335, 97–104. [Google Scholar] [CrossRef]
- Kotarsky, K.; Nilsson, N.E.; Flodgren, E.; Owman, C.; Olde, B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 2003, 301, 406–410. [Google Scholar] [CrossRef]
- Poitout, V. The ins and outs of fatty acids on the pancreatic beta cell. Trends Endocrinol. Metab. 2003, 14, 201–203. [Google Scholar] [CrossRef]
- Gromada, J. The free fatty acid receptor GPR40 generates excitement in pancreatic beta-cells. Endocrinology 2006, 147, 672–673. [Google Scholar] [CrossRef]
- Nolan, C.J.; Madiraju, M.S.; Delghingaro-Augusto, V.; Peyot, M.L.; Prentki, M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 2006, 55 (Suppl. 2), S16–S23. [Google Scholar] [CrossRef]
- Kebede, M.A.; Alquier, T.; Latour, M.G.; Poitout, V. Lipid receptors and islet function: Therapeutic implications? Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 10–20. [Google Scholar] [CrossRef]
- Del Guerra, S.; Bugliani, M.; D’Aleo, V.; Del Prato, S.; Boggi, U.; Mosca, F.; Filipponi, F.; Lupi, R. G-protein-coupled receptor 40 (GPR40) expression and its regulation in human pancreatic islets: The role of type 2 diabetes and fatty acids. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 22–25. [Google Scholar] [CrossRef]
- Vettor, R.; Granzotto, M.; De Stefani, D.; Trevellin, E.; Rossato, M.; Farina, M.G.; Milan, G.; Pilon, C.; Nigro, A.; Federspil, G.; et al. Loss-of-function mutation of the GPR40 gene associates with abnormal stimulated insulin secretion by acting on intracellular calcium mobilization. J. Clin. Endocrinol. Metab. 2008, 93, 3541–3550. [Google Scholar] [CrossRef]
- Garrido, D.M.; Corbett, D.F.; Dwornik, K.A.; Goetz, A.S.; Littleton, T.R.; McKeown, S.C.; Mills, W.Y.; Smalley, T.L.; Briscoe, C.P.; Peat, A.J. Synthesis and activity of small molecule GPR40 agonists. Bioorg. Med. Chem. Lett. 2006, 16, 1840–1845. [Google Scholar] [CrossRef]
- Bharate, S.B.; Rodge, A.; Joshi, R.K.; Kaur, J.; Srinivasan, S.; Kumar, S.S.; Kulkarni-Almeida, A.; Balachandran, S.; Balakrishnan, A.; Vishwakarma, R.A. Discovery of diacylphloroglucinols as a new class of GPR40 (FFAR1) agonists. Bioorg. Med. Chem. Lett. 2008, 18, 6357–6361. [Google Scholar] [CrossRef]
- Christiansen, E.; Urban, C.; Merten, N.; Liebscher, K.; Karlsen, K.K.; Hamacher, A.; Spinrath, A.; Bond, A.D.; Drewke, C.; Ullrich, S.; et al. Discovery of potent and selective agonists for the free fatty acid receptor 1 (FFA(1)/GPR40), a potential target for the treatment of type II diabetes. J. Med. Chem. 2008, 51, 7061–7064. [Google Scholar] [CrossRef]
- Negoro, N.; Sasaki, S.; Mikami, S.; Ito, M.; Suzuki, M.; Tsujihata, Y.; Ito, R.; Harada, A.; Takeuchi, K.; Suzuki, N.; et al. Discovery of TAK-875: A Potent, Selective, and Orally Bioavailable GPR40 Agonist. ACS Med. Chem. Lett. 2010, 1, 290–294. [Google Scholar] [CrossRef]
- Houze, J.B.; Zhu, L.; Sun, Y.; Akerman, M.; Qiu, W.; Zhang, A.J.; Sharma, R.; Schmitt, M.; Wang, Y.; Liu, J.; et al. AMG 837: A potent, orally bioavailable GPR40 agonist. Bioorg. Med. Chem. Lett. 2012, 22, 1267–1270. [Google Scholar] [CrossRef]
- Araki, T.; Hirayama, M.; Hiroi, S.; Kaku, K. GPR40-induced insulin secretion by the novel agonist TAK-875: First clinical findings in patients with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 271–278. [Google Scholar] [CrossRef]
- Vilas-Boas, E.A.; Karabacz, N.; Marsiglio-Librais, G.N.; Valle, M.M.R.; Nalbach, L.; Ampofo, E.; Morgan, B.; Carpinelli, A.R.; Roma, L.P. Chronic activation of GPR40 does not negatively impact upon BRIN-BD11 pancreatic β-cell physiology and function. Pharmacol. Rep. 2020, 72, 1725–1737. [Google Scholar] [CrossRef]
- Carlsson, M.; Wessman, Y.; Almgren, P.; Groop, L. High levels of nonesterified fatty acids are associated with increased familial risk of cardiovascular disease. Arterioscler Thromb. Vasc. Biol. 2000, 20, 1588–1594. [Google Scholar] [CrossRef]
