

Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration

 ,
,
Abstract
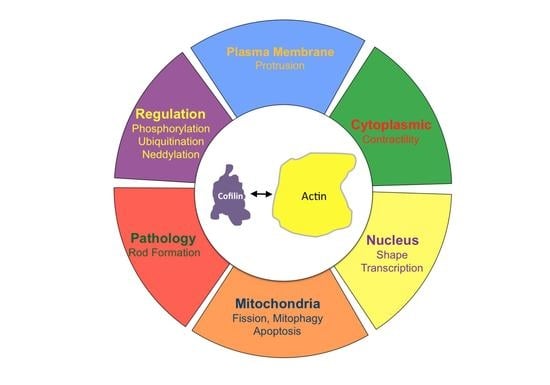
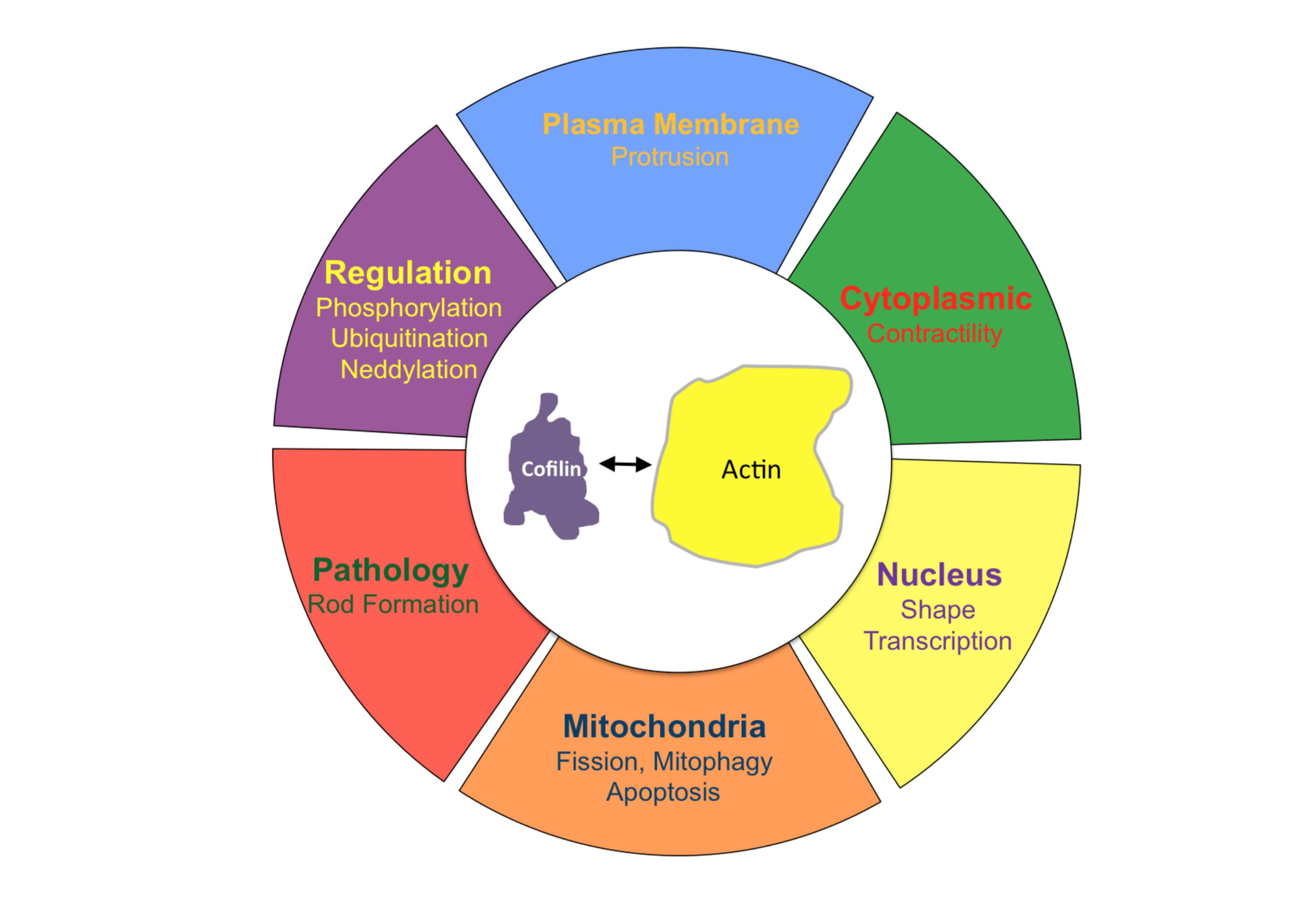{kind=link}
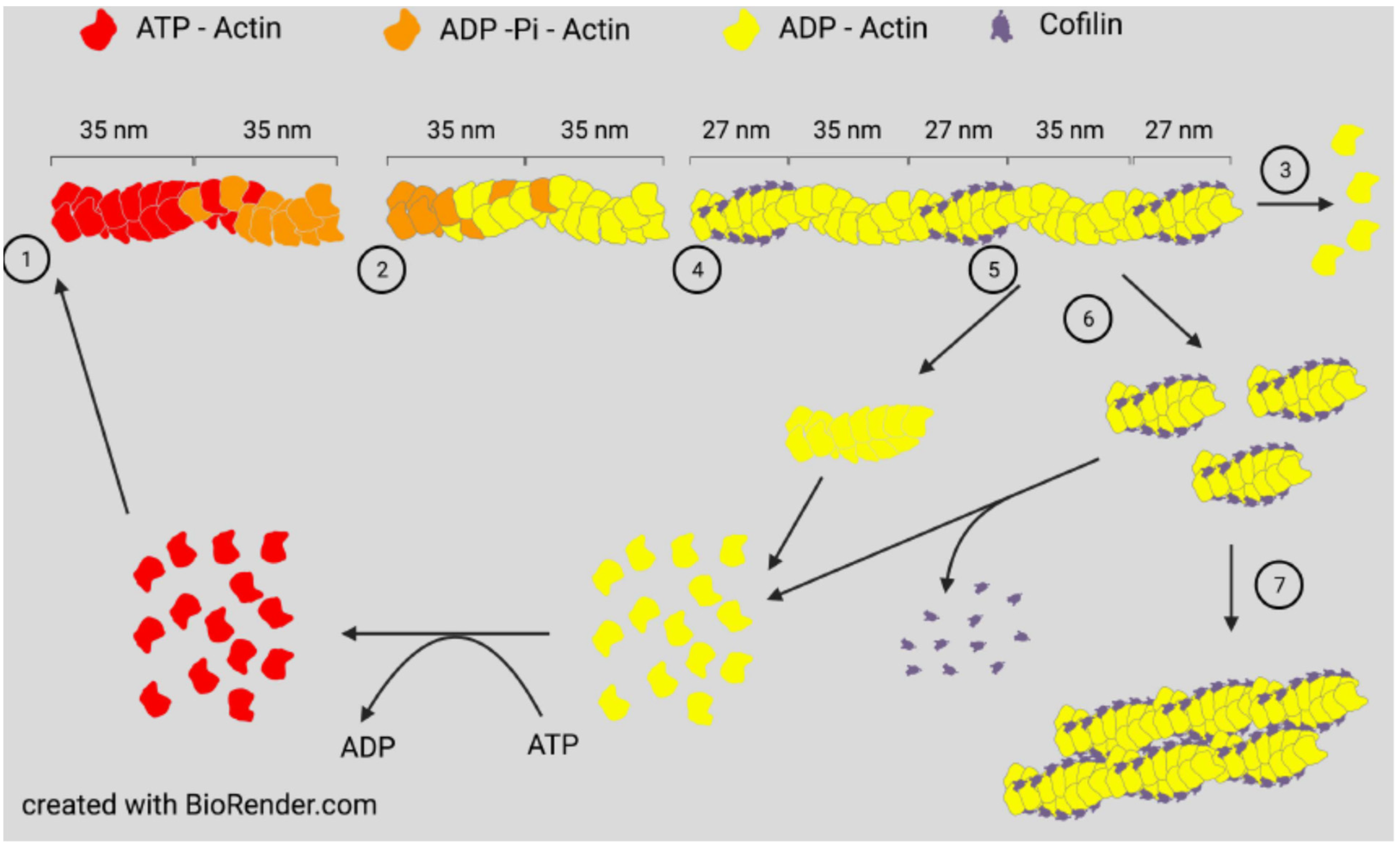{kind=link}
{kind=link}
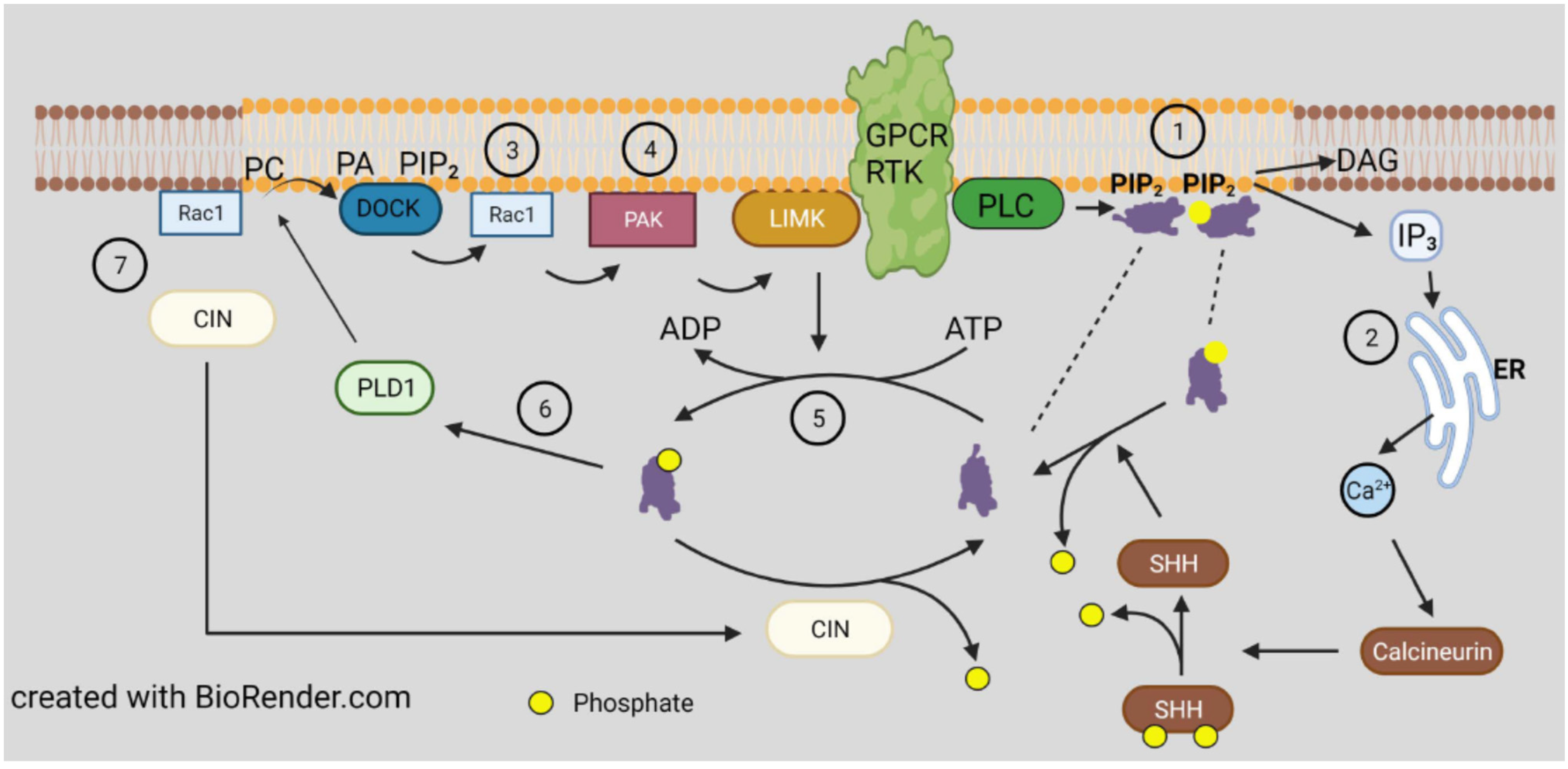{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Actin Dynamics and ADF/Cofilin Basics
3. Dynamic Regulation of Actin Assembly by Cofilin
3.1. Self-Regulation
3.2. Direct Regulation by Lipid Binding
3.3. Post-Translational Regulation of Cofilin
3.4. Phosphorylation
3.5. Ubiquitinylation and Neddylation
3.6. Oxidation/Reduction (Redox) Regulation
3.7. O-GlucNAcylation
4. Other Proteins Modulating Cofilin Activity and Their Regulation

5. Actin Dynamics in Neuritogenesis and Neurite Growth
6. Actin Dynamics in Neurite Consolidation and Branching
7. Cofilin Organelle Localization and Functional Consequences
7.1. Nucleus
7.2. Mitochondria
8. Cofilin-Actin Rods in Neurodegenerative Diseases and Disorders
8.1. Rod Induction and Visualization
8.2. Rod Structure and Composition
8.3. Rods Induced by Excitotoxicity
8.4. Rods Induced by Ischemia
8.5. Rod Pathology in Age-Related Neurodegenerative Diseases
8.6. Rods and Tau Pathology
8.7. Neuronal Death
9. Rod Modulatory Effects of Other Proteins
10. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kang, D.E.; Woo, J.A. Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, S131–S144. [Google Scholar] [CrossRef]
- Hoffmann, L.; Rust, M.B.; Culmsee, C. Actin(g) on Mitochondria—A role for cofilin1 in neuronal cell death pathways. Biol. Chem. 2019, 400, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Ben Zablah, Y.; Merovitch, N.; Jia, Z. The Role of ADF/cofilin in synaptic physiology and Alzheimer’s disease. Front. Cell Dev. Biol. 2020, 8, 594998. [Google Scholar] [CrossRef]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Pollard, T.D. Mechanism of Interaction of Acanthamoeba Actophorin (ADF/Cofilin) with Actin Filaments. J. Biol. Chem. 1999, 274, 15538–15546. [Google Scholar] [CrossRef]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of f-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Galkin, V.E.; Orlova, A.; Lukoyanova, N.; Wriggers, W.; Egelman, E.H. Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J. Cell Biol. 2001, 153, 75–86. [Google Scholar] [CrossRef]
- Andrianantoandro, E.; Pollard, T.D. Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Mol. Cell 2006, 24, 13–23. [Google Scholar] [CrossRef]
- Koffer, A.; Edgar, A.J.; Bamburg, J.R. Identification of Two Species of Actin Depolymerizing Factor in Cultures of BHK Cells. J. Muscle Res. Cell Motil. 1988, 9, 320–328. [Google Scholar] [CrossRef]
- Hayden, S.M.; Miller, P.S.; Brauweiler, A.; Bamburg, J.R. Analysis of the Interactions of Actin Depolymerizing Factor with G- and F-Actin. Biochemistry 1993, 32, 9994–10004. [Google Scholar] [CrossRef]
- Yeoh, S.; Pope, B.; Mannherz, H.G.; Weeds, A. Determining the Differences in Actin Binding by Human ADF and Cofilin. J. Mol. Biol. 2002, 315, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.; Roland, J.; Boujemaa-Paterski, R.; Kang, H.; McCullough, B.R.; Reymann, A.-C.; Guérin, C.; Martiel, J.-L.; De la Cruz, E.M.; Blanchoin, L. Cofilin Tunes the Nucleotide State of Actin Filaments and Severs at Bare and Decorated Segment Boundaries. Curr. Biol. 2011, 21, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Flores, L.R.; Keeling, M.C.; Zhang, X.; Sliogeryte, K.; Gavara, N. Lifeact-GFP Alters F-Actin Organization, Cellular Morphology and Biophysical Behaviour. Sci. Rep. 2019, 9, 3241. [Google Scholar] [CrossRef]
- Bamburg, J.R.; Harris, H.E.; Weeds, A.G. Partial Purification and Characterization of an Actin Depolymerizing Factor from Brain. FEBS Lett. 1980, 121, 178–182. [Google Scholar] [CrossRef]
- Nishida, E.; Maekawa, S.; Muneyuki, E.; Sakai, H. Action of a 19K Protein from Porcine Brain on Actin Polymerization: A New Functional Class of Actin-Binding Proteins. J. Biochem. 1984, 95, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.; Drubin, D.G. The ADF/Cofilin Proteins: Stimulus-Responsive Modulators of Actin Dynamics. Mol. Biol. Cell 1995, 6, 1423–1431. [Google Scholar] [CrossRef]
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential Regulators of Actin Dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. ADF/Cofilin: A Functional Node in Cell Biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef]
- Lappalainen, P.; Kessels, M.M.; Cope, M.J.; Drubin, D.G. The ADF Homology (ADF-H) Domain: A Highly Exploited Actin-Binding Module. Mol. Biol. Cell 1998, 9, 1951–1959. [Google Scholar] [CrossRef]
- Akıl, C.; Tran, L.T.; Orhant-Prioux, M.; Baskaran, Y.; Manser, E.; Blanchoin, L.; Robinson, R.C. Insights into the Evolution of Regulated Actin Dynamics via Characterization of Primitive Gelsolin/Cofilin Proteins from Asgard Archaea. Proc. Natl. Acad. Sci. USA 2020, 117, 19904–19913. [Google Scholar] [CrossRef]
- Nakano, K.; Kuwayama, H.; Kawasaki, M.; Numata, O.; Takaine, M. GMF Is an Evolutionarily Developed Adf/Cofilin-Super Family Protein Involved in the Arp2/3 Complex-Mediated Organization of the Actin Cytoskeleton. Cytoskeleton 2010, 67, 373–382. [Google Scholar] [CrossRef]
- Poukkula, M.; Kremneva, E.; Serlachius, M.; Lappalainen, P. Actin-Depolymerizing Factor Homology Domain: A Conserved Fold Performing Diverse Roles in Cytoskeletal Dynamics. Cytoskeleton 2011, 68, 471–490. [Google Scholar] [CrossRef]
- Ydenberg, C.A.; Padrick, S.B.; Sweeney, M.O.; Gandhi, M.; Sokolova, O.; Goode, B.L. GMF Severs Actin-Arp2/3 Complex Branch Junctions by a Cofilin-like Mechanism. Curr. Biol. 2013, 23, 1037–1045. [Google Scholar] [CrossRef]
- Tedeschi, A.; Dupraz, S.; Curcio, M.; Laskowski, C.J.; Schaffran, B.; Flynn, K.C.; Santos, T.E.; Stern, S.; Hilton, B.J.; Larson, M.J.E.; et al. ADF/Cofilin-Mediated Actin Turnover Promotes Axon Regeneration in the Adult CNS. Neuron 2019, 103, 1073–1085.e6. [Google Scholar] [CrossRef]
- Ressad, F.; Didry, D.; Xia, G.X.; Hong, Y.; Chua, N.H.; Pantaloni, D.; Carlier, M.F. Kinetic Analysis of the Interaction of Actin-Depolymerizing Factor (ADF)/Cofilin with G- and F-Actins. Comparison of Plant and Human ADFs and Effect of Phosphorylation. J. Biol. Chem. 1998, 273, 20894–20902. [Google Scholar] [CrossRef]
- Chen, H.; Bernstein, B.W.; Sneider, J.M.; Boyle, J.A.; Minamide, L.S.; Bamburg, J.R. In Vitro Activity Differences between Proteins of the ADF/Cofilin Family Define Two Distinct Subgroups. Biochemistry 2004, 43, 7127–7142. [Google Scholar] [CrossRef]
- Hocky, G.M.; Sindelar, C.V.; Cao, W.; Voth, G.A.; De La Cruz, E.M. Structural Basis of Fast- and Slow-Severing Actin-Cofilactin Boundaries. J. Biol. Chem. 2021, 296, 100337. [Google Scholar] [CrossRef] [PubMed]
- Tahtamouni, L.H.; Shaw, A.E.; Hasan, M.H.; Yasin, S.R.; Bamburg, J.R. Non-Overlapping Activities of ADF and Cofilin-1 during the Migration of Metastatic Breast Tumor Cells. BMC Cell Biol. 2013, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Devineni, N.; Minamide, L.S.; Niu, M.; Safer, D.; Verma, R.; Bamburg, J.R.; Nachmias, V.T. A Quantitative Analysis of G-Actin Binding Proteins and the G-Actin Pool in Developing Chick Brain. Brain Res. 1999, 823, 129–140. [Google Scholar] [CrossRef]
- Gurniak, C.B.; Perlas, E.; Witke, W. The Actin Depolymerizing Factor N-Cofilin Is Essential for Neural Tube Morphogenesis and Neural Crest Cell Migration. Dev. Biol. 2005, 278, 231–241. [Google Scholar] [CrossRef]
- Smith, R.S.; Hawes, N.L.; Kuhlmann, S.D.; Heckenlively, J.R.; Chang, B.; Roderick, T.H.; Sundberg, J.P. Corn1: A Mouse Model for Corneal Surface Disease and Neovascularization. Investig. Ophthalmol. Vis. Sci. 1996, 37, 397–404. [Google Scholar]
- Ikeda, S.; Cunningham, L.A.; Boggess, D.; Hawes, N.; Hobson, C.D.; Sundberg, J.P.; Naggert, J.K.; Smith, R.S.; Nishina, P.M. Aberrant Actin Cytoskeleton Leads to Accelerated Proliferation of Corneal Epithelial Cells in Mice Deficient for Destrin (Actin Depolymerizing Factor). Hum. Mol. Genet. 2003, 12, 1029–1037. [Google Scholar] [CrossRef]
- Verdoni, A.M.; Aoyama, N.; Ikeda, A.; Ikeda, S. Effect of Destrin Mutations on the Gene Expression Profile in Vivo. Physiol. Genom. 2008, 34, 9–21. [Google Scholar] [CrossRef]
- Verdoni, A.M.; Schuster, K.J.; Cole, B.S.; Ikeda, A.; Kao, W.W.; Ikeda, S. A Pathogenic Relationship between a Regulator of the Actin Cytoskeleton and Serum Response Factor. Genetics 2010, 186, 147–157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morgan, T.E.; Lockerbie, R.O.; Minamide, L.S.; Browning, M.D.; Bamburg, J.R. Isolation and Characterization of a Regulated Form of Actin Depolymerizing Factor. J. Cell Biol. 1993, 122, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Minamide, L.S.; Painter, W.B.; Schevzov, G.; Gunning, P.; Bamburg, J.R. Differential Regulation of Actin Depolymerizing Factor and Cofilin in Response to Alterations in the Actin Monomer Pool. J. Biol. Chem. 1997, 272, 8303–8309. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-Actin Signaling to the MRTF Coactivators Dominates the Immediate Transcriptional Response to Serum in Fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Kawakami-Schulz, S.V.; Verdoni, A.M.; Sattler, S.G.; Jessen, E.; Kao, W.W.-Y.; Ikeda, A.; Ikeda, S. Serum Response Factor: Positive and Negative Regulation of an Epithelial Gene Expression Network in the Destrin Mutant Cornea. Physiol. Genom. 2014, 46, 277–289. [Google Scholar] [CrossRef]
- Bellenchi, G.C.; Gurniak, C.B.; Perlas, E.; Middei, S.; Ammassari-Teule, M.; Witke, W. N-Cofilin Is Associated with Neuronal Migration Disorders and Cell Cycle Control in the Cerebral Cortex. Genes Dev. 2007, 21, 2347–2357. [Google Scholar] [CrossRef]
