Unlike its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Viral Vector Transductions and Generation of Stable Cell Lines
2.2. Protein Purification
2.3. AlphaScreen Assay
2.4. Co-Immunoprecipitation (IP)
2.5. Peptide Synthesis
2.6. NMR Spectroscopy
2.7. HRP-2 Knockout Mouse Mode
2.8. Clonogenic Growth In Vitro
2.9. RNA-Sequencing and Bioinformatics
2.10. Statistical Analysis
3. Results
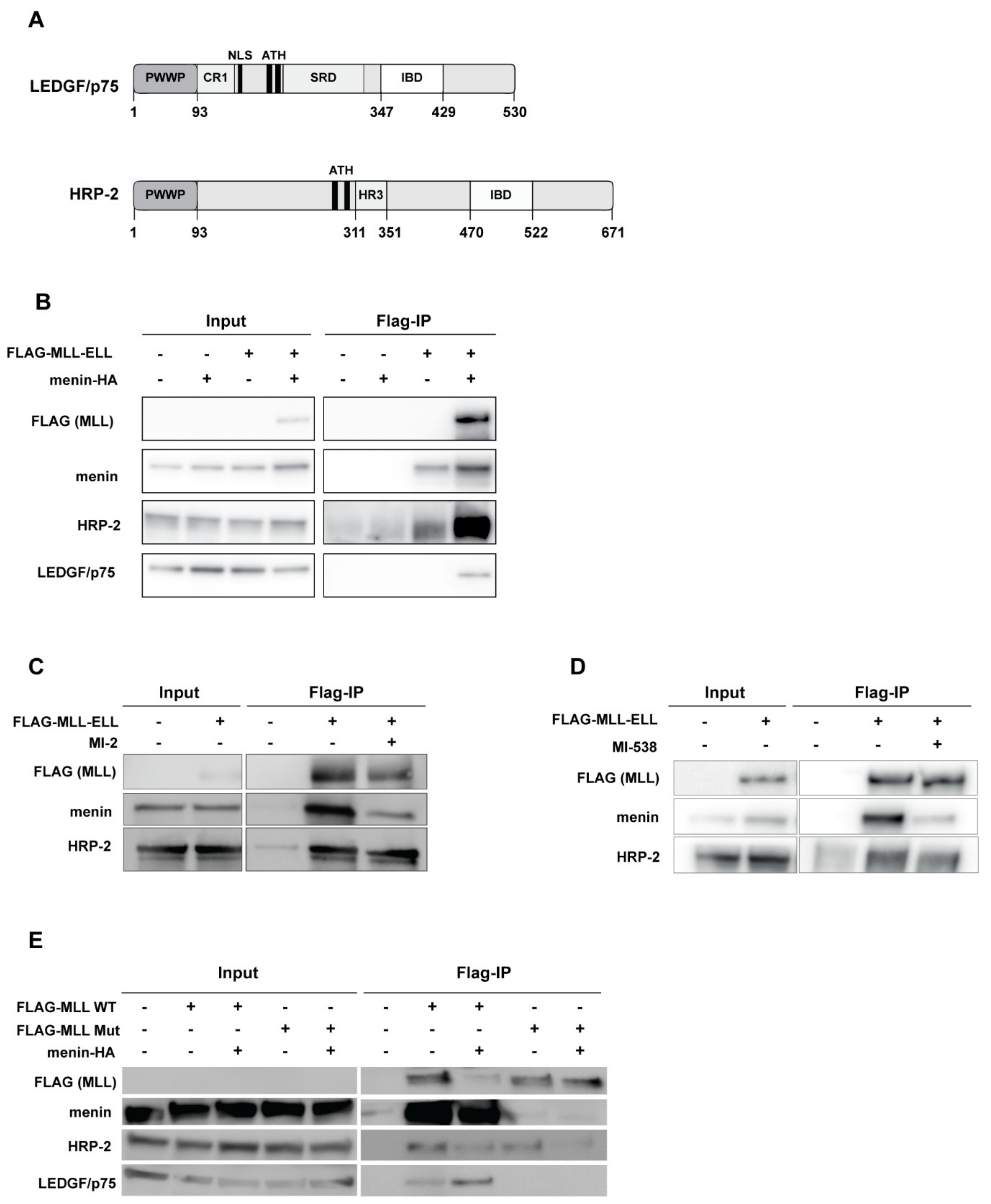
3.1. HRP-2 Interacts with MLL in the Absence of Menin
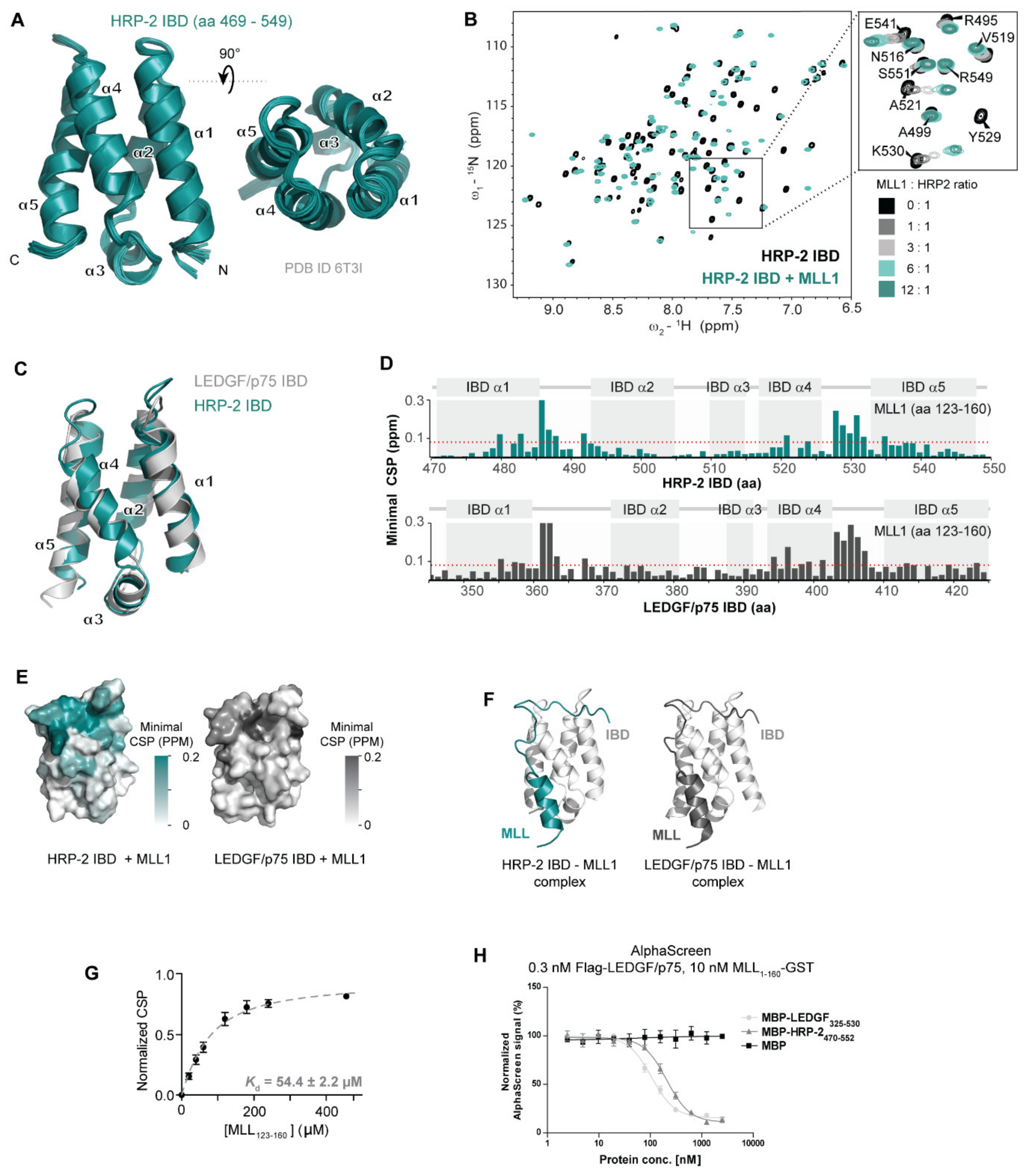
3.2. HRP-2 and LEDGF/p75 Interact with MLL through a Conserved Interface with Similar Affinities
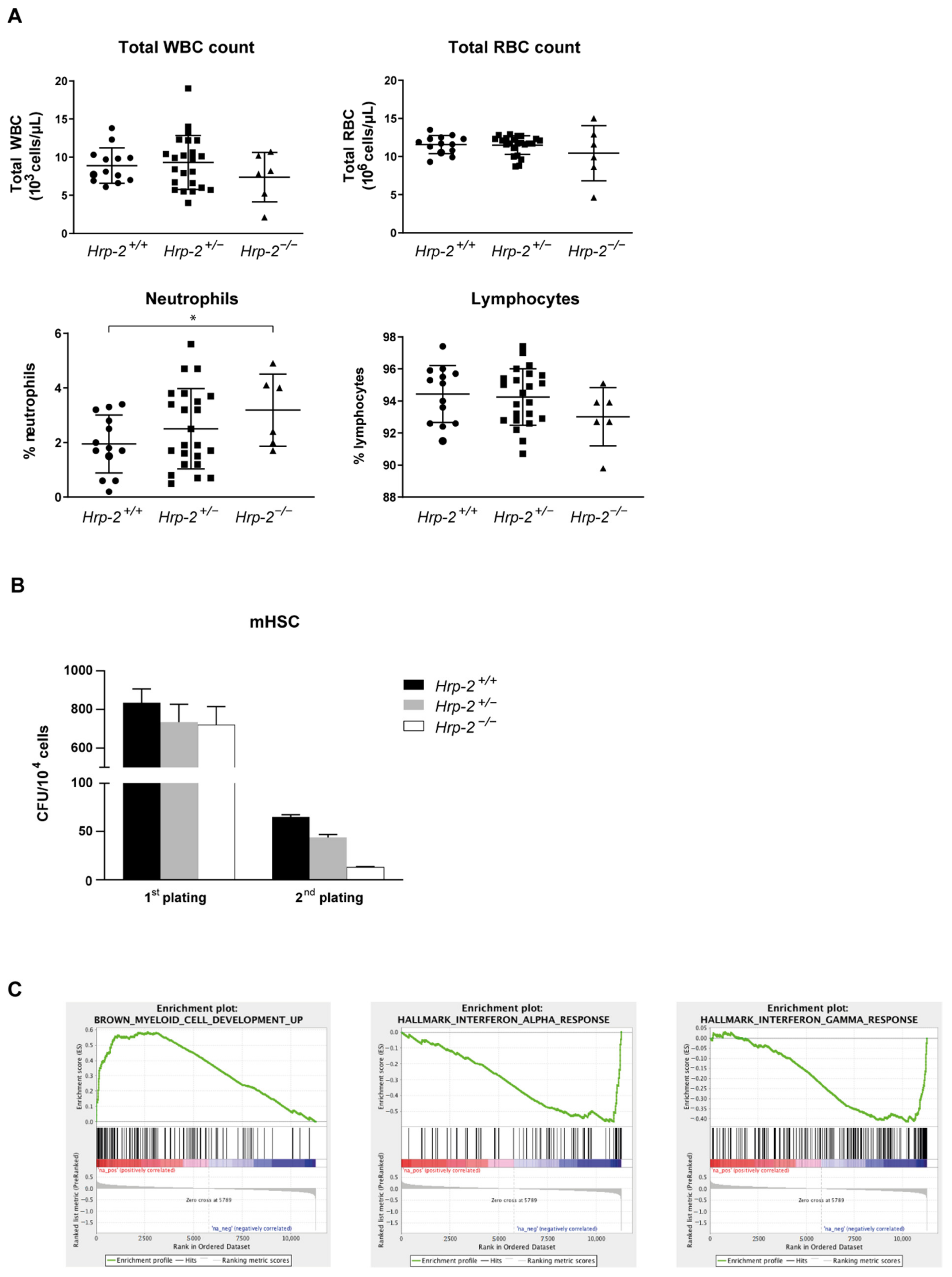
3.3. Systemic HRP-2 Depletion in Mice Leads to Increased Postnatal Mortality and Decreased In Vitro Colony Formation of Hematopoietic Stem Cells
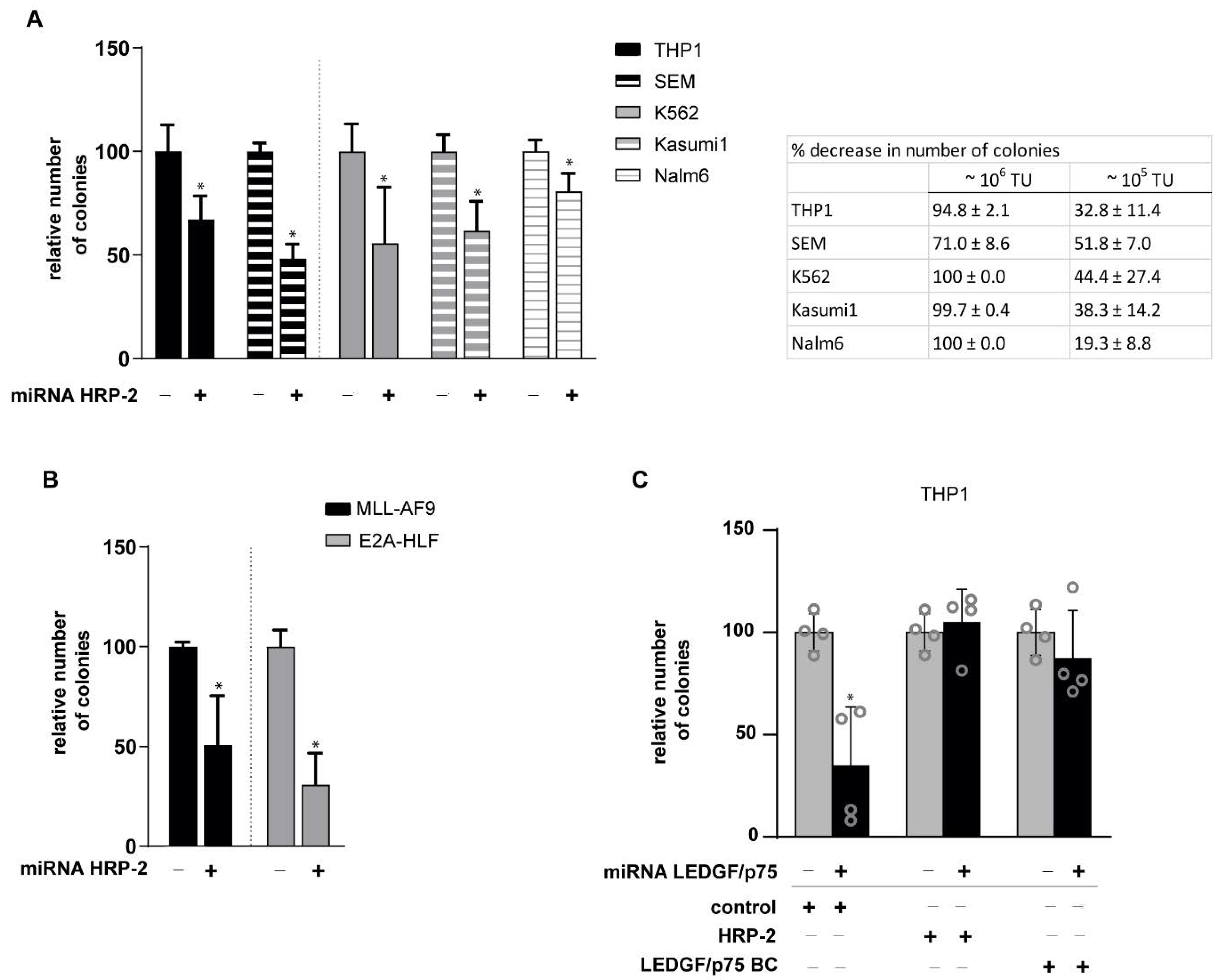
3.4. HRP-2 Depletion Impairs the Clonogenic Growth of Both Human and Mouse Leukemic Cell Lines Independently of MLL Fusions
3.5. HRP-2 Overexpression Rescues MLL-r Clonogenic Growth in LEDGF/p75-Depleted Cells
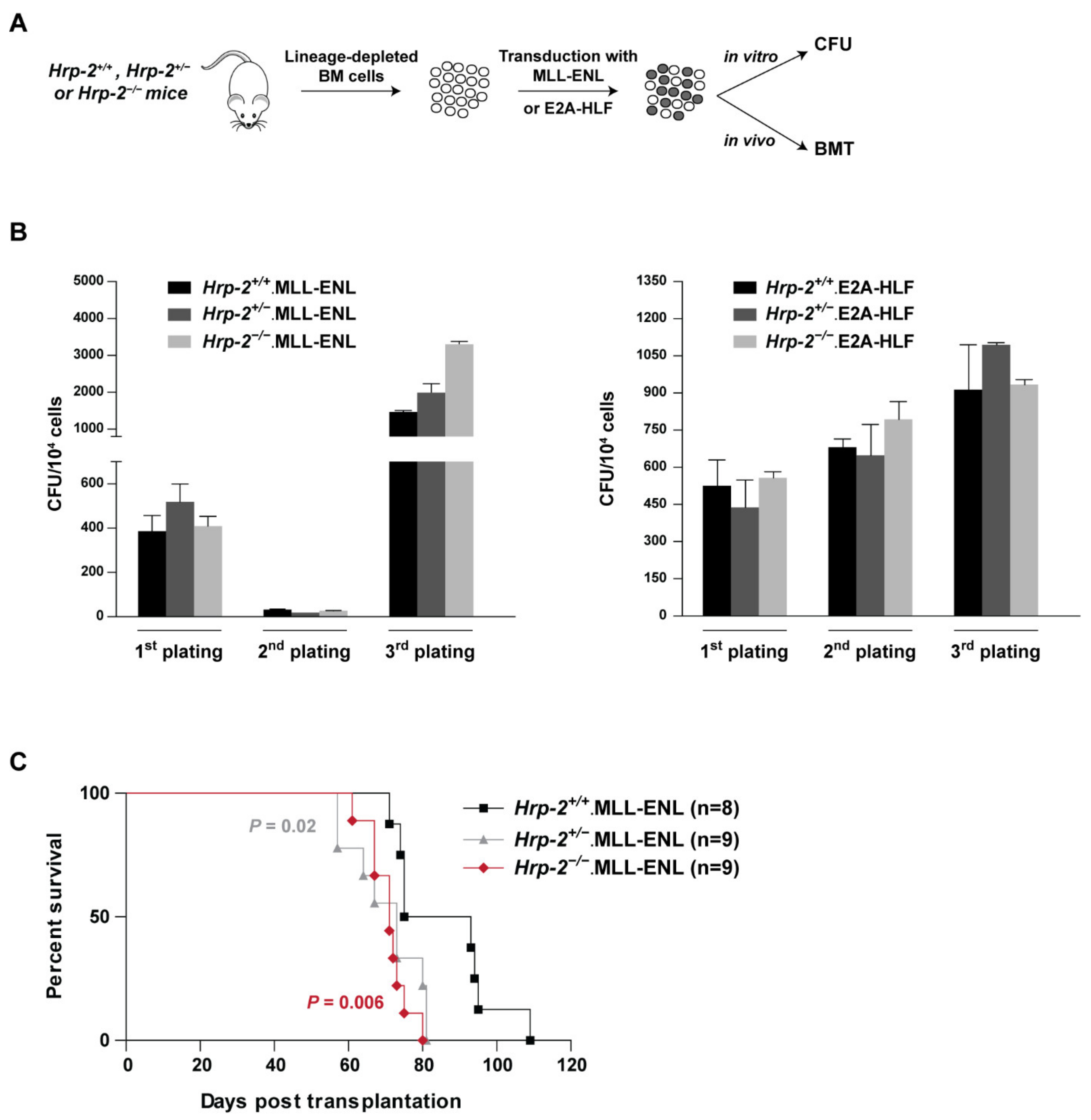
3.6. HRP-2 Is Dispensable for the Initiation of MLL-r Leukemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohan, M.; Lin, C.; Guest, E.; Shilatifard, A. Licensed to elongate: A molecular mechanism for MLL-based leukaemogenesis. Nat. Rev. Cancer 2010, 10, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; Zimmermann, M.; Harbott, J.; Berna Beverloo, H.; von Bergh, A.R.M.; Cloos, J.; Kaspers, G.J.L.; de Haas, V.; et al. Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 2011, 96, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; McGlennen, R.; Smith, F.O.; Woods, W.G.; Salzer, W.L.; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Rondelli, R.; Fagioli, F.; Mastronuzzi, A.; Pierani, P.; Togni, M.; Menna, G.; Pigazzi, M.; Putti, M.C.; Basso, G.; et al. Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 2014, 99, e127. [Google Scholar] [CrossRef] [PubMed]
- Chessells, J.M.; Harrison, C.J.; Kempski, H.; Webb, D.K. Clinical features, cytogenetics and outcome in acute lymphoblastic and myeloid leukaemia of infancy: Report from the MRC Childhood Leukaemia working party. Leukemia 2002, 12, 776–784. [Google Scholar] [CrossRef][Green Version]
- Steinhilber, D.; Marschalek, R. How to effectively treat acute leukemia patients bearing MLL-rearrangements? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef]
- Meyer, C.; Schneider, B.; Jakob, S.; Strehl, S.; Attarbaschi, A.; Schnittger, S.; Schoch, C.; Jansen, M.W.J.C.; van Dongen, J.J.M.; den Boer, M.L.; et al. The MLL recombinome of acute leukemias. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund UK 2006, 20, 777–784. [Google Scholar] [CrossRef]
- Slany, R.K. The molecular mechanics of mixed lineage leukemia. Oncogene 2016, 35, 5215–5223. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.P.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Caslini, C.; Yang, Z.; El-Osta, M.; Milne, T.A.; Slany, R.K.; Hess, J.L. Interaction of MLL Amino Terminal Sequences with Menin Is Required for Transformation. Cancer Res. 2007, 67, 7275–7283. [Google Scholar] [CrossRef]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [CrossRef]