- Salgin, B.; Ong, K.K.; Thankamony, A.; Emmett, P.; Wareham, N.J.; Dunger, D.B. Higher fasting plasma free fatty acid levels are associated with lower insulin secretion in children and adults and a higher incidence of type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3302–3309. [Google Scholar] [CrossRef]
- Mahendran, Y.; Cederberg, H.; Vangipurapu, J.; Kangas, A.J.; Soininen, P.; Kuusisto, J.; Uusitupa, M.; Ala-Korpela, M.; Laakso, M. Glycerol and fatty acids in serum predict the development of hyperglycemia and type 2 diabetes in Finnish men. Diabetes Care 2013, 36, 3732–3738. [Google Scholar] [CrossRef]
- Risérus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef]
- Hodge, A.M.; English, D.R.; O’Dea, K.; Sinclair, A.J.; Makrides, M.; Gibson, R.A.; Giles, G.G. Plasma phospholipid and dietary fatty acids as predictors of type 2 diabetes: Interpreting the role of linoleic acid. Am. J. Clin. Nutr. 2007, 86, 189–197. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Blindauer, C.A.; Stewart, A.J. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef]
- Poitout, V.; Hagman, D.; Stein, R.; Artner, I.; Robertson, R.P.; Harmon, J.S. Regulation of the insulin gene by glucose and fatty acids. J. Nutr. 2006, 136, 873–876. [Google Scholar] [CrossRef]
- Ritz-Laser, B.; Meda, P.; Constant, I.; Klages, N.; Charollais, A.; Morales, A.; Magnan, C.; Ktorza, A.; Philippe, J. Glucose-induced preproinsulin gene expression is inhibited by the free fatty acid palmitate. Endocrinology 1999, 140, 4005–4014. [Google Scholar] [CrossRef]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, P.Y.; Takashi, K. Chronic effects of different non-esterified fatty acids on pancreatic islets of rats. Endocrine 2006, 29, 169–173. [Google Scholar] [CrossRef]
- Maris, M.; Robert, S.; Waelkens, E.; Derua, R.; Hernangomez, M.H.; D’Hertog, W.; Cnop, M.; Mathieu, C.; Overbergh, L. Role of the saturated nonesterified Fatty Acid palmitate in Beta cell dysfunction. J. Proteome Res. 2013, 12, 347–362. [Google Scholar] [CrossRef]
- Dixon, G.; Nolan, J.; McClenaghan, N.H.; Flatt, P.R.; Newsholme, P. Arachidonic acid, palmitic acid and glucose are important for the modulation of clonal pancreatic beta-cell insulin secretion, growth and functional integrity. Clin. Sci. 2004, 106, 191–199. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Keane, D.C.; Takahashi, H.K.; Dhayal, S.; Morgan, N.G.; Curi, R.; Newsholme, P. Arachidonic acid actions on functional integrity and attenuation of the negative effects of palmitic acid in a clonal pancreatic β-cell line. Clin. Sci. 2011, 120, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Eitel, K.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Häring, H.U.; Kellerer, M. Different role of saturated and unsaturated fatty acids in beta-cell apoptosis. Biochem Biophys Res. Commun. 2002, 299, 853–856. [Google Scholar] [CrossRef]
- Keane, D.; Newsholme, P. Saturated and unsaturated (including arachidonic acid) non-esterified fatty acid modulation of insulin secretion from pancreatic beta-cells. Biochem. Soc. Trans. 2008, 36, 955–958. [Google Scholar] [CrossRef]
- Fürstova, V.; Kopska, T.; James, R.F.; Kovar, J. Comparison of the effect of individual saturated and unsaturated fatty acids on cell growth and death induction in the human pancreatic beta-cell line NES2Y. Life Sci. 2008, 82, 684–691. [Google Scholar] [CrossRef]
- Dhayal, S.; Welters, H.J.; Morgan, N.G. Structural requirements for the cytoprotective actions of mono-unsaturated fatty acids in the pancreatic beta-cell line, BRIN-BD11. Br. J. Pharmacol. 2008, 153, 1718–1727. [Google Scholar] [CrossRef]
