Abstract
The PLATZ gene family influences plant growth, development, and responses to both biotic and abiotic stresses. Flax (Linum usitatissimum L.), an important oilseed and fiber crop, has not been extensively studied for its PLATZ genes. In this study, 27 LuPLATZ genes were identified in the recently assembled T2T (Telomere-to-Telomere) flax genome through bioinformatics analyses. Phylogenetic analysis grouped these genes into five subfamilies. Examination of gene structure and motifs showed conserved exon–intron arrangements and similar motif compositions within the same clade. Promoter analysis revealed that most cis-elements are associated with plant hormone responses (such as MeJA and ABA) and abiotic stresses, including anaerobic induction, drought, and low temperature. Duplication analysis identified 33 segmental duplication events, and miRNA target prediction indicated that lus-miR167 is the primary regulator of LuPLATZ genes. Expression profiling based on RNA-seq data showed high expression levels of most LuPLATZ genes in leaves and roots, and qRT-PCR confirmed their stress-responsive expression under cold, drought, and salt conditions, with LuPLATZ14 and LuPLATZ21 significantly upregulated in all treatments. Furthermore, overexpression of these two genes enhanced drought tolerance in yeast transformants.
1. Introduction
PLATZ proteins are plant-specific DNA-binding factors belonging to the zinc finger transcription factor superfamily, which comprises about 15% of all plant transcription factors [1]. Zinc finger proteins are categorized into nine structural types—C2H2, C8, C6, C2HC, C3HC4, C2HC5, C4, C4HC3, and CCCH—according to the number and arrangement of cysteine (Cys) and histidine (His) residues that coordinate zinc ions in their secondary structures [2]. According to their functional characteristics, zinc finger proteins are further divided into several subfamilies, including TFIII, GATA, WRKY, RING, DOF, PHD, and PLATZ [3]. The first PLATZ protein, PLATZ1, was identified in pea (Pisum sativum L.) in 2001. As a zinc-dependent DNA-binding protein, PLATZ1 binds non-specifically to A/T-rich sequences and acts as a transcriptional repressor. Sequence comparisons revealed that PLATZ1 contains two discontinuous zinc finger motifs: C-x2-H-x11-C-x2-C-x(4–5)-C-x2-C-x(3–7)-H-x2-H and C-x2-C-x(10–11)-C-x3-C. These distinct motifs differ from previously described zinc-binding domains, defining PLATZ proteins as a unique class of plant-specific zinc finger transcription factors [4].
In recent years, research on the PLATZ gene family has increased substantially, and members of this family have been identified and analyzed in a range of plant species, including barley [5], tartary buckwheat [6], soybean [7], rice [8], cucumber [9], tomato [10], and Solanum lycopersicum L. [11]. Studies have shown that PLATZ proteins play important roles in plant growth and development by regulating meristem activity, leaf development, and seed development. For example, the PLATZ transcription factor ORESARA15 (ORE15) in Arabidopsis is expressed in leaves, where it promotes the rate and duration of cell proliferation, thereby regulating leaf growth and senescence [12]. Repression of PtaPLATZ18 in poplar increased internode length, xylem thickness, and lignin content, accompanied by upregulation of lignin biosynthesis genes, highlighting their role in growth and secondary cell wall development [13]. A mutation of the conserved histidine in the PLATZ domain to asparagine produces a semi-dominant fl3 mutant that severely disrupts maize endosperm development, likely by reducing tRNA and 5S rRNA transcription and consequently impairing nutrient accumulation [14]. The rice PLATZ transcription factor GL6 interacts with two key components of RNA polymerase III, RPC53 (RNA polymerase III subunit C53) and the class C1 transcription factor (TFC1), to participate in RNA Pol III-mediated transcription, thereby regulating grain length and spikelet number in rice [15].
The PLATZ gene family plays a role not only in regulating plant growth and development but also in mediating responses to abiotic stress [7]. Studies showed that Arabidopsis PLATZ1 and PLATZ2 regulated dehydration tolerance in vegetative tissues and played a key role in seed drought resistance [16]. The Arabidopsis PLATZ family member A1N1 participated in the abscisic acid (ABA) signaling pathway by regulating reactive oxygen species (ROS) homeostasis, thereby affecting root elongation and inhibition in response to drought and salt stress [17]. Heterologous expression of GhPLATZ1 from cotton (Gossypium hirsutum L.) in transgenic Arabidopsis enhanced osmotic tolerance under salt stress and mannitol-induced osmotic stress, and resulted in faster seed germination and higher seedling establishment rates [18]. Arabidopsis AtPLATZ2 suppressed plant salt tolerance by repressing the transcription of CBL4/SOS3 and CBL10/SCaBP8 [19].
The continuous advancement of high-throughput sequencing technologies has driven the in-depth development of genomics, making it possible to assemble complete, gapless telomere-to-telomere (T2T) genomes [20]. A T2T genome not only contains the full genetic information of a species and is regarded as the ultimate form of genome assembly, but also effectively avoids assembly errors, improves the accuracy of variant detection, and facilitates the identification of missing genes as well as the evolutionary study of complex structures such as telomeres and centromeres [21]. In flax research, the most widely used reference genomes were previously derived from the oilseed cultivars CDC Bethune and Longya10, both assembled based on the Illumina platform [22,23]. However, these versions showed significant deficiencies in continuity and completeness. Recently, by integrating PacBio HiFi, Hi-C, and Illumina sequencing technologies, researchers assembled a 482.51 Mb T2T reference genome of fiber flax, with 46,634 genes predicted. This genome achieved an N50 of 33.03 Mb, representing ~17-fold increases compared with Longya10 (1.94 Mb) and CDC Bethune (0.023 Mb) [24]. This genome assembly represents a milestone achievement, laying a solid foundation for further studies in flax genomics.
Flax, an ancient crop species, is cultivated globally, particularly in temperate regions [25]. Based on its usage, flax can be categorized into three agronomic types: linseed (oil-flax), fiber flax, and dual-purpose flax [26,27]. Flaxseeds are packed with nutrients, including lignans and dietary fiber. Notably, alpha-linolenic acid (ALA) is an essential omega-3 fatty acid vital for human health [28]. While PLATZ genes have been well-studied in various model plants, the functions of this gene family in flax, a key fiber crop, remain poorly understood. In this study, we systematically analyzed flax PLATZ genes using bioinformatics approaches, investigating their phylogeny, chromosomal distribution, physicochemical properties, conserved motifs, collinearity, cis-regulatory elements in promoters, subcellular localization, and expression patterns based on RNA-seq data, as well as their responses to cold, salt, and drought stresses. Additionally, we preliminarily confirmed that LuPLATZ14 and LuPLATZ21 could enhance drought tolerance in yeast transformants. This work provides a foundation for further functional characterization of PLATZ genes in flax and represents the first comprehensive survey of the organization and stress-responsive regulation of this gene family in the species.
