
Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
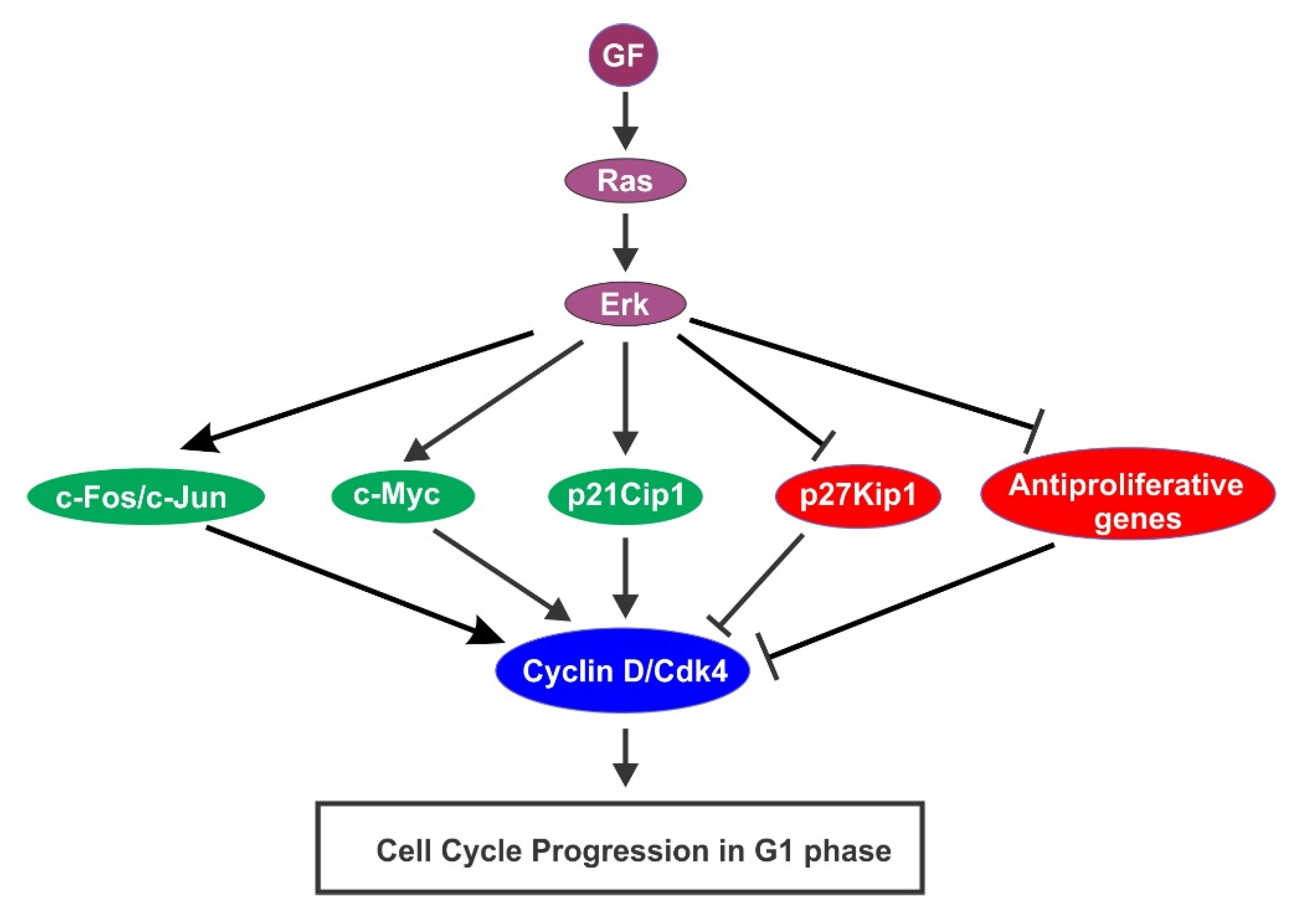{kind=link}
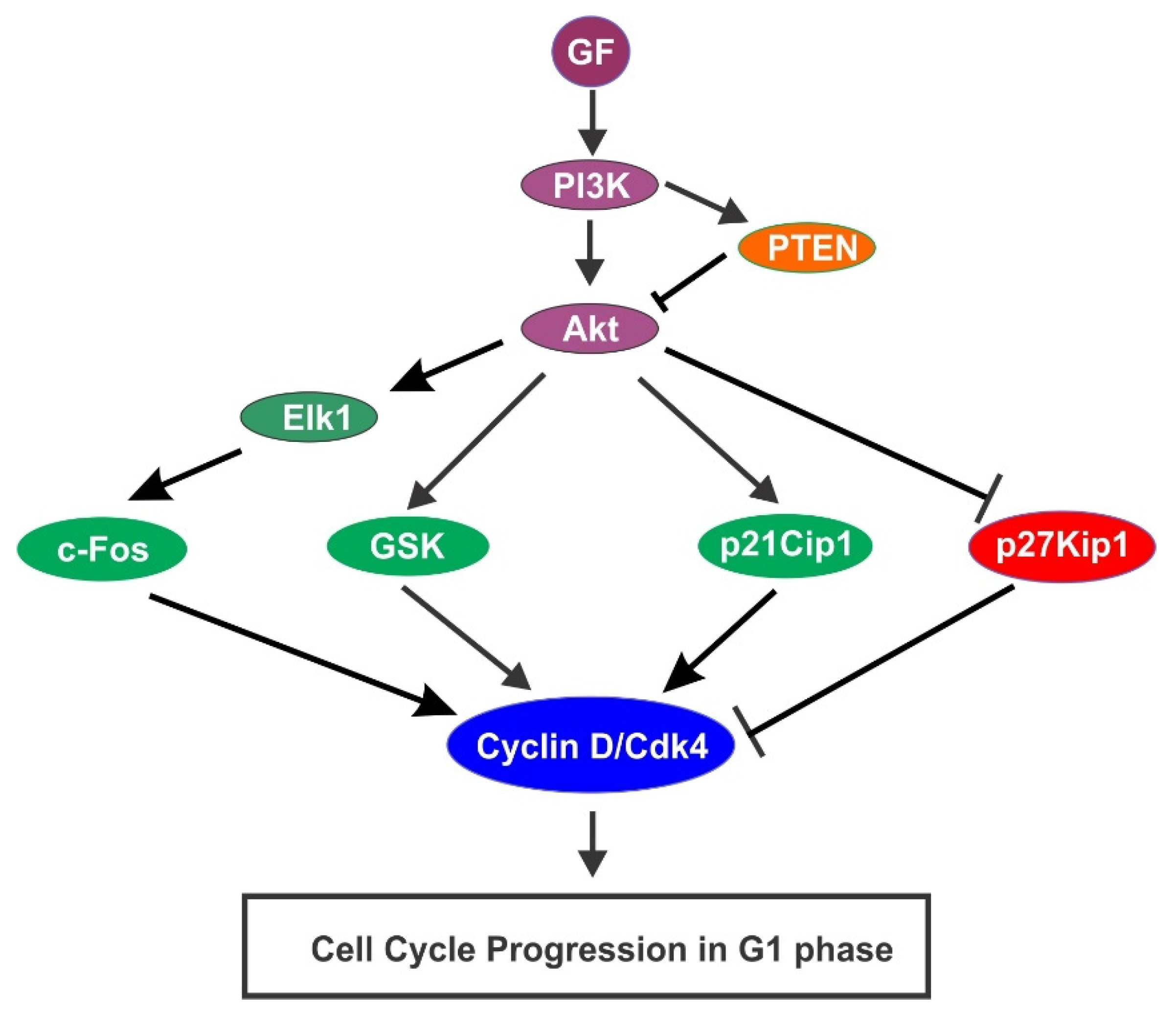{kind=link}
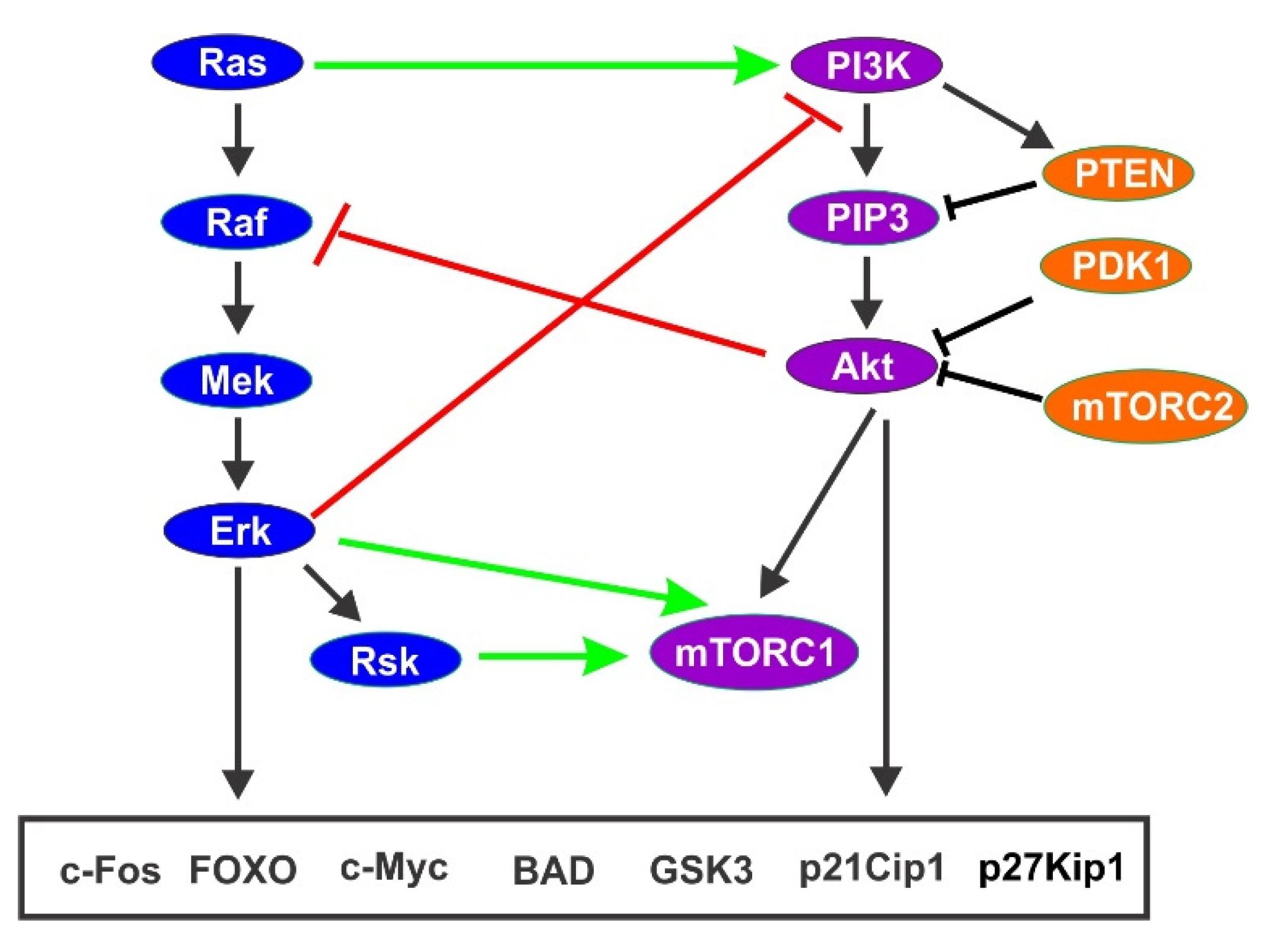{kind=link}
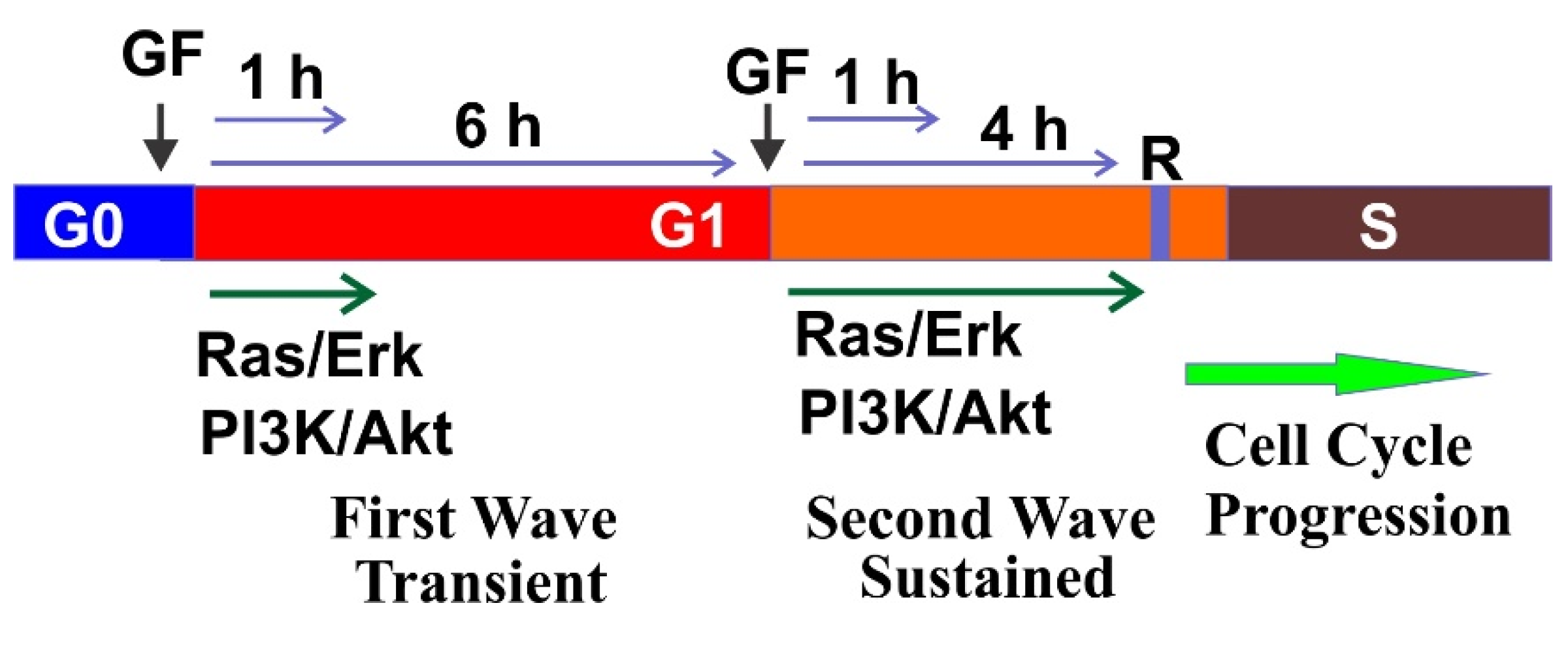{kind=link}
{kind=link}
Abstract
1. Introduction
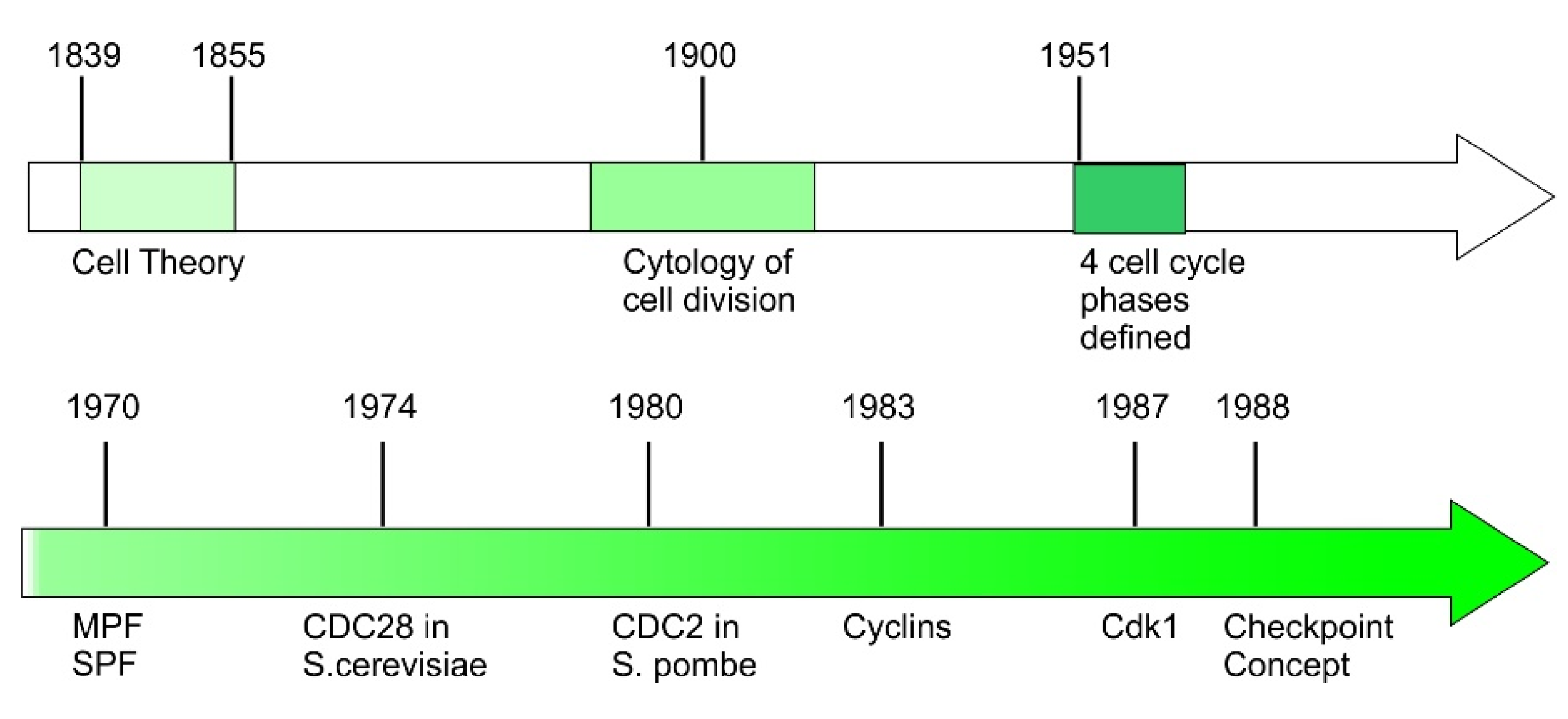
2. Early History of Cell Cycle Discovery
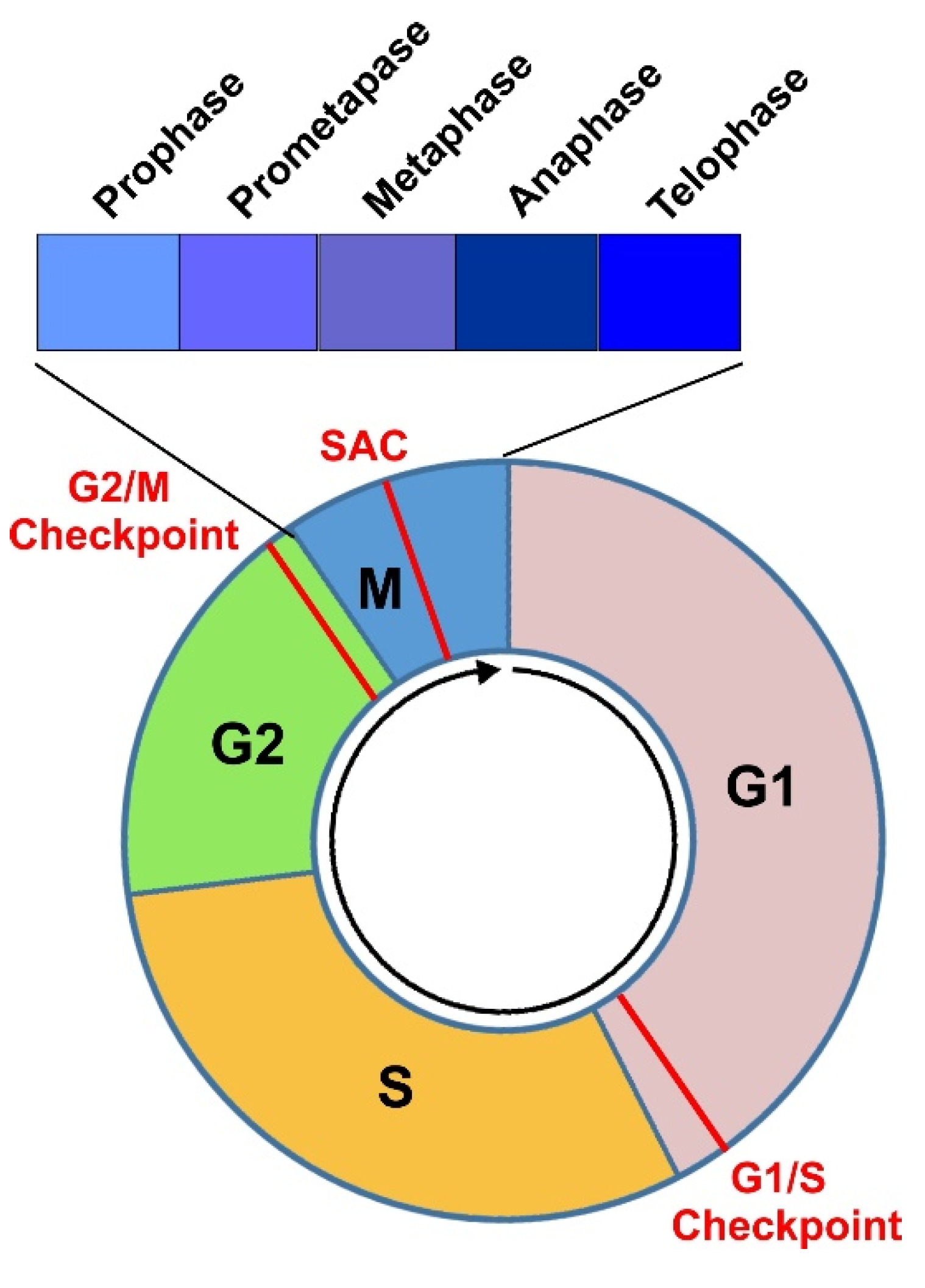
3. Cell Cycle Progression through Various Phases
3.1. G1 Phase
3.2. S Phase
3.3. G2 Phase
3.4. Mitosis and Cytokinesis
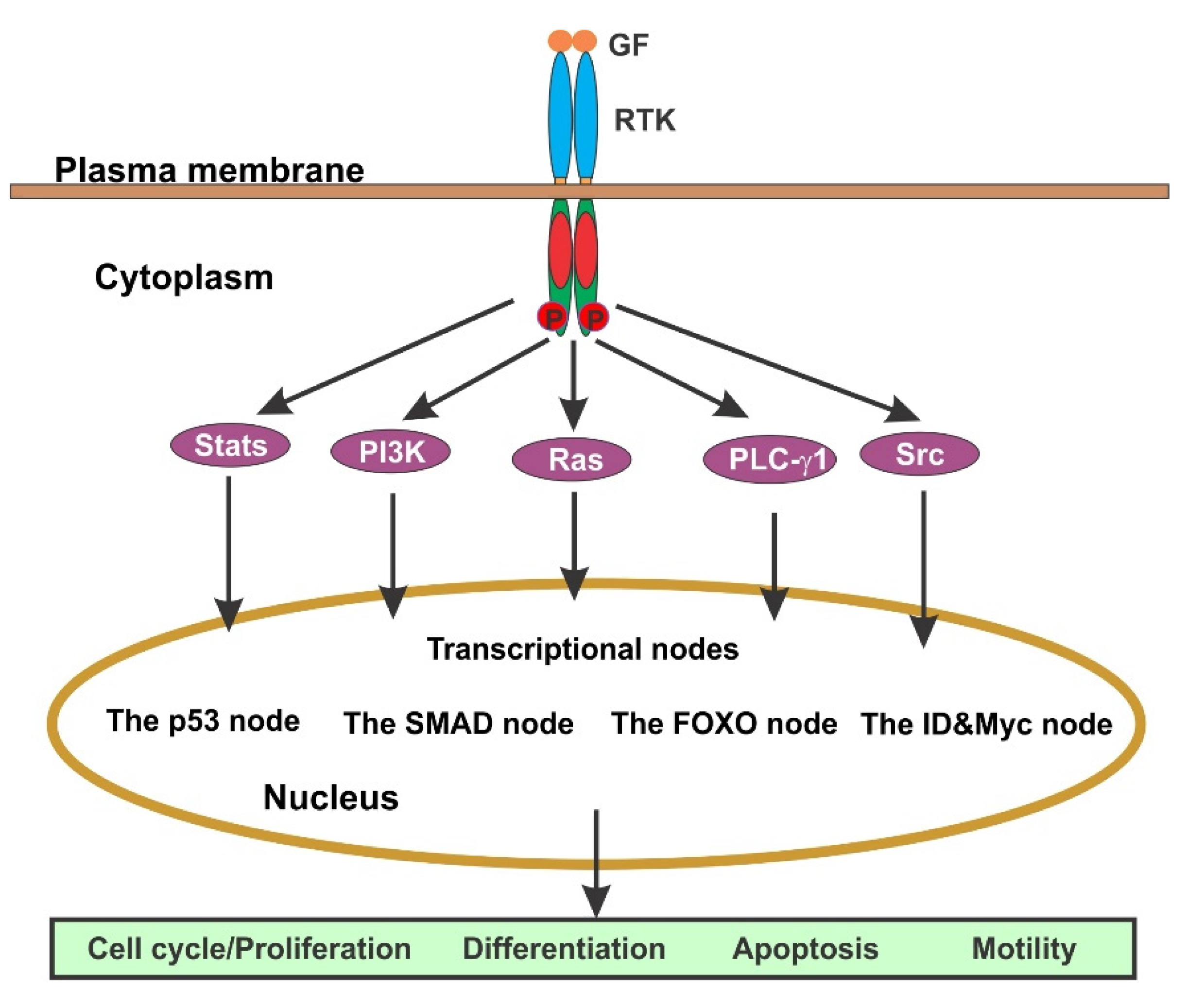
4. The Regulation of Cell Cycle by GF-Initiated Signaling Pathways
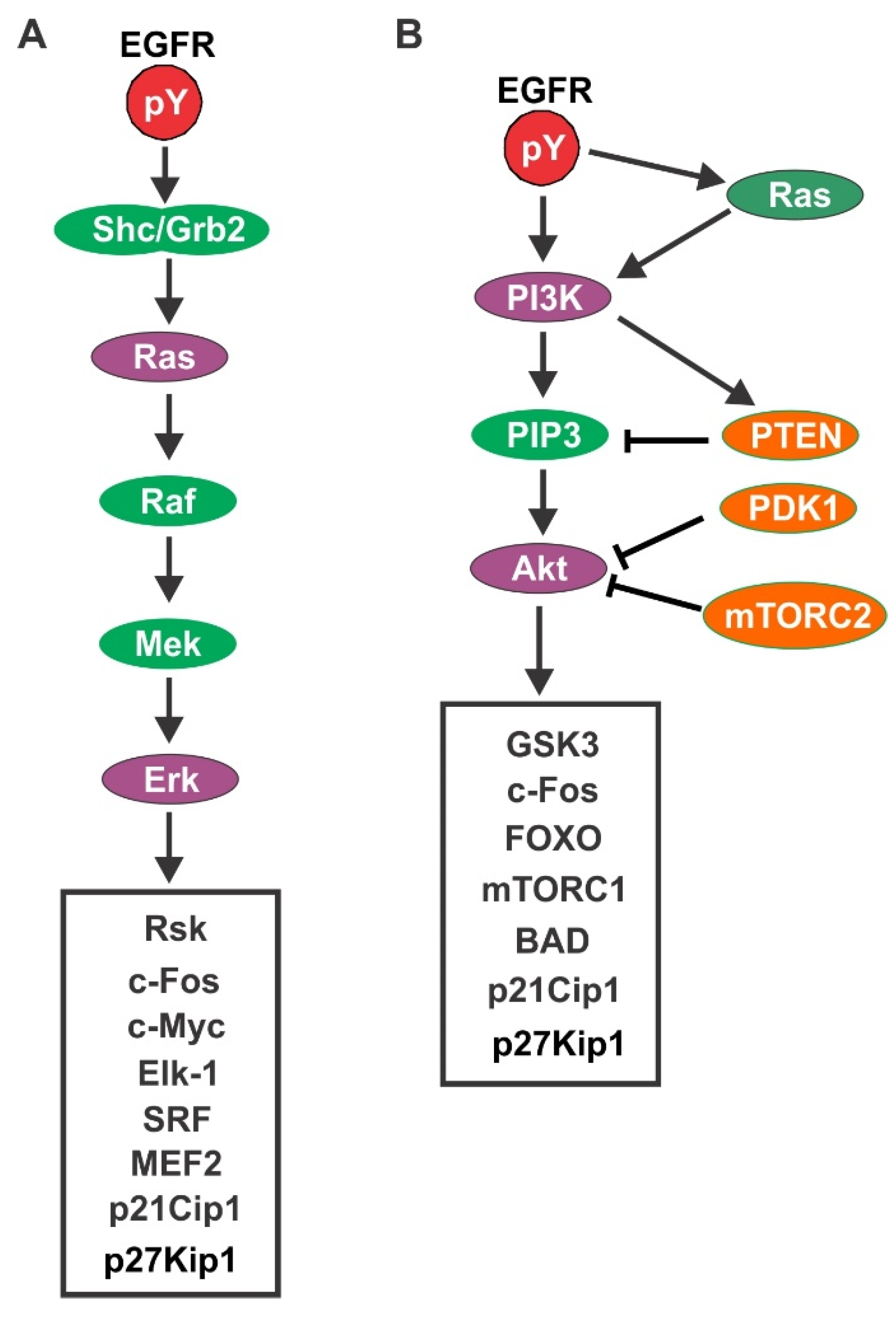
4.1. EGFR-Mediated Signaling Pathways
4.1.1. Ras/Erk Pathway
4.1.2. PI3K/Akt Pathway
4.2. GF-Activated Ras/Erk Pathway Activation in Cell Cycle Progression
4.3. GF-Activated PI3K/Akt Pathway Activation in Cell Cycle Progression
4.4. Co-Regulation of Cell Cycle Progression by PI3K/Akt Pathway and Ras/Erk Pathway
5. Two Waves of GF Signaling Drives Cell Cycle in G1 Phase
6. GF Signaling in S, G2, and M Phase of Cell Cycle
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Panda, S.K.; Ray, S.; Nayak, S.; Behera, S.; Bhanja, S.; Acharya, V. A review on cell cycle checkpoints in relation to cancer. J. Med. Sci. 2019, 5, 88–95. [Google Scholar] [CrossRef]
- Panagopoulos, A.; Altmeyer, M. The hammer and the dance of cell cycle control. Trends Biochem. Sci. 2021, 46, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O′Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Leone, G.W.; Wang, H. Cyclin d-cdk4/6 functions in cancer. Adv. Cancer Res. 2020, 148, 147–169. [Google Scholar] [CrossRef]
- Jones, S.M.; Kazlauskas, A. Connecting signaling and cell cycle progression in growth factor-stimulated cells. Oncogene 2000, 20, 5558–5567. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Cell cycle synchronization of hela cells to assay egfr pathway activation. Methods Mol. Biol. 2017, 1652, 167–181. [Google Scholar] [CrossRef]
- Wolpert, L. Evolution of the cell theory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1995, 349, 227–233. [Google Scholar] [CrossRef]
- Müller-Wille, S. Cell theory, specificity, and reproduction, 1837–1870. Stud. Hist. Philos. Biol. Biomed. Sci. 2010, 41, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Rudolf virchow, the founder of cellular pathology. Rom. J. Morphol. Embryol. 2019, 60, 1381–1382. [Google Scholar]
- Paweletz, N. Walther flemming: Pioneer of mitosis research. Nat. Rev. Mol. Cell Biol. 2001, 2, 72–75. [Google Scholar] [CrossRef]
- Howard, A.; Pelc, S.R. Synthesis of nucleoprotein in bean root cells. Nature 1951, 167, 599–600. [Google Scholar] [CrossRef]
- Nasmyth, K. Viewpoint: Putting the cell cycle in order. Science 1996, 274, 1643–1645. [Google Scholar] [CrossRef]
- Johnson, R.T.; Rao, P.N. Nucleo-cytoplasmic interactions in the acheivement of nuclear synchrony in DNA synthesis and mitosis in multinucleate cells. Biol. Rev. Camb. Philos. Soc. 1971, 46, 97–155. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.N.; Johnson, R.T. Mammalian cell fusion: Studies on the regulation of DNA synthesis and mitosis. Nature 1970, 225, 159–164. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Culotti, J.; Reid, B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. USA 1970, 66, 352–359. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic control of the cell division cycle in yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H. Twenty-five years of cell cycle genetics. Genetics 1991, 129, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P.; Thuriaux, P. Regulatory genes controlling mitosis in the fission yeast schizosaccharomyces pombe. Genetics 1980, 96, 627–637. [Google Scholar] [CrossRef]
- Lee, M.G.; Nurse, P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 1987, 327, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Norbury, C.J.; Spurr, N.K.; Nurse, P. Regulated expression and phosphorylation of a possible mammalian cell-cycle control protein. Nature 1988, 333, 676–679. [Google Scholar] [CrossRef]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mrna in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Pryor, P.R.; Jackson, L.; Gray, S.R.; Edeling, M.A.; Thompson, A.; Sanderson, C.M.; Evans, P.R.; Owen, D.J.; Luzio, J.P. Molecular basis for the sorting of the snare vamp7 into endocytic clathrin-coated vesicles by the arfgap hrb. Cell 2008, 134, 817–827. [Google Scholar] [CrossRef]