- Dietz, F.; Franken, S.; Yoshida, K.; Nakamura, H.; Kappler, J.; Gieselmann, V. The family of hepatoma-derived growth factor proteins: Characterization of a new member HRP-4 and classification of its subfamilies. Biochem. J. 2002, 366, 491–500. [Google Scholar] [CrossRef] [PubMed]
- El-Tahir, H.; Dietz, F.; Dringen, R.; Schwabe, K.; Strenge, K.; Kelm, S.; Abouzied, M.; Gieselmann, V.; Franken, S. Expression of hepatoma-derived growth factor family members in the adult central nervous system. BMC Neurosci. 2006, 7, 6. [Google Scholar] [CrossRef]
- Izumoto, Y.; Kuroda, T.; Harada, H.; Kishimoto, T.; Nakamura, H. Hepatoma-derived growth factor belongs to a gene family in mice showing significant homology in the amino terminus. Biochem. Biophys. Res. Commun. 1997, 238, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zeng, H.; Lam, R.; Tempel, W.; Amaya, M.F.; Xu, C.; Dombrovski, L.; Qiu, W.; Wang, Y.; Min, J. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 2011, 6, e18919. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sørensen, C.S.; Petersen, N.H.T.; Sorensen, P.H.B.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Kirkegaard-Sørensen, T.; Ostenfeld, M.S.; Aaboe, M.; Høyer-Hansen, M.; Ørntoft, T.F.; Rohde, M.; Jäättelä, M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef]
- Wu, X.; Daniels, T.; Molinaro, C.; Lilly, M.B.; Casiano, C.A. Caspase cleavage of the nuclear autoantigen LEDGF/p75 abrogates its pro-survival function: Implications for autoimmunity in atopic disorders. Cell Death Differ. 2002, 9, 915–925. [Google Scholar] [CrossRef][Green Version]