- Kato, T.; Shimano, H.; Yamamoto, T.; Ishikawa, M.; Kumadaki, S.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N.; Nakakuki, M.; Hasty, A.H.; et al. Palmitate impairs and eicosapentaenoate restores insulin secretion through regulation of SREBP-1c in pancreatic islets. Diabetes 2008, 57, 2382–2392. [Google Scholar] [CrossRef]
- Welters, H.J.; Tadayyon, M.; Scarpello, J.H.; Smith, S.A.; Morgan, N.G. Mono-unsaturated fatty acids protect against beta-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett. 2004, 560, 103–108. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 32–42. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef] [PubMed]
- Plötz, T.; von Hanstein, A.S.; Krümmel, B.; Laporte, A.; Mehmeti, I.; Lenzen, S. Structure-toxicity relationships of saturated and unsaturated free fatty acids for elucidating the lipotoxic effects in human EndoC-βH1 beta-cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165525. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic β-cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1929–1942. [Google Scholar] [CrossRef]
- Fang, F.C. Antimicrobial actions of reactive oxygen species. mBio 2011, 2, e00141-11. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Rebelato, E.; Abdulkader, F.; Graciano, M.F.; Oliveira-Emilio, H.R.; Hirata, A.E.; Rocha, M.S.; Bordin, S.; Curi, R.; Carpinelli, A.R. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 2009, 150, 2197–2201. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants 2021, 10, 197. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; Jabůrek, M.; Holendová, B.; Tauber, J.; Pavluch, V.; Berková, Z.; Cahová, M.; Schröder, K.; Brandes, R.P.; Siemen, D.; et al. Glucose-Stimulated Insulin Secretion Fundamentally Requires H. Diabetes 2020, 69, 1341–1354. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Lakey, J.R.; Suarez-Pinzon, W.L.; Strynadka, K.; Korbutt, G.S.; Rajotte, R.V.; Mabley, J.G.; Szabó, C.; Rabinovitch, A. Peroxynitrite is a mediator of cytokine-induced destruction of human pancreatic islet beta cells. Lab. investig. 2001, 81, 1683–1692. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Kaneto, H.; Matsuoka, T.A.; Nakatani, Y.; Kawamori, D.; Miyatsuka, T.; Matsuhisa, M.; Yamasaki, Y. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J. Mol. Med. 2005, 83, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J. Biol. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Grankvist, K.; Marklund, S.L.; Täljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.R.; Funke, M.; Ackermann, W.; Haunhorst, P.; Oesteritz, S.; Capani, F.; Elsässer, H.P.; Lillig, C.H. Redox atlas of the mouse. Immunohistochemical detection of glutaredoxin-, peroxiredoxin-, and thioredoxin-family proteins in various tissues of the laboratory mouse. Biochim. Biophys. Acta 2011, 1810, 2–92. [Google Scholar] [CrossRef] [PubMed]
- Stancill, J.S.; Broniowska, K.A.; Oleson, B.J.; Naatz, A.; Corbett, J.A. Pancreatic β-cells detoxify H. J. Biol. Chem. 2019, 294, 4843–4853. [Google Scholar] [CrossRef] [PubMed]
- Benáková, Š.; Holendová, B.; Plecitá-Hlavatá, L. Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants 2021, 10, 526. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Cui, Q.; Yang, B.; Hou, Y.; Wang, H.; Xu, Y.; Wang, D.; Zhang, Q.; Pi, J. The impairment of glucose-stimulated insulin secretion in pancreatic β-cells caused by prolonged glucotoxicity and lipotoxicity is associated with elevated adaptive antioxidant response. Food Chem. Toxicol. 2017, 100, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ganete, A.; Quiroga-de-Castro, A.; Mateos, R.M.; Medina, F.; Segundo, C.; Lechuga-Sancho, A.M. Toxicity Induced by Cytokines, Glucose, and Lipids Increase Apoptosis and Hamper Insulin Secretion in the 1.1E7 Beta Cell-Line. Int. J. Mol. Sci. 2021, 22, 2559. [Google Scholar] [CrossRef]