- Weinert, T.A.; Hartwell, L.H. The rad9 gene controls the cell cycle response to DNA damage in saccharomyces cerevisiae. Science 1988, 241, 317–322. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Friesen, H.; Andrews, B. Pho85, a multifunctional cyclin-dependent protein kinase in budding yeast. Mol. Microbiol. 2007, 66, 303–314. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Cdk inhibitors: Positive and negative regulators of g1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Dyson, N. The regulation of e2f by prb-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell Biol. 1998, 18, 753–761. [Google Scholar] [CrossRef]
- Petersen, B.O.; Lukas, J.; Sørensen, C.S.; Bartek, J.; Helin, K. Phosphorylation of mammalian cdc6 by cyclin a/cdk2 regulates its subcellular localization. EMBO J. 1999, 18, 396–410. [Google Scholar] [CrossRef]
- Coverley, D.; Pelizon, C.; Trewick, S.; Laskey, R.A. Chromatin-bound cdc6 persists in s and g2 phases in human cells, while soluble cdc6 is destroyed in a cyclin a-cdk2 dependent process. J. Cell Sci. 2000, 113, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Furuno, N.; den Elzen, N.; Pines, J. Human cyclin a is required for mitosis until mid prophase. J. Cell Biol. 1999, 147, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Riabowol, K.; Draetta, G.; Brizuela, L.; Vandre, D.; Beach, D. The cdc2 kinase is a nuclear protein that is essential for mitosis in mammalian cells. Cell 1989, 57, 393–401. [Google Scholar] [CrossRef]
- Duan, L.; Raja, S.M.; Chen, G.; Virmani, S.; Williams, S.H.; Clubb, R.J.; Mukhopadhyay, C.; Rainey, M.A.; Ying, G.; Dimri, M.; et al. Negative regulation of egfr-vav2 signaling axis by cbl ubiquitin ligase controls egf receptor-mediated epithelial cell adherens junction dynamics and cell migration. J. Biol. Chem. 2011, 286, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Limas, J.C.; Cook, J.G. Preparation for DNA replication: The key to a successful s phase. FEBS Lett. 2019, 593, 2853–2867. [Google Scholar] [CrossRef]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef]
- Nelson, D.M.; Ye, X.; Hall, C.; Santos, H.; Ma, T.; Kao, G.D.; Yen, T.J.; Harper, J.W.; Adams, P.D. Coupling of DNA synthesis and histone synthesis in s phase independent of cyclin/cdk2 activity. Mol. Cell Biol. 2002, 22, 7459–7472. [Google Scholar] [CrossRef]
- Ciardo, D.; Goldar, A.; Marheineke, K. On the interplay of the DNA replication program and the intra-s phase checkpoint pathway. Genes 2019, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic s/g(2) checkpoint enforced by atr. Science 2018, 361, 806–810. [Google Scholar] [CrossRef]
- Hannen, R.; Selmansberger, M.; Hauswald, M.; Pagenstecher, A.; Nist, A.; Stiewe, T.; Acker, T.; Carl, B.; Nimsky, C.; Bartsch, J.W. Comparative transcriptomic analysis of temozolomide resistant primary gbm stem-like cells and recurrent gbm identifies up-regulation of the carbonic anhydrase ca2 gene as resistance factor. Cancers 2019, 11, 921. [Google Scholar] [CrossRef]
- Lockhead, S.; Moskaleva, A.; Kamenz, J.; Chen, Y.; Kang, M.; Reddy, A.R.; Santos, S.D.M.; Ferrell, J.E., Jr. The apparent requirement for protein synthesis during g2 phase is due to checkpoint activation. Cell Rep. 2020, 32, 107901. [Google Scholar] [CrossRef] [PubMed]
- Moseley, J.B.; Mayeux, A.; Paoletti, A.; Nurse, P. A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature 2009, 459, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Vukušić, K.; Buđa, R.; Tolić, I.M. Force-generating mechanisms of anaphase in human cells. J. Cell Sci. 2019, 132, jcs231985. [Google Scholar] [CrossRef]
- Afonso, O.; Matos, I.; Pereira, A.J.; Aguiar, P.; Lampson, M.A.; Maiato, H. Feedback control of chromosome separation by a midzone aurora b gradient. Science 2014, 345, 332–336. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A pathway for mitotic chromosome formation. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zickler, D.; Prentiss, M.; Chang, F.S.; Witz, G.; Maeshima, K.; Kleckner, N. Chromosomes progress to metaphase in multiple discrete steps via global compaction/expansion cycles. Cell 2015, 161, 1124–1137. [Google Scholar] [CrossRef]
- Samejima, K.; Samejima, I.; Vagnarelli, P.; Ogawa, H.; Vargiu, G.; Kelly, D.A.; Alves, F.d.; Kerr, A.; Green, L.C.; Hudson, D.F.; et al. Mitotic chromosomes are compacted laterally by kif4 and condensin and axially by topoisomerase iiα. J. Cell Biol. 2012, 199, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Progressive activation of cyclinb1-cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef]
- Santaguida, S.; Musacchio, A. The life and miracles of kinetochores. EMBO J. 2009, 28, 2511–2531. [Google Scholar] [CrossRef]