- Garvalov, B.K.; Flynn, K.C.; Neukirchen, D.; Meyn, L.; Teusch, N.; Wu, X.; Brakebusch, C.; Bamburg, J.R.; Bradke, F. Cdc42 Regulates Cofilin during the Establishment of Neuronal Polarity. J. Neurosci. 2007, 27, 13117–13129. [Google Scholar] [CrossRef]
- Minamide, L.S.; Striegl, A.M.; Boyle, J.A.; Meberg, P.J.; Bamburg, J.R. Neurodegenerative Stimuli Induce Persistent ADF/Cofilin-Actin Rods That Disrupt Distal Neurite Function. Nat. Cell Biol. 2000, 2, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bray, D. Distribution and Cellular Localization of Actin Depolymerizing Factor. J. Cell Biol. 1987, 105, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Theriot, J.A.; Mitchison, T.J.; Tilney, L.G.; Portnoy, D.A. The Rate of Actin-Based Motility of Intracellular Listeria Monocytogenes Equals the Rate of Actin Polymerization. Nature 1992, 357, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Theriot, J.A.; Rosenblatt, J.; Portnoy, D.A.; Goldschmidt-Clermont, P.J.; Mitchison, T.J. Involvement of Profilin in the Actin-Based Motility of L. Monocytogenes in Cells and in Cell-Free Extracts. Cell 1994, 76, 505–517. [Google Scholar] [CrossRef]
- Welch, M.D.; Iwamatsu, A.; Mitchison, T.J. Actin Polymerization Is Induced by Arp2/3 Protein Complex at the Surface of Listeria Monocytogenes. Nature 1997, 385, 265–269. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Agnew, B.J.; Abe, H.; Bamburg, J.R.; Mitchison, T.J. Xenopus Actin Depolymerizing Factor/Cofilin (XAC) Is Responsible for the Turnover of Actin Filaments in Listeria Monocytogenes Tails. J. Cell Biol. 1997, 136, 1323–1332. [Google Scholar] [CrossRef]
- Winkelman, J.D.; Bilancia, C.G.; Peifer, M.; Kovar, D.R. Ena/VASP Enabled Is a Highly Processive Actin Polymerase Tailored to Self-Assemble Parallel-Bundled F-Actin Networks with Fascin. Proc. Natl. Acad. Sci. USA 2014, 111, 4121–4126. [Google Scholar] [CrossRef]
- Loisel, T.P.; Boujemaa, R.; Pantaloni, D.; Carlier, M.F. Reconstitution of Actin-Based Motility of Listeria and Shigella Using Pure Proteins. Nature 1999, 401, 613–616. [Google Scholar] [CrossRef]
- Gouin, E.; Gantelet, H.; Egile, C.; Lasa, I.; Ohayon, H.; Villiers, V.; Gounon, P.; Sansonetti, P.J.; Cossart, P. A Comparative Study of the Actin-Based Motilities of the Pathogenic Bacteria Listeria Monocytogenes, Shigella Flexneri and Rickettsia Conorii. J. Cell Sci. 1999, 112 Pt 11, 1697–1708. [Google Scholar] [CrossRef]
- Bleicher, P.; Sciortino, A.; Bausch, A.R. The Dynamics of Actin Network Turnover Is Self-Organized by a Growth-Depletion Feedback. Sci. Rep. 2020, 10, 6215. [Google Scholar] [CrossRef]
- Zweifel, M.E.; Courtemanche, N. Profilin’s Affinity for Formin Regulates the Availability of Filament Ends for Actin Monomer Binding. J. Mol. Biol. 2020, 432, 166688. [Google Scholar] [CrossRef]
- Moriyama, K.; Yahara, I. Human CAP1 Is a Key Factor in the Recycling of Cofilin and Actin for Rapid Actin Turnover. J. Cell Sci. 2002, 115, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Purde, V.; Busch, F.; Kudryashova, E.; Wysocki, V.H.; Kudryashov, D.S. Oligomerization Affects the Ability of Human Cyclase-Associated Proteins 1 and 2 to Promote Actin Severing by Cofilins. Int. J. Mol. Sci. 2019, 20, 5647. [Google Scholar] [CrossRef] [PubMed]
- Kotila, T.; Kogan, K.; Enkavi, G.; Guo, S.; Vattulainen, I.; Goode, B.L.; Lappalainen, P. Structural Basis of Actin Monomer Re-Charging by Cyclase-Associated Protein. Nat. Commun. 2018, 9, 1892. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Tanaka, K.; Yamashiro, S.; Narita, A.; Watanabe, N. Helical Rotation of the Diaphanous-Related Formin MDia1 Generates Actin Filaments Resistant to Cofilin. Proc. Natl. Acad. Sci. USA 2018, 115, E5000–E5007. [Google Scholar] [CrossRef]
- Yonezawa, N.; Nishida, E.; Iida, K.; Yahara, I.; Sakai, H. Inhibition of the Interactions of Cofilin, Destrin, and Deoxyribonuclease I with Actin by Phosphoinositides. J. Biol. Chem. 1990, 265, 8382–8386. [Google Scholar] [CrossRef]
- Wang, W.; Eddy, R.; Condeelis, J. The Cofilin Pathway in Breast Cancer Invasion and Metastasis. Nat. Rev. Cancer 2007, 7, 429–440. [Google Scholar] [CrossRef]
- Van Rheenen, J.; Song, X.; van Roosmalen, W.; Cammer, M.; Chen, X.; Desmarais, V.; Yip, S.-C.; Backer, J.M.; Eddy, R.J.; Condeelis, J.S. EGF-Induced PIP2 Hydrolysis Releases and Activates Cofilin Locally in Carcinoma Cells. J. Cell Biol. 2007, 179, 1247–1259. [Google Scholar] [CrossRef]
- Podinovskaia, M.; Spang, A. The Endosomal Network: Mediators and Regulators of Endosome Maturation. Prog. Mol. Subcell Biol. 2018, 57, 1–38. [Google Scholar] [CrossRef]
- Han, L.; Stope, M.B.; de Jesús, M.L.; Oude Weernink, P.A.; Urban, M.; Wieland, T.; Rosskopf, D.; Mizuno, K.; Jakobs, K.H.; Schmidt, M. Direct Stimulation of Receptor-Controlled Phospholipase D1 by Phospho-Cofilin. EMBO J. 2007, 26, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Walker, V.; Seo, J.-Y.; Gohla, A.; Fowler, B.; Bohl, B.; DerMardirossian, C. Chronophin Coordinates Cell Leading Edge Dynamics by Controlling Active Cofilin Levels. Proc. Natl. Acad. Sci. USA 2015, 112, E5150–E5159. [Google Scholar] [CrossRef] [PubMed]
- Chua, B.T.; Volbracht, C.; Tan, K.O.; Li, R.; Yu, V.C.; Li, P. Mitochondrial Translocation of Cofilin Is an Early Step in Apoptosis Induction. Nat. Cell Biol. 2003, 5, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Klamt, F.; Zdanov, S.; Levine, R.L.; Pariser, A.; Zhang, Y.; Zhang, B.; Yu, L.-R.; Veenstra, T.D.; Shacter, E. Oxidant-Induced Apoptosis Is Mediated by Oxidation of the Actin-Regulatory Protein Cofilin. Nat. Cell Biol. 2009, 11, 1241–1246. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Shaw, A.E.; Minamide, L.S.; Pak, C.W.; Bamburg, J.R. Incorporation of Cofilin into Rods Depends on Disulfide Intermolecular Bonds: Implications for Actin Regulation and Neurodegenerative Disease. J. Neurosci. 2012, 32, 6670–6681. [Google Scholar] [CrossRef] [PubMed]
- Agnew, B.J.; Minamide, L.S.; Bamburg, J.R. Reactivation of Phosphorylated Actin Depolymerizing Factor and Identification of the Regulatory Site. J. Biol. Chem. 1995, 270, 17582–17587. [Google Scholar] [CrossRef]
- Moriyama, K.; Iida, K.; Yahara, I. Phosphorylation of Ser-3 of Cofilin Regulates Its Essential Function on Actin. Genes Cells 1996, 1, 73–86. [Google Scholar] [CrossRef]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of Actin Dynamics through Phosphorylation of Cofilin by LIM-Kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin Phosphorylation by LIM-Kinase 1 and Its Role in Rac-Mediated Actin Reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Toshima, J.; Toshima, J.Y.; Amano, T.; Yang, N.; Narumiya, S.; Mizuno, K. Cofilin Phosphorylation by Protein Kinase Testicular Protein Kinase 1 and Its Role in Integrin-Mediated Actin Reorganization and Focal Adhesion Formation. Mol. Biol. Cell 2001, 12, 1131–1145. [Google Scholar] [CrossRef]
- Nakano, K.; Kanai-Azuma, M.; Kanai, Y.; Moriyama, K.; Yazaki, K.; Hayashi, Y.; Kitamura, N. Cofilin Phosphorylation and Actin Polymerization by NRK/NESK, a Member of the Germinal Center Kinase Family. Exp. Cell Res. 2003, 287, 219–227. [Google Scholar] [CrossRef]
- George, J.; Soares, C.; Montersino, A.; Beique, J.-C.; Thomas, G.M. Palmitoylation of LIM Kinase-1 Ensures Spine-Specific Actin Polymerization and Morphological Plasticity. Elife 2015, 4, e06327. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Kundu, J.K.; Chae, J.-I.; Shim, J.-H. Targeting ROCK/LIMK/Cofilin Signaling Pathway in Cancer. Arch. Pharm. Res. 2019, 42, 481–491. [Google Scholar] [CrossRef]
- Shamah, S.M.; Lin, M.Z.; Goldberg, J.L.; Estrach, S.; Sahin, M.; Hu, L.; Bazalakova, M.; Neve, R.L.; Corfas, G.; Debant, A.; et al. EphA Receptors Regulate Growth Cone Dynamics through the Novel Guanine Nucleotide Exchange Factor Ephexin. Cell 2001, 105, 233–244. [Google Scholar] [CrossRef]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase Signaling Networks in Cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.C.; Maloney, M.T.; Minamide, L.S.; Flynn, K.C.; Stonebraker, M.A.; Bamburg, J.R. Mapping Cofilin-Actin Rods in Stressed Hippocampal Slices and the Role of Cdc42 in Amyloid-Beta-Induced Rods. J. Alzheimers Dis. 2009, 18, 35–50. [Google Scholar] [CrossRef]
- Sakuma, M.; Shirai, Y.; Yoshino, K.; Kuramasu, M.; Nakamura, T.; Yanagita, T.; Mizuno, K.; Hide, I.; Nakata, Y.; Saito, N. Novel PKCα-Mediated Phosphorylation Site(s) on Cofilin and Their Potential Role in Terminating Histamine Release. Mol. Biol. Cell 2012, 23, 3707–3721. [Google Scholar] [CrossRef]
- Prudent, R.; Demoncheaux, N.; Diemer, H.; Collin-Faure, V.; Kapur, R.; Paublant, F.; Lafanechère, L.; Cianférani, S.; Rabilloud, T. A Quantitative Proteomic Analysis of Cofilin Phosphorylation in Myeloid Cells and Its Modulation Using the LIM Kinase Inhibitor Pyr1. PLoS ONE 2018, 13, e0208979. [Google Scholar] [CrossRef]
- Chatzifrangkeskou, M.; Yadin, D.; Marais, T.; Chardonnet, S.; Cohen-Tannoudji, M.; Mougenot, N.; Schmitt, A.; Crasto, S.; Di Pasquale, E.; Macquart, C.; et al. Cofilin-1 Phosphorylation Catalyzed by ERK1/2 Alters Cardiac Actin Dynamics in Dilated Cardiomyopathy Caused by Lamin A/C Gene Mutation. Hum. Mol. Genet. 2018, 27, 3060–3078. [Google Scholar] [CrossRef]
- Subramanian, K.; Gianni, D.; Balla, C.; Assenza, G.E.; Joshi, M.; Semigran, M.J.; Macgillivray, T.E.; Van Eyk, J.E.; Agnetti, G.; Paolocci, N.; et al. Cofilin-2 Phosphorylation and Sequestration in Myocardial Aggregates: Novel Pathogenetic Mechanisms for Idiopathic Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1199–1214. [Google Scholar] [CrossRef]
