
The Pathophysiology of Farnesoid X Receptor (FXR) in the GI Tract: Inflammation, Barrier Function and Innate Immunity

Abstract
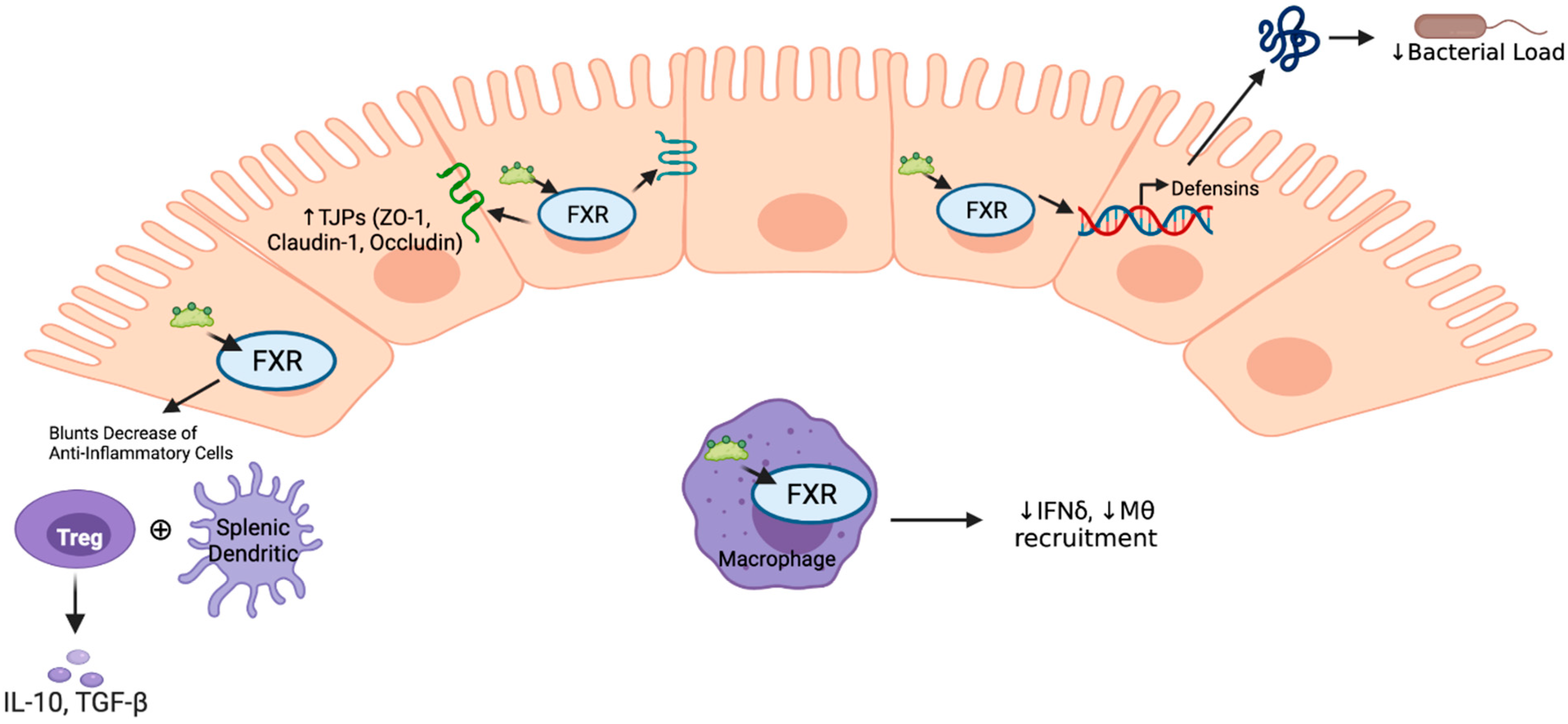
:1. Introduction
2. Inflammation
2.1. Acute Inflammation
2.2. Chronic Inflammation
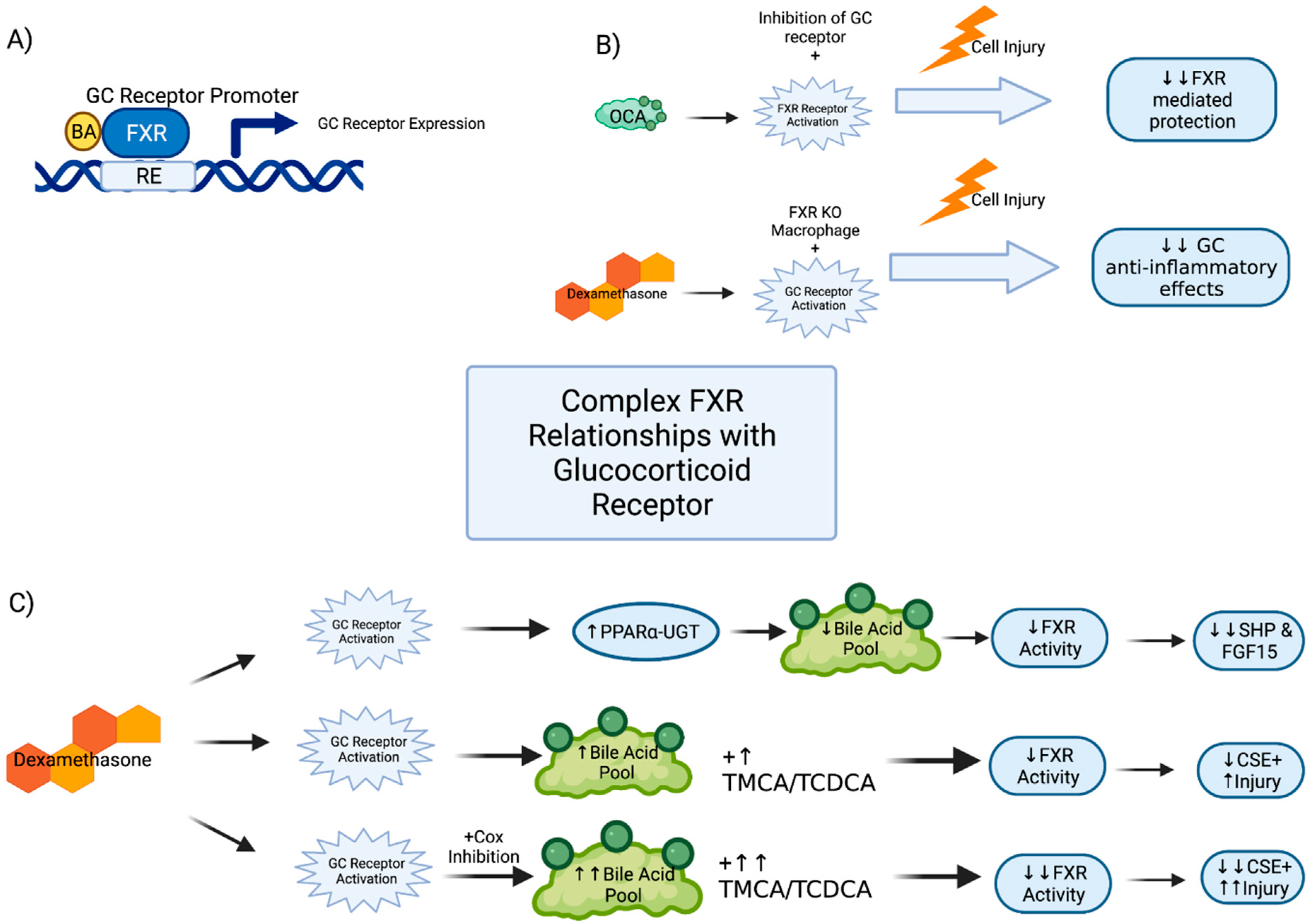
2.3. Glucocorticoid Receptor Interactions
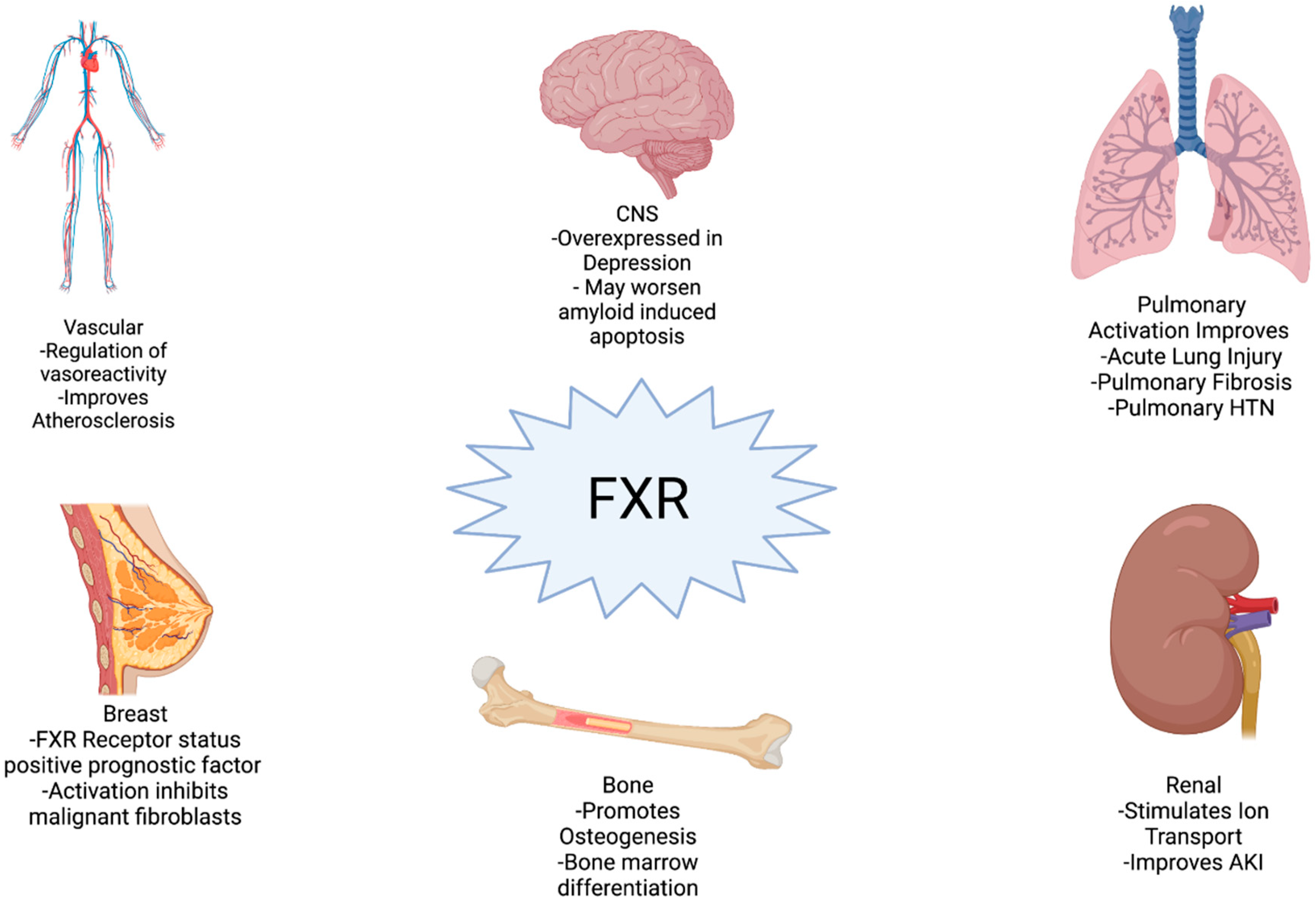
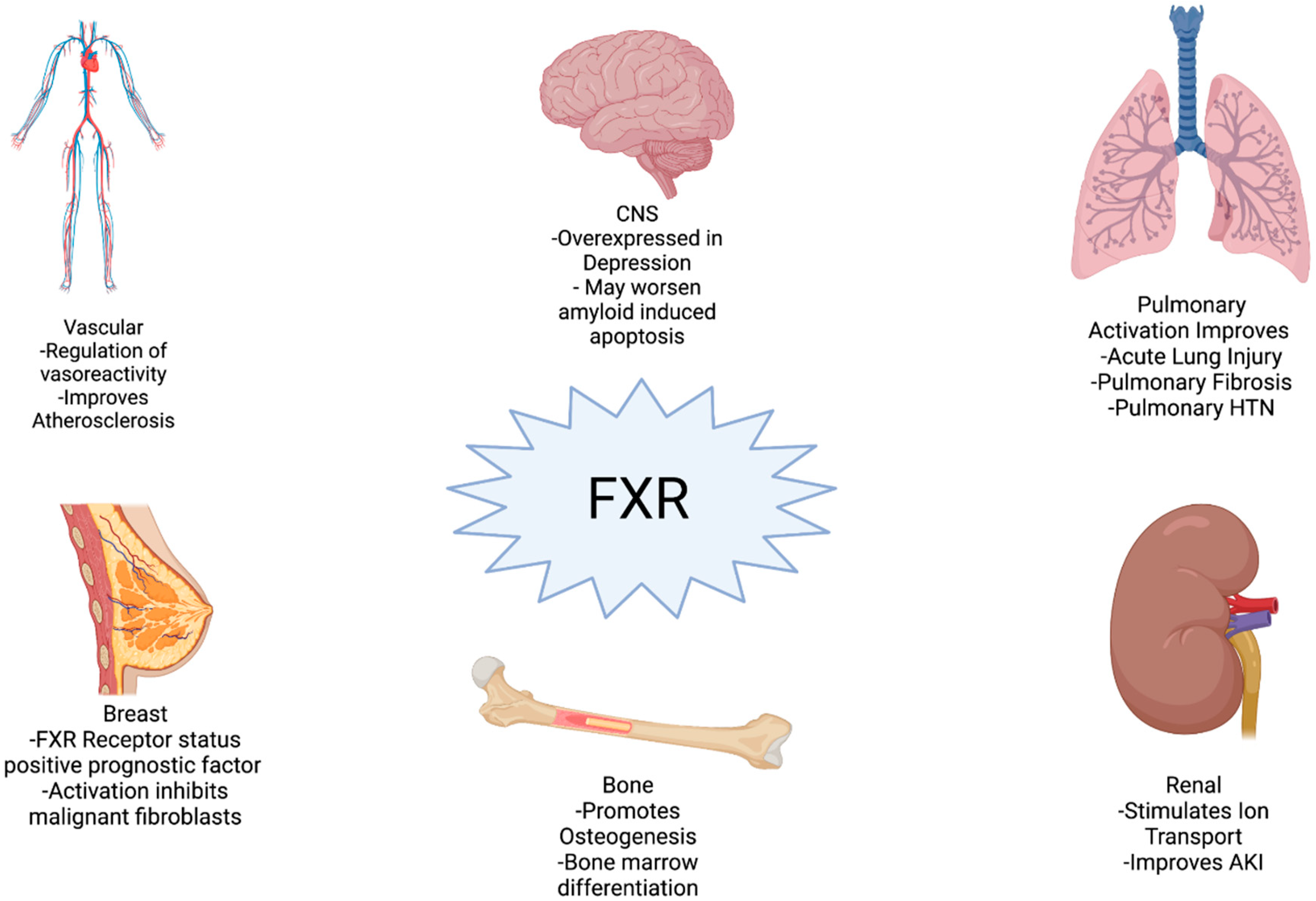
2.4. Clinical Implications
3. Barrier Function
3.1. Histology
3.2. Functional and Homeostatic Effects
3.3. Tight Junctional Proteins
3.4. Proliferative Effects
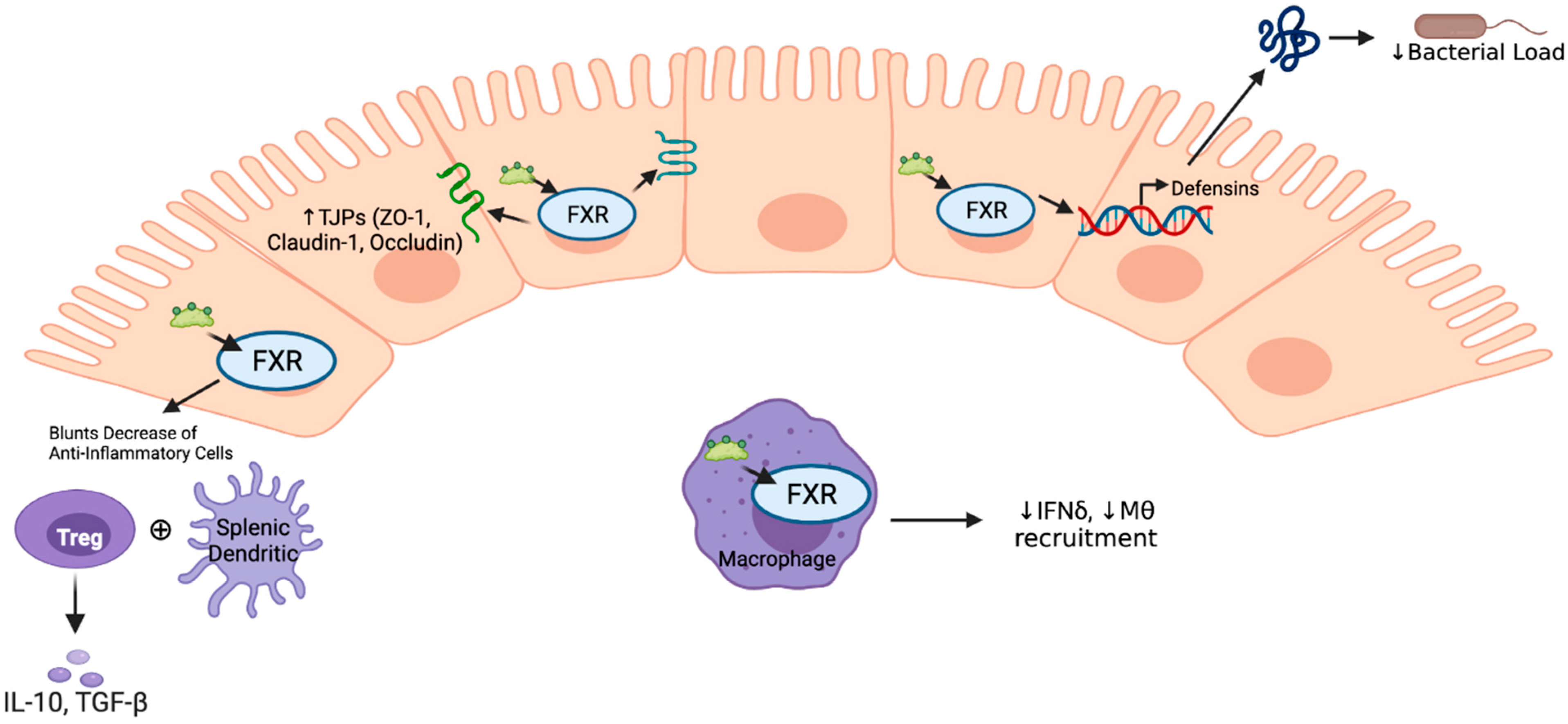
4. Immune Response
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X Receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Hollman, D.A.; Milona, A.; van Erpecum, K.J.; van Mil, S.W. Anti-inflammatory and metabolic actions of FXR: Insights into molecular mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 1443–1452. [Google Scholar] [CrossRef]
- Grober, J.; Zaghini, I.; Fujii, H.; Jones, S.A.; Kliewer, S.A.; Willson, T.M.; Ono, T.; Besnard, P. Identification of a Bile Acid-responsive Element in the Human Ileal Bile Acid-binding Protein Gene. J. Biol. Chem. 1999, 274, 29749–29754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, S.; Gadaleta, R.M.; Moschetta, A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl. Recept. Signal. 2010, 8, e005. [Google Scholar] [CrossRef] [Green Version]
- Otte, K.; Kranz, H.; Kober, I.; Thompson, P.; Hoefer, M.; Haubold, B.; Remmel, B.; Voss, H.; Kaiser, C.; Albers, M.; et al. Identification of Farnesoid X Receptor beta as a novel mammalian nuclear receptor sensing lanosterol. Mol. Cell. Biol. 2003, 23, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.M.; Murphy, K.; Miao, B.; Link, J.R.; Cunningham, M.R.; Rupar, M.J.; Gunyuzlu, P.L.; Haws, T.F.; Kassam, A.; Powell, F.; et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 2002, 290, 35–43. [Google Scholar] [CrossRef]
- Zhang, Y.; Kast-Woelbern, H.R.; Edwards, P.A. Natural Structural Variants of the Nuclear Receptor Farnesoid X Receptor Affect Transcriptional Activation. J. Biol. Chem. 2003, 278, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, G.; Passeri, D.; De Franco, F.; Ciaccioli, G.; Donadio, L.; Rizzo, G.; Orlandi, S.; Sadeghpour, B.; Wang, X.X.; Jiang, T.; et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual Farnesoid X Receptor and TGR5 agonist. Mol. Pharmacol. 2010, 78, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non–Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; van Mil, S.W. Progress and challenges of selective Farnesoid X Receptor modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Gege, C.; Hambruch, E.; Hambruch, N.; Kinzel, O.; Kremoser, C. Nonsteroidal FXR Ligands: Current Status and Clinical Applications. Handb. Exp. Pharm. 2019, 256, 167–205. [Google Scholar] [CrossRef]
