
Calcium Signaling Mediates Cell Death and Crosstalk with Autophagy in Kidney Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Relationship between Ca2+ Signaling and Various Forms of Cell Death in Kidney Cells
2.1. Ca2+ Mediates Necrosis in Kidney Injury
2.2. Ca2+ Signaling-Mediated Apoptosis Contributes to Kidney Disease
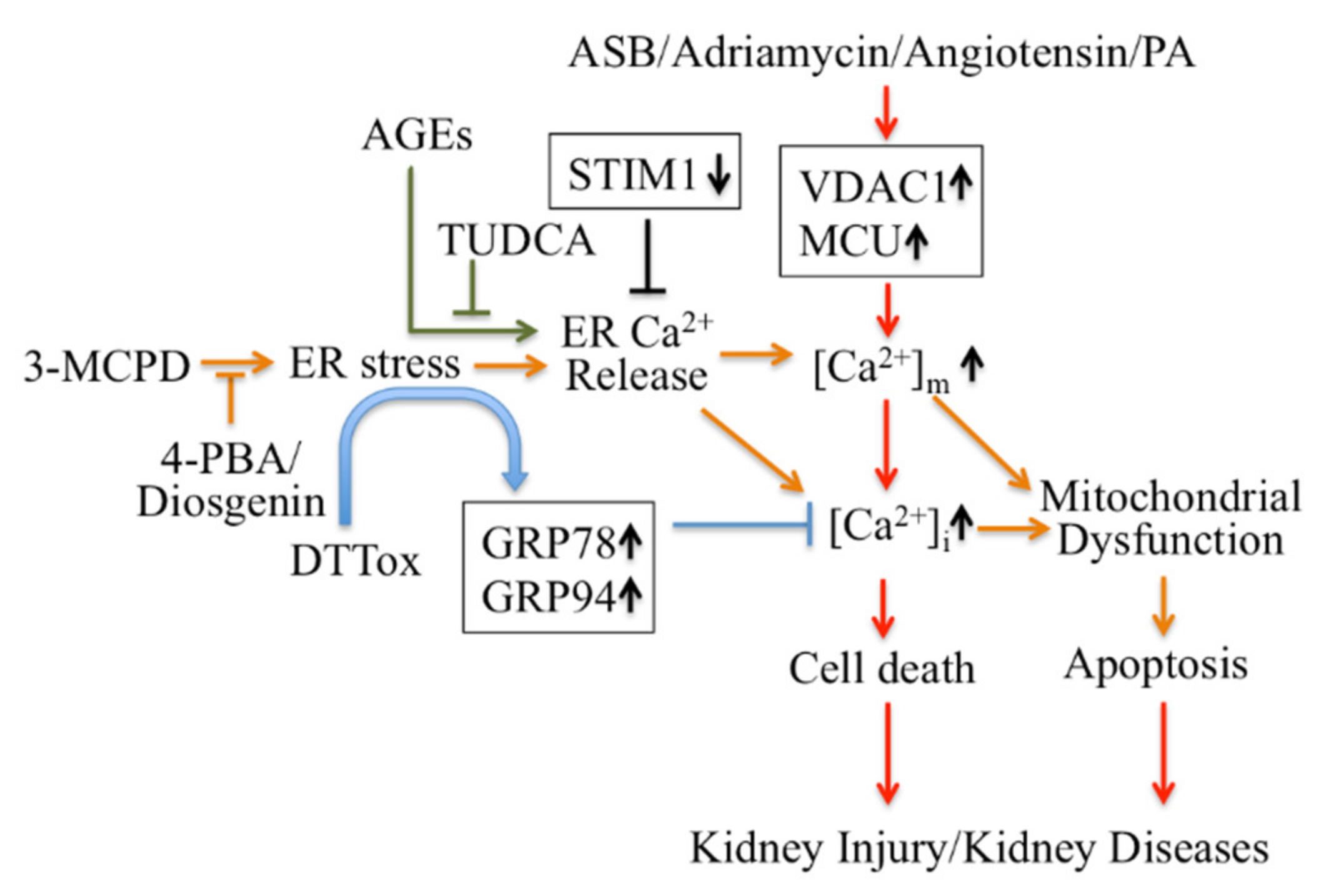
2.2.1. Endoplasmic Reticulum (ER) Ca2+ Signaling Mediates Apoptosis in Kidney Disease
2.2.2. ER-Mitochondrial Ca2+ Signaling Mediates Apoptosis in Kidney Disease
2.3. Ca2+ Mediates Eryptosis in Kidney Injury
2.4. Ca2+ Signaling Regulates Autophagy in Kidney Diseases
2.5. Lysosomal Ca2+ Signaling in Kidney Diseases
3. Ca2+ Signaling Links Cell Death and Autophagy in Kidney Cells
3.1. Induced Autophagy Promotes Cell Death
3.2. Inhibition of Autophagy Promotes against Cell Death
4. Targeted Ca2+ Signaling for Therapy of Kidney Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Pozzan, T.; Rizzuto, R.; Volpe, P.; Meldolesi, J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994, 74, 595–636. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Guerrero-Hernandez, A.; Verkhratsky, A. Calcium signalling in diabetes. Cell Calcium 2014, 56, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca²⁺ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Aromataris, E.; Fernandez, R.; Godfrey, C.M.; Holly, C.; Khalil, H.; Tungpunkom, P. Summarizing systematic reviews: Methodological development, conduct and reporting of an umbrella review approach. Int. J. Evid.-Based Healthc. 2015, 13, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, C.R.; Chen, C.H.; Wang, S.C.; Kuo, S.Y. Effect of methylglyoxal on intracellular calcium levels and viability in renal tubular cells. Cell. Signal. 2005, 17, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Eaddy, A.C.; Cummings, B.S.; McHowat, J.; Schnellmann, R.G. The role of endoplasmic reticulum Ca2+-independent phospholipase a2γ in oxidant-induced lipid peroxidation, Ca2+ release, and renal cell death. Toxicol. Sci. 2012, 128, 544–552. [Google Scholar] [CrossRef] [Green Version]
- De Morais, I.C.; Torres, A.F.; Pereira, G.J.; Pereira, T.P.; De Menezes, R.R.P.B.; Mello, C.P.; Jorge, A.R.C.; Bindá, A.H.; Toyama, M.H.; Monteiro, H.S.; et al. Bothrops leucurus venom induces nephrotoxicity in the isolated perfused kidney and cultured renal tubular epithelia. Toxicon 2013, 61, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.K.; Wu, C.L.; Su, T.C.; Kou, Y.R.; Kor, C.T.; Lee, T.S.; Tarng, D.C. Renal Tubular TRPA1 as a Risk Factor for Recovery of Renal Function from Acute Tubular Necrosis. J. Clin. Med. 2019, 8, 2187. [Google Scholar] [CrossRef] [Green Version]
- Gombedza, F.C.; Shin, S.; Kanaras, Y.L.; Bandyopadhyay, B.C. Abrogation of store-operated Ca(2+) entry protects against crystal-induced ER stress in human proximal tubular cells. Cell Death Discov. 2019, 5, 124. [Google Scholar] [CrossRef] [Green Version]