- Oprescu, A.I.; Bikopoulos, G.; Naassan, A.; Allister, E.M.; Tang, C.; Park, E.; Uchino, H.; Lewis, G.F.; Fantus, I.G.; Rozakis-Adcock, M.; et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: Evidence for a role of oxidative stress in vitro and in vivo. Diabetes 2007, 56, 2927–2937. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef]
- Piro, S.; Anello, M.; Di Pietro, C.; Lizzio, M.N.; Patanè, G.; Rabuazzo, A.M.; Vigneri, R.; Purrello, M.; Purrello, F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: Possible role of oxidative stress. Metabolism 2002, 51, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Michaelis, U.R.; Bredenkötter, D.; Fisslthaler, B.; Dehghani, F.; Brandes, R.P.; Busse, R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 2001, 88, 44–51. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Lenzen, S. The H2O2-sensitive HyPer protein targeted to the endoplasmic reticulum as a mirror of the oxidizing thiol-disulfide milieu. Free Radic. Biol. Med. 2012, 53, 1451–1458. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; del Río, L.A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell Biochem. 2013, 69, 231–255. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH Oxidases. Methods Mol. Biol 2019, 1982, 17–36. [Google Scholar] [CrossRef]
- Berton, G.; Castaldi, M.A.; Cassatella, M.A.; Nauseef, W.M. Editorial: Celebrating the 50th anniversary of the seminal discovery that the phagocyte respiratory burst enzyme is an NADPH oxidase. J. Leukoc. Biol. 2015, 97, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.R.; Verlengia, R.; Carvalho, C.R.; Britto, L.R.; Curi, R.; Carpinelli, A.R. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003, 52, 1457–1463. [Google Scholar] [CrossRef]
- Rebelato, E.; Mares-Guia, T.R.; Graciano, M.F.; Labriola, L.; Britto, L.R.; Garay-Malpartida, H.M.; Curi, R.; Sogayar, M.C.; Carpinelli, A.R. Expression of NADPH oxidase in human pancreatic islets. Life Sci. 2012, 91, 244–249. [Google Scholar] [CrossRef]
- Uchizono, Y.; Takeya, R.; Iwase, M.; Sasaki, N.; Oku, M.; Imoto, H.; Iida, M.; Sumimoto, H. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci. 2006, 80, 133–139. [Google Scholar] [CrossRef]
- Anvari, E.; Wikström, P.; Walum, E.; Welsh, N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Bouzakri, K.; Veyrat-Durebex, C.; Holterman, C.; Arous, C.; Barbieux, C.; Bosco, D.; Altirriba, J.; Alibashe, M.; Tournier, B.B.; Gunton, J.E.; et al. Beta-Cell-Specific Expression of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 5 Aggravates High-Fat Diet-Induced Impairment of Islet Insulin Secretion in Mice. Antioxid Redox Signal 2020, 32, 618–635. [Google Scholar] [CrossRef]
- Kowluru, A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: Lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol. 2011, 81, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Santos, L.R.; Curi, R.; Carpinelli, A.R. NAD(P)H oxidase participates in the palmitate-induced superoxide production and insulin secretion by rat pancreatic islets. J. Cell Physiol. 2011, 226, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Fishwick, D.A. NOX, NOX Who is There? The Contribution of NADPH Oxidase One to Beta Cell Dysfunction. Front. Endocrinol. 2013, 4, 40. [Google Scholar] [CrossRef]
- Sedeek, M.; Montezano, A.C.; Hebert, R.L.; Gray, S.P.; Di Marco, E.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.; Schiffrin, E.L.; Wilkinson-Berka, J.L.; et al. Oxidative stress, Nox isoforms and complications of diabetes—Potential targets for novel therapies. J. Cardiovasc. Transl. Res. 2012, 5, 509–518. [Google Scholar] [CrossRef]