- Singh, D.P.; Ohguro, N.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Increased resistance to thermal and oxidative stresses. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1444–1451. [Google Scholar]
- Basu, A.; Drame, A.; Muñoz, R.; Gijsbers, R.; Debyser, Z.; De Leon, M.; Casiano, C.A. Pathway specific gene expression profiling reveals oxidative stress genes potentially regulated by transcription co-activator LEDGF/p75 in prostate cancer cells. Prostate 2012, 72, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; de León, M.; Casiano, C.A. Expression of the stress response oncoprotein LEDGF/p75 in human cancer: A study of 21 tumor types. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Sapoznik, S.; Cohen, B.; Tzuman, Y.; Meir, G.; Ben-Dor, S.; Harmelin, A.; Neeman, M. Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res. 2009, 69, 9306–9314. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, Ø.; Døskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Li, X.L.; Lin, Y.C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/p75 promotes tumorigenicity in breast cancer cells by promoting the transcription of cell cycle genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef]
- Maertens, G.N.; Cherepanov, P.; Engelman, A. Transcriptional co-activator p75 binds and tethers the Myc-interacting protein JPO2 to chromatin. J. Cell Sci. 2006, 119, 2563–2571. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Christ, F.; Hendrix, J.; Rain, J.-C.; Emiliani, S.; Benarous, R.; Debyser, Z.; Gijsbers, R.; De Rijck, J. Lens epithelium-derived growth factor/p75 interacts with the transposase-derived DDE domain of PogZ. J. Biol. Chem. 2009, 284, 11467–11477. [Google Scholar] [CrossRef]