- Yiu, A.J.; Ibeh, C.L.; Roy, S.K.; Bandyopadhyay, B.C. Melamine induces Ca(2+)-sensing receptor activation and elicits apoptosis in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2017, 313, C27–C41. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Gombedza, F.C.; Bandyopadhyay, B.C. l-ornithine activates Ca(2+) signaling to exert its protective function on human proximal tubular cells. Cell. Signal. 2020, 67, 109484. [Google Scholar] [CrossRef]
- Yamashita, J.; Ogata, M.; Takaoka, M.; Matsumura, Y. KB-R7943, a selective Na+/Ca2+ exchange inhibitor, protects against ischemic acute renal failure in mice by inhibiting renal endothelin-1 overproduction. J. Cardiovasc. Pharmacol. 2001, 37, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yang, D.; Jia, R.; Tan, J. Na+/Ca2+ exchange inhibitor, KB-R7943, attenuates contrast-induced acute kidney injury. J. Nephrol. 2013, 26, 877–885. [Google Scholar] [CrossRef]
- Benesic, A.; Schwerdt, G.; Mildenberger, S.; Freudinger, R.; Gordjani, N.; Gekle, M. Disturbed Ca2+-signaling by chloroacetaldehyde: A possible cause for chronic ifosfamide nephrotoxicity. Kidney Int. 2005, 68, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Pittas, K.; Vrachatis, D.A.; Angelidis, C.; Tsoucala, S.; Giannopoulos, G.; Deftereos, S. The Role of Calcium Handling Mechanisms in Reperfusion Injury. Curr. Pharm. Des. 2018, 24, 4077–4089. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Gava, A.L.; Zhang, D.; Gao, B.; Krepinsky, J.C. Follistatin Protects Against Glomerular Mesangial Cell Apoptosis and Oxidative Stress to Ameliorate Chronic Kidney Disease. Antioxid. Redox Signal. 2019, 31, 551–571. [Google Scholar] [CrossRef]
- Tuffour, A.; Kosiba, A.A.; Zhang, Y.; Peprah, F.A.; Gu, J.; Shi, H. Role of the calcium-sensing receptor (CaSR) in cancer metastasis to bone: Identifying a potential therapeutic target. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188528. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J. Am. Soc. Nephrol. JASN 2018, 29, 1108–1127. [Google Scholar] [CrossRef] [Green Version]
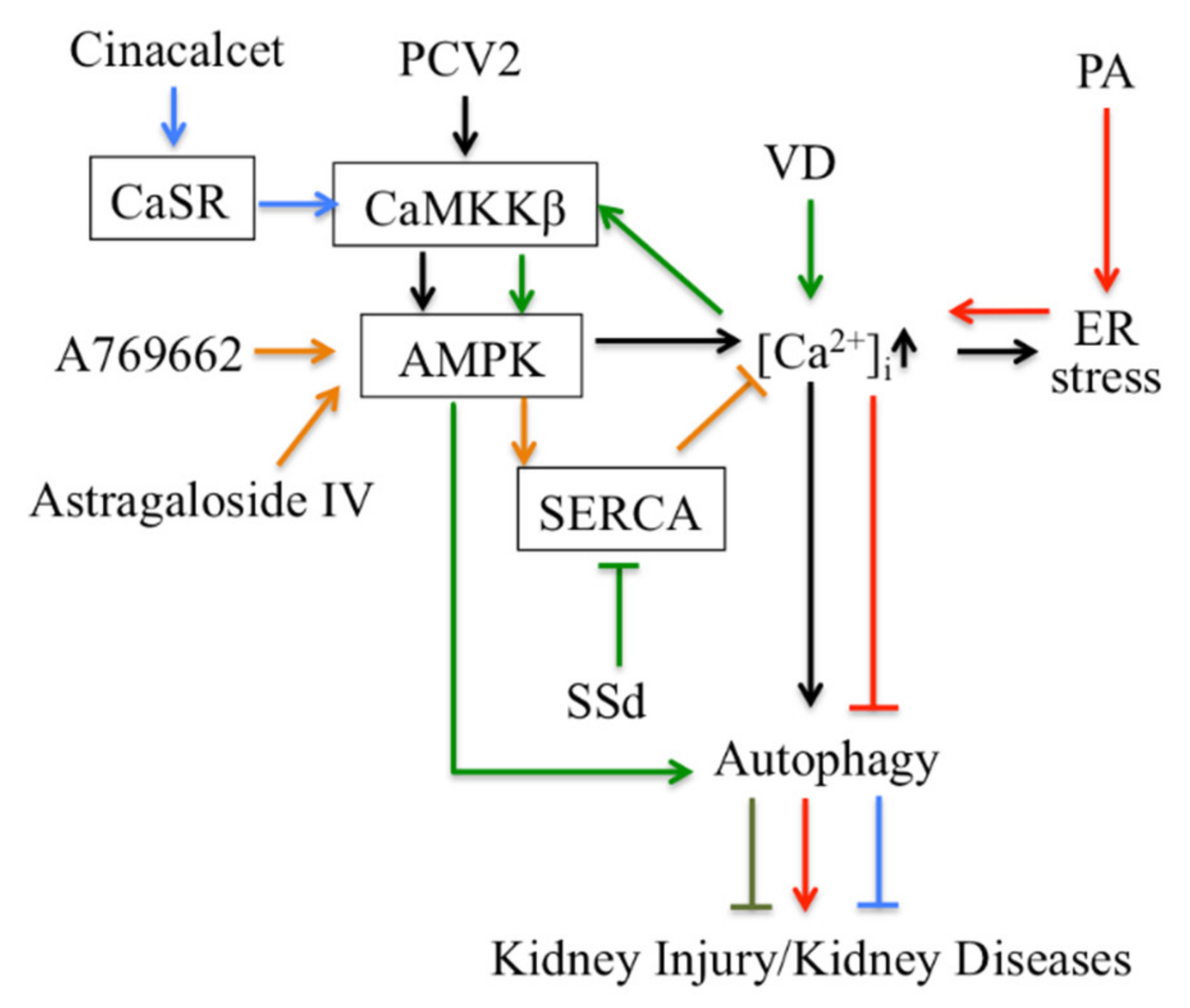
- Lim, J.H.; Kim, H.W.; Kim, M.Y.; Kim, T.W.; Kim, E.N.; Kim, Y.; Chung, S.; Kim, Y.S.; Choi, B.S.; Kim, Y.S.; et al. Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis. 2018, 9, 270. [Google Scholar] [CrossRef]
- Park, S.J.; Li, C.; Chen, Y.M. Endoplasmic Reticulum Calcium Homeostasis in Kidney Disease: Pathogenesis and Therapeutic Targets. Am. J. Pathol. 2021, 191, 256–265. [Google Scholar] [CrossRef]
- Wu, D.; Chen, X.; Ding, R.; Qiao, X.; Shi, S.; Xie, Y.; Hong, Q.; Feng, Z. Ischemia/reperfusion induce renal tubule apoptosis by inositol 1,4,5-trisphosphate receptor and L-type Ca2+ channel opening. Am. J. Nephrol. 2008, 28, 487–499. [Google Scholar] [CrossRef]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Söderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Kim, Y.; Yang, S.M.; Henderson, M.J.; Yang, W.; Lindahl, M.; Urano, F.; Chen, Y.M. Discovery of endoplasmic reticulum calcium stabilizers to rescue ER-stressed podocytes in nephrotic syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 14154–14163. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Cao, A.; Chu, S.; Wang, Y.; Zang, Y.; Mao, X.; Wang, H.; Wang, Y.; Liu, C.; Zhang, X.; et al. Astragaloside IV Attenuates Podocyte Apoptosis Mediated by Endoplasmic Reticulum Stress through Upregulating Sarco/Endoplasmic Reticulum Ca(2+)-ATPase 2 Expression in Diabetic Nephropathy. Front. Pharmacol. 