
Role of MiR-325-3p in the Regulation of CFL2 and Myogenic Differentiation of C2C12 Myoblasts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Differentiation and PA Treatment
2.2. Transfection of Oligonucleotides
2.3. RNA Extraction, PCR and Quantitative Real-Time PCR (qRT-PCR)
2.4. Dual-Luciferase Assay
2.5. Immunoblot Analysis
2.6. Immunofluorescence Analysis
2.7. Cell Proliferation Assays
2.8. Flow Cytometry
2.9. Database and Statistical Analysis
3. Results
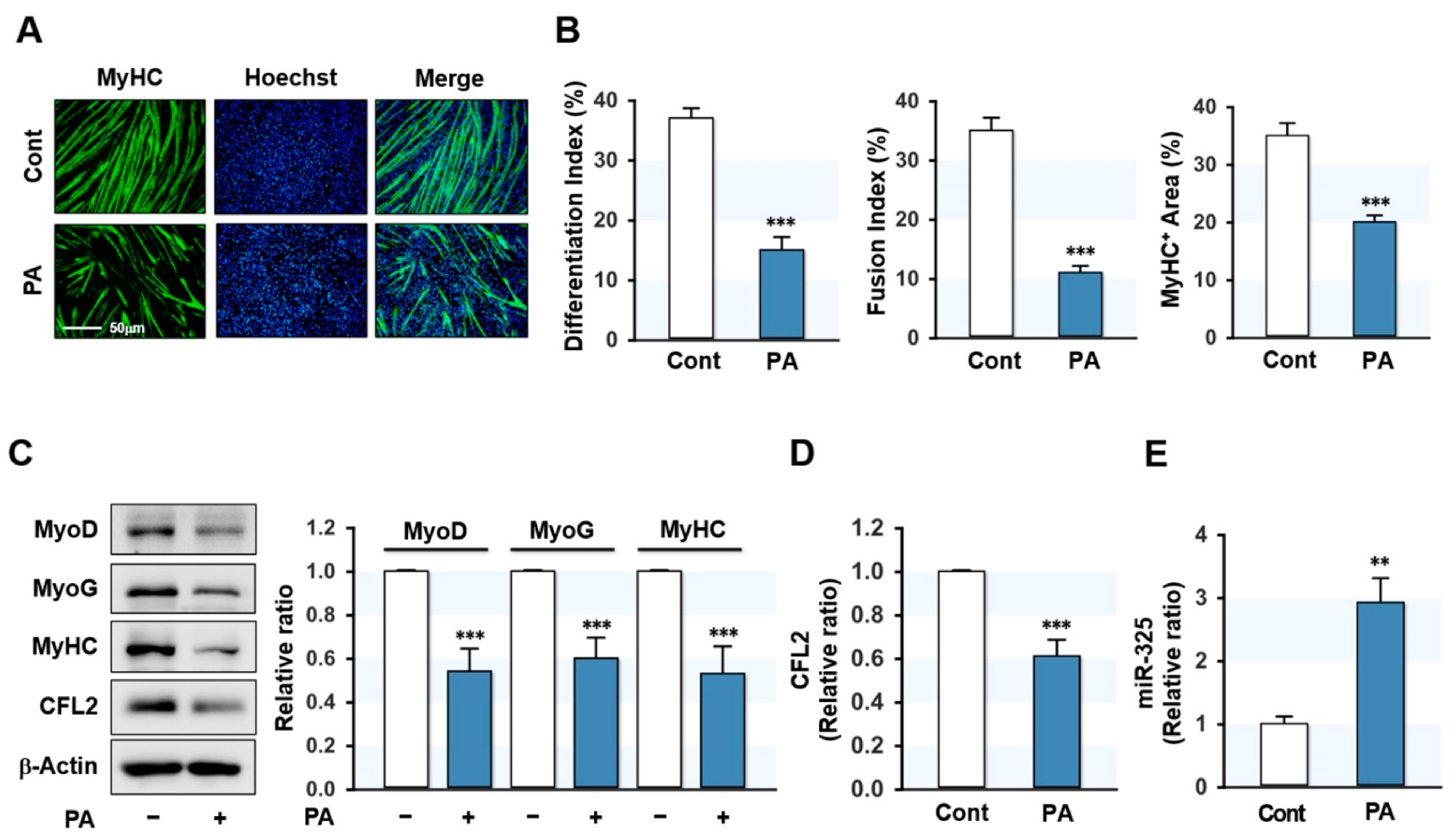
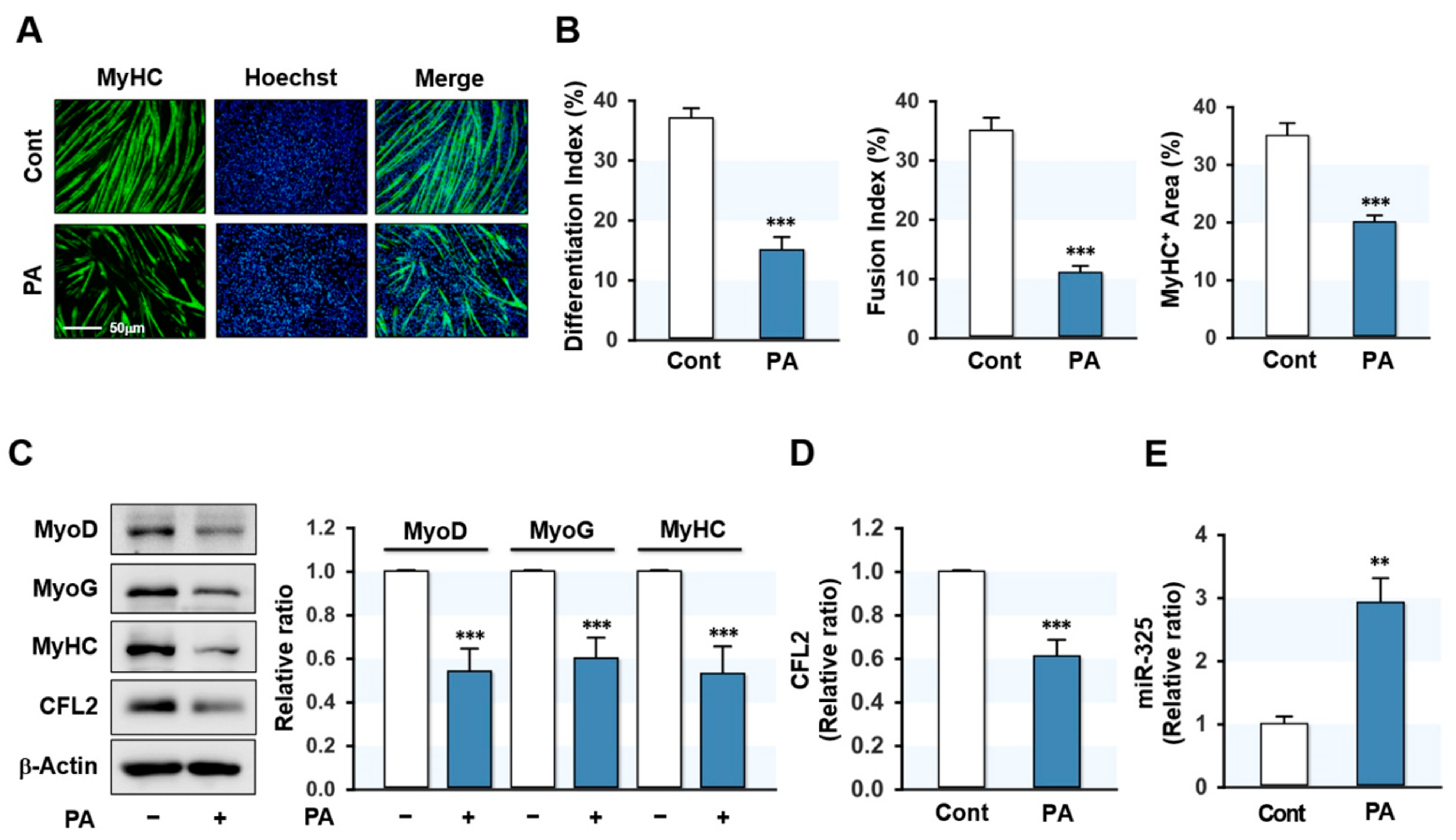
3.1. PA Inhibited Myogenic Differentiation but Elevated miR-325-3p Expression
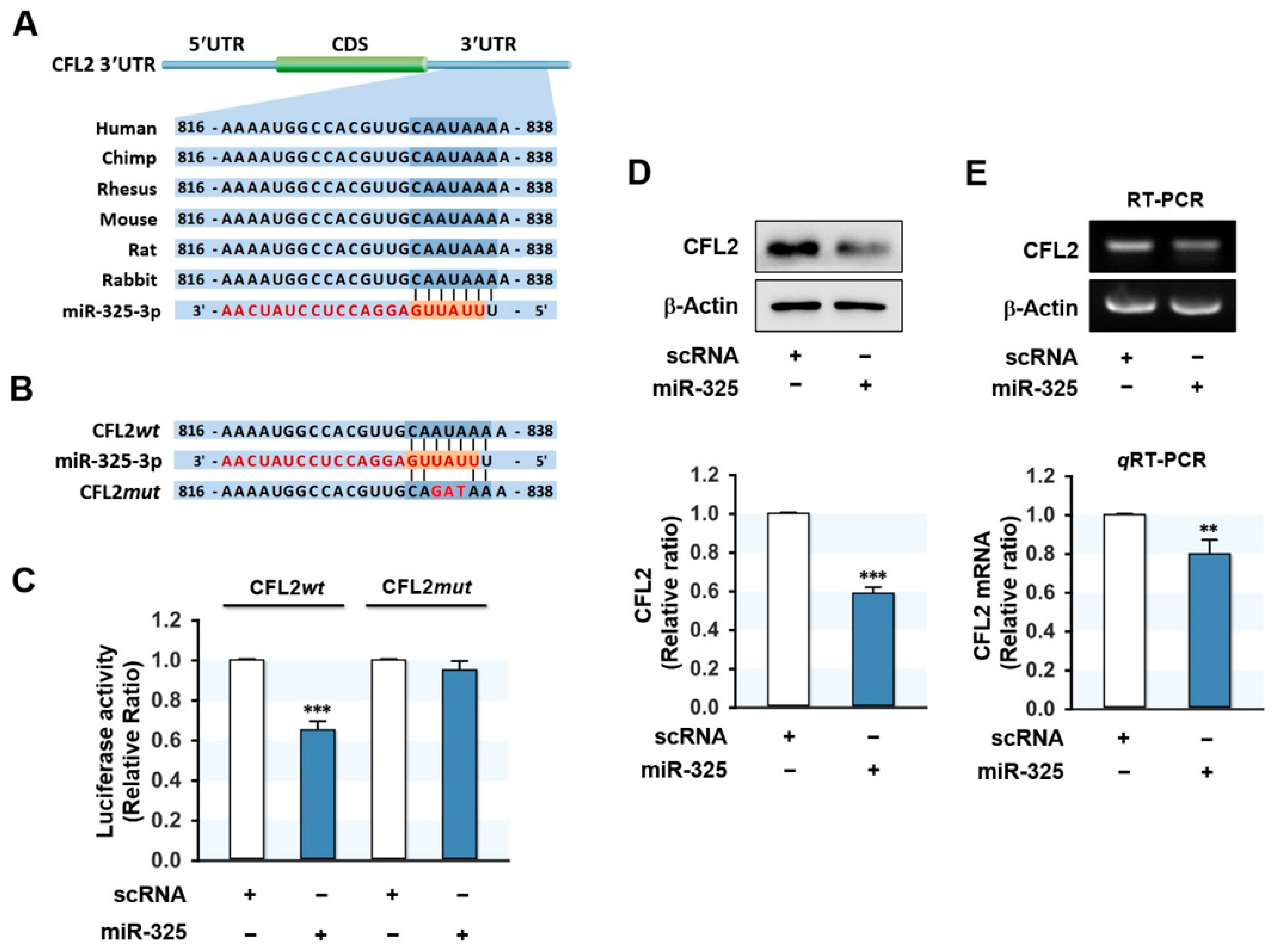
3.2. MiR-325-3p Directly Targeted CFL2 3′UTR
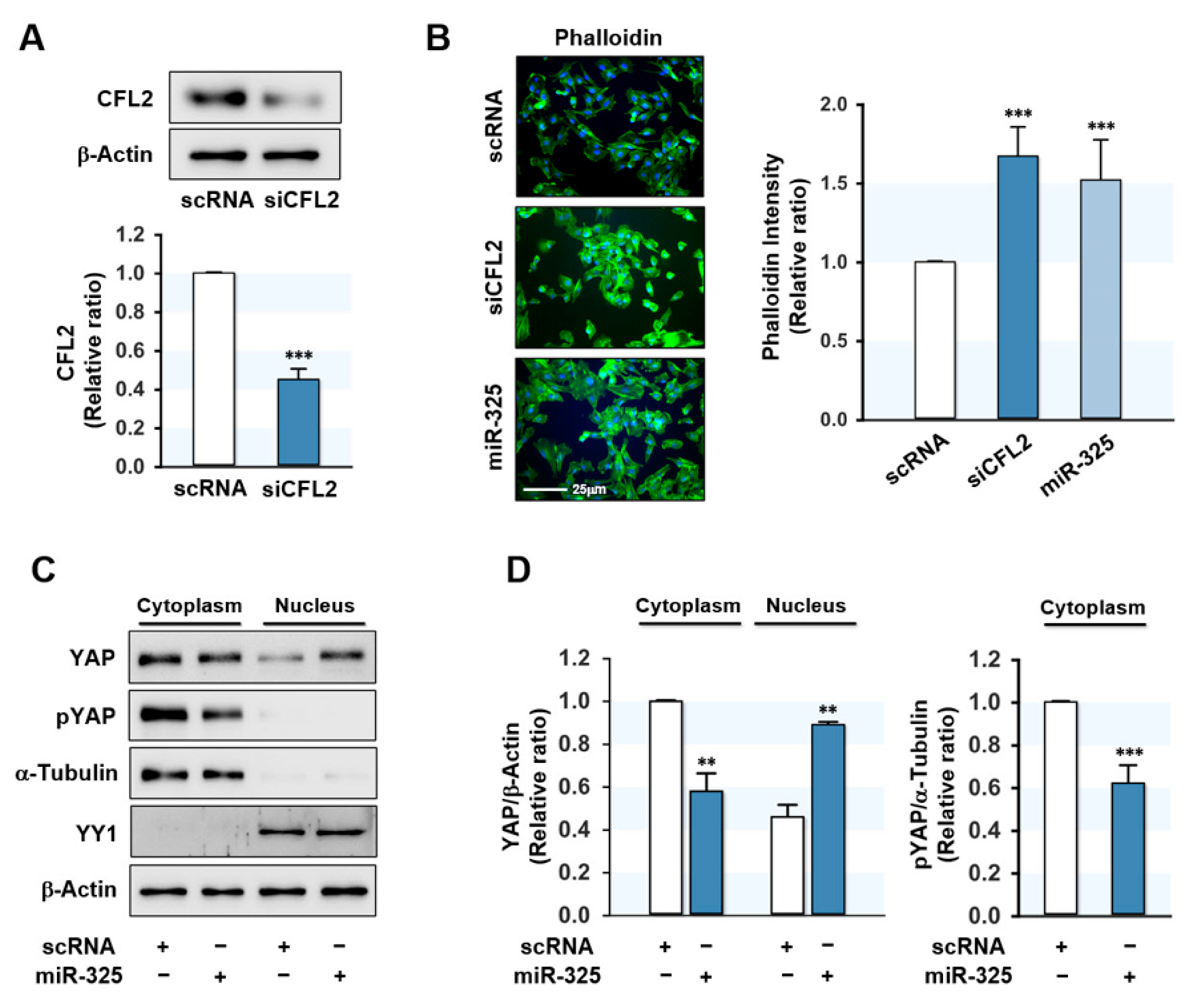
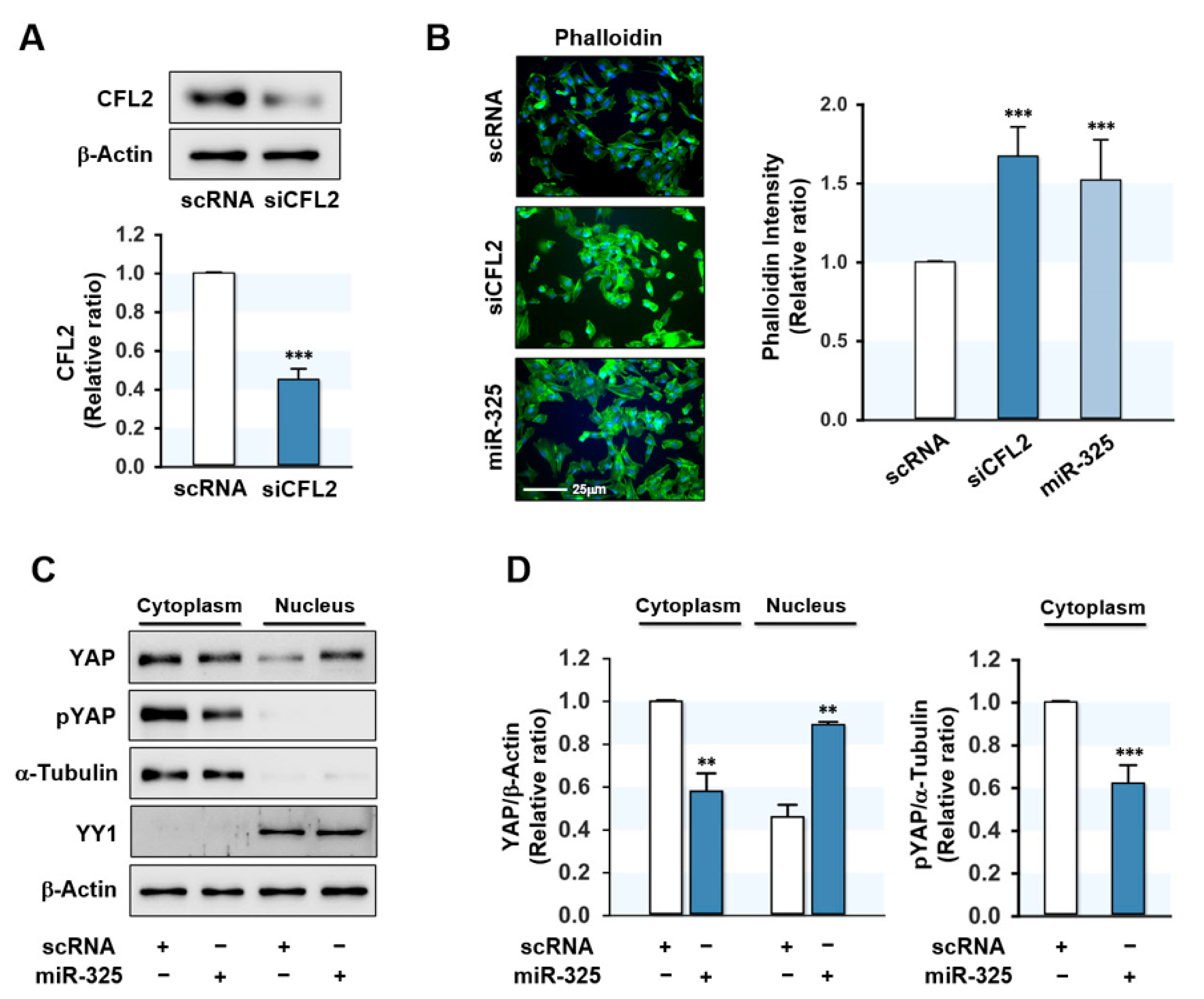
3.3. MiR-325-3p Increased F-Actin and Nuclear Yes-Associated Protein (YAP)
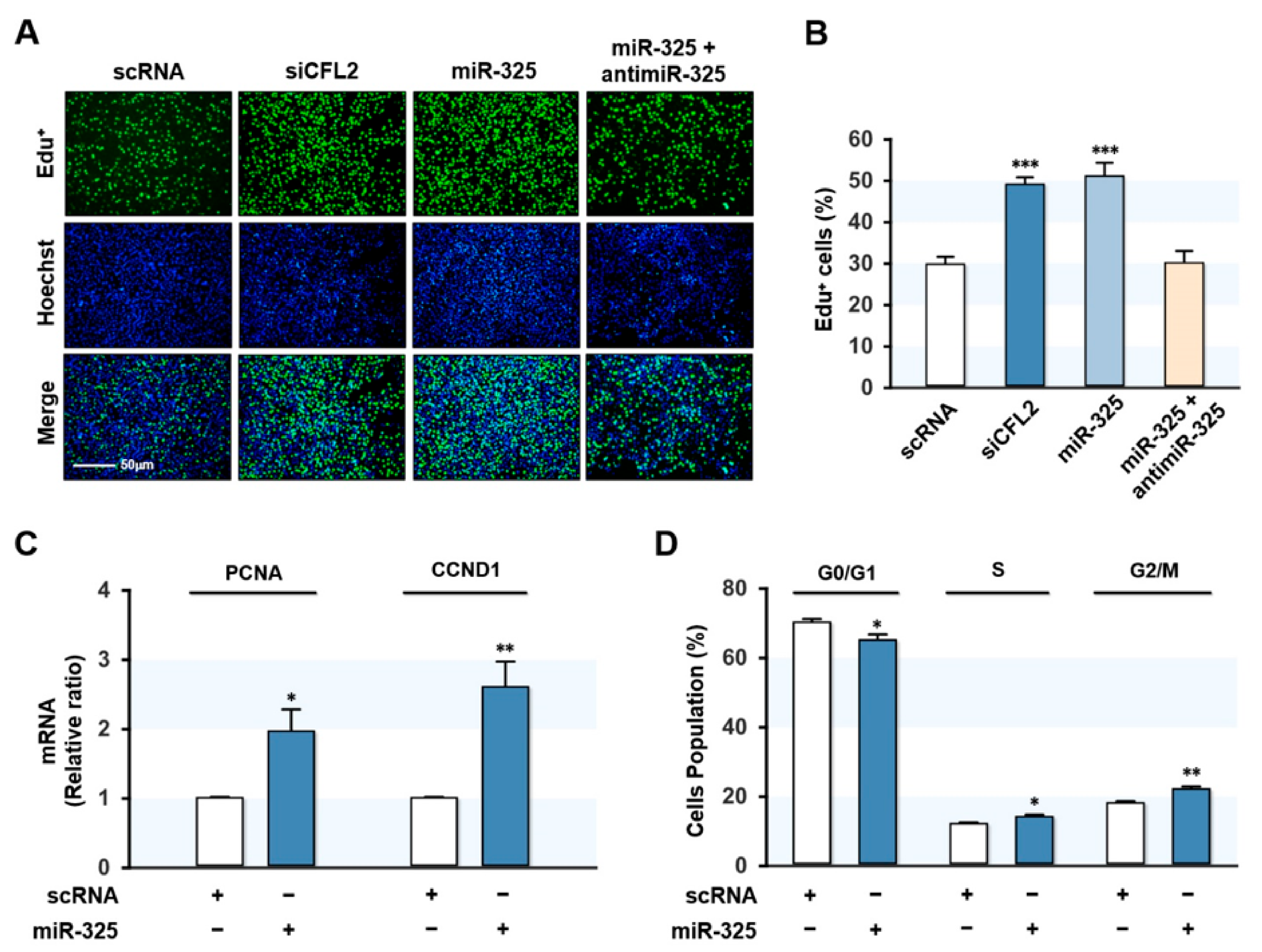
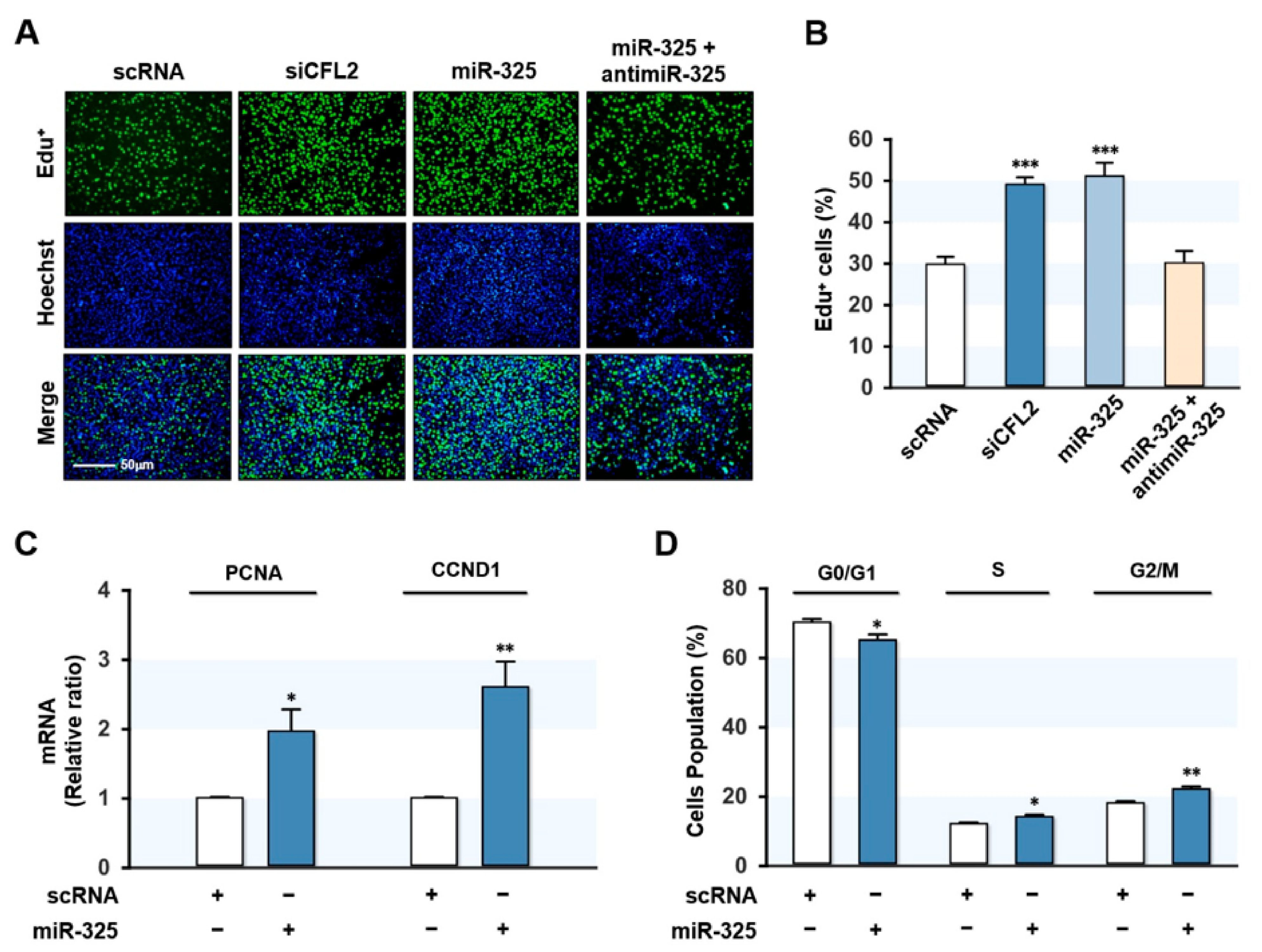
3.4. MiR-325-3p Promoted Myoblast Proliferation
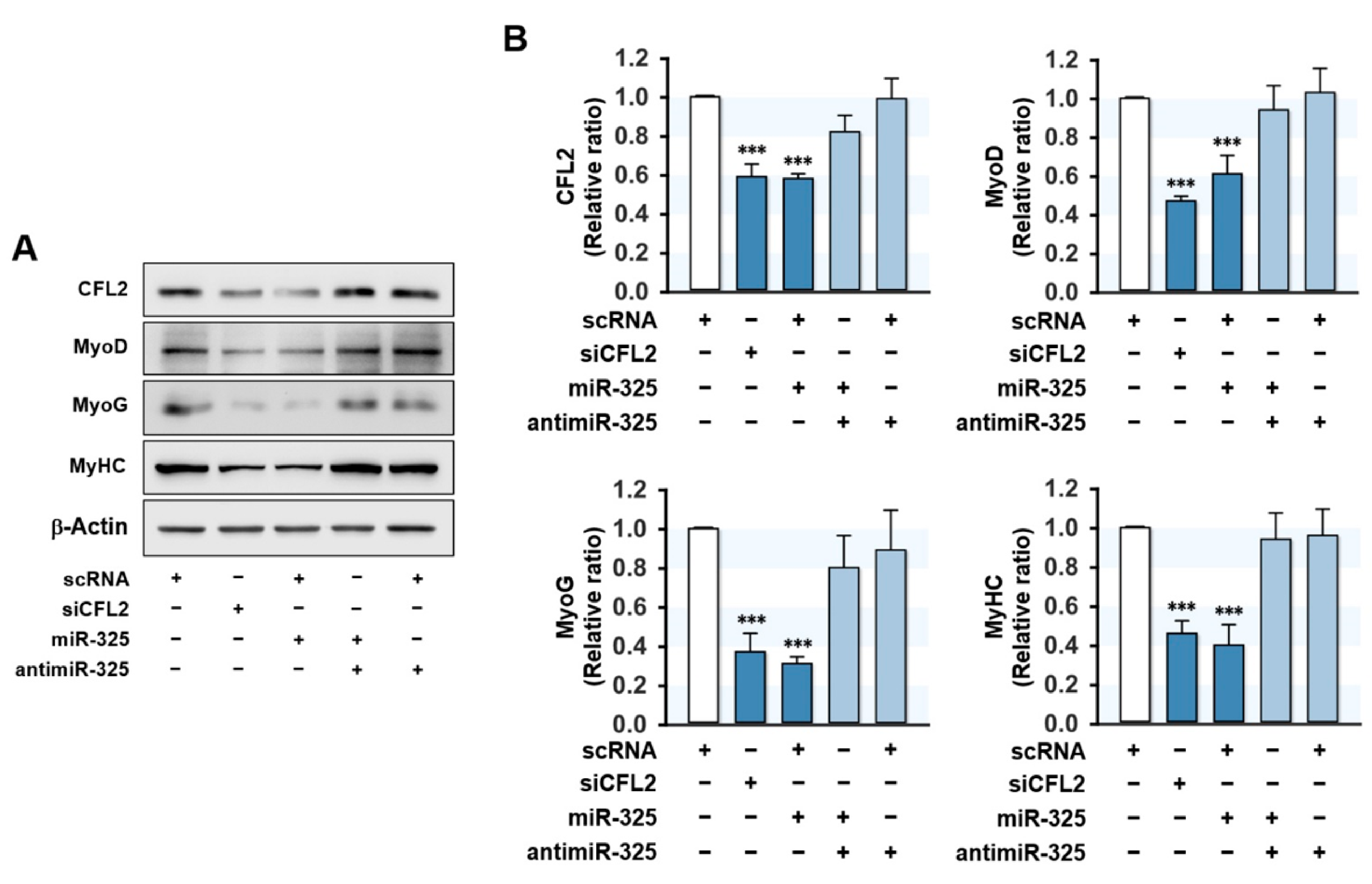
3.5. MiR-325-3p Suppressed the Expressions of Myogenic Factors
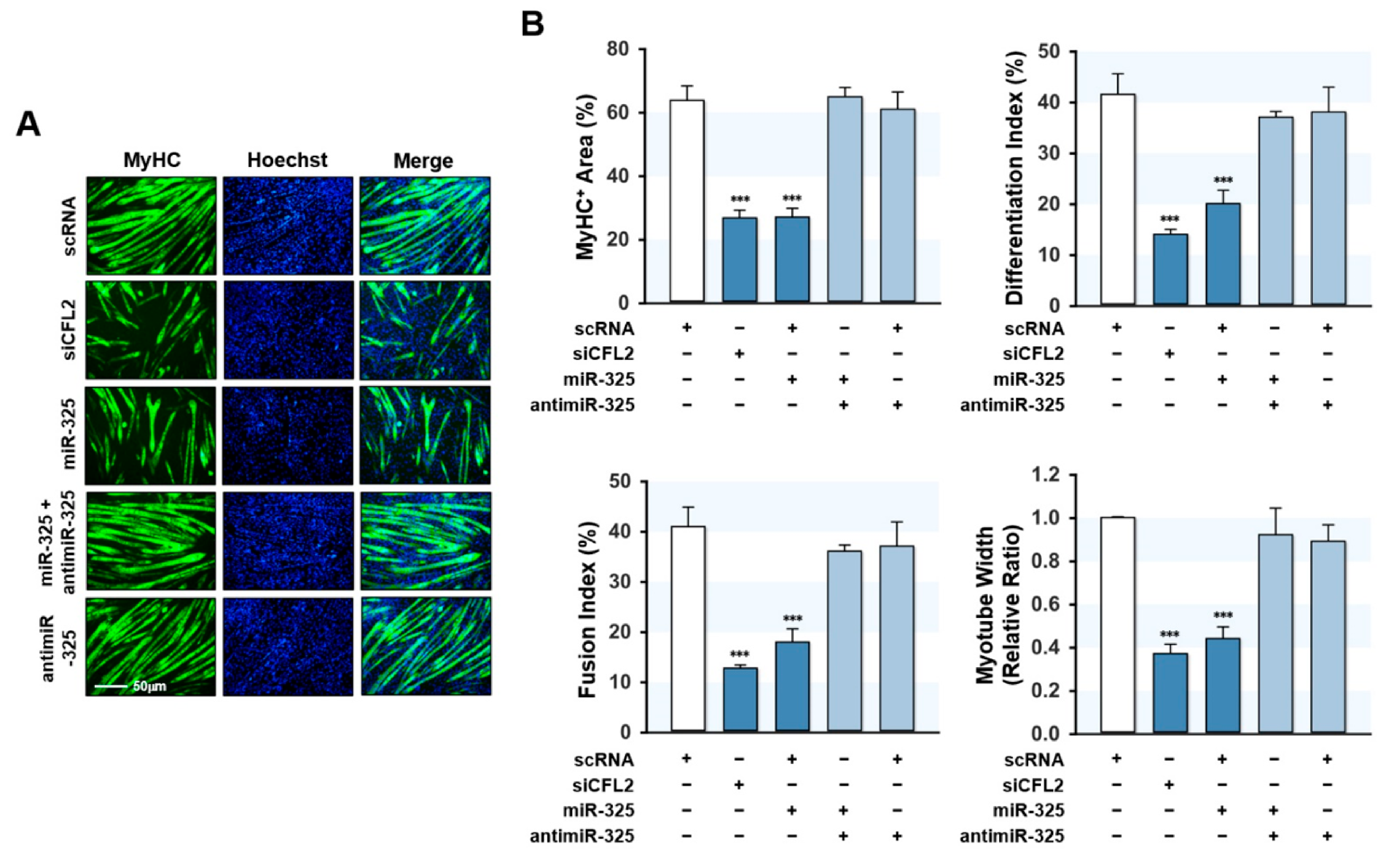
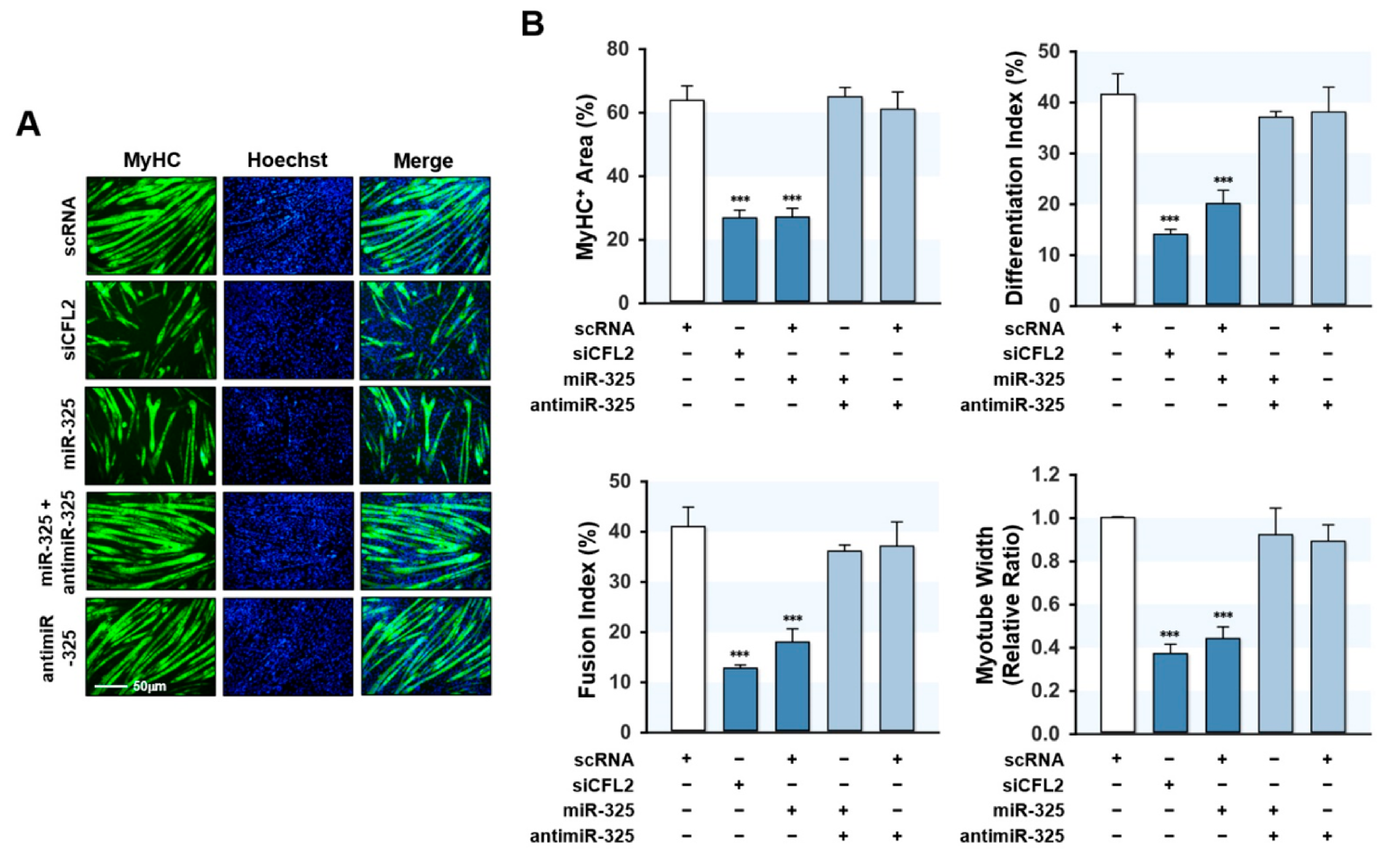