- Hughes, S.; Jenkins, V.; Dar, M.J.; Engelman, A.; Cherepanov, P. Transcriptional co-activator LEDGF interacts with Cdc7-activator of S-phase kinase (ASK) and stimulates its enzymatic activity. J. Biol. Chem. 2010, 285, 541–554. [Google Scholar] [CrossRef]
- Tesina, P.; Čermáková, K.; Hořejší, M.; Procházková, K.; Fábry, M.; Sharma, S.; Christ, F.; Demeulemeester, J.; Debyser, Z.; De Rijck, J.; et al. Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 2015, 6, 7968. [Google Scholar] [CrossRef]
- Sharma, S.; Čermáková, K.; De Rijck, J.; Demeulemeester, J.; Fábry, M.; El Ashkar, S.; Van Belle, S.; Lepšík, M.; Tesina, P.; Duchoslav, V.; et al. Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E7053–E7062. [Google Scholar] [CrossRef]
- Gao, K.; Xu, C.; Jin, X.; Wumaier, R.; Ma, J.; Peng, J.; Wang, Y.; Tang, Y.; Yu, L.; Zhang, P. HDGF-related protein-2 (HRP-2) acts as an oncogene to promote cell growth in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2015, 458, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An Essential Role for LEDGF/p75 in HIV Integration. Science (80-.) 2006, 314, 461–464. [Google Scholar] [CrossRef]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Baude, A.; Aaes, T.L.; Zhai, B.; Al-Nakouzi, N.; Oo, H.Z.; Daugaard, M.; Rohde, M.; Jäättelä, M. Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res. 2016, 44, 2214–2226. [Google Scholar] [CrossRef]