2016, 7, 500. [Google Scholar] [CrossRef] [Green Version]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca(2+) signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12, eaat7397. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Dias, G.F.; Grobe, N.; Rogg, S.; Jörg, D.J.; Pecoits-Filho, R.; Moreno-Amaral, A.N.; Kotanko, P. The Role of Eryptosis in the Pathogenesis of Renal Anemia: Insights From Basic Research and Mathematical Modeling. Front. Cell Dev. Biol. 2020, 8, 598148. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Lupescu, A.; Zbidah, M.; Abed, M.; Shaik, N.; Lang, F. Enhanced apoptotic death of erythrocytes induced by the mycotoxin ochratoxin A. Kidney Blood Press. Res. 2012, 36, 107–118. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Abed, M.; Voelkl, J.; Lang, F. Triggering of suicidal erythrocyte death by uremic toxin indoxyl sulfate. BMC Nephrol. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press. Res. 2013, 37, 158–167. [Google Scholar] [CrossRef]
- Föller, M.; Sopjani, M.; Mahmud, H.; Lang, F. Vanadate-induced suicidal erythrocyte death. Kidney Blood Press. Res. 2008, 31, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Jilani, K.; Bissinger, R.; Rexhepaj, R.; Zelenak, C.; Lupescu, A.; Lang, F.; Qadri, S.M. Vitamin D-Rich Diet in Mice Modulates Erythrocyte Survival. Kidney Blood Press. Res. 2015, 40, 403–412. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, K.; Kong, A.; Zhou, Y.; Chen, D.; Gu, J.; Shi, H. Dysregulation of autophagy acts as a pathogenic mechanism of non-alcoholic fatty liver disease (NAFLD) induced by common environmental pollutants. Ecotoxicol. Environ. Saf. 2021, 217, 112256. [Google Scholar] [CrossRef] [PubMed]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef]
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 2014, 34, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Di Mise, A.; Tamma, G.; Ranieri, M.; Centrone, M.; van den Heuvel, L.; Mekahli, D.; Levtchenko, E.N.; Valenti, G. Activation of Calcium-Sensing Receptor increases intracellular calcium and decreases cAMP and mTOR in PKD1 deficient cells. Sci. Rep. 2018, 8, 5704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Xiao, H.; Zhang, Y.; Zeng, X.; Huang, M.; Chen, X.; Birnbaumer, L.; Liao, Y. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018, 9, 1015. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Huang, M.; Zeng, X.; Zhang, Y.; Sun, A.; Wu, Q.; Zhu, L.; Zhao, H.; Liao, Y. The Role of TRPC6 in Renal Ischemia/Reperfusion and Cellular Hypoxia/Reoxygenation Injuries. Front. Mol. Biosci. 2021, 8, 698975. [Google Scholar] [CrossRef]
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J.; et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 2014, 26, 738–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Wu, D.; Zhao, L.; Zou, W.; Shen, W.; Tu, Q.; He, Q. Effect of autophagy and stromal interaction molecule 1 on podocyte epithelial-mesenchymal transition in diabetic nephropathy. Int. J. Clin. Exp. Pathol. 2018, 11, 2450–2459. [Google Scholar]