- Gohla, A.; Bokoch, G.M. 14-3-3 Regulates Actin Dynamics by Stabilizing Phosphorylated Cofilin. Curr. Biol. 2002, 12, 1704–1710. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of Actin Reorganization by Slingshot, a Family of Phosphatases That Dephosphorylate ADF/Cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef]
- Gohla, A.; Birkenfeld, J.; Bokoch, G.M. Chronophin, a Novel HAD-Type Serine Protein Phosphatase, Regulates Cofilin-Dependent Actin Dynamics. Nat. Cell Biol. 2005, 7, 21–29. [Google Scholar] [CrossRef]
- Li, C.; Liang, Y.-Y.; Feng, X.-H.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. Essential Phosphatases and a Phospho-Degron Are Critical for Regulation of SRC-3/AIB1 Coactivator Function and Turnover. Mol. Cell 2008, 31, 835–849. [Google Scholar] [CrossRef]
- Kim, J.-E.; Lee, D.-S.; Kim, T.-H.; Park, H.; Kim, M.-J.; Kang, T.-C. PLPP/CIN-Mediated NF2-Serine 10 Dephosphorylation Regulates F-Actin Stability and Mdm2 Degradation in an Activity-Dependent Manner. Cell Death Dis. 2021, 12, 37. [Google Scholar] [CrossRef]
- Ohta, Y.; Kousaka, K.; Nagata-Ohashi, K.; Ohashi, K.; Muramoto, A.; Shima, Y.; Niwa, R.; Uemura, T.; Mizuno, K. Differential Activities, Subcellular Distribution and Tissue Expression Patterns of Three Members of Slingshot Family Phosphatases That Dephosphorylate Cofilin. Genes Cells 2003, 8, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Marshall, T.W.; Uetrecht, A.C.; Schafer, D.A.; Bear, J.E. Coronin 1B Coordinates Arp2/3 Complex and Cofilin Activities at the Leading Edge. Cell 2007, 128, 915–929. [Google Scholar] [CrossRef]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between Components of a Novel LIM Kinase-Slingshot Phosphatase Complex Regulates Cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.; Brickner, H.; Delorme-Walker, V.D.; Choi, J.; Saffin, J.-M.; Miller, D.; Panopoulos, A.; DerMardirossian, C.; Fotedar, A.; Margolis, R.L.; et al. WISp39 Binds Phosphorylated Coronin 1B to Regulate Arp2/3 Localization and Cofilin-Dependent Motility. J. Cell Biol. 2015, 208, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Bastea, L.I.; Döppler, H.; Pearce, S.E.; Durand, N.; Spratley, S.J.; Storz, P. Protein Kinase D-Mediated Phosphorylation at Ser99 Regulates Localization of P21-Activated Kinase 4. Biochem. J. 2013, 455, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Döppler, H.; Bastea, L.I.; Borges, S.; Spratley, S.J.; Pearce, S.E.; Storz, P. Protein Kinase d Isoforms Differentially Modulate Cofilin-Driven Directed Cell Migration. PLoS ONE 2014, 9, e98090. [Google Scholar] [CrossRef] [PubMed]
- Nagata-Ohashi, K.; Ohta, Y.; Goto, K.; Chiba, S.; Mori, R.; Nishita, M.; Ohashi, K.; Kousaka, K.; Iwamatsu, A.; Niwa, R.; et al. A Pathway of Neuregulin-Induced Activation of Cofilin-Phosphatase Slingshot and Cofilin in Lamellipodia. J. Cell Biol. 2004, 165, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K. Signaling Mechanisms and Functional Roles of Cofilin Phosphorylation and Dephosphorylation. Cell Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Spratley, S.J.; Bastea, L.I.; Döppler, H.; Mizuno, K.; Storz, P. Protein Kinase D Regulates Cofilin Activity through P21-Activated Kinase 4. J. Biol. Chem. 2011, 286, 34254–34261. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Huang, T.Y.; Bokoch, G.M. Reactive Oxygen Species Regulate a Slingshot-Cofilin Activation Pathway. Mol. Biol. Cell 2009, 20, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Minamide, L.S.; Bamburg, J.R.; Bokoch, G.M. Chronophin Mediates an ATP-Sensing Mechanism for Cofilin Dephosphorylation and Neuronal Cofilin-Actin Rod Formation. Dev. Cell 2008, 15, 691–703. [Google Scholar] [CrossRef]
- Min, J.S.; Kim, J.C.; Kim, J.A.; Kang, I.; Ahn, J.K. SIRT2 Reduces Actin Polymerization and Cell Migration through Deacetylation and Degradation of HSP90. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1230–1238. [Google Scholar] [CrossRef]
- Wang, Y.; Shibasaki, F.; Mizuno, K. Calcium Signal-Induced Cofilin Dephosphorylation Is Mediated by Slingshot via Calcineurin. J. Biol. Chem. 2005, 280, 12683–12689. [Google Scholar] [CrossRef]
- Woo, J.A.; Boggess, T.; Uhlar, C.; Wang, X.; Khan, H.; Cappos, G.; Joly-Amado, A.; De Narvaez, E.; Majid, S.; Minamide, L.S.; et al. RanBP9 at the Intersection between Cofilin and Aβ Pathologies: Rescue of Neurodegenerative Changes by RanBP9 Reduction. Cell Death Dis. 2015, 6, 1676. [Google Scholar] [CrossRef]
- Kestler, C.; Knobloch, G.; Tessmer, I.; Jeanclos, E.; Schindelin, H.; Gohla, A. Chronophin Dimerization Is Required for Proper Positioning of Its Substrate Specificity Loop. J. Biol. Chem. 2014, 289, 3094–3103. [Google Scholar] [CrossRef]
- Bisaria, A.; Hayer, A.; Garbett, D.; Cohen, D.; Meyer, T. Membrane-Proximal F-Actin Restricts Local Membrane Protrusions and Directs Cell Migration. Science 2020, 368, 1205–1210. [Google Scholar] [CrossRef]
- Mseka, T.; Cramer, L.P. Actin Depolymerization-Based Force Retracts the Cell Rear in Polarizing and Migrating Cells. Curr Biol. 2011, 21, 2085–2091. [Google Scholar] [CrossRef]
- Nishida, E.; Maekawa, S.; Sakai, H. Cofilin, a Protein in Porcine Brain That Binds to Actin Filaments and Inhibits Their Interactions with Myosin and Tropomyosin. Biochemistry 1984, 23, 5307–5313. [Google Scholar] [CrossRef]
- Wiggan, O.; Shaw, A.E.; DeLuca, J.G.; Bamburg, J.R. ADF/Cofilin Regulates Actomyosin Assembly through Competitive Inhibition of Myosin II Binding to F-Actin. Dev. Cell 2012, 22, 530–543. [Google Scholar] [CrossRef]
- Wiggan, O.; DeLuca, J.G.; Stasevich, T.J.; Bamburg, J.R. Lamin A/C Deficiency Enables Increased Myosin-II Bipolar Filament Ensembles That Promote Divergent Actomyosin Network Anomalies through Self-Organization. Mol. Biol. Cell 2020, 31, 2363–2378. [Google Scholar] [CrossRef] [PubMed]
- Bowling, F.Z.; Frohman, M.A.; Airola, M.V. Structure and Regulation of Human Phospholipase, D. Adv. Biol. Regul. 2021, 79, 100783. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.; Ho, H.J.; Wang, C.; Guan, J.-L. Tyrosine Phosphorylation of Cofilin at Y68 by V-Src Leads to Its Degradation through Ubiquitin-Proteasome Pathway. Oncogene 2010, 29, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Vogl, A.M.; Phu, L.; Becerra, R.; Giusti, S.A.; Verschueren, E.; Hinkle, T.B.; Bordenave, M.D.; Adrian, M.; Heidersbach, A.; Yankilevich, P.; et al. Global Site-Specific Neddylation Profiling Reveals That NEDDylated Cofilin Regulates Actin Dynamics. Nat. Struct. Mol. Biol. 2020, 27, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.; Peng, Z.; Liu, C.H.; Zhang, L.; Jiang, H. Advances in Cancer Treatment by Targeting the Neddylation Pathway. Front. Cell Dev. Biol. 2021, 9, 653882. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-C.; Guo, Y.-J.; Wang, B.; Wang, C.; Mamun, M.A.A.; Gao, Y.; Liu, H.-M. Targeting Neddylation E2s: A Novel Therapeutic Strategy in Cancer. J. Hematol. Oncol. 2021, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Meberg, P.J.; Ono, S.; Minamide, L.S.; Takahashi, M.; Bamburg, J.R. Actin depolymerizing factor and cofilin phosphorylation dynamics: Response to signals that regulate neurite extension. Cell Motil. Cytoskelet. 1998, 39, 172–190. [Google Scholar] [CrossRef]
- Tanaka, K.; Takeda, S.; Mitsuoka, K.; Oda, T.; Kimura-Sakiyama, C.; Maéda, Y.; Narita, A. Structural basis for cofilin binding and actin filament disassembly. Nat. Commun. 2018, 9, 1860. [Google Scholar] [CrossRef] [PubMed]
- Vogl, A.M.; Brockmann, M.M.; Giusti, S.A.; Maccarrone, G.; Vercelli, C.A.; Bauder, C.A.; Richter, J.S.; Roselli, F.; Hafner, A.-S.; Dedic, N.; et al. Neddylation inhibition impairs spine development, destabilizes synapses and deteriorates cognition. Nat. Neurosci. 2015, 18, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Lee, D.; Song, J.-M.; Park, S.; Park, D.-H.; Lee, S.; Suh, Y.H. Neddylation is required for presynaptic clustering of MGlu7 and maturation of presynaptic terminals. Exp. Mol. Med. 2021, 53, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R.; et al. ADF/Cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 2010, 13, 1208–1215. [Google Scholar] [CrossRef]
- Rust, M.B.; Gurniak, C.B.; Renner, M.; Vara, H.; Morando, L.; Görlich, A.; Sassoè-Pognetto, M.; Banchaabouchi, M.A.; Giustetto, M.; Triller, A.; et al. Learning, AMPA Receptor Mobility and Synaptic Plasticity Depend on n-Cofilin-Mediated Actin Dynamics. EMBO J. 2010, 29, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and Molecular Remodeling of Dendritic Spine Substructures during Long-Term Potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef]
- Pfannstiel, J.; Cyrklaff, M.; Habermann, A.; Stoeva, S.; Griffiths, G.; Shoeman, R.; Faulstich, H. Human Cofilin Forms Oligomers Exhibiting Actin Bundling Activity. J. Biol. Chem. 2001, 276, 49476–49484. [Google Scholar] [CrossRef]
- Salem, F.B.; Bunner, W.P.; Prabhu, V.V.; Kuyateh, A.-B.; O’Bryant, C.T.; Murashov, A.K.; Szatmari, E.M.; Hughes, R.M. CofActor: A Light- and Stress-Gated Optogenetic Clustering Tool to Study Disease-Associated Cytoskeletal Dynamics in Living Cells. J. Biol. Chem. 2020, 295, 11231–11245. [Google Scholar] [CrossRef]
- Luo, S.; Uehara, H.; Shacter, E. Taurine Chloramine-Induced Inactivation of Cofilin Protein through Methionine Oxidation. Free Radic. Biol. Med. 2014, 75, 84–94. [Google Scholar] [CrossRef]
- Zhang, Y.-T.; Ouyang, D.-Y.; Xu, L.-H.; Zha, Q.-B.; He, X.-H. Formation of Cofilin-Actin Rods Following Cucurbitacin-B-Induced Actin Aggregation Depends on Slingshot Homolog 1-Mediated Cofilin Hyperactivation. J. Cell Biochem. 2013, 114, 2415–2429. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsen, M.; Schuldt, M.; Munro, J.; Borucka, D.; Cameron, J.; Baugh, M.; Mleczak, A.; Lilla, S.; Morrice, N.; Olson, M.F. Cucurbitacin Covalent Bonding to Cysteine Thiols: The Filamentous-Actin Severing Protein Cofilin1 as an Exemplary Target. Cell Commun. Signal 2013, 11, 58. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Rossi, R.; Colombo, R.; Milzani, A. Reversible S-Glutathionylation of Cys 374 Regulates Actin Filament Formation by Inducing Structural Changes in the Actin Molecule. Free Radic. Biol. Med. 2003, 34, 23–32. [Google Scholar] [CrossRef]