- Newsholme, P.; Keane, D.; Welters, H.J.; Morgan, N.G. Life and death decisions of the pancreatic beta-cell: The role of fatty acids. Clin. Sci. 2007, 112, 27–42. [Google Scholar] [CrossRef]
- Blanchetot, C.; Boonstra, J. The ROS-NOX connection in cancer and angiogenesis. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 35–45. [Google Scholar] [CrossRef]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal 2014, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Hébert, R.L.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M. Molecular mechanisms of hypertension: Role of Nox family NADPH oxidases. Curr. Opin. Nephrol. Hypertens 2009, 18, 122–127. [Google Scholar] [CrossRef]
- Hecker, L.; Cheng, J.; Thannickal, V.J. Targeting NOX enzymes in pulmonary fibrosis. Cell Mol. Life Sci. 2012, 69, 2365–2371. [Google Scholar] [CrossRef]
- You, Y.H.; Okada, S.; Ly, S.; Jandeleit-Dahm, K.; Barit, D.; Namikoshi, T.; Sharma, K. Role of Nox2 in diabetic kidney disease. Am. J. Physiol. Renal. Physiol. 2013, 304, F840–F848. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Inoguchi, T.; Sonta, T.; Maeda, Y.; Sasaki, S.; Sawada, F.; Tsubouchi, H.; Sonoda, N.; Kobayashi, K.; Sumimoto, H.; et al. Increased expression of NAD(P)H oxidase in islets of animal models of Type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochem. Biophys. Res. Commun. 2005, 332, 927–933. [Google Scholar] [CrossRef]
- Syed, I.; Kyathanahalli, C.N.; Jayaram, B.; Govind, S.; Rhodes, C.J.; Kowluru, R.A.; Kowluru, A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: Role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 2011, 60, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Lu, Y.; Huang, X.; He, Q.; Man, Y.; Zhou, Y.; Wang, S.; Li, J. Suppression of NADPH oxidase 2 substantially restores glucose-induced dysfunction of pancreatic NIT-1 cells. FEBS J. 2010, 277, 5061–5071. [Google Scholar] [CrossRef]
- Mohammed, A.M.; Kowluru, A. Activation of apocynin-sensitive NADPH oxidase (Nox2) activity in INS-1 832/13 cells under glucotoxic conditions. Islets 2013, 5, 129–131. [Google Scholar] [CrossRef]
- Elumalai, S.; Karunakaran, U.; Lee, I.K.; Moon, J.S.; Won, K.C. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017, 11, 126–134. [Google Scholar] [CrossRef]
- de Souza, A.H.; Santos, L.R.; Roma, L.P.; Bensellam, M.; Carpinelli, A.R.; Jonas, J.C. NADPH oxidase-2 does not contribute to β-cell glucotoxicity in cultured pancreatic islets from C57BL/6J mice. Mol. Cell Endocrinol. 2017, 439, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, B.; Brun, T.; Deffert-Delbouille, C.; Mahiout, Z.; Daali, Y.; Ma, X.J.; Krause, K.H.; Maechler, P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012, 61, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, G.; Walther, R.; Newsholme, P. Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic β-cell dysfunction. Biosci. Rep. 2010, 30, 445–453. [Google Scholar] [CrossRef]
- Nunes Marsiglio-Librais, G.; Aparecida Vilas-Boas, E.; Carlein, C.; Hoffmann, M.D.A.; Roma, L.P.; Carpinelli, A.R. Evidence for NADPH oxidase activation by GPR40 in pancreatic β-cells. Redox Rep. 2020, 25, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Elksnis, A.; Wikström, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, E.A.; Nalbach, L.; Ampofo, E.; Lucena, C.F.; Naudet, L.; Ortis, F.; Carpinelli, A.R.; Morgan, B.; Roma, L.P. Transient NADPH oxidase 2-dependent H2O2 production drives early palmitate-induced lipotoxicity in pancreatic islets. Free Radic. Biol. Med. 2020, 162, 1–13. [Google Scholar] [CrossRef]