- Paulson, J.R.; Laemmli, U.K. The structure of histone-depleted metaphase chromosomes. Cell 1977, 12, 817–828. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Mattingly, M.; Gerton, J.L. Regulation of kinetochore configuration during mitosis. Curr. Genet. 2018, 64, 1197–1203. [Google Scholar] [CrossRef]
- Vukušić, K.; Tolić, I.M. Anaphase b: Long-standing models meet new concepts. Semin. Cell Dev. Biol. 2021, 117, 127–139. [Google Scholar] [CrossRef]
- Su, K.C.; Barry, Z.; Schweizer, N.; Maiato, H.; Bathe, M.; Cheeseman, I.M. A regulatory switch alters chromosome motions at the metaphase-to-anaphase transition. Cell Rep. 2016, 17, 1728–1738. [Google Scholar] [CrossRef]
- Green, R.A.; Paluch, E.; Oegema, K. Cytokinesis in animal cells. Annu. Rev. Cell Dev. Biol. 2012, 28, 29–58. [Google Scholar] [CrossRef]
- Fededa, J.P.; Gerlich, D.W. Molecular control of animal cell cytokinesis. Nat. Cell Biol. 2012, 14, 440–447. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef]
- Mierzwa, B.; Gerlich, D.W. Cytokinetic abscission: Molecular mechanisms and temporal control. Dev. Cell 2014, 31, 525–538. [Google Scholar] [CrossRef]
- Gromley, A.; Yeaman, C.; Rosa, J.; Redick, S.; Chen, C.T.; Mirabelle, S.; Guha, M.; Sillibourne, J.; Doxsey, S.J. Centriolin anchoring of exocyst and snare complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell 2005, 123, 75–87. [Google Scholar] [CrossRef]
- Schiel, J.A.; Park, K.; Morphew, M.K.; Reid, E.; Hoenger, A.; Prekeris, R. Endocytic membrane fusion and buckling-induced microtubule severing mediate cell abscission. J. Cell Sci. 2011, 124, 1411–1424. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Wang, Z. Erbb receptors and cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef]
- Carpenter, G.; Lembach, K.J.; Morrison, M.M.; Cohen, S. Characterization of the binding of 125-i-labeled epidermal growth factor to human fibroblasts. J. Biol. Chem. 1975, 250, 4297–4304. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J. Human epidermal growth factor receptor cdna sequence and aberrant expression of the amplified gene in a431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (map) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Avruch, J. Map kinase pathways: The first twenty years. Biochim. Biophys. Acta-Mol. Cell Res. 2007, 1773, 1150–1160. [Google Scholar] [CrossRef]
- Marshall, C.J. Map kinase kinase kinase, map kinase kinase and map kinase. Curr. Opin. Genet. Dev. 1994, 4, 82–89. [Google Scholar] [CrossRef]
- Marshall, C.J. Cell signalling. Raf gets it together. Nature 1996, 383, 127–128. [Google Scholar] [CrossRef]
- Pawson, T. Protein modules and signalling networks. Nature 1995, 373, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wee, P.; Jiang, J.; Chen, X.; Wang, Z. Differential regulation of transcription factors by location-specific egf receptor signaling via a spatio-temporal interplay of erk activation. PLoS ONE 2012, 7, e41354. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 1998, 8, 49–54. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. Ras interaction with pi3k: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Tran, T.T.; Kondo, T.; Luo, J.; Ueki, K.; Cantley, L.C.; Kahn, C.R. Phosphoinositide 3-kinase regulatory subunit p85alpha suppresses insulin action via positive regulation of pten. Proc. Natl. Acad. Sci. USA 2006, 103, 12093–12097. [Google Scholar] [CrossRef]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Anderson, D.H. Direct positive regulation of pten by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 5471–5476. [Google Scholar] [CrossRef]
- Burgering, B.M.; Coffer, P.J. Protein kinase b (c-akt) in phosphatidylinositol-3-oh kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Mechanisms and consequences of activation of protein kinase b/akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Okano, J.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/protein kinase b isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef]
- Nakamura, K.D.; Martinez, R.; Weber, M.J. Tyrosine phosphorylation of specific proteins after mitogen stimulation of chicken embryo fibroblasts. Mol. Cell Biol. 1983, 3, 380–390. [Google Scholar] [CrossRef]
- Pagès, G.; Lenormand, P.; L’Allemain, G.; Chambard, J.C.; Meloche, S.; Pouysségur, J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. USA 1993, 90, 8319–8323. [Google Scholar] [CrossRef]
- Cheng, M.; Sexl, V.; Sherr, C.J.; Roussel, M.F. Assembly of cyclin d-dependent kinase and titration of p27kip1 regulated by mitogen-activated protein kinase kinase (mek1). Proc. Natl. Acad. Sci. USA 1998, 95, 1091–1096. [Google Scholar] [CrossRef]