- Kruyer, A.; Ball, L.E.; Townsend, D.M.; Kalivas, P.W.; Uys, J.D. Post-Translational S-Glutathionylation of Cofilin Increases Actin Cycling during Cocaine Seeking. PLoS ONE 2019, 14, e0223037. [Google Scholar] [CrossRef] [PubMed]
- Döppler, H.; Storz, P. Mitochondrial and Oxidative Stress-Mediated Activation of Protein Kinase D1 and Its Importance in Pancreatic Cancer. Front. Oncol. 2017, 7, 41. [Google Scholar] [CrossRef]
- Torres, C.R.; Hart, G.W. Topography and Polypeptide Distribution of Terminal N-Acetylglucosamine Residues on the Surfaces of Intact Lymphocytes. Evidence for O-Linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-Linked Beta-N-Acetylglucosamine on Nucleocytoplasmic Proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Olivier-Van Stichelen, S. The Human O-GlcNAcome Database and Meta-Analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef]
- Shafi, R.; Iyer, S.P.; Ellies, L.G.; O’Donnell, N.; Marek, K.W.; Chui, D.; Hart, G.W.; Marth, J.D. The O-GlcNAc Transferase Gene Resides on the X Chromosome and Is Essential for Embryonic Stem Cell Viability and Mouse Ontogeny. Proc. Natl. Acad. Sci. USA 2000, 97, 5735–5739. [Google Scholar] [CrossRef]
- Cheng, J.; Wu, Y.; Chen, L.; Li, Y.; Liu, F.; Shao, J.; Huang, M.; Fan, M.; Wu, H. Loss of O-GlcNAc Transferase in Neural Stem Cells Impairs Corticogenesis. Biochem. Biophys. Res. Commun. 2020, 532, 541–547. [Google Scholar] [CrossRef]
- Chen, J.; Dong, X.; Cheng, X.; Zhu, Q.; Zhang, J.; Li, Q.; Huang, X.; Wang, M.; Li, L.; Guo, W.; et al. Ogt Controls Neural Stem/Progenitor Cell Pool and Adult Neurogenesis through Modulating Notch Signaling. Cell Rep. 2021, 34, 108905. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, E.G.; Albarran, E.; White, C.W.; Bieri, G.; Sanchez-Diaz, C.; Pratt, K.; Snethlage, C.E.; Ding, J.B.; Villeda, S.A. Neuronal O-GlcNAcylation Improves Cognitive Function in the Aged Mouse Brain. Curr. Biol. 2019, 29, 3359–3369.e4. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Vosseller, K.; Cole, R.N.; Cronshaw, J.M.; Matunis, M.J.; Hart, G.W. Mapping Sites of O-GlcNAc Modification Using Affinity Tags for Serine and Threonine Post-Translational Modifications. Mol. Cell Proteom. 2002, 1, 791–804. [Google Scholar] [CrossRef]
- Huang, X.; Pan, Q.; Sun, D.; Chen, W.; Shen, A.; Huang, M.; Ding, J.; Geng, M. O-GlcNAcylation of Cofilin Promotes Breast Cancer Cell Invasion. J. Biol. Chem. 2013, 288, 36418–36425. [Google Scholar] [CrossRef]
- Monsky, W.L.; Kelly, T.; Lin, C.Y.; Yeh, Y.; Stetler-Stevenson, W.G.; Mueller, S.C.; Chen, W.T. Binding and Localization of M(r) 72,000 Matrix Metalloproteinase at Cell Surface Invadopodia. Cancer Res. 1993, 53, 3159–3164. [Google Scholar]
- Santiago-Medina, M.; Gregus, K.A.; Nichol, R.H.; O’Toole, S.M.; Gomez, T.M. Regulation of ECM Degradation and Axon Guidance by Growth Cone Invadosomes. Development 2015, 142, 486–496. [Google Scholar] [CrossRef]
- Lee, B.E.; Kim, H.Y.; Kim, H.-J.; Jeong, H.; Kim, B.-G.; Lee, H.-E.; Lee, J.; Kim, H.B.; Lee, S.E.; Yang, Y.R.; et al. O-GlcNAcylation Regulates Dopamine Neuron Function, Survival and Degeneration in Parkinson Disease. Brain 2020, 143, 3699–3716. [Google Scholar] [CrossRef]
- Park, J.; Ha, H.-J.; Chung, E.S.; Baek, S.H.; Cho, Y.; Kim, H.K.; Han, J.; Sul, J.H.; Lee, J.; Kim, E.; et al. O-GlcNAcylation Ameliorates the Pathological Manifestations of Alzheimer’s Disease by Inhibiting Necroptosis. Sci. Adv. 2021, 7, eabd3207. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Spasojevic, I.; Lu, L.; Shen, Y.; Qu, X.; Hoffmann, U.; Warner, D.S.; Paschen, W.; Sheng, H.; et al. Increasing O-GlcNAcylation Is Neuroprotective in Young and Aged Brains after Ischemic Stroke. Exp. Neurol. 2021, 339, 113646. [Google Scholar] [CrossRef]
- Hsieh, Y.-L.; Su, F.-Y.; Tsai, L.-K.; Huang, C.-C.; Ko, Y.-L.; Su, L.-W.; Chen, K.-Y.; Shih, H.-M.; Hu, C.-M.; Lee, W.-H. NPGPx-Mediated Adaptation to Oxidative Stress Protects Motor Neurons from Degeneration in Aging by Directly Modulating O-GlcNAcase. Cell Rep. 2019, 29, 2134–2143.e7. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Sanchez, R.G.; Jarome, T.J.; Webb, W.M.; Lubin, F.D. O-GlcNAc and EZH2-Mediated Epigenetic Regulation of Gene Expression during Consolidation of Fear Memories. Learn Mem. 2019, 26, 373–379. [Google Scholar] [CrossRef]
- Cantrelle, F.-X.; Loyens, A.; Trivelli, X.; Reimann, O.; Despres, C.; Gandhi, N.S.; Hackenberger, C.P.R.; Landrieu, I.; Smet-Nocca, C. Phosphorylation and O-GlcNAcylation of the PHF-1 Epitope of Tau Protein Induce Local Conformational Changes of the C-terminus and modulate tau self-assembly into fibrillar aggregates. Front. Mol. Neurosci. 2021, 14, 661368. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.H.; Cho, Y.Y.; Kim, T.-W.; Chung, S. O-GlcNAcylation of Amyloid-β Protein Precursor by Insulin Signaling Reduces Amyloid-β Production. J. Alzheimers Dis. 2019, 69, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, N.; Ghosh, S.; Sept, D.; Cooper, J.A. Binding of Myotrophin/V-1 to Actin-Capping Protein: Implications for How Capping Protein Binds to the Filament Barbed End. J. Biol. Chem. 2006, 281, 31021–31030. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Pernier, J.; Carlier, M.-F. Regulators of actin filament barbed ends at a glance. J. Cell Sci. 2016, 129, 1085–1091. [Google Scholar] [CrossRef]
- Henty-Ridilla, J.L.; Juanes, M.A.; Goode, B.L. Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr. Biol. 2017, 27, 3535–3543.e4. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Costa, R.; Sousa, M.M. Profilin as a dual regulator of actin and microtubule dynamics. Cytoskeleton 2020, 77, 76–83. [Google Scholar] [CrossRef]
- Shekhar, S.; Hoeprich, G.J.; Gelles, J.; Goode, B.L. Twinfilin Bypasses Assembly Conditions and Actin Filament Aging to Drive Barbed End Depolymerization. J. Cell Biol. 2021, 220, e202006022. [Google Scholar] [CrossRef]
- Hakala, M.; Wioland, H.; Tolonen, M.; Kotila, T.; Jegou, A.; Romet-Lemonne, G.; Lappalainen, P. Twinfilin Uncaps filament barbed ends to promote turnover of lamellipodial actin networks. Nat. Cell Biol. 2021, 23, 147–159. [Google Scholar] [CrossRef]
- Hilton, D.M.; Aguilar, R.M.; Johnston, A.B.; Goode, B.L. Species-specific functions of twinfilin in actin filament depolymerization. J. Mol. Biol. 2018, 430, 3323–3336. [Google Scholar] [CrossRef]
- Okada, K.; Ravi, H.; Smith, E.M.; Goode, B.L. Aip1 and cofilin promote rapid turnover of yeast actin patches and cables: A coordinated mechanism for severing and capping filaments. Mol. Biol. Cell 2006, 17, 2855–2868. [Google Scholar] [CrossRef]
- Hayakawa, K.; Sekiguchi, C.; Sokabe, M.; Ono, S.; Tatsumi, H. Real-Time Single-Molecule Kinetic Analyses of AIP1-Enhanced actin filament severing in the presence of cofilin. J. Mol. Biol. 2019, 431, 308–322. [Google Scholar] [CrossRef]
- López-Coral, A.; Striz, A.C.; Tuma, P.L. A Serine/threonine kinase 16-based phospho-proteomics screen identifies WD Repeat Protein-1 as a regulator of constitutive secretion. Sci. Rep. 2018, 8, 13049. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Breitsprecher, D.; Little, K.; Sharov, G.; Sokolova, O.; Goode, B.L. Srv2/Cyclase-associated protein forms hexameric shurikens that directly catalyze actin filament severing by cofilin. Mol. Biol. Cell 2013, 24, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Collins, A.; Golden, L.; Sokolova, O.; Goode, B.L. Structure and Mechanism of Mouse Cyclase-Associated Protein (CAP1) in Regulating Actin Dynamics. J. Biol. Chem. 2014, 289, 30732–30742. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Duong, T.-A.; Metz, I.; Winkelmeier, J.; Hübner, C.A.; Endesfelder, U.; Rust, M.B. Mutual Functional Dependence of Cyclase-Associated Protein 1 (CAP1) and Cofilin1 in Neuronal Actin Dynamics and Growth Cone Function. Prog. Neurobiol. 2021, 202, 102050. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Paeger, L.; Kosmas, K.; Kloppenburg, P.; Noegel, A.A.; Peche, V.S. Neuronal Actin Dynamics, Spine Density and Neuronal Dendritic Complexity Are Regulated by CAP2. Front. Cell. Neurosci. 2016, 10, 180. [Google Scholar] [CrossRef]
- Pelucchi, S.; Vandermeulen, L.; Pizzamiglio, L.; Aksan, B.; Yan, J.; Konietzny, A.; Bonomi, E.; Borroni, B.; Padovani, A.; Rust, M.B.; et al. Cyclase-Associated Protein 2 Dimerization Regulates Cofilin in Synaptic Plasticity and Alzheimer’s Disease. Brain Commun. 2020, 2, fcaa086. [Google Scholar] [CrossRef]
- Gandhi, M.; Achard, V.; Blanchoin, L.; Goode, B.L. Coronin switches roles in actin disassembly depending on the nucleotide state of actin. Mol. Cell 2009, 34, 364–374. [Google Scholar] [CrossRef]
- Chan, K.T.; Creed, S.J.; Bear, J.E. Unraveling the Enigma: Progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011, 21, 481–488. [Google Scholar] [CrossRef]
- Ge, P.; Durer, Z.A.O.; Kudryashov, D.; Zhou, Z.H.; Reisler, E. Cryo-EM reveals different coronin binding modes for ADP- and ADP-BeFx Actin Filaments. Nat. Struct. Mol. Biol. 2014, 21, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- BoseDasgupta, S.; Moes, S.; Jenoe, P.; Pieters, J. Cytokine-induced macropinocytosis in macrophages is regulated by 14-3-3ζ through its interaction with serine-phosphorylated coronin 1. FEBS J. 2015, 282, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Gateva, G.; Kremneva, E.; Reindl, T.; Kotila, T.; Kogan, K.; Gressin, L.; Gunning, P.W.; Manstein, D.J.; Michelot, A.; Lappalainen, P. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr Biol. 2017, 27, 705–713. [Google Scholar] [CrossRef]