- Schaap, F.; Trauner, M.; Jansen, P.L.M. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef]
- Shaik, F.B.; Prasad, D.V.R.; Narala, V.R. Role of Farnesoid X Receptor in inflammation and resolution. Inflamm. Res. 2014, 64, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, H.; Zhang, M.; Guo, G.L. Fatty liver diseases, bile acids, and FXR. Acta Pharm. Sin. B 2016, 6, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, D.; Giaginis, C.; Tsourouflis, G.; Theocharis, S. Farnesoid-X Receptor (FXR) as a Promising Pharmaceutical Target in Atherosclerosis. Curr. Med. Chem. 2017, 24, 1147–1157. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-B.; Xu, S.; Li, J.; Song, J.; Luo, B.; Song, Y.-P.; Zhang, Z.-H.; Chen, Y.-H.; Xie, D.-D.; Yu, D.-X.; et al. Farnesoid X Receptor agonist obeticholic acid inhibits renal inflammation and oxidative stress during lipopolysaccharide-induced acute kidney injury. Eur. J. Pharmacol. 2018, 838, 60–68. [Google Scholar] [CrossRef]
- Fei, J.; Fu, L.; Hu, B.; Chen, Y.-H.; Zhao, H.; Xu, D.-X.; Li, J.-B. Obeticholic acid alleviate lipopolysaccharide-induced acute lung injury via its anti-inflammatory effects in mice. Int. Immunopharmacol. 2019, 66, 177–184. [Google Scholar] [CrossRef]
- Giaginis, C.; Karandrea, D.; Alexandrou, P.; Giannopoulou, I.; Tsourouflis, G.; Troungos, C.; Danas, E.; Keramopoulos, A.; Patsouris, E.; Nakopoulou, L.; et al. High Farnesoid X Receptor (FXR) expression is a strong and independent prognosticator in invasive breast carcinoma. Neoplasma 2017, 64, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Ran, H.; Peng, L.; Zhang, Y.; Shen, W.; Sun, T.; Cao, F.; Chen, Y. Farnesoid X Receptor regulates vasoreactivity via Angiotensin II type 2 receptor and the kallikrein-kinin system in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 327–334. [Google Scholar] [CrossRef]
- Chen, W.-G.; Zheng, J.-X.; Xu, X.; Hu, Y.-M.; Ma, Y.-M. Hippocampal FXR plays a role in the pathogenesis of depression: A preliminary study based on lentiviral gene modulation. Psychiatry Res. 2018, 264, 374–379. [Google Scholar] [CrossRef]
- Huang, C.; Wang, J.; Hu, W.; Wang, C.; Lu, X.; Tong, L.; Wu, F.; Zhang, W. Identification of functional Farnesoid X Receptors in brain neurons. FEBS Lett. 2016, 590, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ma, H.; Guo, X.; Liu, J.; Gui, T.; Gai, Z. Farnesoid X Receptor (FXR) Aggravates Amyloid-β-Triggered Apoptosis by Modulating the cAMP-Response Element-Binding Protein (CREB)/Brain-Derived Neurotrophic Factor (BDNF) Pathway In Vitro. Med. Sci. Monit. 2019, 25, 9335–9345. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef] [PubMed]