- Boletta, A. Emerging evidence of a link between the polycystins and the mTOR pathways. PathoGenetics 2009, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef] [Green Version]
- Yanda, M.K.; Liu, Q.; Cebotaru, V.; Guggino, W.B.; Cebotaru, L. Role of calcium in adult onset polycystic kidney disease. Cell. Signal. 2019, 53, 140–150. [Google Scholar] [CrossRef]
- Peña-Oyarzun, D.; Rodriguez-Peña, M.; Burgos-Bravo, F.; Vergara, A.; Kretschmar, C.; Sotomayor-Flores, C.; Ramirez-Sarmiento, C.A.; De Smedt, H.; Reyes, M.; Perez, W.; et al. PKD2/polycystin-2 induces autophagy by forming a complex with BECN1. Autophagy 2021, 17, 1714–1728. [Google Scholar] [CrossRef]
- Pays, E. The function of apolipoproteins L (APOLs): Relevance for kidney disease, neurotransmission disorders, cancer and viral infection. FEBS J. 2021, 288, 360–381. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Saido, T.C.; Sorimachi, H. Calpain research for drug discovery: Challenges and potential. Nat. Rev. Drug Discov. 2016, 15, 854–876. [Google Scholar] [CrossRef] [PubMed]
- Peintner, L.; Venkatraman, A.; Waeldin, A.; Hofherr, A.; Busch, T.; Voronov, A.; Viau, A.; Kuehn, E.W.; Köttgen, M.; Borner, C. Loss of PKD1/polycystin-1 impairs lysosomal activity in a CAPN (calpain)-dependent manner. Autophagy 2021, 17, 2384–2400. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Elmonem, M.A.; Bongaerts, I.; Luyten, T.; Missiaen, L.; van den Heuvel, L.P.; Levtchenko, E.N.; Bultynck, G. Ca(2+) signalling in human proximal tubular epithelial cells deficient for cystinosin. Cell Calcium 2016, 60, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Yang, W.; Sun, L. Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Prospective Roles in Kidney Disease. Oxidative Med. Cell. Longev. 2020, 2020, 3120539. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Z.K.; Wang, Z.Y.; Yang, D.B.; Liu, Z.P.; Wang, L. Mitochondrial permeability transition and its regulatory components are implicated in apoptosis of primary cultures of rat proximal tubular cells exposed to lead. Arch. Toxicol. 2016, 90, 1193–1209. [Google Scholar] [CrossRef]
- Song, X.B.; Liu, G.; Liu, F.; Yan, Z.G.; Wang, Z.Y.; Liu, Z.P.; Wang, L. Autophagy blockade and lysosomal membrane permeabilization contribute to lead-induced nephrotoxicity in primary rat proximal tubular cells. Cell Death Dis. 2017, 8, e2863. [Google Scholar] [CrossRef]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP(3) Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca(2+) to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef] [Green Version]
- López-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Forster, C.; Kane, P.M. Cytosolic Ca2+ homeostasis is a constitutive function of the V-ATPase in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 38245–38253. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.K.; Probst, S.; Santoyo-Sánchez, M.P.; Al-Hamdani, W.; Diebels, I.; von Sivers, J.K.; Kerek, E.; Prenner, E.J.; Thévenod, F. Initial autophagic protection switches to disruption of autophagic flux by lysosomal instability during cadmium stress accrual in renal NRK-52E cells. Arch. Toxicol. 2017, 91, 3225–3245. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, X.; Dai, S.; Liu, H.; Yan, D.; Zhou, Y.; Gu, J.; Shi, H. Cadmium induced redistribution of cholesterol by upregulating ABCA1 and downregulating OSBP. J. Inorg. Biochem. 2018, 189, 199–207. [Google Scholar] [CrossRef]