- Sharma, S.; Grintsevich, E.E.; Hsueh, C.; Reisler, E.; Gimzewski, J.K. Molecular Cooperativity of Drebrin1-300 Binding and Structural Remodeling of F-Actin. Biophys. J. 2012, 103, 275–283. [Google Scholar] [CrossRef]
- Takahashi, H.; Yamazaki, H.; Hanamura, K.; Sekino, Y.; Shirao, T. Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. J. Cell Sci. 2009, 122, 1211–1219. [Google Scholar] [CrossRef]
- Sekino, Y.; Koganezawa, N.; Mizui, T.; Shirao, T. Role of drebrin in synaptic plasticity. Adv. Exp. Med. Biol. 2017, 1006, 183–201. [Google Scholar] [CrossRef]
- Geraldo, S.; Khanzada, U.K.; Parsons, M.; Chilton, J.K.; Gordon-Weeks, P.R. Targeting of the F-Actin-Binding Protein Drebrin by the Microtubule plus-Tip Protein EB3 Is Required for Neuritogenesis. Nat. Cell Biol. 2008, 10, 1181–1189. [Google Scholar] [CrossRef]
- Gordon-Weeks, P.R. Phosphorylation of Drebrin and Its Role in Neuritogenesis. Adv. Exp. Med. Biol. 2017, 1006, 49–60. [Google Scholar] [CrossRef]
- Meiring, J.C.M.; Bryce, N.S.; Wang, Y.; Taft, M.H.; Manstein, D.J.; Liu Lau, S.; Stear, J.; Hardeman, E.C.; Gunning, P.W. Co-polymers of actin and tropomyosin account for a major fraction of the human actin cytoskeleton. Curr. Biol. 2018, 28, 2331–2337.e5. [Google Scholar] [CrossRef]
- Hardeman, E.C.; Bryce, N.S.; Gunning, P.W. Impact of the actin cytoskeleton on cell development and function mediated via tropomyosin isoforms. Semin. Cell Dev. Biol. 2020, 102, 122–131. [Google Scholar] [CrossRef]
- Geeves, M.A.; Hitchcock-DeGregori, S.E.; Gunning, P.W. A systematic nomenclature for mammalian tropomyosin isoforms. J. Muscle Res. Cell Motil. 2015, 36, 147–153. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Schevzov, G.; Hotulainen, P.; Naumanen, P.; Martin, C.; Gunning, P.W.; Lappalainen, P. A Molecular Pathway for Myosin II Recruitment to Stress Fibers. Curr. Biol. 2011, 21, 539–550. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. Tropomyosin Binding to F-Actin Protects the F-Actin from disassembly by brain actin-depolymerizing factor (ADF). Cell Motil. 1982, 2, 1–8. [Google Scholar] [CrossRef]
- Manstein, D.J.; Meiring, J.C.M.; Hardeman, E.C.; Gunning, P.W. Actin-Tropomyosin Distribution in Non-Muscle Cells. J. Muscle Res. Cell Motil. 2020, 41, 11–22. [Google Scholar] [CrossRef]
- Wiggan, O.; Schroder, B.; Krapf, D.; Bamburg, J.R.; DeLuca, J.G. Cofilin Regulates Nuclear Architecture through a Myosin-II Dependent Mechanotransduction Module. Sci. Rep. 2017, 7, 40953. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Mabuchi, I. Actin-Depolymerizing Protein Adf1 Is Required for Formation and Maintenance of the Contractile Ring during Cytokinesis in Fission Yeast. Mol. Biol. Cell 2006, 17, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Tomanić, T.; Martin, C.; Stefen, H.; Parić, E.; Gunning, P.; Fath, T. Deletion of the Actin-Associated Tropomyosin Tpm3 Leads to Reduced Cell Complexity in Cultured Hippocampal Neurons-New Insights into the Role of the C-Terminal Region of Tpm3.1. Cells 2021, 10, 715. [Google Scholar] [CrossRef]
- Parreno, J.; Fowler, V.M. Multifunctional Roles of Tropomodulin-3 in Regulating Actin Dynamics. Biophys. Rev. 2018, 10, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- A, M.; Latario, C.J.; Pickrell, L.E.; Higgs, H.N. Lysine Acetylation of Cytoskeletal Proteins: Emergence of an Actin Code. J. Cell Biol. 2020, 219, e202006151. [Google Scholar] [CrossRef]
- Vedula, P.; Kurosaka, S.; Leu, N.A.; Wolf, Y.I.; Shabalina, S.A.; Wang, J.; Sterling, S.; Dong, D.W.; Kashina, A. Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. Elife 2017, 6, e31661. [Google Scholar] [CrossRef]
- Vedula, P.; Kurosaka, S.; MacTaggart, B.; Ni, Q.; Papoian, G.; Jiang, Y.; Dong, D.W.; Kashina, A. Different Translation Dynamics of β- and γ-Actin Regulates Cell Migration. Elife 2021, 10, e68712. [Google Scholar] [CrossRef]
- Condeelis, J.; Singer, R.H. How and Why Does Beta-Actin MRNA Target? Biol Cell 2005, 97, 97–110. [Google Scholar] [CrossRef]
- Chen, L.; Kashina, A. Post-Translational Modifications of the Protein Termini. Front. Cell Dev. Biol. 2021, 9, 719590. [Google Scholar] [CrossRef] [PubMed]
- Kanellos, G.; Frame, M.C. Cellular Functions of the ADF/Cofilin Family at a Glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Terman, J.R.; Kashina, A. Post-Translational Modification and Regulation of Actin. Curr. Opin. Cell Biol. 2013, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Balta, E.; Kramer, J.; Samstag, Y. Redox Regulation of the Actin Cytoskeleton in Cell Migration and Adhesion: On the Way to a Spatiotemporal View. Front. Cell Dev. Biol. 2020, 8, 618261. [Google Scholar] [CrossRef]
- Karatsai, O.; Stasyk, O.; Redowicz, M.J. Effects of Arginine and Its Deprivation on Human Glioblastoma Physiology and Signaling. Adv. Exp. Med. Biol. 2020, 1202, 243–258. [Google Scholar] [CrossRef]
- A, M.; Fung, T.S.; Francomacaro, L.M.; Huynh, T.; Kotila, T.; Svindrych, Z.; Higgs, H.N. Regulation of INF2-mediated actin polymerization through site-specific lysine acetylation of actin itself. Proc. Natl. Acad. Sci. USA 2020, 117, 439–447. [Google Scholar] [CrossRef]
- Hung, R.-J.; Yazdani, U.; Yoon, J.; Wu, H.; Yang, T.; Gupta, N.; Huang, Z.; van Berkel, W.J.H.; Terman, J.R. Mical Links Semaphorins to F-Actin Disassembly. Nature 2010, 463, 823–827. [Google Scholar] [CrossRef]
- Hung, R.-J.; Pak, C.W.; Terman, J.R. Direct Redox Regulation of F-Actin Assembly and Disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef]
- Grintsevich, E.E.; Ge, P.; Sawaya, M.R.; Yesilyurt, H.G.; Terman, J.R.; Zhou, Z.H.; Reisler, E. Catastrophic Disassembly of Actin Filaments via Mical-Mediated Oxidation. Nat. Commun. 2017, 8, 2183. [Google Scholar] [CrossRef]
- Tamura, M.; Itoh, K.; Akita, H.; Takano, K.; Oku, S. Identification of an Actin-Binding Site in P47phox an Organizer Protein of NADPH Oxidase. FEBS Lett. 2006, 580, 261–267. [Google Scholar] [CrossRef]
- Flynn, K.C.; Hellal, F.; Neukirchen, D.; Jacob, S.; Tahirovic, S.; Dupraz, S.; Stern, S.; Garvalov, B.K.; Gurniak, C.; Shaw, A.E.; et al. ADF/cofilin-mediated actin retrograde flow directs neurite formation in the developing brain. Neuron 2012, 76, 1091–1107. [Google Scholar] [CrossRef]
- Moriyama, K.; Yahara, I. Two activities of cofilin, severing and accelerating directional depolymerization of actin filaments, are affected differentially by mutations around the actin-binding helix. EMBO J. 1999, 18, 6752–6761. [Google Scholar] [CrossRef]
- Moriyama, K.; Yahara, I. The actin-severing activity of cofilin is exerted by the interplay of three distinct sites on cofilin and essential for cell viability. Biochem. J. 2002, 365, 147–155. [Google Scholar] [CrossRef]
- Leite, S.C.; Pinto-Costa, R.; Sousa, M.M. Actin Dynamics in the Growth Cone: A Key Player in Axon Regeneration. Curr. Opin. Neurobiol. 2021, 69, 11–18. [Google Scholar] [CrossRef]
- Flynn, K.C.; Pak, C.W.; Bamburg, J.R. Regulation of Growth Cone Initiation and Actin Dynamics by ADF/cofilin. In Intracellular Mechanisms for Neuritogenesis; Springer: New York, NY, USA, 2007; pp. 25–56. ISBN 0-387-33128-X. [Google Scholar]
- Marsick, B.M.; Flynn, K.C.; Santiago-Medina, M.; Bamburg, J.R.; Letourneau, P.C. Activation of ADF/Cofilin Mediates Attractive Growth Cone Turning toward Nerve Growth Factor and Netrin-1. Dev. Neurobiol. 2010, 70, 565–588. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Han, L.; Bamburg, J.R.; Shim, S.; Ming, G.; Zheng, J.Q. BMP Gradients Steer Nerve Growth Cones by a Balancing Act of LIM Kinase and Slingshot Phosphatase on ADF/Cofilin. J. Cell Biol. 2007, 178, 107–119. [Google Scholar] [CrossRef]
- Yang, C.; Svitkina, T. Filopodia Initiation: Focus on the Arp2/3 Complex and Formins. Cell Adh. Migr. 2011, 5, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell functions of a multifaceted actin-binding protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Spillane, M.; Ketschek, A.; Donnelly, C.J.; Pacheco, A.; Twiss, J.L.; Gallo, G. Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 Complex. J. Neurosci. 2012, 32, 17671–17689. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Bai, X.; Bowen, J.R.; Dolat, L.; Korobova, F.; Yu, W.; Baas, P.W.; Svitkina, T.; Gallo, G.; Spiliotis, E.T. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Curr. Biol. 2012, 22, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Efimova, N.; Yang, C.; Chia, J.X.; Li, N.; Lengner, C.J.; Neufeld, K.L.; Svitkina, T.M. Branched actin networks are assembled on microtubules by adenomatous polyposis coli for targeted membrane protrusion. J. Cell Biol. 2020, 219, e202003091. [Google Scholar] [CrossRef] [PubMed]
- Armijo-Weingart, L.; Gallo, G. It takes a village to raise a branch: Cellular mechanisms of the initiation of axon collateral branches. Mol. Cell. Neurosci. 2017, 84, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Ruthel, G.; Banker, G. Actin-dependent anterograde movement of growth-cone-like structures along growing hippocampal axons: A novel form of axonal transport? Cell Motil. Cytoskeleton 1998, 40, 160–173. [Google Scholar] [CrossRef]
- Ruthel, G.; Banker, G. Role of moving growth cone-like “wave” structures in the outgrowth of cultured hippocampal axons and dendrites. J. Neurobiol. 1999, 39, 97–106. [Google Scholar] [CrossRef]
- Flynn, K.C.; Pak, C.W.; Shaw, A.E.; Bradke, F.; Bamburg, J.R. Growth cone-like waves transport actin and promote axonogenesis and neurite branching. Dev. Neurobiol. 2009, 69, 761–779. [Google Scholar] [CrossRef]