- LeRoy, G.; Oksuz, O.; Descostes, N.; Aoi, Y.; Ganai, R.A.; Kara, H.O.; Yu, J.-R.; Lee, C.-H.; Stafford, J.; Shilatifard, A.; et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019, 5, eaay3068. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Pan, J.; Liu, X.; Chen, H.; Zhou, X.; Yuan, Z.; Wang, X.; Mo, D. iTRAQ-based quantitative proteomic analysis reveals the distinct early embryo myofiber type characteristics involved in landrace and miniature pig. BMC Genomics 2016, 17, 137. [Google Scholar] [CrossRef]
- Hu, Y.; He, C.; Liu, J.P.; Li, N.S.; Peng, C.; Yang-Ou, Y.B.; Yang, X.Y.; Lu, N.H.; Zhu, Y. Analysis of key genes and signaling pathways involved in Helicobacter pylori-associated gastric cancer based on The Cancer Genome Atlas database and RNA sequencing data. Helicobacter 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Geraerts, M.; Michiels, M.; Baekelandt, V.; Debyser, Z.; Gijsbers, R. Upscaling of lentiviral vector production by tangential flow filtration. J. Gene Med. 2005, 7, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; Vandekerckhove, L.; Gijsbers, R.; Hombrouck, A.; Hendrix, J.; Vercammen, J.; Engelborghs, Y.; Christ, F.; Debyser, Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 2006, 80, 11498–11509. [Google Scholar] [CrossRef]
- Méreau, H.; De Rijck, J.; Čermáková, K.; Kutz, A.; Juge, S.; Demeulemeester, J.; Gijsbers, R.; Christ, F.; Debyser, Z.; Schwaller, J. Impairing MLL-fusion gene-mediated transformation by dissecting critical interactions with the lens epithelium-derived growth factor (LEDGF/p75). Leukemia 2013, 27, 1245–1253. [Google Scholar] [CrossRef]