- Kong, A.; Zhang, Y.; Ning, B.; Li, K.; Ren, Z.; Dai, S.; Chen, D.; Zhou, Y.; Gu, J.; Shi, H. Cadmium induces triglyceride levels via microsomal triglyceride transfer protein (MTTP) accumulation caused by lysosomal deacidification regulated by endoplasmic reticulum (ER) Ca(2+) homeostasis. Chem.-Biol. Interact. 2021, 348, 109649. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, G.H.; Bhattarai, K.R.; Lee, M.S.; Back, S.H.; Kim, H.R.; Chae, H.J. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy through regulation of lysosomal calcium. Autophagy 2021, 17, 761–778. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.Y.; Fan, R.F.; Yang, D.B.; Zhang, D.; Wang, L. Puerarin reverses cadmium-induced lysosomal dysfunction in primary rat proximal tubular cells via inhibiting Nrf2 pathway. Biochem. Pharmacol. 2019, 162, 132–141. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Z.F.; Zhang, D.; Wang, Z.Y.; Wang, L. Quercetin alleviates Cadmium-induced autophagy inhibition via TFEB-dependent lysosomal restoration in primary proximal tubular cells. Ecotoxicol. Environ. Saf. 2021, 208, 111743. [Google Scholar] [CrossRef]
- Fan, R.F.; Tang, K.K.; Wang, Z.Y.; Wang, L. Persistent activation of Nrf2 promotes a vicious cycle of oxidative stress and autophagy inhibition in cadmium-induced kidney injury. Toxicology 2021, 464, 152999. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Huang, D.; Bhat, O.M.; Poklis, J.L.; Zhang, A.; Zou, Y.; Kidd, J.; Gehr, T.W.B.; Li, P.L. Abnormal podocyte TRPML1 channel activity and exosome release in mice with podocyte-specific Asah1 gene deletion. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158856. [Google Scholar] [CrossRef]
- Smaili, S.S.; Pereira, G.J.; Costa, M.M.; Rocha, K.K.; Rodrigues, L.; do Carmo, L.G.; Hirata, H.; Hsu, Y.T. The role of calcium stores in apoptosis and autophagy. Curr. Mol. Med. 2013, 13, 252–265. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, T.; Sun, L.; Luo, Y.; Liu, D.H.; Xie, S.T.; Song, X.Y.; Wang, G.F.; Chen, X.L.; Zhou, B.C.; et al. Calpain, Atg5 and Bak play important roles in the crosstalk between apoptosis and autophagy induced by influx of extracellular calcium. Apoptosis Int. J. Program. Cell Death 2013, 18, 435–451. [Google Scholar] [CrossRef]
- Mammano, F.; Bortolozzi, M. Ca(2+) signaling, apoptosis and autophagy in the developing cochlea: Milestones to hearing acquisition. Cell Calcium 2018, 70, 117–126. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, W.; Shi, H.; Hou, Y.; Xu, Q. Calcium homeostasis disruption—A bridge connecting cadmium-induced apoptosis, autophagy and tumorigenesis. Oncol. Res. Treat. 