- Stojancevic, M.; Stankov, K.; Mikov, M. The Impact of Farnesoid X Receptor Activation on Intestinal Permeability in Inflammatory Bowel Disease. Can. J. Gastroenterol. 2012, 26, 631–637. [Google Scholar] [CrossRef]
- Mazuy, C.; Helleboid, A.; Staels, B.; Lefebvre, P. Nuclear bile acid signaling through the Farnesoid X Receptor. Cell. Mol. Life Sci. 2014, 72, 1631–1650. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; An, J.H.; Park, H.; Yang, J.-Y.; Choi, H.J.; Kim, S.W.; Park, Y.J.; Kim, S.Y.; Yim, M.; Baek, W.-Y.; et al. Positive regulation of osteogenesis by bile acid through FXR. J. Bone Miner. Res. 2013, 28, 2109–2121. [Google Scholar] [CrossRef]
- Giordano, C.; Barone, I.; Vircillo, V.; Panza, S.; Malivindi, R.; Gelsomino, L.; Pellegrino, M.; Rago, V.; Mauro, L.; Lanzino, M.; et al. Activated FXR Inhibits Leptin Signaling and Counteracts Tumor-promoting Activities of Cancer-Associated Fibroblasts in Breast Malignancy. Sci. Rep. 2016, 6, 21782. [Google Scholar] [CrossRef]
- Mencarelli, A.; Fiorucci, S. FXR an emerging therapeutic target for the treatment of atherosclerosis. J. Cell. Mol. Med. 2010, 14, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, H.-M.; Zang, M.; Zhang, Q.; Shi, R.-B.; Shi, X.-J.; Mamtilahun, M.; Liu, C.; Luo, L.-L.; Tian, X.; Zhang, Z.; et al. Farnesoid X Receptor knockout protects brain against ischemic injury through reducing neuronal apoptosis in mice. J. Neuroinflamm. 2020, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Wongwan, T.; Chatsudthipong, V.; Soodvilai, S. Farnesoid X Receptor Activation Stimulates Organic Cations Transport in Human Renal Proximal Tubular Cells. Int. J. Mol. Sci. 2020, 21, 6078. [Google Scholar] [CrossRef]
- Comeglio, P.; Filippi, S.; Sarchielli, E.; Morelli, A.; Cellai, I.; Corno, C.; Adorini, L.; Vannelli, G.B.; Maggi, M.; Vignozzi, L. Therapeutic effects of the selective Farnesoid X Receptor agonist obeticholic acid in a monocrotaline-induced pulmonary hypertension rat model. J. Endocrinol. Investig. 2019, 42, 951–965. [Google Scholar] [CrossRef]
- Comeglio, P.; Filippi, S.; Sarchielli, E.; Morelli, A.; Cellai, I.; Corcetto, F.; Corno, C.; Maneschi, E.; Pini, A.; Adorini, L.; et al. Anti-fibrotic effects of chronic treatment with the selective FXR agonist obeticholic acid in the bleomycin-induced rat model of pulmonary fibrosis. J. Steroid Biochem. Mol. Biol. 2017, 168, 26–37. [Google Scholar] [CrossRef]