2. Materials and Methods
2.1. Plant Material
Experiments were performed using the fiber-type flax cultivar “Gaosi,” obtained from the College of Agriculture, Jilin Agricultural University [24]. Seeds were surface-sterilized with 75% ethanol for 10 min, thoroughly rinsed with sterile water, and sown in soil. Plants were grown in a controlled growth chamber under a 16 h light/8 h dark photoperiod at 26/18 °C, with a relative humidity maintained at approximately 60%. Three-week-old seedlings were used for all experiments. Stress treatments were imposed once seedlings reached 6–7 cm in height. For drought and salt stress treatments, plants were gently removed from soil, washed with distilled water, and transferred to conical flasks containing 10% polyethylene glycol (PEG) or 100 mM NaCl, respectively, as these concentrations have been commonly used in previous studies to simulate drought and salt stress in flax and were shown to induce clear physiological responses [29,30]. Control plants were maintained in distilled water, while low-temperature treatment plants were transferred to a 4 °C chamber. Leaf samples were harvested simultaneously from all groups at 0, 3, 6, 12, and 24 h, with three biological replicates per time point, to avoid time-of-day effects. All collected samples were immediately frozen in liquid nitrogen and then stored at −80 °C to preserve RNA integrity for subsequent analyses.
2.2. Identification of PLATZ Gene in Flax
The T2T flax (Gsosi) genome assembly and annotation data, including genome, CDS, and protein sequences, were obtained from the China National Center for Bioinformation (PRJCA037526, https://ngdc.cncb.ac.cn/, accessed 3 April 2025). Twelve Arabidopsis PLATZ protein sequences were obtained from the TAIR database (https://www.arabidopsis.org/, accessed 3 April 2025) [31]. Candidate PLATZ genes in flax were initially identified via BLASTp (BLAST+ v2.15.0) against the complete flax proteome using an E-value threshold of 1 × 10−5. In parallel, the Hidden Markov Model (HMM) for the PLATZ domain (PF04640) was downloaded from the Pfam database (http://pfam.xfam.org/, accessed 3 April 2025) and employed with HMMER3.0’s hmmsearch program to further predict PLATZ genes [32]. To ensure accuracy, only sequences containing an intact and complete PLATZ domain (PF04640) were considered as true PLATZ genes. The presence of the domain was verified using both SMART (https://smart.embl-heidelberg.de, accessed 3 April 2025) and the NCBI Conserved Domain Database (CDD, https://www.ncbi.nlm.nih.gov/cdd/, accessed 3 April 2025). After removing redundant sequences, a total of 27 non-redundant LuPLATZ genes were identified. The genome and protein sequence files of maize and wheat were obtained from the publicly available Phytozome database (https://phytozome-next.jgi.doe.gov/, accessed on 25 April 2025). The identification of PLATZ gene family members in these species was performed using the same approach as applied for flax. These genes were named with the prefix “Lu” and numbered according to their chromosomal locations. Physicochemical properties of the LuPLATZ proteins, including coding sequence length, amino acid number, molecular weight (MW), theoretical isoelectric point (pI), and grand average of hydropathicity (GRAVY), were calculated using ExPASy ProtParam (https://web.expasy.org/protparam/, accessed 3 April 2025). Subcellular localization predictions were obtained through the BUSCA web server (https://busca.biocomp.unibo.it, accessed 3 April 2025).
2.3. Phylogeny, Chromosome Location, Conserved Domain and Conserved Motif of LuPLATZ Gene
PLATZ amino acid sequences from rice, soybean, maize, Arabidopsis, and flax were aligned using ClustalW in MEGA version 11 with default parameters. A maximum likelihood (ML) phylogenetic tree was then constructed in MEGA 11 under default settings (neighbor-joining; bootstrap = 1000) and visualized using iTOL version 6 (https://itol.embl.de/, accessed 10 April 2025). The chromosomal positions of LuPLATZ genes were determined from the flax genome FASTA and GFF3 annotation files. Conserved domains were predicted using NCBI’s CD-Search tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed 10 April 2025), and motif analysis of LuPLATZ proteins was performed with MEME (https://meme-suite.org/meme/tools/meme, accessed 10 April 2025), with results visualized in TBtools (v2.069) [33].
2.4. Genome-Wide Replication and Collinear Analysis of LuPLATZ Gene
Genome sequences and annotation files for Arabidopsis, maize, and wheat were obtained from the public Phytozome database (https://phytozome-next.jgi.doe.gov/, accessed 25 April 2025) [34]. Collinearity analyses were performed using the MCScanX toolkit (version 1.0). LuPLATZ genes arising from whole-genome duplications (WGDs) were identified, while tandem duplicates were defined as two or more homologous genes on the same chromosome separated by ≤100 kb with no intervening genes. Segmental duplications were detected using nucleotide BLAST (BLAST+ v2.12.0) with an E-value cutoff of <1 × 10−5 within a 100 kb window surrounding the coding sequences (50 kb upstream and downstream). Duplicated genes were confirmed based on a minimum alignment length of 200 bp and sequence identity exceeding 85% [35].
2.5. MiRNA Prediction and Cis-Acting Element Analysis
Flax miRNA sequences were obtained from previous studies [36]. Potential targets of these miRNAs were predicted by comparing them against the 5′ and 3′ untranslated regions (UTRs) and coding sequences (CDS) of all LuPLATZ genes using the Plant MicroRNA Target Analysis Server (psRNATarget) with default settings (https://www.zhaolab.org/psRNATarget/analysis?function=3, accessed 25 April 2025). The 2000 bp upstream genomic sequences of LuPLATZ genes were extracted using TBtools. To explore their possible regulatory roles under environmental stress, cis-regulatory elements within the 2.0 kb promoter regions were identified via the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed 25 April 2025) and visualized using TBtools.
2.6. Analysis of Expression Pattern of LuPLATZ Gene Family
In this study, we performed a hypothetical analysis of the expression patterns of the LuPLATZ gene family using publicly available RNA-seq data from five flax transcriptome datasets: (i) pistil, stamen, fruit, and apical stem tissues (PRJNA1002756; https://www.ncbi.nlm.nih.gov/sra/?term=, accessed 5 May 2025); (ii) floral tissues at 30, 20, 10, and 5 days after anthesis (PRJNA833557); (iii) embryo, anther, and seed tissues at different developmental stages (PRJNA663265); (iv) roots and leaves under salt stress (PRJNA977728); and (v) stems exposed to heat stress (PRJNA874329). Raw sequencing reads were processed with fastp v0.23.2, mapped to the T2T reference genome of the Gaosi flax variety, and gene expression levels were quantified using the R packages tidyverse v1.3.1 [37], Rsubread v2.10.5 [38] and limma v3.54.2 [39]. Log2-transformed FPKM values were then visualized as heatmaps using TBtools.
2.7. RNA Extraction and Fluorescence Quantitative PCR Analysis
The total RNA of flax leaves was extracted by Trizol method (Invitrogen, Carlsbad, USA). First-strand cDNA was synthesized from the total RNA using the M-MLV reverse transcription kit (Promega, Madison, WI, USA). Leaf samples were collected at 0, 3, 6, 9, 12, and 24 h after stress treatments for RNA extraction and subsequent cDNA synthesis. Oligo 7 was used to design upstream and downstream primers (Table S3), and TB Green ®Pemix ExTaqTMII (Takara Bio, Kyoto, Japan) was used for RT-PCR test. Each sample included three biological replicates and three technical replicates. The experimental results were statistically analyzed by 2−ΔΔCT method [40]. Using GAPDH as the internal control gene, each sample was repeated 3 times to calculate the CT value [41].