3.6. MiR-325-3p Impeded Myogenic Differentiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; Villareal, D.T. Sarcopenic obesity in older adults: Aetiology, epidemiology and treatment strategies. Nat. Rev. Endocrinol. 2018, 14, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Bischoff, S.; Boirie, Y.; Busetto, L.; Cederholm, T.; Dicker, D.; Toplak, H.; Van Gossum, A.; Yumuk, V.; Vettor, R. Sarcopenic Obesity: Time to Meet the Challenge. Obes. Facts 2018, 11, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, D.; Berdeaux, R. The effects of obesity on skeletal muscle regeneration. Front. Physiol. 2013, 4, 371. [Google Scholar] [CrossRef] [Green Version]
- Teng, S.; Huang, P. The effect of type 2 diabetes mellitus and obesity on muscle progenitor cell function. Stem. Cell Res. Ther. 2019, 10, 103. [Google Scholar] [CrossRef]
- Ji, C.; Guo, X. The clinical potential of circulating microRNAs in obesity. Nat. Rev. Endocrinol. 2019, 15, 731–743. [Google Scholar] [CrossRef]
- Ortiz-Dosal, A.; Rodil-Garcia, P.; Salazar-Olivo, L.A. Circulating microRNAs in human obesity: A systematic review. Biomarkers 2019, 24, 499–509. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, M.; Lian, D.; Li, Y.; Li, Y.; Wang, J.; Deng, S.; Yu, K.; Lian, Z. Non-Coding RNA Regulates the Myogenesis of Skeletal Muscle Satellite Cells, Injury Repair and Diseases. Cells 2019, 8, 988. [Google Scholar] [CrossRef] [Green Version]
- Mok, G.F.; Lozano-Velasco, E.; Munsterberg, A. MicroRNAs in skeletal muscle development. Semin. Cell Dev. Biol. 2017, 72, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Sannicandro, A.J.; Soriano-Arroquia, A.; Goljanek-Whysall, K. Micro(RNA)-managing muscle wasting. J. Appl. Physiol. 2019, 127, 619–632. [Google Scholar] [CrossRef]
- Guerin, C.M.; Kramer, S.G. Cytoskeletal remodeling during myotube assembly and guidance: Coordinating the actin and microtubule networks. Commun. Integr. Biol. 2009, 2, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Heng, Y.W.; Koh, C.G. Actin cytoskeleton dynamics and the cell division cycle. Int. J. Biochem. Cell Biol. 2010, 42, 1622–1633. [Google Scholar] [CrossRef]
- Kanellos, G.; Frame, M.C. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.B.; Joshi, M.; Savic, T.; Chen, Z.; Beggs, A.H. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 2012, 21, 2341–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Winter, J.M.; Ottenheijm, C.A.C. Sarcomere Dysfunction in Nemaline Myopathy. J. Neuromuscul. Dis. 2017, 4, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.; Min, K.H.; Kim, D.; Park, S.Y.; Lee, W. CFL2 is an essential mediator for myogenic differentiation in C2C12 myoblasts. Biochem. Biophys. Res. Commun. 2020, 533, 710–716. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Min, K.H.; Lee, W. MiR-96-5p Induced by Palmitic Acid Suppresses the Myogenic Differentiation of C2C12 Myoblasts by Targeting FHL1. Int. J. Mol. Sci. 2020, 21, 9445. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Jeong, H.J.; Park, S.W.; Lee, W. Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol. Nutr. Food Res. 2015, 59, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.S.; Park, S.Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343. [Google Scholar] [CrossRef]
- Tang, Z.; Yang, Y.; Wang, Z.; Zhao, S.; Mu, Y.; Li, K. Integrated analysis of miRNA and mRNA paired expression profiling of prenatal skeletal muscle development in three genotype pigs. Sci. Rep. 2015, 5, 15544. [Google Scholar] [CrossRef] [Green Version]
- Van Pelt, D.W.; Vechetti, I.J., Jr.; Lawrence, M.M.; Van Pelt, K.L.; Patel, P.; Miller, B.F.; Butterfield, T.A.; Dupont-Versteegden, E.E. Serum extracellular vesicle miR-203a-3p content is associated with skeletal muscle mass and protein turnover during disuse atrophy and regrowth. Am. J. Physiol. Cell Physiol. 2020, 319, C419–C431. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Wang, H.; Hu, X.; Yang, F.; Xiao, H. MiR-325-3p promotes the proliferation, invasion and EMT of breast cancer cells by directly targeting S100A2. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2021, 28, 731–744. [Google Scholar] [CrossRef]
- Song, C.; Wang, X.; Zhao, X.; Ai, J.; Qi, Y.; Chen, A. MicroRNA-325-3p contributes to colorectal carcinoma by targeting cytokeratin 18. Oncol. Lett. 2021, 21, 248. [Google Scholar] [CrossRef]
- Deng, Q.; Hu, H.; Yu, X.; Liu, S.; Wang, L.; Chen, W.; Zhang, C.; Zeng, Z.; Cao, Y.; Xu-Monette, Z.Y.; et al. Tissue-specific microRNA expression alters cancer susceptibility conferred by a TP53 noncoding variant. Nat. Commun. 2019, 10, 5061. [Google Scholar] [CrossRef] [Green Version]