- Massafra, V.; Ijssennagger, N.; Plantinga, M.; Milona, A.; Pittol, J.M.R.; Boes, M.; van Mil, S.W. Splenic dendritic cell involvement in FXR-mediated amelioration of DSS colitis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, M.; Lario, M.; Muñoz, L.; Borrero, M.-J.; Serrano, E.M.R.; Sánchez-Díaz, A.M.; del Campo, R.; Lledo, L.; Pastor, O.; García-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef]
- Ceulemans, L.J.; Verbeke, L.; Decuypere, J.-P.; Farre, R.; De Hertogh, G.; Lenaerts, K.; Jochmans, I.; Monbaliu, D.; Nevens, F.; Tack, J.; et al. Farnesoid X Receptor Activation Attenuates Intestinal Ischemia Reperfusion Injury in Rats. PLoS ONE 2017, 12, e0169331. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, S.; Chen, M.; Liu, J.; Dong, R.; Wang, H.; Zhu, S. Activation of the Nuclear Receptor Fxr Improves Intestinal Cell Tolerance to Ischemia–Reperfusion Injury. Shock 2018, 50, 316–323. [Google Scholar] [CrossRef]
- Fiorucci, S.; Mencarelli, A.; Cipriani, S.; Renga, B.; Palladino, G.; Santucci, L.; Distrutti, E. Activation of the Farnesoid-X Receptor protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs in mice. Br. J. Pharmacol. 2011, 164, 1929–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renga, B.; Mencarelli, A.; Migliorati, M.; Distrutti, E.; Fiorucci, S. Bile-acid-activated Farnesoid X Receptor regulates hydrogen sulfide production and hepatic microcirculation. World J. Gastroenterol. 2009, 15, 2097–2108. [Google Scholar] [CrossRef]
- Horikawa, T.; Oshima, T.; Li, M.; Kitayama, Y.; Eda, H.; Nakamura, K.; Tamura, A.; Ogawa, T.; Yamasaki, T.; Okugawa, T.; et al. Chenodeoxycholic Acid Releases Proinflammatory Cytokines from Small Intestinal Epithelial Cells Through the Farnesoid X Receptor. Digestion 2019, 100, 286–294. [Google Scholar] [CrossRef]
- Kim, M.S.; Shigenaga, J.; Moser, A.; Feingold, K.; Grunfeld, C. Repression of Farnesoid X Receptor during the Acute Phase Response. J. Biol. Chem. 2003, 278, 8988–8995. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.; Siersema, P.D.; Van Erpecum, K.J.; Van Mil, S.W. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim. Biophys. Acta 2011, 1812, 851–858. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, L.; Jiang, C.; Xie, Y.; Cheng, X.; Krausz, K.W.; Qi, Y.; Sun, L.; Shah, Y.M.; Gonzalez, F.J.; et al. PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat. Commun. 2014, 5, 4573. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.; Jiang, Y.; Thymann, T.; Sangild, P.; Maj, M.; Manjarin, R.; Burrin, D. Rapid Postnatal Upregulation of Intestinal Farnesoid X Receptor-Fibroblast Growth Factor 19 Signaling in Premature Pigs. J. Pediatr. Gastroenterol. Nutr. 2020, 70, e94–e99. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xiang, X.; Li, Y.; Ji, R.; Xu, H.; Mai, K.; Ai, Q. Molecular cloning and characterization of Farnesoid X Receptor from large yellow croaker (Larimichthys crocea) and the effect of dietary CDCA on the expression of inflammatory genes in intestine and spleen. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2018, 216, 10–17. [Google Scholar] [CrossRef]
- Xu, M.; Cen, M.; Shen, Y.; Zhu, Y.; Cheng, F.; Tang, L.; Hu, W.; Dai, N. Deoxycholic Acid-Induced Gut Dysbiosis Disrupts Bile Acid Enterohepatic Circulation and Promotes Intestinal Inflammation. Dig. Dis. Sci. 2020, 66, 568–576. [Google Scholar] [CrossRef]
- Maran, R.R.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X Receptor Deficiency in Mice Leads to Increased Intestinal Epithelial Cell Proliferation and Tumor Development. J. Pharmacol. Exp. Ther. 2008, 328, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [Green Version]
- Renga, B.; D’Amore, C.; Cipriani, S.; Mencarelli, A.; Carino, A.; Sepe, V.; Zampella, A.; Distrutti, E.; Fiorucci, S. FXR mediates a chromatin looping in the GR promoter thus promoting the resolution of colitis in rodents. Pharmacol. Res. 2013, 77, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Cariello, M.; Scialpi, N.; Peres, C.; Vetrano, S.; Fiorino, G.; Danese, S.; Ko, B.; Luo, J.; et al. Fibroblast Growth Factor 19 modulates intestinal microbiota and inflammation in presence of Farnesoid X Receptor. EBioMedicine 2020, 54, 102719. [Google Scholar] [CrossRef]
- Rau, M.; Stieger, B.; Monte, M.J.; Schmitt, J.; Jahn, D.; Frey-Wagner, I.; Raselli, T.; Marin, J.J.G.; Müllhaupt, B.; Rogler, G.; et al. Alterations in Enterohepatic Fgf15 Signaling and Changes in Bile Acid Composition Depend on Localization of Murine Intestinal Inflammation. Inflamm. Bowel Dis. 2016, 22, 2382–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adcock, I.M.; Mumby, S. Glucocorticoids. Handb. Exp. Pharmacol. 2017, 237, 171–196. [Google Scholar]
- Lu, Z.; Lu, Y.; Wang, X.; Wang, F.; Zhang, Y. Activation of intestinal GR-FXR and PPARα-UGT signaling exacerbates ibuprofen-induced enteropathy in mice. Arch. Toxicol. 2018, 92, 1249–1265. [Google Scholar] [CrossRef]
- Torres, J.; Palmela, C.; De Sena, P.G.; Santos, M.P.C.; Gouveia, C.; Oliveira, M.H.; Henriques, A.R.; Rodrigues, C.; Cravo, M.; Borralho, P. Farnesoid X Receptor Expression in Microscopic Colitis: A Potential Role in Disease Etiopathogenesis. GE-Port. J. Gastroenterol. 2018, 25, 30–37. [Google Scholar] [CrossRef]