- Cermakova, K.; Tesina, P.; Demeulemeester, J.; El Ashkar, S.; Méreau, H.; Schwaller, J.; \vRezáčová, P.; Veverka, V.; De Rijck, J. Validation and structural characterization of the LEDGF/p75-MLL interface as a new target for the treatment of MLL-dependent leukemia. Cancer Res. 2014, 74, 5139–5151. [Google Scholar] [CrossRef]
- Renshaw, P.S.; Veverka, V.; Kelly, G.; Frenkiel, T.A.; Williamson, R.A.; Gordon, S.V.; Hewinson, R.G.; Carr, M.D. Sequence-specific assignment and secondary structure determination of the 195-residue complex formed by the Mycobacterium tuberculosis proteins CFP-10 and ESAT-6. J. Biomol. NMR 2004, 30, 225–226. [Google Scholar] [CrossRef]
- Veverka, V.; Lennie, G.; Crabbe, T.; Bird, I.; Taylor, R.J.; Carr, M.D. NMR assignment of the mTOR domain responsible for rapamycin binding. J. Biomol. NMR 2006, 36 (Suppl. 1), 3. [Google Scholar] [CrossRef]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef]
- Harjes, E.; Harjes, S.; Wohlgemuth, S.; Müller, K.-H.; Krieger, E.; Herrmann, C.; Bayer, P. GTP-Ras Disrupts the Intramolecular Complex of C1 and RA Domains of Nore1. Structure 2006, 14, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Yokogawa, M.; Kobashigawa, Y.; Yoshida, N.; Ogura, K.; Harada, K.; Inagaki, F. NMR Analyses of the Interaction between the FYVE Domain of Early Endosome Antigen 1 (EEA1) and Phosphoinositide Embedded in a Lipid Bilayer. J. Biol. Chem. 2012, 287, 34936–34945. [Google Scholar] [CrossRef] [PubMed]
- NorCOMM2-Phenotyping-Project. Available online: http://www.norcomm2.org/norcomm2/index.php (accessed on 18 January 2020).