- Treinies, I.; Paterson, H.F.; Hooper, S.; Wilson, R.; Marshall, C.J. Activated mek stimulates expression of ap-1 components independently of phosphatidylinositol 3-kinase (pi3-kinase) but requires a pi3-kinase signal to stimulate DNA synthesis. Mol. Cell Biol. 1999, 19, 321–329. [Google Scholar] [CrossRef]
- Jones, S.M.; Kazlauskas, A. Growth factor-dependent signaling and cell cycle progression. FEBS Lett. 2001, 490, 110–116. [Google Scholar] [CrossRef]
- Meloche, S.; Pouysségur, J. The erk1/2 mitogen-activated protein kinase pathway as a master regulator of the g1- to s-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef]
- Filmus, J.; Robles, A.I.; Shi, W.; Wong, M.J.; Colombo, L.L.; Conti, C.J. Induction of cyclin d1 overexpression by activated ras. Oncogene 1994, 9, 3627–3633. [Google Scholar] [PubMed]
- Arber, N.; Sutter, T.; Miyake, M.; Kahn, S.M.; Venkatraj, V.S.; Sobrino, A.; Warburton, D.; Holt, P.R.; Weinstein, I.B. Increased expression of cyclin d1 and the rb tumor suppressor gene in c-k-ras transformed rat enterocytes. Oncogene 1996, 12, 1903–1908. [Google Scholar] [PubMed]
- Winston, J.T.; Coats, S.R.; Wang, Y.Z.; Pledger, W.J. Regulation of the cell cycle machinery by oncogenic ras. Oncogene 1996, 12, 127–134. [Google Scholar] [PubMed]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Müller, R.; Pouysségur, J. Cyclin d1 expression is regulated positively by the p42/p44mapk and negatively by the p38/hogmapk pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Cook, S.J. Sustained map kinase activation is required for the expression of cyclin d1, p21cip1 and a subset of ap-1 proteins in ccl39 cells. Oncogene 1999, 18, 3085–3097. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Treisman, R. Transcriptional regulation by extracellular signals: Mechanisms and specificity. Cell 1995, 80, 199–211. [Google Scholar] [CrossRef]
- Strudwick, S.; Borden, K.L. The emerging roles of translation factor eif4e in the nucleus. Differentiation 2002, 70, 10–22. [Google Scholar] [CrossRef]
- Dey, A.; She, H.; Kim, L.; Boruch, A.; Guris, D.L.; Carlberg, K.; Sebti, S.M.; Woodley, D.T.; Imamoto, A.; Li, W. Colony-stimulating factor-1 receptor utilizes multiple signaling pathways to induce cyclin d2 expression. Mol. Biol. Cell 2000, 11, 3835–3848. [Google Scholar] [CrossRef][Green Version]
- Tetsu, O.; McCormick, F. Proliferation of cancer cells despite cdk2 inhibition. Cancer Cell 2003, 3, 233–245. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. C-myc: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple ras-dependent phosphorylation pathways regulate myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of cdk4 as a target of c-myc. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef]
- Coller, H.A.; Grandori, C.; Tamayo, P.; Colbert, T.; Lander, E.S.; Eisenman, R.N.; Golub, T.R. Expression analysis with oligonucleotide microarrays reveals that myc regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3260–3265. [Google Scholar] [CrossRef]
- Galaktionov, K.; Chen, X.; Beach, D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature 1996, 382, 511–517. [Google Scholar] [CrossRef]
- Li, Y.; Jenkins, C.W.; Nichols, M.A.; Xiong, Y. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene 1994, 9, 2261–2268. [Google Scholar]
- Liu, Y.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. Regulation of p21waf1/cip1 expression through mitogen-activated protein kinase signaling pathway. Cancer Res. 1996, 56, 31–35. [Google Scholar] [PubMed]
- Bottazzi, M.E.; Zhu, X.; Böhmer, R.M.; Assoian, R.K. Regulation of p21(cip1) expression by growth factors and the extracellular matrix reveals a role for transient erk activity in g1 phase. J. Cell Biol. 1999, 146, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of cdk inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef]
- Rank, K.B.; Evans, D.B.; Sharma, S.K. The n-terminal domains of cyclin-dependent kinase inhibitory proteins block the phosphorylation of cdk2/cyclin e by the cdk-activating kinase. Biochem. Biophys. Res. Commun. 2000, 271, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Kawada, M.; Yamagoe, S.; Murakami, Y.; Suzuki, K.; Mizuno, S.; Uehara, Y. Induction of p27kip1 degradation and anchorage independence by ras through the map kinase signaling pathway. Oncogene 1997, 15, 629–637. [Google Scholar] [CrossRef]