- Braicu, O.L.; Budisan, L.; Buiga, R.; Jurj, A.; Achimas-Cadariu, P.; Pop, L.A.; Braicu, C.; Irimie, A.; Berindan-Neagoe, I. MiRNA expression profiling in formalin-fixed paraffin-embedded endometriosis and ovarian cancer samples. Onco Targets Ther. 2017, 10, 4225–4238. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Wang, X.; Du, M.; Dong, Y. LncRNA MSC-AS1 Promotes Colorectal Cancer Progression by Regulating miR-325/TRIM14 Axis. J. Oncol. 2021, 2021, 9954214. [Google Scholar] [CrossRef]
- Wang, F.; Wang, F.; Zhang, S.; Xu, X. MicroRNA-325 inhibits the proliferation and induces the apoptosis of T cell acute lymphoblastic leukemia cells in a BAG2-dependent manner. Exp. Ther. Med. 2021, 21, 631. [Google Scholar] [CrossRef]
- Li, L.; Ji, Y.; Chen, Y.C.; Zhen, Z.J. MiR-325-3p mediate the CXCL17/CXCR8 axis to regulate angiogenesis in hepatocellular carcinoma. Cytokine 2021, 141, 155436. [Google Scholar] [CrossRef]
- Mendez, M.G.; Janmey, P.A. Transcription factor regulation by mechanical stress. Int. J. Biochem. Cell Biol. 2012, 44, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Xu, Y.; Feng, Y.; Shen, M.; Yuan, F.; Yuan, Y. YAP nuclear-cytoplasmic translocation is regulated by mechanical signaling, protein modification, and metabolism. Cell Biol. Int. 2020, 44, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jo, H.; Hong, H.; Kim, M.H.; Kim, J.M.; Lee, J.K.; Heo, W.D.; Kim, J. Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat. Commun. 2015, 6, 6781. [Google Scholar] [CrossRef]
- Torrini, C.; Cubero, R.J.; Dirkx, E.; Braga, L.; Ali, H.; Prosdocimo, G.; Gutierrez, M.I.; Collesi, C.; Licastro, D.; Zentilin, L.; et al. Common Regulatory Pathways Mediate Activity of MicroRNAs Inducing Cardiomyocyte Proliferation. Cell Rep. 2019, 27, 2759–2771.e2755. [Google Scholar] [CrossRef] [Green Version]
- Denechaud, P.D.; Fajas, L.; Giralt, A. E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. 2017, 8, 311. [Google Scholar] [CrossRef]
- Bo, L.; Su-Ling, D.; Fang, L.; Lu-Yu, Z.; Tao, A.; Stefan, D.; Kun, W.; Pei-Feng, L. Autophagic program is regulated by miR-325. Cell Death Differ. 2014, 21, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Haim, Y.; Blüher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef]
- Choi, Y.; Jang, S.; Choi, M.-S.; Ryoo, Z.Y.; Park, T. Increased expression of FGF1-mediated signaling molecules in adipose tissue of obese mice. J. Physiol. Biochem. 2016, 72, 157–167. [Google Scholar] [CrossRef]
- Pervin, S.; Singh, V.; Tucker, A.; Collazo, J.; Singh, R. Modulation of transforming growth factor-β/follistatin signaling and white adipose browning: Therapeutic implications for obesity related disorders. Horm. Mol. Biol. Clin. Investig. 2017, 31. [Google Scholar] [CrossRef]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from Obesity and Diabetes by Blockade of TGF-β/Smad3 Signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.T.; Lee, W. Role of MiR-325-3p in the Regulation of CFL2 and Myogenic Differentiation of C2C12 Myoblasts. Cells 2021, 10, 2725. https://doi.org/10.3390/cells10102725
Nguyen MT, Lee W. Role of MiR-325-3p in the Regulation of CFL2 and Myogenic Differentiation of C2C12 Myoblasts. Cells. 2021; 10(10):2725. https://doi.org/10.3390/cells10102725
Chicago/Turabian StyleNguyen, Mai Thi, and Wan Lee. 2021. "Role of MiR-325-3p in the Regulation of CFL2 and Myogenic Differentiation of C2C12 Myoblasts" Cells 10, no. 10: 2725. https://doi.org/10.3390/cells10102725
APA StyleNguyen, M. T., & Lee, W. (2021). Role of MiR-325-3p in the Regulation of CFL2 and Myogenic Differentiation of C2C12 Myoblasts. Cells, 10(10), 2725. https://doi.org/10.3390/cells10102725