- Winans, A.M.; Collins, S.R.; Meyer, T. Waves of actin and microtubule polymerization drive microtubule-based transport and neurite growth before single axon formation. Elife 2016, 5, e12387. [Google Scholar] [CrossRef]
- Mortal, S.; Iseppon, F.; Perissinotto, A.; D’Este, E.; Cojoc, D.; Napolitano, L.M.R.; Torre, V. Actin waves do not boost neurite outgrowth in the early stages of neuron maturation. Front. Cell. Neurosci. 2017, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, T.; Uesugi, Y.; Kaneko, N.; Yoshida, W.; Sawamoto, K.; Inagaki, N. Shootin1b mediates a mechanical clutch to produce force for neuronal migration. Cell Rep. 2018, 25, 624–639.e6. [Google Scholar] [CrossRef]
- Minegishi, T.; Inagaki, N. Forces to drive neuronal migration steps. Front. Cell Dev. Biol. 2020, 8, 863. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, N.; Dubey, P.; Tang, Y.; Ganguly, A.; Ladt, K.; Leterrier, C.; Jung, P.; Roy, S. Processive flow by biased polymerization mediates the slow axonal transport of actin. J. Cell Biol. 2019, 218, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef]
- Unsain, N.; Stefani, F.D.; Cáceres, A. The actin/spectrin membrane-associated periodic skeleton in neurons. Front. Synaptic Neurosci. 2018, 10, 10. [Google Scholar] [CrossRef]
- Vassilopoulos, S.; Gibaud, S.; Jimenez, A.; Caillol, G.; Leterrier, C. Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. Nat. Commun. 2019, 10, 5803. [Google Scholar] [CrossRef]
- Han, B.; Zhou, R.; Xia, C.; Zhuang, X. Structural organization of the actin-spectrin-based membrane skeleton in dendrites and soma of neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E6678–E6685. [Google Scholar] [CrossRef]
- Albrecht, D.; Winterflood, C.M.; Sadeghi, M.; Tschager, T.; Noé, F.; Ewers, H. Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J. Cell Biol. 2016, 215, 37–46. [Google Scholar] [CrossRef]
- Costa, A.R.; Sousa, S.C.; Pinto-Costa, R.; Mateus, J.C.; Lopes, C.D.; Costa, A.C.; Rosa, D.; Machado, D.; Pajuelo, L.; Wang, X.; et al. The membrane periodic skeleton is an actomyosin network that regulates axonal diameter and conduction. Elife 2020, 9, e55471. [Google Scholar] [CrossRef]
- Lavoie-Cardinal, F.; Bilodeau, A.; Lemieux, M.; Gardner, M.-A.; Wiesner, T.; Laramée, G.; Gagné, C.; De Koninck, P. Neuronal activity remodels the f-actin based submembrane lattice in dendrites but not axons of hippocampal neurons. Sci. Rep. 2020, 10, 11960. [Google Scholar] [CrossRef]
- Leterrier, C. Putting the Axonal Periodic Scaffold in Order. Curr. Opin. Neurobiol. 2021, 69, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Han, B.; Xia, C.; Zhuang, X. Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science 2019, 365, 929–934. [Google Scholar] [CrossRef]
- González-Tapia, D.; González-Tapia, D.C.; Vázquez-Hernández, N.; Martínez-Torres, N.I.; Flores-Soto, M.; González-Burgos, I. Modifications to cytoskeleton-associated proteins in dendritic spines underlie the adaptive plasticity involved in long term reference memory. Neurobiol. Learn. Mem. 2020, 172, 107247. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Bardai, F.; Olsen, A.L.; Lohr, K.M.; Zhang, Y.-Y.; Feany, M.B. Oligomerization of Lrrk Controls actin severing and α-synuclein neurotoxicity in vivo. Mol. Neurodegener. 2021, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Leverenz, J.B.; Schneider, J.S.; Adler, C.H. The neurobiological basis of cognitive impairment in Parkinson’s Disease. Mov. Disord. 2014, 29, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Iida, K.; Yonezawa, N.; Koyasu, S.; Yahara, I.; Sakai, H. Cofilin is a component of intranuclear and cytoplasmic actin rods induced in cultured cells. Proc. Natl. Acad. Sci. USA 1987, 84, 5262–5266. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa-Ankerhold, H.C.; Daszkiewicz, W.; Schleicher, M.; Müller-Taubenberger, A. Actin-interacting protein 1 contributes to intranuclear rod assembly in dictyostelium discoideum. Sci. Rep. 2017, 7, 40310. [Google Scholar] [CrossRef]
- Matsuzaki, F.; Matsumoto, S.; Yahara, I.; Yonezawa, N.; Nishida, E.; Sakai, H. Cloning and characterization of porcine brain cofilin CDNA. Cofilin contains the nuclear transport signal sequence. J. Biol. Chem. 1988, 263, 11564–11568. [Google Scholar] [CrossRef]
- Munsie, L.N.; Desmond, C.R.; Truant, R. Cofilin nuclear-cytoplasmic shuttling affects cofilin-actin rod formation during stress. J. Cell Sci. 2012, 125, 3977–3988. [Google Scholar] [CrossRef]
- Ono, S.; Abe, H.; Nagaoka, R.; Obinata, T. Colocalization of ADF and cofilin in intranuclear actin rods of cultured muscle cells. J. Muscle Res. Cell Motil. 1993, 14, 195–204. [Google Scholar] [CrossRef]
- Minamide, L.S.; Maiti, S.; Boyle, J.A.; Davis, R.C.; Coppinger, J.A.; Bao, Y.; Huang, T.Y.; Yates, J.; Bokoch, G.M.; Bamburg, J.R. Isolation and characterization of cytoplasmic cofilin-actin rods. J. Biol. Chem. 2010, 285, 5450–5460. [Google Scholar] [CrossRef]
- Lappalainen, P.; Drubin, D.G. Cofilin promotes rapid actin filament turnover in vivo. Nature 1997, 388, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, A.; Pope, B.; Weeds, A.; Koffer, A. Latrunculin B or ATP depletion induces cofilin-dependent translocation of actin into nuclei of mast cells. J. Biol. Chem. 2003, 278, 14394–14400. [Google Scholar] [CrossRef]
- Stüven, T.; Hartmann, E.; Görlich, D. Exportin 6: A novel nuclear export receptor that is specific for profilin.actin complexes. EMBO J. 2003, 22, 5928–5940. [Google Scholar] [CrossRef] [PubMed]
- Dopie, J.; Skarp, K.-P.; Rajakylä, E.K.; Tanhuanpää, K.; Vartiainen, M.K. Active maintenance of nuclear actin by importin 9 supports transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E544–E552. [Google Scholar] [CrossRef] [PubMed]
- Egly, J.M.; Miyamoto, N.G.; Moncollin, V.; Chambon, P. Is Actin a Transcription Initiation Factor for RNA Polymerase B? EMBO J. 1984, 3, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. Actin and Myosin as Transcription Factors. Curr. Opin. Genet. Dev. 2006, 16, 191–196. [Google Scholar] [CrossRef]
- Willhoft, O.; Wigley, D.B. INO80 and SWR1 Complexes: The non-identical twins of chromatin remodelling. Curr. Opin. Struct. Biol. 2020, 61, 50–58. [Google Scholar] [CrossRef]
- Posern, G.; Sotiropoulos, A.; Treisman, R. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol. Biol. Cell 2002, 13, 4167–4178. [Google Scholar] [CrossRef]
- Kloc, M.; Chanana, P.; Vaughn, N.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. New insights into cellular functions of nuclear actin. Biology 2021, 10, 304. [Google Scholar] [CrossRef]
- Baarlink, C.; Plessner, M.; Sherrard, A.; Morita, K.; Misu, S.; Virant, D.; Kleinschnitz, E.-M.; Harniman, R.; Alibhai, D.; Baumeister, S.; et al. A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 2017, 19, 1389–1399. [Google Scholar] [CrossRef]
- Koy, A.; Ilkovski, B.; Laing, N.; North, K.; Weis, J.; Neuen-Jacob, E.; Mayatepek, E.; Voit, T. Nemaline myopathy with exclusively intranuclear rods and a novel mutation in ACTA1 (Q139H). Neuropediatrics 2007, 38, 282–286. [Google Scholar] [CrossRef]
- Wang, L.; Wang, M.; Wang, S.; Qi, T.; Guo, L.; Li, J.; Qi, W.; Ampah, K.K.; Ba, X.; Zeng, X. Actin polymerization negatively regulates P53 function by impairing its nuclear import in response to DNA Damage. PLoS ONE 2013, 8, e60179. [Google Scholar] [CrossRef]
- Liu, T.; Wang, F.; LePochat, P.; Woo, J.-A.A.; Bukhari, M.Z.; Hong, K.W.; Trotter, C.; Kang, D.E. Cofilin-mediated neuronal apoptosis via P53 Translocation and PLD1 Regulation. Sci. Rep. 2017, 7, 11532. [Google Scholar] [CrossRef]
- Wang, Y.; Sherrard, A.; Zhao, B.; Melak, M.; Trautwein, J.; Kleinschnitz, E.-M.; Tsopoulidis, N.; Fackler, O.T.; Schwan, C.; Grosse, R. GPCR-induced calcium transients trigger nuclear actin assembly for chromatin dynamics. Nat. Commun. 2019, 10, 5271. [Google Scholar] [CrossRef]
- Krippner, S.; Winkelmeier, J.; Knerr, J.; Brandt, D.T.; Virant, D.; Schwan, C.; Endesfelder, U.; Grosse, R. Postmitotic expansion of cell nuclei requires nuclear actin filament bundling by α-actinin 4. EMBO Rep. 2020, 21, e50758. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Chakraborty, S.; Cheng, X.; Su, Y.-T.; Kao, H.-Y. The actin-binding protein, actinin alpha 4 (ACTN4), is a nuclear receptor coactivator that promotes proliferation of MCF-7 breast cancer cells. J. Biol. Chem. 2011, 286, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Ito, Y.; Miura, N.; Nagamura, Y.; Nakabo, A.; Fukami, K.; Honda, K.; Sakai, R. Actinin-1 and actinin-4 play essential but distinct roles in invadopodia formation by carcinoma cells. Eur. J. Cell Biol. 2017, 96, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Tentler, D.; Lomert, E.; Novitskaya, K.; Barlev, N.A. Role of ACTN4 in tumorigenesis, metastasis, and EMT. Cells 2019, 8, 1427. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kang, M.; Kim, S.; An, H.-T.; Gettemans, J.; Ko, J. α-actinin-4 promotes the progression of prostate cancer through the Akt/GSK-3β/β-catenin signaling pathway. Front. Cell Dev. Biol. 2020, 8, 588544. [Google Scholar] [CrossRef] [PubMed]
- Domazetovska, A.; Ilkovski, B.; Cooper, S.T.; Ghoddusi, M.; Hardeman, E.C.; Minamide, L.S.; Gunning, P.W.; Bamburg, J.R.; North, K.N. Mechanisms underlying intranuclear rod formation. Brain 2007, 130, 3275–3284. [Google Scholar] [CrossRef] [PubMed]
- Goebel, H.H.; Warlo, I. Nemaline myopathy with intranuclear rods-Intranuclear rod myopathy. Neuromuscul. Disord. 1997, 7, 13–19. [Google Scholar] [CrossRef]