2015, 38, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Kosiba, A.A.; Wang, Y.; Chen, D.; Wong, C.K.C.; Gu, J.; Shi, H. The roles of calcium-sensing receptor (CaSR) in heavy metals-induced nephrotoxicity. Life Sci. 2020, 242, 117183. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J.B. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 2011, 1813, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacquier, B.; Combettes, L.; Van Nhieu, G.T.; Dupont, G. Interplay Between Intracellular Ca(2+) Oscillations and Ca(2+)-stimulated Mitochondrial Metabolism. Sci. Rep. 2016, 6, 19316. [Google Scholar] [CrossRef]
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [Green Version]
- Pi, H.; Xu, S.; Reiter, R.J.; Guo, P.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy 2015, 11, 1037–1051. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Wang, Y.; Liu, Y.; Shi, M.; Yin, L.; Hou, Y.; Zhou, Y.; Wong, C.K.C.; Chen, D.; Guo, Z.; et al. Inhibition of Autophagy Alleviates Cadmium-Induced Mouse Spleen and Human B Cells Apoptosis. Toxicol. Sci. 2019, 170, 109–122. [Google Scholar] [CrossRef]
- So, K.Y.; Lee, B.H.; Oh, S.H. The critical role of autophagy in cadmium-induced immunosuppression regulated by endoplasmic reticulum stress-mediated calpain activation in RAW264.7 mouse monocytes. Toxicology 2018, 393, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.M.; Allen, D.A.; Raftery, M.J.; Yaqoob, M.M. High glucose initiates calpain-induced necrosis before apoptosis in LLC-PK1 cells. Kidney Int. 2007, 71, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. life Sci. CMLS 2008, 65, 3640–3652. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, H.; Liu, Y.; Ahmadi, B.; Templeton, D.M. Protective effect of cadmium-induced autophagy in rat renal mesangial cells. Arch. Toxicol. 2018, 92, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.X.; Zhu, H.L.; Shi, X.T.; Nan, Y.; Liu, W.B.; Dai, L.M.; Xiong, Y.W.; Yi, S.J.; Cao, X.L.; Xu, D.X.; et al. Autophagy in Sertoli cell protects against environmental cadmium-induced germ cell apoptosis in mouse testes. Environ. Pollut. 2021, 270, 116241. [Google Scholar] [CrossRef]
- Zhu, H.L.; Xu, X.F.; Shi, X.T.; Feng, Y.J.; Xiong, Y.W.; Nan, Y.; Zhang, C.; Gao, L.; Chen, Y.H.; Xu, D.X.; et al. Activation of autophagy inhibits cadmium-triggered apoptosis in human placental trophoblasts and mouse placenta. Environ. Pollut. 2019, 254, 112991. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Sun, X.; Kong, A.; Ma, H.; Xie, Y.; Cheng, D.; Wong, C.K.C.; Zhou, Y.; Gu, J. Cadmium induces epithelial-mesenchymal transition and migration of renal cancer cells by increasing PGE2 through a cAMP/PKA-COX2 dependent mechanism. Ecotoxicol. Environ. Saf. 2021, 207, 111480. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Jiang, T.; Yuan, X.; Ren, Z.; Tuffour, A.; Liu, H.; Zhou, Y.; Gu, J.; Shi, H. Nephrotoxicity Profile of Cadmium Revealed by Proteomics in Mouse Kidney. Biol. Trace Elem. Res. 2021, 199, 1929–1940. [Google Scholar] [CrossRef]