- Kleniewska, P.; Piechota, A.; Skibska, B.; Gorąca, A. The NADPH oxidase family and its inhibitors. Arch. Immunol. Ther. Exp. 2012, 60, 277–294. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, E.; Meijles, D.N.; Pagano, P.J. The quest for selective nox inhibitors and therapeutics: Challenges, triumphs and pitfalls. Antioxid. Redox Signal 2014, 20, 2741–2754. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Cnop, M.; Igoillo-Esteve, M.; Cunha, D.A.; Ladrière, L.; Eizirik, D.L. An update on lipotoxic endoplasmic reticulum stress in pancreatic beta-cells. Biochem. Soc. Trans 2008, 36, 909–915. [Google Scholar] [CrossRef]
- Cnop, M.; Ladrière, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 76–82. [Google Scholar] [CrossRef]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell. Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Meyerovich, K.; Ortis, F.; Allagnat, F.; Cardozo, A.K. Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J. Mol. Endocrinol. 2016, 57, R1–R17. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Ortis, F.; Cunha, D.A.; Gosset, G.; Li, M.; Cardozo, A.K.; Eizirik, D.L. Signaling by IL-1beta+IFN-gamma and ER stress converge on DP5/Hrk activation: A novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ. 2009, 16, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Germano, C.M.; Cunha, D.A.; Ortis, F.; Vanderwinden, J.M.; Marchetti, P.; Zhang, L.; Eizirik, D.L. p53 up-regulated modulator of apoptosis (PUMA) activation contributes to pancreatic beta-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J. Biol. Chem. 2010, 285, 19910–19920. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium efflux from the endoplasmic reticulum leads to β-cell death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef]
- Preston, A.M.; Gurisik, E.; Bartley, C.; Laybutt, D.R.; Biden, T.J. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia 2009, 52, 2369–2373. [Google Scholar] [CrossRef]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [CrossRef]
- Marmugi, A.; Parnis, J.; Chen, X.; Carmichael, L.; Hardy, J.; Mannan, N.; Marchetti, P.; Piemonti, L.; Bosco, D.; Johnson, P.; et al. Sorcin Links Pancreatic β-Cell Lipotoxicity to ER Ca2+ Stores. Diabetes 2016, 65, 1009–1021. [Google Scholar] [CrossRef]
- Lytrivi, M.; Ghaddar, K.; Lopes, M.; Rosengren, V.; Piron, A.; Yi, X.; Johansson, H.; Lehtiö, J.; Igoillo-Esteve, M.; Cunha, D.A.; et al. Combined transcriptome and proteome profiling of the pancreatic β-cell response to palmitate unveils key pathways of β-cell lipotoxicity. BMC Genom. 2020, 21, 590. [Google Scholar] [CrossRef]
- Hansen, H.G.; Schmidt, J.D.; Søltoft, C.L.; Ramming, T.; Geertz-Hansen, H.M.; Christensen, B.; Sørensen, E.S.; Juncker, A.S.; Appenzeller-Herzog, C.; Ellgaard, L. Hyperactivity of the Ero1α oxidase elicits endoplasmic reticulum stress but no broad antioxidant response. J. Biol. Chem. 2012, 287, 39513–39523. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Avezov, E.; Jörns, A.; Lenzen, S. ER-resident antioxidative GPx7 and GPx8 enzyme isoforms protect insulin-secreting INS-1E β-cells against lipotoxicity by improving the ER antioxidative capacity. Free Radic. Biol. Med. 2017, 112, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; MacIntosh, G.; Preston, A.M.; Bartley, C.; Busch, A.K.; Fuller, M.; Laybutt, D.R.; Meikle, P.J.; Biden, T.J. A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 2011, 435, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef]
- Acosta-Montaño, P.; Rodríguez-Velázquez, E.; Ibarra-López, E.; Frayde-Gómez, H.; Mas-Oliva, J.; Delgado-Coello, B.; Rivero, I.A.; Alatorre-Meda, M.; Aguilera, J.; Guevara-Olaya, L.; et al. Fatty Acid and Lipopolysaccharide Effect on Beta Cells Proteostasis and its Impact on Insulin Secretion. Cells 2019, 8, 884. [Google Scholar] [CrossRef]
- Giacca, A.; Xiao, C.; Oprescu, A.I.; Carpentier, A.C.; Lewis, G.F. Lipid-induced pancreatic β-cell dysfunction: Focus on in vivo studies. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E255–E262. [Google Scholar] [CrossRef]