- Lenferink, A.E.; Simpson, J.F.; Shawver, L.K.; Coffey, R.J.; Forbes, J.T.; Arteaga, C.L. Blockade of the epidermal growth factor receptor tyrosine kinase suppresses tumorigenesis in mmtv/neu + mmtv/tgf-alpha bigenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 9609–9614. [Google Scholar] [CrossRef]
- Gysin, S.; Lee, S.H.; Dean, N.M.; McMahon, M. Pharmacologic inhibition of RAF→MEK→ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27kip1. Cancer Res. 2005, 65, 4870–4880. [Google Scholar] [CrossRef]
- Mirza, A.M.; Gysin, S.; Malek, N.; Nakayama, K.; Roberts, J.M.; McMahon, M. Cooperative regulation of the cell division cycle by the protein kinases raf and akt. Mol. Cell Biol. 2004, 24, 10868–10881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous erk activation downregulates antiproliferative genes throughout g1 phase to allow cell-cycle progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef]
- Sewing, A.; Wiseman, B.; Lloyd, A.C.; Land, H. High-intensity raf signal causes cell cycle arrest mediated by p21cip1. Mol. Cell Biol. 1997, 17, 5588–5597. [Google Scholar] [CrossRef]
- Woods, D.; Parry, D.; Cherwinski, H.; Bosch, E.; Lees, E.; McMahon, M. Raf-induced proliferation or cell cycle arrest is determined by the level of raf activity with arrest mediated by p21cip1. Mol. Cell Biol. 1997, 17, 5598–5611. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoff, E.; Rapp, U.R. High-intensity raf signals convert mitotic cell cycling into cellular growth. Cancer Res. 1998, 58, 1636–1640. [Google Scholar]
- Tombes, R.M.; Auer, K.L.; Mikkelsen, R.; Valerie, K.; Wymann, M.P.; Marshall, C.J.; McMahon, M.; Dent, P. The mitogen-activated protein (map) kinase cascade can either stimulate or inhibit DNA synthesis in primary cultures of rat hepatocytes depending upon whether its activation is acute/phasic or chronic. Biochem. J. 1998, 330, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Valius, M.; Bazenet, C.; Kazlauskas, A. Tyrosines 1021 and 1009 are phosphorylation sites in the carboxy terminus of the platelet-derived growth factor receptor beta subunit and are required for binding of phospholipase c gamma and a 64-kilodalton protein, respectively. Mol.Cell Biol. 1993, 13, 133–143. [Google Scholar] [PubMed]
- Choudhury, G.G.; Karamitsos, C.; Hernandez, J.; Gentilini, A.; Bardgette, J.; Abboud, H.E. Pi-3-kinase and mapk regulate mesangial cell proliferation and migration in response to pdgf. Am. J. Physiol. 1997, 273, 931–938. [Google Scholar] [CrossRef]
- Jones, S.M.; Klinghoffer, R.; Prestwich, G.D.; Toker, A.; Kazlauskas, A. Pdgf induces an early and a late wave of pi 3-kinase activity, and only the late wave is required for progression through g1. Curr. Biol. 1999, 9, 512–521. [Google Scholar] [CrossRef]
- Gille, H.; Downward, J. Multiple ras effector pathways contribute to g(1) cell cycle progression [in process citation]. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef]
- Sun, H.; Lesche, R.; Li, D.M.; Liliental, J.; Zhang, H.; Gao, J.; Gavrilova, N.; Mueller, B.; Liu, X.; Wu, H. Pten modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and akt/protein kinase b signaling pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 6199–6204. [Google Scholar] [CrossRef]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. Afx-like forkhead transcription factors mediate cell-cycle regulation by ras and pkb through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Choudhury, G.G. Akt serine threonine kinase regulates platelet-derived growth factor-induced DNA synthesis in glomerular mesangial cells: Regulation of c-fos and p27(kip1) gene expression. J. Biol. Chem. 2001, 276, 35636–35643. [Google Scholar] [CrossRef]
- Mitsuuchi, Y.; Johnson, S.W.; Selvakumaran, M.; Williams, S.J.; Hamilton, T.C.; Testa, J.R. The phosphatidylinositol 3-kinase/akt signal transduction pathway plays a critical role in the expression of p21waf1/cip1/sdi1 induced by cisplatin and paclitaxel. Cancer Res. 2000, 60, 5390–5394. [Google Scholar]
- Rössig, L.; Jadidi, A.S.; Urbich, C.; Badorff, C.; Zeiher, A.M.; Dimmeler, S. Akt-dependent phosphorylation of p21(cip1) regulates pcna binding and proliferation of endothelial cells. Mol. Cell Biol. 2001, 21, 5644–5657. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dowbenko, D.; Lasky, L.A. Akt/pkb phosphorylation of p21cip/waf1 enhances protein stability of p21cip/waf1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef]
- Hu, Q.; Klippel, A.; Muslin, A.J.; Fantl, W.J.; Williams, L.T. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 1995, 268, 100–102. [Google Scholar] [CrossRef]