- Moll, P.; Ante, M.; Seitz, A.; Reda, T. QuantSeq 3′ mRNA sequencing for RNA quantification. Nat. Methods 2014, 11, i–iii. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef]
- Borkin, D.; Pollock, J.; Kempinska, K.; Purohit, T.; Li, X.; Wen, B.; Zhao, T.; Miao, H.; Shukla, S.; He, M.; et al. Property Focused Structure-Based Optimization of Small Molecule Inhibitors of the Protein-Protein Interaction between Menin and Mixed Lineage Leukemia (MLL). J. Med. Chem. 2016, 59, 892–913. [Google Scholar] [CrossRef]
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef]
- Blokken, J.; De Rijck, J.; Christ, F.; Debyser, Z. Protein–protein and protein–chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 2017, 24, 25–31. [Google Scholar] [CrossRef]
- Wang, H.; Shun, M.-C.; Dickson, A.K.; Engelman, A.N. Embryonic Lethality Due to Arrested Cardiac Development in Psip1/Hdgfrp2 Double-Deficient Mice. PLoS ONE 2015, 10, e0137797. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In~Vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef]
- Aguilar, A.; Zheng, K.; Xu, T.; Xu, S.; Huang, L.; Fernandez-Salas, E.; Liu, L.; Bernard, D.; Harvey, K.P.; Foster, C.; et al. Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein–Protein Interaction. J. Med. Chem. 2019, 62, 6015–6034. [Google Scholar] [CrossRef]
- Sutherland, H.G.; Newton, K.; Brownstein, D.G.; Holmes, M.C.; Kress, C.; Semple, C.A.; Bickmore, W.A. Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol. Cell. Biol. 2006, 26, 7201–7210. [Google Scholar] [CrossRef]
- Zhu, X.; Lan, B.; Yi, X.; He, C.; Dang, L.; Zhou, X.; Lu, Y.; Sun, Y.; Liu, Z.; Bai, X.; et al. HRP2-DPF3a-BAF complex coordinates histone modification and chromatin remodeling to regulate myogenic gene transcription. Nucleic Acids Res. 2020, 48, 6563–6582. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number (%) of Mice | |||
|---|---|---|---|
| Age | HRP-2+/+ | HRP-2+/− | HRP-2−/− |
| 1 day | 36 (23.2) | 88 (56.7) | 31 (20.0) |
| 6–8 weeks | 31 (29.5) | 64 (61.0) | 10 (9.5) |
| Expected values | 25% | 50% | 25% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Belle, S.; El Ashkar, S.; Čermáková, K.; Matthijssens, F.; Goossens, S.; Canella, A.; Hodges, C.H.; Christ, F.; De Rijck, J.; Van Vlierberghe, P.; et al. Unlike its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival. Cells 2021, 10, 192. https://doi.org/10.3390/cells10010192
Van Belle S, El Ashkar S, Čermáková K, Matthijssens F, Goossens S, Canella A, Hodges CH, Christ F, De Rijck J, Van Vlierberghe P, et al. Unlike its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival. Cells. 2021; 10(1):192. https://doi.org/10.3390/cells10010192
Chicago/Turabian StyleVan Belle, Siska, Sara El Ashkar, Kateřina Čermáková, Filip Matthijssens, Steven Goossens, Alessandro Canella, Courtney H. Hodges, Frauke Christ, Jan De Rijck, Pieter Van Vlierberghe, and et al. 2021. "Unlike its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival" Cells 10, no. 1: 192. https://doi.org/10.3390/cells10010192
APA StyleVan Belle, S., El Ashkar, S., Čermáková, K., Matthijssens, F., Goossens, S., Canella, A., Hodges, C. H., Christ, F., De Rijck, J., Van Vlierberghe, P., Veverka, V., & Debyser, Z. (2021). Unlike its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival. Cells, 10(1), 192. https://doi.org/10.3390/cells10010192