2.8. Subcellular Localization of LuPLATZ14 and LuPLATZ21 Genes
The full-length coding sequence of LuPLATZ was amplified by PCR (Table S3) using specific primers designed with Oligo 7 (Thermo Fisher Scientific, Waltham, MA, USA), and cloned into the pGM-T vector (Promega, Madison, MA, USA) before transformation into Escherichia coli (DH5α, Thermo Fisher Scientific, Waltham, MA, USA). The target fragment was excised from pGM-T-LuPLATZ14 and pGM-T-LuPLATZ21 using XhoI and SalI restriction enzymes (New England Biolabs, Ipswich, MA, USA) and ligated into the similarly digested pCAMBIA1301 vector (Clontech, Mountain View, CA, USA) to generate pCAMBIA1301-LuPLATZ14-GFP and pCAMBIA1301-LuPLATZ21-GFP. Both the empty pCAMBIA1301 vector and recombinant plasmid were separately introduced into Agrobacterium tumefaciens GV3101 (strain, Agilent Technologies, Santa Clara, CA, USA) via the freeze–thaw method. The bacterial culture was resuspended in infiltration buffer (10 mM MES, pH 5.6, 10 mM MgCl2, and 150 µM acetosyringone, Sigma-Aldrich, St. Louis, MO, USA), and after 3 h of induction at 28 °C, the culture was injected into Nicotiana benthamiana leaves. After 3 days of dark incubation, GFP fluorescence signals were observed using a confocal microscope (Leica TCS SP8, Leica Microsystems, Wetzlar, Germany).
2.9. Heterologous Expression of LuPLATZ14 and LuPLATZ21 Genes in the Saccharomyces cerevisiae Strain INVSc1
Using a directional cloning approach with HindIII and BamHI restriction enzymes (Takara Bio, Kyoto, Japan), LuPLATZ14 and LuPLATZ21 were inserted into the multiple cloning site of the Saccharomyces cerevisiae expression vector pYES2 (Invitrogen, Carlsbad, CA, USA), producing the recombinant plasmids pYES2-LuPLATZ14 and pYES2-LuPLATZ21. These plasmids were subsequently transformed into S. cerevisiae INVSc1 (Thermo Fisher Scientific, Waltham, MA, USA). The empty pYES2 vector served as a negative control. Recombinant plasmids pYES2-LuPLATZ14, pYES2-LuPLATZ21, and the empty vector were serially diluted 10−1 to 10−4, and 2 µL of each dilution was spotted onto SD-ura plates (BD Biosciences, Franklin Lakes, NJ, USA) and SD-ura plates containing 300 mM mannitol (Sigma-Aldrich, St. Louis, MO, USA), followed by observation of their phenotypic responses.
3. Results
3.1. Identification and Phylogenetic Analysis of PLATZ Gene Family in Flax
Based on the hidden Markov model (HMM) of the PLATZ structural domain (PF04640), a total of 27 PLATZ genes were identified in the flax T2T genome (Gaosi) and were named LuPLATZ1 to LuPLATZ27 according to their chromosomal locations (Table 1). Analysis of the physicochemical properties of the proteins encoded by the LuPLATZ gene family revealed that LuPLATZ12 encodes the longest protein, containing 547 amino acids, whereas LuPLATZ4 encodes the shortest protein, with only 124 amino acids. The molecular weights of LuPLATZ family proteins ranged from 13.19 kDa to 61.67 kDa, showing considerable variation. Isoelectric point (pI) analysis indicated that most proteins in this family are basic, with 81.5% having a pI greater than 8, suggesting enrichment in basic amino acids. The instability index ranged from 46.13 to 85.33, with all proteins exceeding 40, indicating that LuPLATZ family proteins are generally unstable. The aliphatic index varied from 49.01 to 75.99, suggesting relatively small differences in thermal stability and an overall moderate stability. GRAVY analysis showed that all LuPLATZ proteins had negative values (−0.002 to −1.045), indicating hydrophilicity, with LuPLATZ12 being the most hydrophilic (−1.045). Subcellular localization prediction indicated that all LuPLATZ proteins are located in the nucleus, suggesting that they may function as transcription factors within the nucleus.

Table 1.
Prediction and characterization of PLATZ Gene in Flax.
To gain deeper insight into the phylogenetic relationships of the flax PLATZ gene family, we constructed a phylogenetic tree using the Maximum Likelihood (ML) method with 1000 bootstrap replicates, incorporating PLATZ proteins from five species: Arabidopsis, Oryza sativa, Zea mays, Glycine max, and Linum usitatissimum (Figure 1) (Table S1). Bootstrap support values are shown at the nodes to assess the reliability of the clades. Based on the phylogenetic analysis, the 27 LuPLATZ proteins were classified into five major evolutionary groups (Group I–V). Group I contained the largest number of LuPLATZ members, totaling nine, followed by Group IV and Group V, which contained six and five LuPLATZ genes, respectively. Group II and Group III included four and three members, respectively. This distribution pattern suggests that the LuPLATZ family has undergone significant gene expansion during evolution. Several LuPLATZ genes clustered together with PLATZ proteins from different species, indicating strong conservation and suggesting that they may perform similar functions in plant growth, development, and environmental responses.
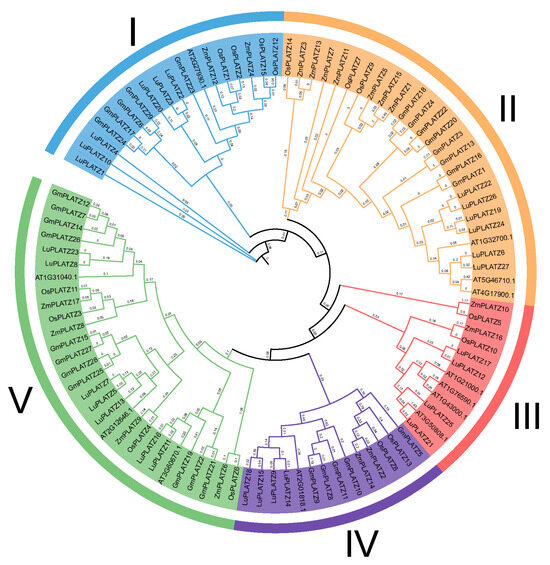
Figure 1.
Phylogenetic tree of PLATZ proteins. The prefixes At, Lu, Gm, and Zm represent genes from Arabidopsis, flax, soybean, and maize, respectively. All PLATZ genes were classified into five subfamilies, which are indicated by different colors and letters.
3.2. Analysis of Gene Structure and Conservative Motif of LuPLATZ
To elucidate the domain composition of the LuPLATZ family proteins, we performed a systematic analysis of conserved motifs for all 27 LuPLATZ genes. The phylogenetic tree constructed based on multiple sequence alignment (Figure 2A) showed a clustering pattern consistent with that in Figure 1. Ten conserved motifs were predicted and named motif1 to motif10 (Figure 2B). All members contained motifs 1, 3, and 6; all members except LuPLATZ4 contained motif8; all members except LuPLATZ4 and LuPLATZ10 contained motif4; and all members except LuPLATZ4 and LuPLATZ13 contained motif2. All members except four genes (LuPLATZ1 and LuPLATZ4) contained motif5. We further found that only LuPLATZ12, LuPLATZ17, LuPLATZ21, and LuPLATZ25 contained motif7, which is a unique motif of the III subfamily. Overall, the types, numbers, and distribution patterns of motifs were comparable among LuPLATZ proteins within each branch. Exon–intron structure analysis of LuPLATZ genes showed that exon numbers varied from 2 to 11 and intron numbers from 1 to 10 (Figure 2C). LuPLATZ11 possessed the most complex structure, with 11 exons and 10 introns, followed by LuPLATZ19, which contained 5 exons and 4 introns. LuPLATZ25 had the fewest, with only 2 exons and 1 intron. Sixteen genes (59.26% of the total) contained three exons. We also observed that genes in the Group V branch all contained four exons, suggesting functional similarity among these genes.