- Viglietto, G.; Motti, M.L.; Bruni, P.; Melillo, R.M.; D′Alessio, A.; Califano, D.; Vinci, F.; Chiappetta, G.; Tsichlis, P.; Bellacosa, A.; et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(kip1) by pkb/akt-mediated phosphorylation in breast cancer. Nat. Med. 2002, 8, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. Pkb/akt mediates cell-cycle progression by phosphorylation of p27(kip1) at threonine 157 and modulation of its cellular localization. Nat. Med. 2002, 8, 1145–1152. [Google Scholar] [CrossRef]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. Pkb/akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated g1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin d1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Van Weeren, P.C.; de Bruyn, K.M.; de Vries-Smits, A.M.; van Lint, J.; Burgering, B.M. Essential role for protein kinase b (pkb) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of pkb. J. Biol. Chem. 1998, 273, 13150–13156. [Google Scholar] [CrossRef]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of pi3k/akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef]
- Dong, J.; Peng, J.; Zhang, H.; Mondesire, W.H.; Jian, W.; Mills, G.B.; Hung, M.C.; Meric-Bernstam, F. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005, 65, 1961–1972. [Google Scholar] [CrossRef]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell cycle control by pten. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, pten/mmac1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lin, J.R.; Cimprich, K.A.; Meyer, T. A two-dimensional erk-akt signaling code for an ngf-triggered cell-fate decision. Mol. Cell 2012, 45, 196–209. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The ras-erk and pi3k-mtor pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kodaki, T.; Woscholski, R.; Hallberg, B.; Rodriguez-Viciana, P.; Downward, J.; Parker, P.J. The activation of phosphatidylinositol 3-kinase by ras. Curr. Biol. 1994, 4, 798–806. [Google Scholar] [CrossRef]
- Yu, C.F.; Liu, Z.X.; Cantley, L.G. Erk negatively regulates the epidermal growth factor-mediated interaction of gab1 and the phosphatidylinositol 3-kinase. J. Biol. Chem. 2002, 277, 19382–19388. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo antitumor activity of mek and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef]
- Lehr, S.; Kotzka, J.; Avci, H.; Sickmann, A.; Meyer, H.E.; Herkner, A.; Muller-Wieland, D. Identification of major erk-related phosphorylation sites in gab1. Biochemistry 2004, 43, 12133–12140. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Chen, Z.; Pratap, J. Role of runx2 in crosstalk between mek/erk and pi3k/akt signaling in mcf-10a cells. J. Cell Biochem. 2014, 115, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Zmajkovicova, K.; Jesenberger, V.; Catalanotti, F.; Baumgartner, C.; Reyes, G.; Baccarini, M. Mek1 is required for pten membrane recruitment, akt regulation, and the maintenance of peripheral tolerance. Mol. Cell 2013, 50, 43–55. [Google Scholar] [CrossRef]
- Moelling, K.; Schad, K.; Bosse, M.; Zimmermann, S.; Schweneker, M. Regulation of raf-akt cross-talk. J. Biol. Chem. 2002, 277, 31099–31106. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of raf by akt (protein kinase b). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.; Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Akt3 and mutant v600e b-raf cooperate to promote early melanoma development. Cancer Res. 2008, 68, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef]
- Pledger, W.J.; Stiles, C.D.; Antoniades, H.N.; Scher, C.D. Induction of DNA synthesis in balb/c 3t3 cells by serum components: Reevaluation of the commitment process. Proc. Natl. Acad. Sci. USA 1977, 74, 4481–4485. [Google Scholar] [CrossRef] [PubMed]
- Pledger, W.J.; Stiles, C.D.; Antoniades, H.N.; Scher, C.D. An ordered sequence of events is required before balb/c-3t3 cells become committed to DNA synthesis. Proc. Natl. Acad. Sci. USA 1978, 75, 2839–2843. [Google Scholar] [CrossRef]
- Zwang, Y.; Sas-Chen, A.; Drier, Y.; Shay, T.; Avraham, R.; Lauriola, M.; Shema, E.; Lidor-Nili, E.; Jacob-Hirsch, J.; Amariglio, N.; et al. Two phases of mitogenic signaling unveil roles for p53 and egr1 in elimination of inconsistent growth signals. Mol. Cell 2011, 42, 524–535. [Google Scholar] [CrossRef]
- Gross, S.M.; Rotwein, P. Unraveling growth factor signaling and cell cycle progression in individual fibroblasts. J. Biol. Chem. 2016, 291, 14628–14638. [Google Scholar] [CrossRef]
- Stiles, C.D.; Capone, G.T.; Scher, C.D.; Antoniades, H.N.; van Wyk, J.J.; Pledger, W.J. Dual control of cell growth by somatomedins and platelet-derived growth factor. Proc. Natl. Acad. Sci. USA 1979, 76, 1279–1283. [Google Scholar] [CrossRef]