- Dopie, J.; Rajakylä, E.K.; Joensuu, M.S.; Huet, G.; Ferrantelli, E.; Xie, T.; Jäälinoja, H.; Jokitalo, E.; Vartiainen, M.K. Genome-Wide RNAi screen for nuclear actin reveals a network of cofilin regulators. J. Cell Sci. 2015, 128, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Leu, J.-D.; Lee, Y.-J. The Actin Depolymerizing Factor (ADF)/Cofilin signaling pathway and DNA damage responses in cancer. Int. J. Mol. Sci. 2015, 16, 4095–4120. [Google Scholar] [CrossRef]
- Cheng, C.; Seen, D.; Zheng, C.; Zeng, R.; Li, E. Role of Small GTPase RhoA in DNA damage response. Biomolecules 2021, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Pandey, D.; Siess, W. Phosphorylation-dependent regulation of unique nuclear and nucleolar localization signals of LIM kinase 2 in endothelial cells. J. Biol. Chem. 2006, 281, 25223–25230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, A.; Shi, J.; Fang, Y.; Gu, C.; Cai, J.; Lin, C.; Zhao, L.; Liu, S. Imbalanced LIMK1 and LIMK2 expression leads to human colorectal cancer progression and metastasis via promoting β-catenin nuclear translocation. Cell Death Dis. 2018, 9, 749. [Google Scholar] [CrossRef]
- Clark, A.; Burleson, M. SPOP and cancer: A systematic review. Am. J. Cancer Res. 2020, 10, 704–726. [Google Scholar] [PubMed]
- Nikhil, K.; Haymour, H.S.; Kamra, M.; Shah, K. Phosphorylation-dependent regulation of SPOP by LIMK2 Promotes castration-resistant prostate cancer. Br. J. Cancer 2021, 124, 995–1008. [Google Scholar] [CrossRef]
- Johnson, E.O.; Chang, K.-H.; Ghosh, S.; Venkatesh, C.; Giger, K.; Low, P.S.; Shah, K. LIMK2 Is a crucial regulator and effector of aurora-a-kinase-mediated malignancy. J. Cell Sci. 2012, 125, 1204–1216. [Google Scholar] [CrossRef] [PubMed]
- Dantas, T.J.; Carabalona, A.; Hu, D.J.K.; Vallee, R.B. Emerging roles for motor proteins in progenitor cell behavior and neuronal migration during brain development. Cytoskeleton 2016, 73, 566–576. [Google Scholar] [CrossRef]
- Forero-Quintero, L.S.; Raymond, W.; Handa, T.; Saxton, M.N.; Morisaki, T.; Kimura, H.; Bertrand, E.; Munsky, B.; Stasevich, T.J. Live-cell imaging reveals the spatiotemporal organization of endogenous RNA Polymerase II phosphorylation at a single gene. Nat. Commun. 2021, 12, 3158. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ying, J.; Wang, X.; Zhao, T.; Yoon, S.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Hua, F. Mitochondrial Dynamics: A Key role in neurodegeneration and a potential target for neurodegenerative disease. Front. Neurosci. 2021, 15, 654785. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Ji, W.-K.; Higgs, H.N.; Chakrabarti, R. Two distinct actin filament populations have effects on mitochondria, with differences in stimuli and assembly factors. J. Cell Sci. 2019, 132, jcs234435. [Google Scholar] [CrossRef] [PubMed]
- Hatch, A.L.; Ji, W.-K.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments as dynamic reservoirs for Drp1 recruitment. Mol. Biol. Cell 2016, 27, 3109–3121. [Google Scholar] [CrossRef]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L.F. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef]
- Wanderoy, S.; Hees, J.T.; Klesse, R.; Edlich, F.; Harbauer, A.B. Kill one or kill the many: Interplay between mitophagy and apoptosis. Biol. Chem. 2020, 402, 73–88. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. P62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Fang, C.; Woo, J.-A.A.; Liu, T.; Zhao, X.; Cazzaro, S.; Yan, Y.; Matlack, J.; Kee, T.; LePochat, P.; Kang, D.E. SSH1 Impedes SQSTM1/P62 Flux and MAPT/Tau clearance independent of CFL (Cofilin) activation. Autophagy 2020, 1–22. [Google Scholar] [CrossRef]
- Posadas, I.; Pérez-Martínez, F.C.; Guerra, J.; Sánchez-Verdú, P.; Ceña, V. Cofilin Activation Mediates Bax Translocation to Mitochondria during Excitotoxic Neuronal Death. J. Neurochem. 2012, 120, 515–527. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, G.-L.; Vedantam, S.; Li, P.; Field, J. Mitochondrial Shuttling of CAP1 Promotes Actin- and Cofilin-Dependent Apoptosis. J. Cell Sci. 2008, 121, 2913–2920. [Google Scholar] [CrossRef]
- Rehklau, K.; Hoffmann, L.; Gurniak, C.B.; Ott, M.; Witke, W.; Scorrano, L.; Culmsee, C.; Rust, M.B. Cofilin1-Dependent Actin Dynamics Control DRP1-Mediated Mitochondrial Fission. Cell Death Dis. 2017, 8, e3063. [Google Scholar] [CrossRef]
- Phillips, D.M. The Presence of Acetyl Groups of Histones. Biochem. J. 1963, 87, 258–263. [Google Scholar] [CrossRef]
- Gallwitz, D.; Sekeris, C.E. The Acetylation of Histones of Rat Liver Nuclei in Vitro by Acetyl-CoA. Hoppe Seylers Z. Physiol. Chem. 1969, 350, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Fujimoto, D. Histone deacetylase from calf thymus. Biochim. Biophys Acta 1970, 220, 307–316. [Google Scholar] [CrossRef]
- Li, S.; Shi, B.; Liu, X.; An, H.-X. Acetylation and deacetylation of DNA repair proteins in cancers. Front. Oncol. 2020, 10, 573502. [Google Scholar] [CrossRef]
- Tsushima, H.; Emanuele, M.; Polenghi, A.; Esposito, A.; Vassalli, M.; Barberis, A.; Difato, F.; Chieregatti, E. HDAC6 and RhoA are novel players in abeta-driven disruption of neuronal polarity. Nat. Commun. 2015, 6, 7781. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.-J.; Huang, F.-I.; Liou, J.-P.; Yang, C.-R. The Novel Histone de Acetylase 6 Inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.T.; Minamide, L.S.; Kinley, A.W.; Boyle, J.A.; Bamburg, J.R. Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: A feedforward mechanism for Alzheimer’s disease. J. Neurosci. 2005, 25, 11313–11321. [Google Scholar] [CrossRef]
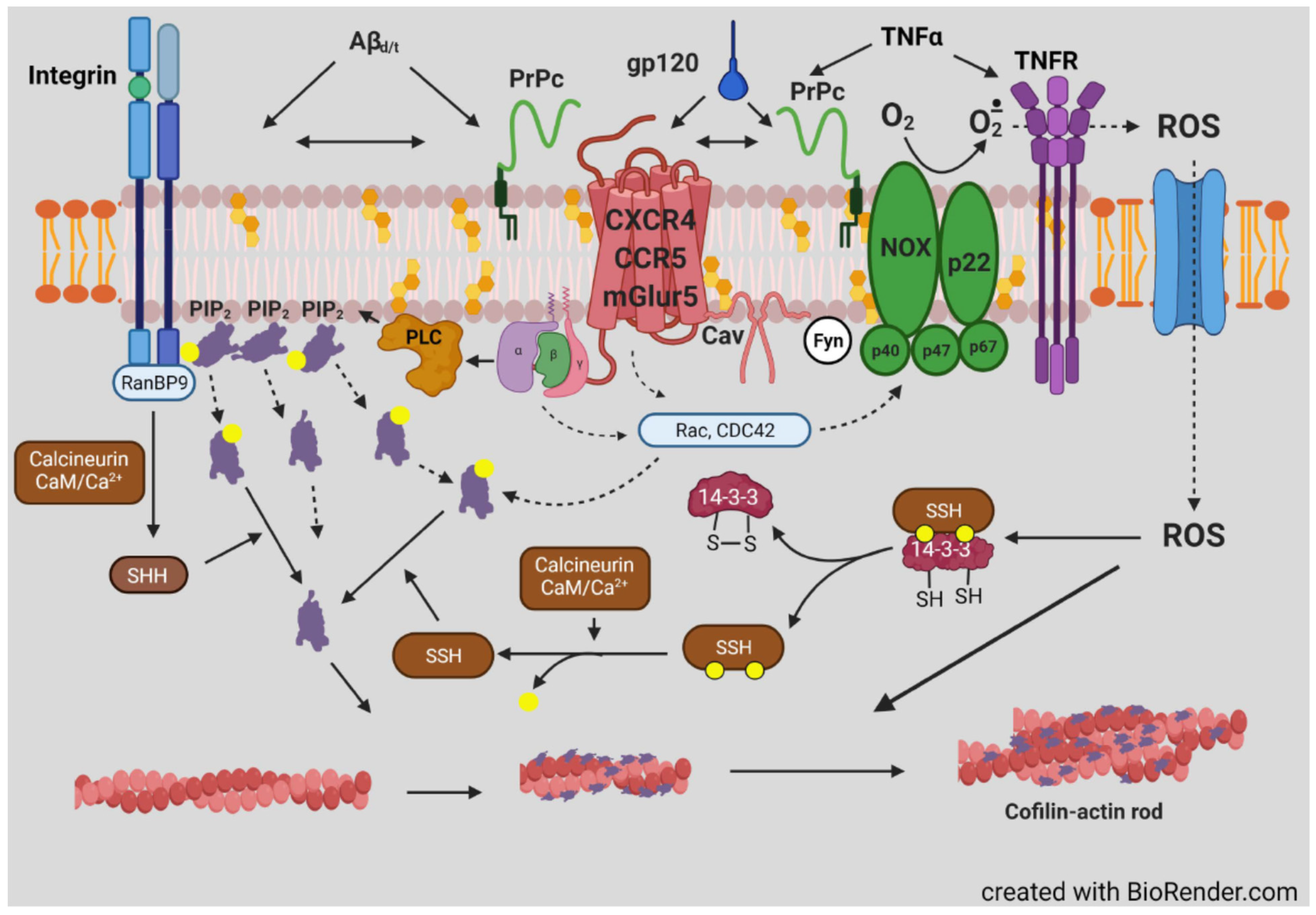
- Walsh, K.P.; Minamide, L.S.; Kane, S.J.; Shaw, A.E.; Brown, D.R.; Pulford, B.; Zabel, M.D.; Lambeth, J.D.; Kuhn, T.B.; Bamburg, J.R. Amyloid-β and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS ONE 2014, 9, e95995. [Google Scholar] [CrossRef]
- Whiteman, I.T.; Gervasio, O.L.; Cullen, K.M.; Guillemin, G.J.; Jeong, E.V.; Witting, P.K.; Antao, S.T.; Minamide, L.S.; Bamburg, J.R.; Goldsbury, C. Activated actin-depolymerizing factor/cofilin sequesters phosphorylated microtubule-associated protein during the assembly of alzheimer-like neuritic cytoskeletal striations. J. Neurosci. 2009, 29, 12994–13005. [Google Scholar] [CrossRef]
- Whiteman, I.T.; Minamide, L.S.; Goh, D.L.; Bamburg, J.R.; Goldsbury, C. Rapid Changes in Phospho-MAP/tau epitopes during neuronal stress: Cofilin-actin rods primarily recruit microtubule binding domain epitopes. PLoS ONE 2011, 6, e20878. [Google Scholar] [CrossRef] [PubMed]
- Munsie, L.; Caron, N.; Atwal, R.S.; Marsden, I.; Wild, E.J.; Bamburg, J.R.; Tabrizi, S.J.; Truant, R. Mutant huntingtin causes defective actin remodeling during stress: Defining a new role for transglutaminase 2 in neurodegenerative disease. Hum. Mol. Genet. 2011, 20, 1937–1951. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Lasso, D.C.; Mollá, B.; Calap-Quintana, P.; García-Giménez, J.L.; Pallardo, F.V.; Palau, F.; Gonzalez-Cabo, P. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci. Rep. 2020, 10, 5207. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, S.; Verheijen, B.M.; Hensel, N.; Peters, M.; Bora, G.; Brandes, G.; Vieira de Sá, R.; Heidrich, N.; Fischer, S.; Brinkmann, H.; et al. Metalloprotease-Mediated Cleavage of PlexinD1 and Its Sequestration to Actin Rods in the Motoneuron Disease Spinal Muscular Atrophy (SMA). Hum. Mol. Genet. 2017, 26, 3946–3959. [Google Scholar] [CrossRef]
- Walter, L.M.; Rademacher, S.; Pich, A.; Claus, P. Profilin2 regulates actin rod assembly in neuronal cells. Sci. Rep. 2021, 11, 10287. [Google Scholar] [CrossRef]
- Walter, L.M.; Franz, P.; Lindner, R.; Tsiavaliaris, G.; Hensel, N.; Claus, P. Profilin2a-phosphorylation as a regulatory mechanism for actin dynamics. FASEB J. 2020, 34, 2147–2160. [Google Scholar] [CrossRef]
- Mi, J.; Shaw, A.E.; Pak, C.W.; Walsh, K.P.; Minamide, L.S.; Bernstein, B.W.; Kuhn, T.B.; Bamburg, J.R. A Genetically encoded reporter for real-time imaging of cofilin-actin rods in living neurons. PLoS ONE 2013, 8, e83609. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, M.; Zhou, M.; Chen, L.; Liu, X.; Wang, X.; Wang, Y. Both NMDA and Non-NMDA receptors mediate glutamate stimulation induced cofilin rod formation in cultured hippocampal neurons. Brain Res. 2012, 1486, 1–13. [Google Scholar] [CrossRef]
- Won, S.J.; Minnella, A.M.; Wu, L.; Eun, C.H.; Rome, E.; Herson, P.S.; Shaw, A.E.; Bamburg, J.R.; Swanson, R.A. Cofilin-actin rod formation in neuronal processes after brain ischemia. PLoS ONE 2018, 13, e0198709. [Google Scholar] [CrossRef]
- Shu, L.; Chen, B.; Chen, B.; Xu, H.; Wang, G.; Huang, Y.; Zhao, Y.; Gong, H.; Jiang, M.; Chen, L.; et al. Brain ischemic insult induces cofilin rod formation leading to synaptic dysfunction in neurons. J. Cereb. Blood Flow Metab. 2019, 39, 2181–2195. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lin, W.; Qi, W.; Li, S.; Hong, Z.; Zhao, H. Cofilin inhibition by limk1 reduces rod formation and cell apoptosis after ischemic stroke. Neuroscience 2020, 444, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, S.L.; Sandoval, R.M.; Hosford, M.; Bamburg, J.R.; Molitoris, B.A. Ischemic Injury Induces ADF relocalization to the apical domain of rat proximal tubule cells. Am. J. Physiol. Renal Physiol. 2001, 280, F886–F894. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An english translation of Alzheimer’s 1907 paper, “uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Cuveillier, C.; Boulan, B.; Ravanello, C.; Denarier, E.; Deloulme, J.-C.; Gory-Fauré, S.; Delphin, C.; Bosc, C.; Arnal, I.; Andrieux, A. Beyond neuronal microtubule stabilization: MAP6 and CRMPS, two converging stories. Front. Mol. Neurosci. 2021, 14, 665693. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-Type Dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Minatani, S.; Takeuchi, J.; Takeda, A.; Kawabe, J.; Wada, Y.; Mawatari, A.; Watanabe, Y.; Shimada, H.; Higuchi, M.; et al. Heavy tau burden with subtle amyloid β accumulation in the cerebral cortex and cerebellum in a case of familial Alzheimer’s Disease with APP osaka mutation. Int. J. Mol. Sci. 2020, 21, 4443. [Google Scholar] [CrossRef]