- Nijmeijer, R.M.; Gadaleta, R.M.; van Mil, S.W.C.; van Bodegraven, A.A.; Crusius, J.B.A.; Dijkstra, G.; Hommes, D.W.; de Jong, D.J.; Stokkers, P.C.F.; Verspaget, H.W.; et al. Farnesoid X Receptor (FXR) Activation and FXR Genetic Variation in Inflammatory Bowel Disease. PLoS ONE 2011, 6, e23745. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Almousa, A.; Teft, W.A.; Kim, R.B. Attenuation of bile acid-mediated FXR and PXR activation in patients with Crohn’s disease. Sci. Rep. 2020, 10, 1866. [Google Scholar] [CrossRef] [PubMed]
- Negroni, A.; Fiaschini, N.; Palone, F.; Vitali, R.; Colantoni, E.; Laudadio, I.; Oliva, S.; Aloi, M.; Cucchiara, S.; Stronati, L. Intestinal Inflammation Alters the Expression of Hepatic Bile Acid Receptors Causing Liver Impairment. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, T.; Mullish, B.H.; Patterson, J.; Wong, G.K.; Marchesi, J.R.; Xu, H.; Jilani, T.; Kao, D. Effective fecal microbiota transplantation for recurrent Clostridioides difficile infection in humans is associated with increased signalling in the bile acid-Farnesoid X Receptor-fibroblast growth factor pathway. Gut Microbes 2019, 10, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Winston, J.A.; Rivera, A.J.; Cai, J.; Thanissery, R.; Montgomery, S.A.; Patterson, A.D.; Theriot, C.M. Ursodeoxycholic Acid (UDCA) Mitigates the Host Inflammatory Response during Clostridioides difficile Infection by Altering Gut Bile Acids. Infect. Immun. 2020, 88, e00045-20. [Google Scholar] [CrossRef] [PubMed]
- Attinkara, R.; Mwinyi, J.; Truninger, K.; Regula, J.; Gaj, P.; Rogler, G.; Kullak-Ublick, G.A.; Eloranta, J.J. The Swiss IBD Cohort Study Group Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor FXR, with inflammatory bowel disease. BMC Res. Notes 2012, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Wang, Q.; Almousa, A.A.; Jansen, L.E.; Choi, Y.-H.; Schwarz, U.I.; Kim, R.B. Genetic variation in the farnesoid X-receptor predicts Crohn’s disease severity in female patients. Sci. Rep. 2020, 10, 11725. [Google Scholar] [CrossRef]
- Zahiri, H.R.; Perrone, E.E.; Strauch, E.D. Bile salt supplementation acts via the Farnesoid X Receptor to alleviate lipopolysaccharide-induced intestinal injury. Surgery 2011, 150, 480–489. [Google Scholar] [CrossRef]
- Liu, H.-M.; Liao, J.-F.; Lee, T.-Y. Farnesoid X Receptor agonist GW4064 ameliorates lipopolysaccharide-induced ileocolitis through TLR4/MyD88 pathway related mitochondrial dysfunction in mice. Biochem. Biophys. Res. Commun. 2017, 490, 841–848. [Google Scholar] [CrossRef]
- Park, P.O.; Haglund, U.; Bulkley, G.B.; Fält, K. The sequence of development of intestinal tissue injury after strangulation ischemia and reperfusion. Surgery 1990, 107, 574–580. [Google Scholar]