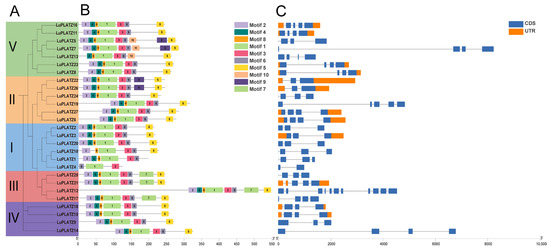
Figure 2.
Gene structure, motif composition, and chromosomal distribution of LuPLATZ genes. (A) Phylogenetic relationships among LuPLATZ genes. (B) Motif composition of LuPLATZ genes. Gray lines indicate the full gene length, while distinct colors correspond to different conserved motifs. (C) Structural organization of LuPLATZ genes. Orange boxes denote UTR, dark blue boxes represent coding sequences (CDS), and black lines indicate introns.
3.3. Chromosome Mapping and Collinearity Analysis of LuPLATZ Gene
Using the flax reference genome, we determined the chromosomal distribution of LuPLATZ genes. The 27 genes were unevenly mapped to 15 chromosomes (Figure 3). Chromosome 1 harbored the most members (four, 14.81%), followed by chromosomes 13 and 15 with three each (11.11%), while chromosomes 3–6, 11–12, and 14 contained only one gene (3.7%).
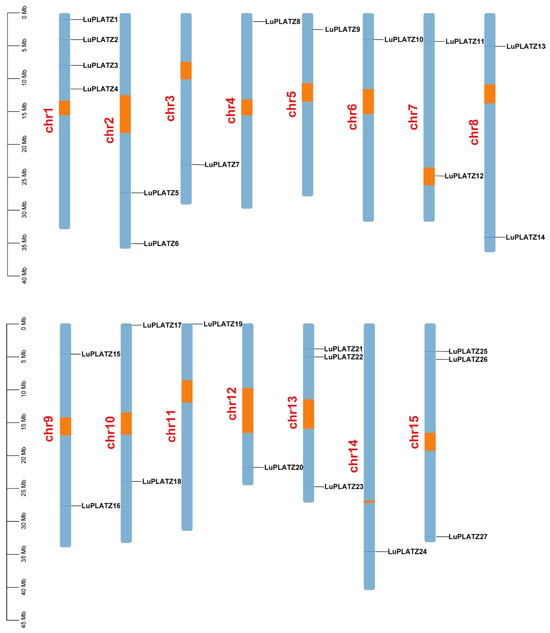
Figure 3.
The chromosome distribution map of LuPLATZ genes. Orange represents the centromere region.
We performed a gene duplication analysis of the LuPLATZ gene family using BLAST and MCScanX. No tandem duplication events were identified within the LuPLATZ family. To further investigate gene duplication, we constructed a Circos map (Figure 4A), which revealed 33 pairs of duplicated LuPLATZ genes in the flax genome, indicating a substantial expansion of this gene family. We also found that all collinear gene pairs of LuPLATZ have Ka/Ks ratios less than 1, indicating that they evolved under purifying selection (Table 2). To clarify the evolutionary relationships of flax, we analyzed its collinearity with three representative species—Arabidopsis, maize, and wheat (Figure 4B). The results showed 15, 3, and 1 collinear gene pairs, respectively. Among them, all five Arabidopsis chromosomes displayed collinearity with flax chromosomes; maize PLATZ collinear pairs were located on flax chromosomes 2, 11, and 15; and the wheat PLATZ collinear pair was located on flax chromosome 13. Taken together, these results indicate that most PLATZ genes exhibit conserved collinearity among flax and the three representative species, while species-specific variations in gene pairs also exist, highlighting the important role of LuPLATZ genes in the evolutionary history of the family.
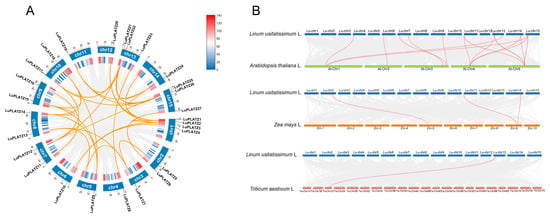
Figure 4.
Synteny analysis of the LuPLATZ gene family. (A) Intra-species syntenic relationships among LuPLATZ members. (B) Cross-species synteny comparisons of PLATZ genes between flax and three representative species: Arabidopsis, maize and wheat. In both panels, gray connections indicate genome-wide collinear gene pairs, while orange linkages specifically highlight syntenic pairs involving LuPLATZ genes.
Table 2.
Ka, Ks and Ka/Ks of replication pairs of PLATZ gene family in flax.
3.4. Analysis of Cis-Acting Elements and MiRNA Prediction
To investigate the regulatory roles of LuPLATZ genes under abiotic stress, we predicted and visualized cis-acting elements within the 2000 bp upstream promoter regions (Figure 5A). Excluding common motifs such as the TATA-box and CAAT-box, we identified 763 elements, which were classified into four categories: light-responsive, hormone-responsive, stress-related, and development-related (Figure 5B,C) (Table S2). Light-responsive elements were the most abundant, accounting for 355 elements (46.52%), and included G-box, Box 4, GT1-motif, and MRE. Hormone-responsive elements represented the second largest category, totaling 234 elements (30.67%), and included MeJA-responsive motifs (CGTCA- and TGACG-motifs), abscisic acid-responsive elements (ABRE), auxin-responsive motifs (TGA-element and AuxRR-core), gibberellin-responsive elements (GARE-motif and P-box), and salicylic acid-responsive motifs (TCA-element). Among these, MeJA- and ABA-related elements were the most abundant within LuPLATZ promoters. Stress-related elements accounted for 127 motifs (16.64%) and comprised anaerobic induction elements (ARE and GC-motif), low-temperature-responsive elements (LTR), MYBHv1 binding sites (CCAAT-box), drought-inducible MYB binding sites (MBS), defense- and stress-responsive elements (TC-rich repeats), and wound-responsive motifs (WUN-motif). The smallest category was development-related elements, with 47 elements (6.16%), such as meristem expression elements (CAT-box), seed-specific regulatory elements (RY-element), zein metabolism regulatory elements (O2-site), cell cycle regulation elements (MSA-like), circadian-responsive elements (circadian), and endosperm expression elements (GCN4_motif).