- Bennett, A.M.; Hausdorff, S.F.; O’Reilly, A.M.; Freeman, R.M.; Neel, B.G. Multiple requirements for shptp2 in epidermal growth factor- mediated cell cycle progression. Mol.Cell Biol. 1996, 16, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Pennock, S.; Wang, Z. Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling. Mol.Cell Biol. 2003, 23, 5803–5815. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Shalloway, D. Cell cycle-dependent activation of ras. Curr. Biol. 1996, 6, 1621–1627. [Google Scholar] [CrossRef]
- Mulcahy, L.S.; Smith, M.R.; Stacey, D.W. Requirement for ras proto-oncogene function during serum- stimulated growth of nih 3t3 cells. Nature 1985, 313, 241–243. [Google Scholar] [CrossRef]
- Dobrowolski, S.; Harter, M.; Stacey, D.W. Cellular ras activity is required for passage through multiple points of the g0/g1 phase in balb/c 3t3 cells. Mol. Cell Biol. 1994, 14, 5441–5449. [Google Scholar] [CrossRef]
- Takuwa, N.; Takuwa, Y. Ras activity late in g1 phase required for p27kip1 downregulation, passage through the restriction point, and entry into s phase in growth factor-stimulated nih 3t3 fibroblasts. Mol. Cell Biol. 1997, 17, 5348–5358. [Google Scholar] [CrossRef]
- Roche, S.; Koegl, M.; Courtneidge, S.A. The phosphatidylinositol 3-kinase alpha is required for DNA synthesis induced by some, but not all, growth factors. Proc. Natl. Acad. Sci. USA 1994, 91, 9185–9189. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, F.; Bacqueville, D.; Pillaire, M.J.; Malecaze, F.; Manenti, S.; Breton-Douillon, M.; Darbon, J.M. G1 phase arrest by the phosphatidylinositol 3-kinase inhibitor ly 294002 is correlated to up-regulation of p27kip1 and inhibition of g1 cdks in choroidal melanoma cells. FEBS Lett. 1998, 422, 385–390. [Google Scholar] [CrossRef]
- Wang, Y.; Pennock, S.; Chen, X.; Wang, Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol.Cell Biol. 2002, 22, 7279–7290. [Google Scholar] [CrossRef]
- Wang, Y.; Pennock, S.D.; Chen, X.; Kazlauskas, A.; Wang, Z. Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J. Biol. Chem. 2004, 279, 8038–8046. [Google Scholar] [CrossRef]
- Vasjari, L.; Bresan, S.; Biskup, C.; Pai, G.; Rubio, I. Ras signals principally via erk in g1 but cooperates with pi3k/akt for cyclin d induction and s-phase entry. Cell Cycle 2019, 18, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.J.; Kim, S.; Lee, M.W.; Lee, B.S.; Hara, T.; Saya, H.; Cho, H.; Lee, J.H. The erk-rsk1 activation by growth factors at g2 phase delays cell cycle progression and reduces mitotic aberrations. Cell Signal. 2008, 20, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.Y.; Nam, H.J.; Lee, J.H. Hepatocyte growth factor at s phase induces g2 delay through sustained erk activation. Biochem. Biophys. Res. Commun. 2007, 356, 300–305. [Google Scholar] [CrossRef]
- Dangi, S.; Chen, F.M.; Shapiro, P. Activation of extracellular signal-regulated kinase (erk) in g(2) phase delays mitotic entry through p21(cip1). Cell Prolif. 2006, 39, 261–279. [Google Scholar] [CrossRef]
- Mardin, B.R.; Isokane, M.; Cosenza, M.R.; Kramer, A.; Ellenberg, J.; Fry, A.M.; Schiebel, E. Egf-induced centrosome separation promotes mitotic progression and cell survival. Dev. Cell 2013, 25, 229–240. [Google Scholar] [CrossRef]
- Astuti, P.; Pike, T.; Widberg, C.; Payne, E.; Harding, A.; Hancock, J.; Gabrielli, B. Mapk pathway activation delays g2/m progression by destabilizing cdc25b. J. Biol. Chem. 2009, 284, 33781–33788. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J. P42-map kinase is activated in egf-stimulated interphase but not in metaphase-arrested hela cells. FEBS Lett. 1999, 443, 126–130. [Google Scholar] [CrossRef]
- Dangi, S.; Shapiro, P. Cdc2-mediated inhibition of epidermal growth factor activation of the extracellular signal-regulated kinase pathway during mitosis. J. Biol. Chem. 2005, 280, 24524–24531. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, N.; Lee, E.K.; Karunagaran, D.; Lin, S.Y.; Hung, M.C. Mitosis-specific negative regulation of epidermal growth factor receptor, triggered by a decrease in ligand binding and dimerization, can be overcome by overexpression of receptor. J. Biol. Chem. 1997, 272, 18656–18665. [Google Scholar] [CrossRef]
- Liu, L.; Shi, H.; Chen, X.; Wang, Z. Regulation of egf-stimulated egf receptor endocytosis during m phase. Traffic 2011, 12, 201–217. [Google Scholar] [CrossRef]