- Chiu, C.-J.; McArdle, A.H.; Brown, R.; Scott, H.J.; Gurd, F.N. Intestinal Mucosal Lesion in Low-Flow States. Arch. Surg. 1970, 101, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Ye, J.; Zhang, F.; Su, H.; Yang, X.; He, H.; Liu, F.; Zhu, X.; Wang, L.; Gao, P.; et al. Chenodeoxycholic Acid (CDCA) Protects against the Lipopolysaccharide-Induced Impairment of the Intestinal Epithelial Barrier Function via the FXR-MLCK Pathway. J. Agric. Food Chem. 2019, 67, 8868–8874. [Google Scholar] [CrossRef] [PubMed]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Mroz, M.S.; Lajczak, N.K.; Goggins, B.J.; Keely, S.; Keely, S.J. The bile acids, deoxycholic acid and ursodeoxycholic acid, regulate colonic epithelial wound healing. Am. J. Physiol. Liver Physiol. 2018, 314, G378–G387. [Google Scholar] [CrossRef]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR Agonist Obeticholic Acid Prevents Gut Barrier Dysfunction and Bacterial Translocation in Cholestatic Rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Peng, Z.; Raufman, J.-P.; Xie, G. Src-Mediated Cross-Talk between Farnesoid X and Epidermal Growth Factor Receptors Inhibits Human Intestinal Cell Proliferation and Tumorigenesis. PLoS ONE 2012, 7, e48461. [Google Scholar] [CrossRef] [Green Version]
- Dossa, A.Y.; Escobar, O.; Golden, J.; Frey, M.R.; Ford, H.R.; Gayer, C.P. Bile acids regulate intestinal cell proliferation by modulating EGFR and FXR signaling. Am. J. Physiol. Liver Physiol. 2016, 310, G81–G92. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Cariello, M.; Morgano, A.; Gross, I.; Vegliante, M.C.; Murzilli, S.; Salvatore, L.; Freund, J.-N.; Sabba, C.; Moschetta, A. Transcriptional Regulation of the Intestinal Nuclear Bile Acid Farnesoid X Receptor (FXR) by the caudal-related Homeobox 2 (CDX2). J. Biol. Chem. 2014, 289, 28421–28432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Allaire, J.; Crowley, S.M.; Law, H.T.; Chang, S.-Y.; Ko, H.-J.; Vallance, B.A. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 2018, 39, 677–696. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The Bile Acid Sensor FXR Is Required for Immune-Regulatory Activities of TLR-9 in Intestinal Inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef] [Green Version]
- Renga, B.; Migliorati, M.; Mencarelli, A.; Fiorucci, S. Reciprocal regulation of the bile acid-activated receptor FXR and the interferon-gamma-STAT-1 pathway in macrophages. Biochim. Biophys. Acta 2009, 1792, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Guo, C.; Chi, Z.; Huang, B.; Wu, Y.; Wang, D.; Xia, D. A rapid administration of GW 4064 inhibits the NLRP 3 inflammasome activation independent of Farnesoid X Receptor agonism. FEBS Lett. 2017, 591, 2836–2847. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Experimental Conditions | Intervention + Observed Effects |