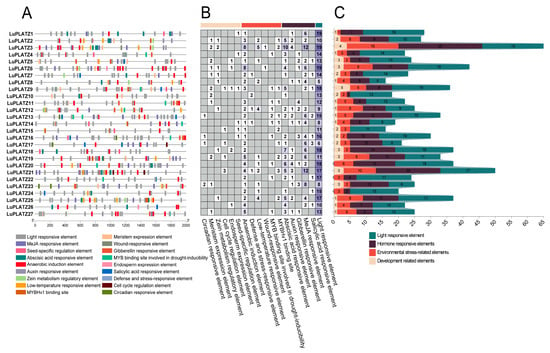
Figure 5.
Prediction of cis-acting elements in LuPLATZ genes. (A) Distribution of cis-elements among the various LuPLATZ subfamilies. (B,C) Quantitative analysis of cis-element types in LuPLATZ genes. Light yellow indicates development-related elements, dark orange represents stress-related elements, dark purple corresponds to hormone-responsive elements, and dark green marks light-responsive elements.
MiRNA prediction analysis revealed that only 6 of the 27 LuPLATZ genes (22.22%) were targeted by 12 putative miRNAs (Table 3). Among them, LuPLATZ1 and LuPLATZ4 had the largest number of predicted miRNA targets, each with nine, including lus-miR167a/b/c/d/e/f/g/h/i. In contrast, LuPLATZ9, LuPLATZ15, and LuPLATZ18 had the fewest, with only one predicted miRNA target each. We also observed that different LuPLATZ genes could be targeted by the same miRNA. For example, lus-miR167 simultaneously targeted LuPLATZ1 and LuPLATZ4, while lus-miR408 targeted LuPLATZ9, LuPLATZ15, and LuPLATZ18. Taken together, these findings indicate that lus-miR167 is the predominant miRNA regulator of the LuPLATZ gene family.
Table 3.
Potential MiRNA targets of LuPLATZ gene.
3.5. Analysis of LuPLATZ Gene Expression Pattern Based on RNA-Seq Data
To investigate the potential functions of LuPLATZ genes in abiotic stress responses, publicly available transcriptome data were analyzed under salt and heat stress (Figure 6A). No major transcriptional changes were observed under heat stress, except for LuPLATZ15, which was downregulated in stem tissue. In contrast, most LuPLATZ genes exhibited marked expression changes under salt stress in both leaves and roots. Compared with the control, six genes (LuPLATZ4, 15, 17, 18, 22, and 26) were upregulated in roots, with LuPLATZ15 showing the strongest induction, while another six genes (LuPLATZ2, 3, 5, 7, 13, and 23) were downregulated. In leaves, eight genes (LuPLATZ11, 15, 16, 17, 20, 24, 25, and 27) were significantly upregulated, with LuPLATZ16 displaying the highest induction.
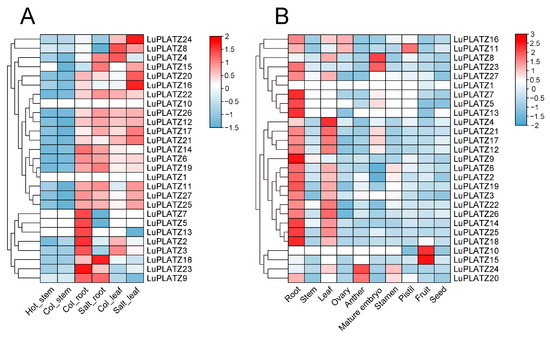
Figure 6.
Expression patterns of the LuPLATZ gene family. (A) LuPLATZ gene expression under salt and heat treatments. (B) Expression of LuPLATZ genes across different flax tissues. FPKM values are log2-transformed, with colors representing relative expression levels from high (red) to low (blue).
To investigate their involvement in plant growth and development, RNA-seq data from 10 tissues were analyzed (Figure 6B). The results revealed clear tissue-specific expression patterns, with most LuPLATZ genes showing high expression in roots and leaves, but low levels in seeds. Specifically, all members except five (LuPLATZ1, 8, 10, 15, and 24) were strongly expressed in roots. LuPLATZ11 and LuPLATZ16 were predominantly expressed in the ovary, LuPLATZ20 and LuPLATZ24 in the anther and stamen, LuPLATZ8 and LuPLATZ23 in the mature embryo, LuPLATZ11 in the pistil, and LuPLATZ10 and LuPLATZ15 in the fruit. Overall, these findings indicate that most LuPLATZ genes are preferentially expressed in roots and leaves, suggesting that these tissues serve as major sites for reactive oxygen species (ROS) regulation and represent the primary antioxidant defense system in flax. The LuPLATZ genes are therefore likely to play critical regulatory roles in stress adaptation and developmental processes.
3.6. qRT-PCR Analysis of LuPLATZ Gene Expression in Flax Under Abiotic Stress
To capture the diversity of the LuPLATZ gene family, ten genes were selected from five subfamilies, all exhibiting high homology with known Arabidopsis genes. The relative expression of these genes was evaluated by qRT-PCR under abiotic stresses in leaves sampled at 0, 3, 6, 9, 12, and 24 h, with 0 h as the control (Figure 7). Under cold stress, all LuPLATZ genes responded significantly over time (Figure 7A). LuPLATZ6 and LuPLATZ13 peaked at 3 h, showing approximately 12.4- and 8.2-fold increases compared with the control, then rapidly declined. LuPLATZ3, LuPLATZ14, and LuPLATZ25 peaked at 9 h, increasing by ~1.8-, 3.7-, and 6.2-fold, respectively. Five genes (LuPLATZ9, 11, 20, 21, and 27) were downregulated, with expression suppressed after 6 h. Under salt stress, significant changes were observed over time (Figure 7B). Seven genes (LuPLATZ3, 6, 11, 13, 14, 21, and 25) peaked at 3 h, with fold changes of 4.9-, 5.8-, 7.9-, 8.8-, 7.5-, 5.6-, and 7.3-fold, respectively, before declining. LuPLATZ27 was upregulated at 12 h (~1.8-fold), while LuPLATZ9 and LuPLATZ20 were downregulated, reaching minima at 9 h and 3 h, respectively. Under drought stress, LuPLATZ genes also showed marked responses (Figure 7C). LuPLATZ3 and LuPLATZ25 peaked at 3 h (~3.3- and 5.2-fold). Six genes (LuPLATZ6, 9, 11, 14, 21, and 27) peaked at 12 h, with fold changes of 3.8-, 9.1-, 1.9-, 6.1-, 9.8-, and 5.2-fold, respectively. LuPLATZ20 was significantly upregulated at 24 h (~9.5-fold). Overall, these results indicate that 6 h under salt stress and 12 h under drought stress may represent critical time points for LuPLATZ gene responses.
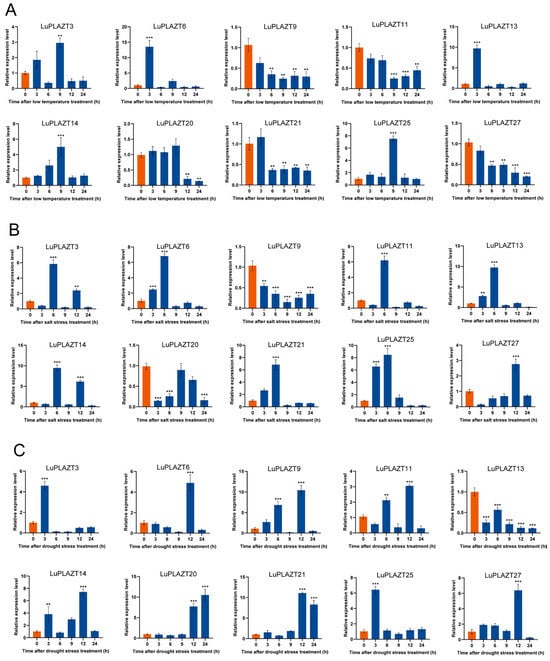
Figure 7.