- Gu, J.; Ren, Z.; Zhao, J.; Peprah, F.A.; Xie, Y.; Cheng, D.; Wang, Y.; Liu, H.; Chu Wong, C.K.; Zhou, Y.; et al. Calcimimetic compound NPS R-467 protects against chronic cadmium-induced mouse kidney injury by restoring autophagy process. Ecotoxicol. Environ. Saf. 2020, 189, 110052. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Dai, S.; Liu, Y.; Liu, H.; Zhang, Y.; Ji, X.; Yu, F.; Zhou, Y.; Chen, L.; Tse, W.K.F.; et al. Activation of Ca(2+)-sensing receptor as a protective pathway to reduce Cadmium-induced cytotoxicity in renal proximal tubular cells. Sci. Rep. 2018, 8, 1092. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.X.; Fan, R.F.; Lin, S.Q.; Yang, D.B.; Wang, Z.Y.; Wang, L. Interplay between autophagy and apoptosis in lead(II)-induced cytotoxicity of primary rat proximal tubular cells. J. Inorg. Biochem. 2018, 182, 184–193. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.K.; Jiao, P.; Zhou, X.P.; Yang, D.B.; Wang, Z.Y.; Wang, L. Redistribution of subcellular calcium and its effect on apoptosis in primary cultures of rat proximal tubular cells exposed to lead. Toxicology 2015, 333, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, Z.F.; Wang, Z.Y.; Wang, L. Role of subcellular calcium redistribution in regulating apoptosis and autophagy in cadmium-exposed primary rat proximal tubular cells. J. Inorg. Biochem. 2016, 164, 99–109. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.Y.; Zhou, X.P.; Liu, Z.P.; Song, X.B.; Wang, Z.Y.; Wang, L. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca(2+)-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 2017, 383, 13–23. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yang, H.; Wang, M.G.; Yang, D.B.; Wang, Z.Y.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef]
- Liu, H.; Bowes, R.C., 3rd; van de Water, B.; Sillence, C.; Nagelkerke, J.F.; Stevens, J.L. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 1997, 272, 21751–21759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmellash, S.; Stevens, J.L.; Ichimura, T. Modulating the endoplasmic reticulum stress response with trans-4,5-dihydroxy-1,2-dithiane prevents chemically induced renal injury in vivo. Toxicol. Sci. 2005, 88, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, C.P.; Xu, K.F.; Mao, X.D.; Lu, Y.B.; Fang, L.; Yang, J.W.; Liu, C. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am. J. Nephrol. 2008, 28, 1014–1022. [Google Scholar] [CrossRef]
- Zhong, Y.; Jin, C.; Han, J.; Zhu, J.; Liu, Q.; Sun, D.; Xia, X.; Peng, X. Inhibition of ER stress attenuates kidney injury and apoptosis induced by 3-MCPD via regulating mitochondrial fission/fusion and Ca(2+) homeostasis. Cell Biol. Toxicol. 2021, 37, 795–809. [Google Scholar] [CrossRef]
- Zhong, Y.; Jin, C.; Han, J.; Zhu, J.; Liu, Q.; Sun, D.; Xia, X.; Zhang, Y.; Peng, X. Diosgenin Protects Against Kidney Injury and Mitochondrial Apoptosis Induced by 3-MCPD Through the Regulation of ER Stress, Ca(2+) Homeostasis, and Bcl2 Expression. Mol. Nutr. Food Res. 2021, 65, e2001202. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, R.; Geng, S.; Shan, Y.; Li, X.; Fang, W. Porcine Circovirus Type 2 Induces ORF3-Independent Mitochondrial Apoptosis via PERK Activation and Elevation of Cytosolic Calcium. J. Virol. 