|---|---|
| DSS Colitis | FXR Activation: ↑ IL10, SHP, FGF15, ↓IL1β ↑PPARα-UGT: ↓ FXR activity +worse injury PPARα-UGT KO mice: ↑FXR activity, ↓Injury Treatment with FGF15 analogue: ↓ IL1β, IL6, TNFα No effect in FXR KO mice |
| Ischemia Reperfusion Injury | FXR Activation Blunts increase Lactate, LDH, IL6, IL1β, IFNγ, Limits autophagic influx |
| Cirrhotic Rats | FXR Activation ↓ IFNγ, TNFα, MadCam-1, IL17, IL10 |
| O/G Deprivation | FXR Activation ↑cell viability, ↓NFκB, TNFα, IL6 |
| CDCA treatment in CACO-2 | FXR Activation ↑IL8 IL6, TNFα, VegF FXR Inhibition Blunts Increases in IL8, IL6 |
| High Fat Diet in Fish | ↓FXR activity leads to increase in pro-inflammatory signaling CDCA dietary supplementation ↓IL1β, TNFα, COX-2, IL6 ↑ IL10 |
| Disease Process | Proposed FXR Role |
|---|---|
| Microscopic Colitis | Loss of normal proximal-to-distal FXR expression gradient |
| Crohn’s Disease | No observed difference in FXR expression relative to healthy control Decreased ileal SHP and FGF19 Minor alleles within FXR gene → Increased surgery risk? SNP at FXR gene (NR1H4) → Increased Risk of CD |
| Pediatric Crohn’s and Ulcerative Colitis | Decreased FXR expression |
| C. difficile colitis | FMT leads to increased FGF-19 expression (FMT→ ↑total bile acids? → ↑ FXR activity? → ↑FGF-19) |
| Malignancy | APC inactivating mutations: decreased FXR expression Decreased FXR mRNA expression in colitis associated neoplasia Decreased FXR expression CRC and pre-malignant polyps |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, K.M.; Gayer, C.P. The Pathophysiology of Farnesoid X Receptor (FXR) in the GI Tract: Inflammation, Barrier Function and Innate Immunity. Cells 2021, 10, 3206. https://doi.org/10.3390/cells10113206
Anderson KM, Gayer CP. The Pathophysiology of Farnesoid X Receptor (FXR) in the GI Tract: Inflammation, Barrier Function and Innate Immunity. Cells. 2021; 10(11):3206. https://doi.org/10.3390/cells10113206
Chicago/Turabian StyleAnderson, Kemp M., and Christopher P. Gayer. 2021. "The Pathophysiology of Farnesoid X Receptor (FXR) in the GI Tract: Inflammation, Barrier Function and Innate Immunity" Cells 10, no. 11: 3206. https://doi.org/10.3390/cells10113206
APA StyleAnderson, K. M., & Gayer, C. P. (2021). The Pathophysiology of Farnesoid X Receptor (FXR) in the GI Tract: Inflammation, Barrier Function and Innate Immunity. Cells, 10(11), 3206. https://doi.org/10.3390/cells10113206