Expression profiles of LuPLATZ genes under abiotic stresses. (A) Cold stress. (B) Salt stress. (C) Drought stress. Control samples are shown in orange, and treated samples in blue. Statistical significance was determined by Student’s t-test, with asterisks denoting ** p < 0.01 and *** p < 0.001.
3.7. Subcellular Localization of LuPLATZ14 and LuPLATZ21 Genes
The subcellular localization of LuPLATZ14 and LuPLATZ21 was determined by transiently expressing GFP-tagged fusion constructs, pCAMBIA1301-LuPLATZ14-GFP and pCAMBIA1301-LuPLATZ21-GFP, in young tobacco leaves using Agrobacterium tumefaciens GV3101. The empty vector control (pCAMBIA1301-GFP) showed GFP fluorescence throughout the cell, whereas the GFP signals of LuPLATZ14 and LuPLATZ21 were specifically localized to the nucleus (Figure 8), consistent with the predictions listed in Table 1.
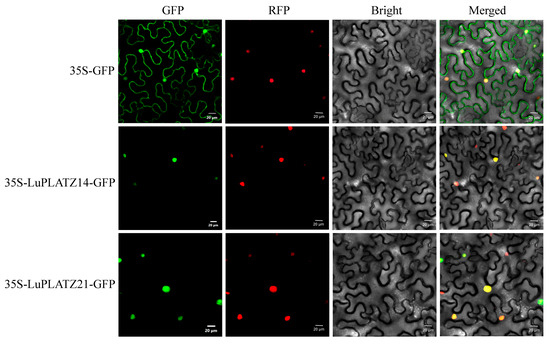
Figure 8.
Subcellular localization analysis of LuPLATZ14 and LuPLATZ21 proteins in tobacco cells. RFP (red fluorescence protein) used as a marker for subcellular localization. Scale bar = 20 µm.
3.8. Drought Tolerance Assay of Yeast Transformants
Based on previous experiments, LuPLATZ14 and LuPLATZ21, which exhibited the strongest drought tolerance, were selected as candidate genes for further study. To determine their effects on the survival of yeast recombinants under osmotic stress, yeast strains carrying pYES2-LuPLATZ14, pYES2-LuPLATZ21, or the empty vector (pYES2) were assessed for colony growth under 300 mM mannitol treatment (Figure 9). Under optimal growth conditions, no significant differences were observed between the transformants and control strains. Under 300 mM mannitol-induced drought stress, the transformants exhibited enhanced growth compared with the control. These results indicate that overexpression of LuPLATZ14 and LuPLATZ21 can improve drought tolerance in yeast transformants.
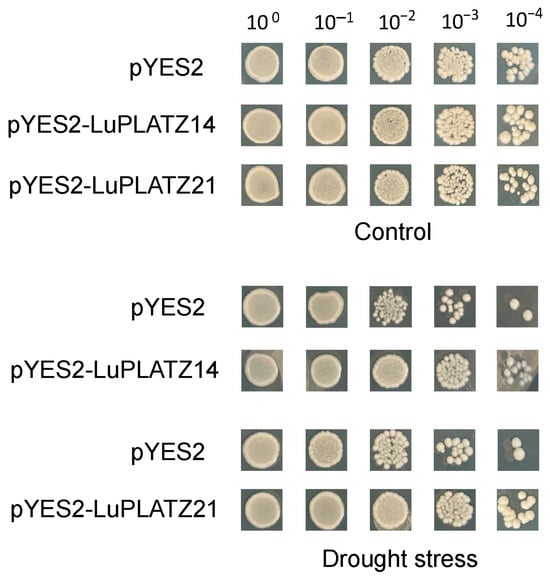
Figure 9.
Overexpression of LuPLATZ14 and LuPLATZ21 enhanced drought tolerance in yeast transformants. Control yeast strains were grown at 30 °C for 48 h under normal conditions, while the treatment strains were cultured under the same temperature and duration in the presence of 300 mM mannitol to simulate osmotic stress.
4. Discussion
As plant-specific transcription factors with zinc-dependent DNA-binding ability, PLATZ proteins participate in regulating plant growth, developmental processes, and adaptation to environmental stresses by recognizing particular DNA motifs. [19]. Although PLATZ transcription factors have been reported in other plant species, information on LuPLATZ in flax remains limited. To date, no studies have comprehensively identified the LuPLATZ gene family using genomic approaches. In the present study, 27 LuPLATZ genes were identified in the flax T2T genome (Table 1). Our analysis showed that the number of LuPLATZ genes in flax (27) was slightly higher than that in Arabidopsis (12) [1], rice (15) [8], maize (17) [1], and cucumber (12) [9], but was comparable to that in soybean (29) [7] and tomato (24) [42]. Phylogenetic analysis further revealed that the LuPLATZ genes were classified into five subfamilies (Figure 1), which was largely consistent with previous studies [5].
Conserved motif analysis revealed that most LuPLATZ proteins share a similar motif arrangement (Figure 2B), consisting of motifs 2, 4, 8, 1, 3, 6, and 5, which is consistent with observations in tomato [42]. The exon–intron analysis of LuPLATZ genes revealed that exon numbers varied between 2 and 11, while intron counts ranged from 1 to 10 (Figure 2C), with most genes exhibiting structural variations. Such differences in PLATZ gene structures may result from exon/intron insertion or deletion events [43], may provide the genomic basis for alternative splicing under stress conditions, thereby contributing to functional diversification. Moreover, genes within the same LuPLATZ subfamily displayed similar exon–intron patterns and motif compositions, implying conserved functions in abiotic stress responses, as also reported in pecan [44]. Compared with Arabidopsis and maize, flax is regarded as a temperate crop that is frequently exposed to abiotic stresses such as cold and drought, particularly during its vegetative growth stage. The adaptation of flax to these conditions may have been facilitated by the expansion and diversification of LuPLATZ genes, through which stress tolerance mechanisms could have been enhanced early in development. Moreover, conserved motif composition and exon–intron structures have been observed between LuPLATZ genes and their counterparts in tomato, another dicot species, suggesting that core regulatory functions may be retained by PLATZ genes across species while flax-specific roles associated with its growth habit and environmental resilience may also have been evolved [42].