- Ly, L.D.; Ly, D.D.; Nguyen, N.T.; Kim, J.H.; Yoo, H.; Chung, J.; Lee, M.S.; Cha, S.K.; Park, K.S. Mitochondrial Ca2+ Uptake Relieves Palmitate-Induced Cytosolic Ca2+ Overload in MIN6 Cells. Mol. Cells 2020, 43, 66–75. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Araujo, T.L.; Abrahão, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal 2014, 20, 2755–2775. [Google Scholar] [CrossRef]
- Gehrmann, W.; Würdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism Between Saturated and Unsaturated Fatty Acids in ROS Mediated Lipotoxicity in Rat Insulin-Producing Cells. Cell Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Scheuner, D.; Han, J.; Song, B.; Ribick, M.; Wang, J.; Gildersleeve, R.D.; Pennathur, S.; Kaufman, R.J. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009, 10, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, H.; Shen, E.; Wan, L.; Arnold, J.M.; Peng, T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010, 59, 2033–2042. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, W.M.; Zhang, L.; Schuiki, I.; Curi, R.; Volchuk, A.; Alba-Loureiro, T.C. NADPH oxidase-dependent production of reactive oxygen species induces endoplasmatic reticulum stress in neutrophil-like HL60 cells. PLoS ONE 2015, 10, e0116410. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Contreras-Ferrat, A.; Llanos, P.; Vásquez, C.; Espinosa, A.; Osorio-Fuentealba, C.; Arias-Calderon, M.; Lavandero, S.; Klip, A.; Hidalgo, C.; Jaimovich, E. Insulin elicits a ROS-activated and an IP3-dependent Ca2+ release, which both impinge on GLUT4 translocation. J. Cell Sci. 2014, 127, 1911–1923. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, E.A.; Carlein, C.; Nalbach, L.; Almeida, D.C.; Ampofo, E.; Carpinelli, A.R.; Roma, L.P.; Ortis, F. Early Cytokine-Induced Transient NOX2 Activity Is ER Stress-Dependent and Impacts β-Cell Function and Survival. Antioxidants 2021, 10, 1305. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NOX Influence on ER Stress | Model | Details | Ref |
|---|---|---|---|
| NOX inhibition → ER stress inhibition | Cardiomyocytes | NOX inhibition with apocynin or Rac1 KO mice | [184] |
| NOX inhibition → ER stress inhibition | Macrophages | NOX2 siRNA or NOX2 KO mice | [185] |
| NOX inhibition → ER stress inhibition | Neutrophils | HL60 cell line; NOX inhibition with DPI | [186] |
| NOX activation → ER stress activation | Neutrophils | HL60 cell line; NOX activation with PMA | [186] |
| NOX inhibition → no effect in ER stress | Mice islets | NOX inhibition with VAS2870 | [144] |
| ER Stress Influence on NOX | Model | Details | Ref |
| ER stress induction → NOX increased expression | Macrophages | ER stress induction by cholesterol or 7-ketocholesterol | [185] |
| ER stress attenuation → NOX inhibition | Mice islets | ER stress attenuation with 4-PBA; NOX2-derived H2O2 peak was delayed | [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; Ortis, F.; Carpinelli, A.R. Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells 2021, 10, 3328. https://doi.org/10.3390/cells10123328
Vilas-Boas EA, Almeida DC, Roma LP, Ortis F, Carpinelli AR. Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells. 2021; 10(12):3328. https://doi.org/10.3390/cells10123328
Chicago/Turabian StyleVilas-Boas, Eloisa Aparecida, Davidson Correa Almeida, Leticia Prates Roma, Fernanda Ortis, and Angelo Rafael Carpinelli. 2021. "Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress" Cells 10, no. 12: 3328. https://doi.org/10.3390/cells10123328
APA StyleVilas-Boas, E. A., Almeida, D. C., Roma, L. P., Ortis, F., & Carpinelli, A. R. (2021). Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells, 10(12), 3328. https://doi.org/10.3390/cells10123328