- Wee, P.; Shi, H.; Jiang, J.; Wang, Y.; Wang, Z. Egf stimulates the activation of egf receptors and the selective activation of major signaling pathways during mitosis. Cell Signal. 2015, 27, 638–651. [Google Scholar] [CrossRef]
- Bruns, A.F.; Herbert, S.P.; Odell, A.F.; Jopling, H.M.; Hooper, N.M.; Zachary, I.C.; Walker, J.H.; Ponnambalam, S. Ligand-stimulated vegfr2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 2010, 11, 161–174. [Google Scholar] [CrossRef]
- Walker, F.; Zhang, H.H.; Burgess, A.W. Identification of a novel egf-sensitive cell cycle checkpoint. Exp. Cell Res. 2007, 313, 511–526. [Google Scholar] [CrossRef]
- Hitomi, M.; Stacey, D.W. Cellular ras and cyclin d1 are required during different cell cycle periods in cycling nih 3t3 cells. Mol. Cell Biol. 1999, 19, 4623–4632. [Google Scholar] [CrossRef]
- Stacey, D.W.; Hitomi, M.; Chen, G. Influence of cell cycle and oncogene activity upon topoisomerase iialpha expression and drug toxicity. Mol. Cell Biol. 2000, 20, 9127–9137. [Google Scholar] [CrossRef]
- Hitomi, M.; Stacey, D.W. Ras-dependent cell cycle commitment during g2 phase. FEBS Lett. 2001, 490, 123–131. [Google Scholar] [CrossRef]
- Guo, Y.; Stacey, D.W.; Hitomi, M. Post-transcriptional regulation of cyclin d1 expression during g2 phase. Oncogene 2002, 21, 7545–7556. [Google Scholar] [CrossRef]
- Sa, G.; Hitomi, M.; Harwalkar, J.; Stacey, A.W.; Gc, G.C.; Stacey, D.W. Ras is active throughout the cell cycle, but is able to induce cyclin d1 only during g2 phase. Cell Cycle 2002, 1, 50–58. [Google Scholar] [CrossRef]
- Knauf, J.A.; Ouyang, B.; Knudsen, E.S.; Fukasawa, K.; Babcock, G.; Fagin, J.A. Oncogenic ras induces accelerated transition through g2/m and promotes defects in the g2 DNA damage and mitotic spindle checkpoints. J. Biol. Chem. 2006, 281, 3800–3809. [Google Scholar] [CrossRef]
- Wright, J.H.; Munar, E.; Jameson, D.R.; Andreassen, P.R.; Margolis, R.L.; Seger, R.; Krebs, E.G. Mitogen-activated protein kinase kinase activity is required for the g(2)/m transition of the cell cycle in mammalian fibroblasts. Proc. Natl. Acad. Sci. USA 1999, 96, 11335–11340. [Google Scholar] [CrossRef]
- Rahmouni, S.; Cerignoli, F.; Alonso, A.; Tsutji, T.; Henkens, R.; Zhu, C.; Louis-dit-Sully, C.; Moutschen, M.; Jiang, W.; Mustelin, T. Loss of the vhr dual-specific phosphatase causes cell-cycle arrest and senescence. Nat. Cell Biol. 2006, 8, 524–531. [Google Scholar] [CrossRef]
- Zhang, Z.; Miao, L.; Lv, C.; Sun, H.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone b induces g2/m phase arrest and apoptosis via the ras/raf/mapk signaling pathway in human hepatoma smmc-7721 cells. Cell Death Dis. 2013, 4, e657. [Google Scholar] [CrossRef]
- Kandel, E.S.; Skeen, J.; Majewski, N.; di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of akt/protein kinase b overcomes a g(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef]
- Saito, Y.; Swanson, X.; Mhashilkar, A.M.; Oida, Y.; Schrock, R.; Branch, C.D.; Chada, S.; Zumstein, L.; Ramesh, R. Adenovirus-mediated transfer of the pten gene inhibits human colorectal cancer growth in vitro and in vivo. Gene Ther. 2003, 10, 1961–1969. [Google Scholar] [CrossRef]
- Shtivelman, E.; Sussman, J.; Stokoe, D. A role for pi 3-kinase and pkb activity in the g2/m phase of the cell cycle. Curr. Biol. 2002, 12, 919–924. [Google Scholar] [CrossRef]
- Blasina, A.; de Weyer, I.V.; Laus, M.C.; Luyten, W.H.; Parker, A.E.; McGowan, C.H. A human homologue of the checkpoint kinase cds1 directly inhibits cdc25 phosphatase. Curr. Biol. 1999, 9, 1–10. [Google Scholar] [CrossRef]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of pten sequesters chk1 and initiates genetic instability. Cancer Cell 2005, 7, 193–204. [Google Scholar] [CrossRef]
- Dangi, S.; Cha, H.; Shapiro, P. Requirement for phosphatidylinositol-3 kinase activity during progression through s-phase and entry into mitosis. Cell. Signal. 2003, 15, 667–675. [Google Scholar] [CrossRef]
- Benary, M.; Bohn, S.; Lüthen, M.; Nolis, I.K.; Blüthgen, N.; Loewer, A. Disentangling pro-mitotic signaling during cell cycle progression using time-resolved single-cell imaging. Cell Rep. 2020, 31, 107514. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells 2021, 10, 3327. https://doi.org/10.3390/cells10123327
Wang Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells. 2021; 10(12):3327. https://doi.org/10.3390/cells10123327
Chicago/Turabian StyleWang, Zhixiang. 2021. "Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling" Cells 10, no. 12: 3327. https://doi.org/10.3390/cells10123327
APA StyleWang, Z. (2021). Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells, 10(12), 3327. https://doi.org/10.3390/cells10123327