Gene duplication events play a critical role in the evolution of PLATZ genes, as duplicated genes can acquire new functions, thereby enhancing plant responses to environmental stresses [45]. In this study, chromosomal mapping revealed that the 27 LuPLATZ genes are unevenly distributed across 15 chromosomes, with no tandem duplications detected (Figure 3). However, collinearity analysis identified 33 pairs of segmentally duplicated genes (Figure 4A), indicating that segmental duplication is the main driver of LuPLATZ gene expansion [46]. Collinearity analysis is a powerful approach for investigating gene evolutionary trajectories [47]. Flax was an ancient diploid plant that underwent three whole-genome duplication (WGD) events approximately 11.5, 53.5, and 114 million years ago (MYA) [24]. WGD was likely one of the primary driving forces behind the expansion of the LuPLATZ gene family, providing a large number of redundant gene copies. Comparative analysis revealed 15, 3, and 1 collinear gene pairs between flax and Arabidopsis, maize, and wheat, respectively (Figure 4B), suggesting that these homologous gene pairs may have originated from a common ancestor prior to species divergence [48]. These findings demonstrate that LuPLATZ genes are highly conserved in flax, and their expansion is likely driven primarily by segmental duplication. The expansion of the LuPLATZ gene family may have contributed to flax’s adaptation to temperate environments by enhancing its tolerance to abiotic stresses, such as cold and drought. Moreover, the functional diversification of duplicated genes might have facilitated the divergence between fiber and oilseed flax types by influencing traits related to growth, development, and stress responses.
Promoter cis-elements are key regulators of gene expression through their interactions with transcription factors [49]. Analysis of LuPLATZ promoters identified numerous elements involved in hormone signaling (MeJA and ABA) and abiotic stress responses, including anaerobic induction, drought, and cold (Figure 5). In detail, LTR elements were associated with cold response, ARE with anaerobic induction, and TC-rich repeats with drought and defense mechanisms [50]. The promoters also harbored ABA- and MeJA-responsive motifs (ABRE, TGACG-motif, CGTCA-motif) as well as MYB binding sites related to drought inducibility (MBS), indicating that these conserved cis-elements are likely pivotal for regulating both hormone-mediated and stress-related responses [51]. Notably, several drought-inducible genes (LuPLATZ6, 14, 21) contained MBSs, cold-responsive genes (LuPLATZ3, 25) carried LTR elements, and salt-induced genes (LuPLATZ11, 15) harbored ABRE motifs, suggesting that these cis-elements may underlie their stress-responsive expression.
MicroRNAs are key regulators that coordinate plant development and interactions with the environment [52]. In this study, lus-miR167 was identified as the primary miRNA targeting the LuPLATZ gene family (Table 3). The conserved miR167 has been shown to regulate plant growth and stress responses mainly through the targeting of auxin response factors (ARF6 and ARF8). In Arabidopsis, floral organ development was reported to be controlled by miR167, and misregulation of its expression was associated with defects in anther dehiscence and ovule formation [53]. In addition, miR167 expression has been found to respond to multiple stresses, through which auxin signaling was fine-tuned to enhance stress adaptation [54]. In Tamarix chinensis, the miR167/ARF6 module was observed to be negatively correlated under salt stress, suggesting that salt tolerance was promoted by this regulatory pathway [55]. In addition, LuPLATZ1 and LuPLATZ4 may be regulated by miR167, implying a potential post-transcriptional regulatory layer acting on these genes. Such regulation could contribute to fine-tuning their roles in growth and stress response pathways, adding further complexity to the regulatory network of the LuPLATZ gene family in flax. The regulatory mechanisms of PLATZ genes and the miR167–PLATZ module require further investigation.
Analysis of RNA-seq data revealed that LuPLATZ genes exhibit tissue-specific expression patterns. Most LuPLATZ genes were highly expressed in roots and leaves, whereas their expression levels were generally low in seed tissues (Figure 6). This pattern contrasts with observations in other species. For example, in Arabidopsis, AtPLATZ1 and AtPLATZ2 have been shown to contribute to seed desiccation tolerance, while AtPLATZ2 negatively regulates salt tolerance [56]. In maize, Floury3 (FL3; ZmPLATZ12) interacts with the RNA polymerase III subunit RPC53 and the transcription factor TFC1, playing a crucial role in the transcription of tRNAs and 5S rRNA in the endosperm, which is essential for proper seed development [14]. These observations suggested that PLATZ proteins have diversified functions across species. In flax, the predominant expression of LuPLATZ genes in vegetative tissues points to roles in growth and stress responses, while their low expression in seeds indicates limited involvement in reproduction. Overall, their tissue-specific expression highlights functional specialization in flax.
PLATZ transcription factors constitute a broadly conserved and versatile gene family in plants. Beyond their roles in growth and development, these genes are also key players in stress responses. In this study, several LuPLATZ genes showed marked transcriptional activation in both leaves and roots under salt stress, indicating their potential involvement in stress-responsive regulatory networks. Notably, the strong induction of LuPLATZ15 in roots and LuPLATZ16 in leaves suggests that specific members may play tissue-preferential roles in modulating stress responses (Figure 6). In this study, qRT-PCR analysis revealed dynamic and stress-specific expression profiles of several LuPLATZ genes. Under cold and salt stresses, many genes (LuPLATZ6, LuPLATZ13, LuPLATZ14, LuPLATZ25) responded rapidly with early expression peaks, whereas drought stress triggered a delayed but pronounced induction (LuPLATZ6, LuPLATZ11, LuPLATZ21, LuPLATZ27) (Figure 7). Phylogenetic analysis showed that several of these stress-inducible LuPLATZ genes are closely related to functionally validated orthologs in other crops. For example, LuPLATZ14 and LuPLATZ9 (cold/salt-responsive) clustered with GmPLATZ11, LuPLATZ2 and LuPLATZ3 (drought-responsive) grouped near GmPLATZ17, and LuPLATZ22, LuPLATZ26, LuPLATZ8, and LuPLATZ23 were close to OsPLATZ7/9/11 (stress- and hormone-responsive in rice). These relationships suggest that stress-related functions may be conserved between flax and other species. In rice, OsPLATZ9 exhibited consistently high expression across diverse tissues, implying its multifaceted role in plant development, whereas OsPLATZ7 and OsPLATZ11 displayed variable expression under different abiotic stresses and hormone treatments, underscoring their significance in stress response and hormonal signaling [57]. Overexpression of GhPLATZ1 in Arabidopsis enhanced seedling and seed tolerance to salt and osmotic stress and reduced sensitivity to ABA and GA/PAC treatments [18]. Functional validation of SiPLATZ12 in foxtail millet indicated that it repressed salt-tolerance genes, thereby reducing salt resistance [58]. In Arabidopsis, AtPLATZ2 was transcriptionally upregulated under salt stress, and overexpression increased salt sensitivity, whereas AtPLATZ2/PLATZ7 double mutants exhibited markedly reduced salt tolerance [19]. Further yeast transformation assays in this study demonstrated that overexpression of LuPLATZ14 and LuPLATZ21 enhanced drought tolerance (Figure 9). Other studies supported similar mechanisms: AtPLATZ4 improved drought tolerance by directly binding the A/T-rich promoter of PIP2;8, a plasma membrane aquaporin gene, repressing its expression to promote stomatal closure and reduce water loss [59]. In soybean, GmPLATZ17 negatively regulated drought tolerance through interaction with GmDREB5 [60]. Overexpression of bamboo PhePLATZ1 in Arabidopsis enhanced drought resistance and improved physiological traits such as water retention and reduced oxidative damage, supporting a positive regulatory role via the ABA pathway [61]. Collectively, these findings provided strong evidence that PLATZ genes played crucial roles in abiotic stress responses, and characterization of the LuPLATZ gene family laidv the foundation for further understanding their molecular functions in flax stress tolerance. However, as this study is primarily based on computational and expression analyses, the specific functions of LuPLATZ genes have not yet been experimentally validated, which represents a limitation that should be addressed in future work.