2019, 93, e01784-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, W.M.; Yuan, T.J.; Xu, J.D.; Gu, L.L.; Liang, P.; Lu, H. Proteomic identification of mitochondrial targets involved in andrographolide sodium bisulfite-induced nephrotoxicity in a rat model. Environ. Toxicol. Pharmacol. 2015, 40, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cao, A.; Liu, H.; Guo, H.; Zang, Y.; Wang, Y.; Wang, Y.; Wang, H.; Yin, P.; Peng, W. Calcium Uptake via Mitochondrial Uniporter Contributes to Palmitic Acid-Induced Apoptosis in Mouse Podocytes. J. Cell. Biochem. 2017, 118, 2809–2818. [Google Scholar] [CrossRef]
- Zang, Y.; Liu, S.; Cao, A.; Shan, X.; Deng, W.; Li, Z.; Wang, H.; Wang, Y.; Wang, L.; Peng, W. Astragaloside IV inhibits palmitic acid-induced apoptosis through regulation of calcium homeostasis in mice podocytes. Mol. Biol. Rep. 2021, 48, 1453–1464. [Google Scholar] [CrossRef]
- Kong, Y.; Zhao, X.; Qiu, M.; Lin, Y.; Feng, P.; Li, S.; Liang, B.; Zhu, Q.; Huang, H.; Li, C.; et al. Tubular Mas receptor mediates lipid-induced kidney injury. Cell Death Dis. 2021, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP(3)R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Gu, Y.; Qi, B.; Zhou, Y.; Jiang, X.; Zhang, X.; Li, X.; Fang, W. Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium. Viruses 2016, 8, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Yi, B.; Han, H.; Yang, S.; Hu, Z.; Zheng, L.; Wang, J.; Liao, Q.; Zhang, H. Vitamin D-VDR (vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy 2021, 1–14. [Google Scholar] [CrossRef]
- Rodríguez, C.; Contreras, C.; Sáenz-Medina, J.; Muñoz, M.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernandez, M.; López, M.; Rivera, L.; et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates Nox-derived reactive oxygen species (ROS) generation. Redox Biol. 2020, 34, 101575. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Y.; Zhang, X.; Zang, Y.; Zhang, Y.; Wang, L.; Wang, H.; Wang, Y.; Cao, A.; Peng, W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKα-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci. Rep. 2017, 7, 6852. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J.; et al. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKβ-AMPK-mTOR pathway in ADPKD cells. Mol. Cell. Biochem. 2018, 449, 219–226. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, B.; Guo, C.; Kong, A.; Li, K.; Xie, Y.; Shi, H.; Gu, J. Calcium Signaling Mediates Cell Death and Crosstalk with Autophagy in Kidney Disease. Cells 2021, 10, 3204. https://doi.org/10.3390/cells10113204
Ning B, Guo C, Kong A, Li K, Xie Y, Shi H, Gu J. Calcium Signaling Mediates Cell Death and Crosstalk with Autophagy in Kidney Disease. Cells. 2021; 10(11):3204. https://doi.org/10.3390/cells10113204
Chicago/Turabian StyleNing, Bo, Chuanzhi Guo, Anqi Kong, Kongdong Li, Yimin Xie, Haifeng Shi, and Jie Gu. 2021. "Calcium Signaling Mediates Cell Death and Crosstalk with Autophagy in Kidney Disease" Cells 10, no. 11: 3204. https://doi.org/10.3390/cells10113204
APA StyleNing, B., Guo, C., Kong, A., Li, K., Xie, Y., Shi, H., & Gu, J. (2021). Calcium Signaling Mediates Cell Death and Crosstalk with Autophagy in Kidney Disease. Cells, 10(11), 3204. https://doi.org/10.3390/cells10113204