- Rahman, T.; Davies, D.S.; Tannenberg, R.K.; Fok, S.; Shepherd, C.; Dodd, P.R.; Cullen, K.M.; Goldsbury, C. Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1443–1460. [Google Scholar] [CrossRef]
- Cichon, J.; Sun, C.; Chen, B.; Jiang, M.; Chen, X.A.; Sun, Y.; Wang, Y.; Chen, G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J. Biol. Chem. 2012, 287, 3919–3929. [Google Scholar] [CrossRef]
- Havekes, R.; Park, A.J.; Tudor, J.C.; Luczak, V.G.; Hansen, R.T.; Ferri, S.L.; Bruinenberg, V.M.; Poplawski, S.G.; Day, J.P.; Aton, S.J.; et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife 2016, 5, e13424. [Google Scholar] [CrossRef]
- Bolsius, Y.G.; Meerlo, P.; Kas, M.J.; Abel, T.; Havekes, R. Sleep deprivation reduces the density of individual spine subtypes in a branch-specific fashion in CA1 Neurons. J. Sleep Res. 2021, e13438. [Google Scholar] [CrossRef]
- Woo, J.A.; Zhao, X.; Khan, H.; Penn, C.; Wang, X.; Joly-Amado, A.; Weeber, E.; Morgan, D.; Kang, D.E. Slingshot-cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to Β1-integrin conformers. Cell Death Differ. 2015, 22, 1069–1070. [Google Scholar] [CrossRef]
- Deng, Y.; Wei, J.; Cheng, J.; Zhong, P.; Xiong, Z.; Liu, A.; Lin, L.; Chen, S.; Yan, Z. Partial amelioration of synaptic and cognitive deficits by inhibiting cofilin dephosphorylation in an animal model of Alzheimer’s disease. J. Alzheimers Dis. 2016, 53, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Shankar, G.M.; Townsend, M.; Fadeeva, J.V.; Betts, V.; Podlisny, M.B.; Cleary, J.P.; Ashe, K.H.; Rowan, M.J.; et al. The role of cell-derived oligomers of abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1087–1090. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-Beta Oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002, 30, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. Medical research council cognitive function and ageing study the presence of sodium dodecyl sulphate-stable abeta dimers is strongly associated with Alzheimer-type dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef]
- Davis, R.C.; Marsden, I.T.; Maloney, M.T.; Minamide, L.S.; Podlisny, M.; Selkoe, D.J.; Bamburg, J.R. Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol. Neurodegener. 2011, 6, 10. [Google Scholar] [CrossRef]
- Smith, L.K.; Babcock, I.W.; Minamide, L.S.; Shaw, A.E.; Bamburg, J.R.; Kuhn, T.B. Direct Interaction of HIV Gp120 with neuronal CXCR4 and CCR5 receptors induces cofilin-actin rod pathology via a cellular prion protein- and NOX-dependent mechanism. PLoS ONE 2021, 16, e0248309. [Google Scholar] [CrossRef]
- Smith, L.K.; Kuhn, T.B.; Chen, J.; Bamburg, J.R. HIV Associated Neurodegenerative Disorders: A new perspective on the role of lipid rafts in Gp120-mediated neurotoxicity. Curr. HIV Res. 2018, 16, 258–269. [Google Scholar] [CrossRef]
- Ordonez, D.G.; Lee, M.K.; Feany, M.B. α-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 2018, 97, 108–124.e6. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; De Strooper, B. Neuroscience. Promiscuous Alzheimer’s amyloid: Yet another partner. Science 2013, 341, 1354–1355. [Google Scholar] [CrossRef] [PubMed]
- Wiatrak, B.; Piasny, J.; Kuźniarski, A.; Gąsiorowski, K. Interactions of amyloid-β with membrane proteins. Int. J. Mol. Sci. 2021, 22, 6075. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- Cannarozzo, C.; Fred, S.M.; Girych, M.; Biojone, C.; Enkavi, G.; Róg, T.; Vattulainen, I.; Casarotto, P.C.; Castrén, E. Cholesterol-recognition motifs in the transmembrane domain of the tyrosine kinase receptor family: The Case of TRKB. Eur. J. Neurosci. 2021, 53, 3311–3322. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic. Biol. Med. 2011, 51, 171–178. [Google Scholar] [CrossRef]
- Hong, Z.; Staiculescu, M.C.; Hampel, P.; Levitan, I.; Forgacs, G. How Cholesterol regulates endothelial biomechanics. Front. Physiol. 2012, 3, 426. [Google Scholar] [CrossRef]
- Dhawan, S.; Myers, P.; Bailey, D.M.D.; Ostrovsky, A.D.; Evers, J.F.; Landgraf, M. Reactive oxygen species mediate activity-regulated dendritic plasticity through NADPH oxidase and aquaporin regulation. Front. Cell. Neurosci. 2021, 15, 641802. [Google Scholar] [CrossRef]
- Tomasi, V. Signal Transduction in Neurons: Effects of cellular prion protein on fyn kinase and ERK1/2 Kinase. Immun. Ageing 2010, 7, S5. [Google Scholar] [CrossRef]
- Linden, R. The biological function of the prion protein: A cell surface scaffold of signaling modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Pérez, E.; López-Vázquez, S.; Núñez, J.; Villalobos, C.; Núñez, L. Remodeling of intracellular Ca2+ homeostasis in rat hippocampal neurons aged in vitro. Int. J. Mol. Sci. 2020, 21, 1549. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Mori, H. A new bioluminescence-based tool for modulating target proteins in live cells. Sci. Rep. 2019, 9, 18239. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, C.; Jiao, X.; Ashton, A.; Patel, K.; Kossenkov, A.V.; Pestell, R.G. The G protein coupled receptor CCR5 in Cancer. Adv. Cancer Res. 2020, 145, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Gavriel, Y.; Rabinovich-Nikitin, I.; Ezra, A.; Barbiro, B.; Solomon, B. Subcutaneous Administration of AMD3100 into mice models of Alzheimer’s disease ameliorated cognitive impairment, reduced neuroinflammation, and improved pathophysiological markers. J. Alzheimers Dis. 2020, 78, 653–671. [Google Scholar] [CrossRef]
- Friedman-Levi, Y.; Liraz-Zaltsman, S.; Shemesh, C.; Rosenblatt, K.; Kesner, E.L.; Gincberg, G.; Carmichael, S.T.; Silva, A.J.; Shohami, E. Pharmacological blockers of CCR5 and CXCR4 improve recovery after traumatic brain injury. Exp. Neurol. 2021, 338, 113604. [Google Scholar] [CrossRef] [PubMed]
- Liraz-Zaltsman, S.; Friedman-Levi, Y.; Shabashov-Stone, D.; Gincberg, G.; Atrakcy-Baranes, D.; Joy, M.T.; Carmichael, S.T.; Silva, A.J.; Shohami, E. Chemokine Receptors CC Chemokine Receptor 5 and C-X-C motif chemokine receptor 4 are new therapeutic targets for brain recovery after traumatic brain injury. J. Neurotrauma 2021, 38, 2003–2017. [Google Scholar] [CrossRef]
- Joy, M.T.; Ben Assayag, E.; Shabashov-Stone, D.; Liraz-Zaltsman, S.; Mazzitelli, J.; Arenas, M.; Abduljawad, N.; Kliper, E.; Korczyn, A.D.; Thareja, N.S.; et al. CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 2019, 176, 1143–1157.e13. [Google Scholar] [CrossRef]
- Gates, T.M.; Cysique, L.A.; Siefried, K.J.; Chaganti, J.; Moffat, K.J.; Brew, B.J. Maraviroc-Intensified Combined Antiretroviral Therapy Improves Cognition in Virally Suppressed HIV-Associated Neurocognitive Disorder. AIDS 2016, 30, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-F.; Fu, A.K.Y.; Ip, N.Y. Cytokine signaling convergence regulates the microglial state transition in Alzheimer’s Disease. Cell. Mol. Life Sci. 2021, 78, 4703–4712. [Google Scholar] [CrossRef]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Götz, J.; Feany, M.B. Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol. 2015, 25, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Steinhilb, M.L.; Dias-Santagata, D.; Fulga, T.A.; Felch, D.L.; Feany, M.B. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 2007, 18, 5060–5068. [Google Scholar] [CrossRef] [PubMed]
- Madineni, A.; Alhadidi, Q.; Shah, Z.A. Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of ischemia. Mol. Neurobiol. 2016, 53, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Chen, H.; Boyle, J.A.; Bamburg, J.R. Formation of Actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am. J. Physiol. Cell Physiol. 2006, 291, C828–C839. [Google Scholar] [CrossRef]
- Hui, L.; Abbas, T.; Pielak, R.M.; Joseph, T.; Bargonetti, J.; Foster, D.A. Phospholipase D Elevates the Level of MDM2 and Suppresses DNA Damage-Induced Increases in P53. Mol. Cell Biol. 2004, 24, 5677–5686. [Google Scholar] [CrossRef]
- Wolf, M.; Zimmermann, A.-M.; Görlich, A.; Gurniak, C.B.; Sassoè-Pognetto, M.; Friauf, E.; Witke, W.; Rust, M.B. ADF/cofilin controls synaptic actin dynamics and regulates synaptic vesicle mobilization and exocytosis. Cereb. Cortex 2015, 25, 2863–2875. [Google Scholar] [CrossRef]
- Shaw, A.E.; Bamburg, J.R. Peptide Regulation of Cofilin Activity in the CNS: A novel therapeutic approach for treatment of multiple neurological disorders. Pharmacol. Ther. 2017, 175, 17–27. [Google Scholar] [CrossRef]
- Gordon, A.; Gousset, K. Utilization of laser capture microdissection coupled to mass spectrometry to uncover the proteome of cellular protrusions. Methods Mol. Biol. 2021, 2259, 25–45. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamburg, J.R.; Minamide, L.S.; Wiggan, O.; Tahtamouni, L.H.; Kuhn, T.B. Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration. Cells 2021, 10, 2726. https://doi.org/10.3390/cells10102726
Bamburg JR, Minamide LS, Wiggan O, Tahtamouni LH, Kuhn TB. Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration. Cells. 2021; 10(10):2726. https://doi.org/10.3390/cells10102726
Chicago/Turabian StyleBamburg, James R., Laurie S. Minamide, O’Neil Wiggan, Lubna H. Tahtamouni, and Thomas B. Kuhn. 2021. "Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration" Cells 10, no. 10: 2726. https://doi.org/10.3390/cells10102726
APA StyleBamburg, J. R., Minamide, L. S., Wiggan, O., Tahtamouni, L. H., & Kuhn, T. B. (2021). Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration. Cells, 10(10), 2726. https://doi.org/10.3390/cells10102726