5. Conclusions
This study provides the first genome-wide characterization of the PLATZ gene family in flax, identifying 27 members based on the T2T gap-free genome. LuPLATZ genes are phylogenetically diverse and exhibit subfamily-specific responses to hormonal and abiotic stresses, with LuPLATZ14 and LuPLATZ21 conferring drought tolerance in yeast. These findings establish a molecular framework for understanding stress signaling in flax and highlight candidate genes for enhancing stress resilience, yield stability, and productivity in this dual-purpose oilseed and fiber crop. Future work should focus on functional validation of additional LuPLATZ genes and their incorporation into breeding strategies to improve flax performance under variable environments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy15092233/s1, Table S1. The PLATZ family genes in Linum usitatissimum L.; Table S2. Information of hormone- and stress-related cis-elements detected in the promoters regions of LuPLATZs.; Table S3. The list primer was used for gene expression analysis.
Author Contributions
Conceptualization, J.L. (Jianyu Lu) and H.W. (Hanlu Wu); methodology, J.L. (Jianyu Lu) and H.W. (Hanlu Wu); validation, H.W. (Hang Wang); writing—original draft preparation, J.L. (Jianyu Lu), H.W. (Hanlu Wu), H.W. (Hang Wang), J.L. (Jinxi Li) and Z.Z.; writing—review and editing, J.L. (Jianyu Lu); visualization, Z.Z.; supervision, G.W. and J.Z.; project administration, G.W. and J.Z.; funding acquisition, G.W. and J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the Jilin Agricultural University high-level researcher grant JLAUHLRG20102006 and Jilin Provincial Department of Human Resources and Social Security Grant: No. 201020012.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, J.; Ji, C.; Li, Q.; Zhou, Y.; Wu, Y. Genome-wide analysis of the plant-specific PLATZ proteins in maize and identification of their general role in interaction with RNA polymerase III complex. BMC Plant Biol. 2018, 18, 221. [Google Scholar] [CrossRef]
- Berg, J.M.; Shi, Y. The galvanization of biology: A growing appreciation for the roles of zinc. Science 1996, 271, 1081–1085. [Google Scholar] [CrossRef]
- Takatsuji, H. Zinc-finger transcription factors in plants. Cell. Mol. Life Sci. 1998, 54, 582–596. [Google Scholar] [CrossRef]
- Nagano, Y.; Furuhashi, H.; Inaba, T.; Sasaki, Y. A novel class of plant-specific zinc-dependent DNA-binding protein that binds to A/T-rich DNA sequences. Nucleic Acids Res. 2001, 29, 4097–4105. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhu, G.; Meng, Q.; Zeng, J.; He, X.; Liu, W. Comprehensive analysis of PLATZ family genes and their responses to abiotic stresses in Barley. BMC Plant Biol. 2024, 24, 982. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, S.; Zhang, Y.; Xu, L.; Luo, Y.; Yuan, Y.; Yang, Q.; Feng, B. Genome-wide identification and expression analysis of the plant-specific PLATZ gene family in Tartary buckwheat (Fagopyrum tataricum). BMC Plant Biol. 2022, 22, 160. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Guan, Z.; Wu, S.; Zhang, J.; Lin, C.; Sun, Y.; Shen, M.; Zhang, C. Genome-Wide Identification and Salt Stress-Responsive Expression Analysis of the GmPLATZ Gene Family in Soybean (Glycine max L.). Plants 2025, 14, 2004. [Google Scholar] [CrossRef]
- Yang, T.; Xu, X.-T.; Tang, L.-J.; Wei, W.-T.; Zhao, Y.-Y.; Liu, J.-X.; Yao, X.-F.; Zhao, H.; Liu, C.-M.; Bai, A.N. Genome-Wide Study of Plant-Specific PLATZ Transcription Factors and Functional Analysis of OsPLATZ1 in Regulating Caryopsis Development of Rice (Oryza sativa L.). Plants 2025, 14, 151. [Google Scholar] [CrossRef]
- Wang, P.; Teng, H.; Qiao, D.; Liang, F.; Zhu, K.; Miao, M.; Hua, B. The Role of PLATZ6 in Raffinose Family Oligosaccharides Loading of Leaves via PLATZ Family Characterization in Cucumber. Plants 2024, 13, 2825. [Google Scholar] [CrossRef]
- Fan, B.; Ren, M.; Chen, G.; Zhou, X.; Cheng, G.; Yang, J.; Sun, H. Exploring the Roles of the Plant AT-Rich Sequence and Zinc-Binding (PLATZ) Gene Family in Tomato (Solanum lycopersicum L.) Under Abiotic Stresses. Int. J. Mol. Sci. 2025, 26, 1682. [Google Scholar] [CrossRef]
- Wai, A.H.; Rahman, M.M.; Waseem, M.; Cho, L.H.; Naing, A.H.; Jeon, J.S.; Lee, D.J.; Kim, C.K.; Chung, M.Y. Comprehensive Genome-Wide Analysis and Expression Pattern Profiling of PLATZ Gene Family Members in Solanum Lycopersicum L. under Multiple Abiotic Stresses. Plants 2022, 11, 3112. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, J.; Jun, S.E.; Park, S.; Timilsina, R.; Kwon, D.S.; Kim, Y.; Park, S.J.; Hwang, J.Y.; Nam, H.G.; et al. ORESARA15, a PLATZ transcription factor, mediates leaf growth and senescence in Arabidopsis. New Phytol. 2018, 220, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Guérin, C.; Behr, M.; Sait, J.; Mol, A.; El Jaziri, M.; Baucher, M. Evidence for poplar PtaPLATZ18 in the regulation of plant growth and vascular tissues development. Front. Plant Sci. 2023, 14, 1302536. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, J.; Ye, J.; Zheng, X.; Xiang, X.; Li, C.; Fu, M.; Wang, Q.; Zhang, Z.; Wu, Y. The Maize Imprinted Gene Floury3 Encodes a PLATZ Protein Required for tRNA and 5S rRNA Transcription through Interaction with RNA Polymerase III. Plant Cell 2017, 29, 2661–2675. [Google Scholar] [CrossRef]
- Wang, A.; Hou, Q.; Si, L.; Huang, X.; Luo, J.; Lu, D.; Zhu, J.; Shangguan, Y.; Miao, J.; Xie, Y.; et al. The PLATZ Transcription Factor GL6 Affects Grain Length and Number in Rice. Plant Physiol. 2019, 180, 2077–2090. [Google Scholar] [CrossRef]
- Gonzalez-Morales, S.I.; Chavez-Montes, R.A.; Hayano-Kanashiro, C.; Alejo-Jacuinde, G.; Rico-Cambrón, T.Y.; de Folter, S.; Herrera-Estrella, L. Regulatory network analysis reveals novel regulators of seed desiccation tolerance in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2016, 113, E5232–E5241. [Google Scholar] [CrossRef]
- Dong, T.; Yin, X.; Wang, H.; Lu, P.; Liu, X.; Gong, C.; Wu, Y. ABA-INDUCED expression 1 is involved in ABA-inhibited primary root elongation via modulating ROS homeostasis in Arabidopsis. Plant Sci. 2021, 304, 110821. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, R.; Huo, Y.; Liu, S.; Yang, G.; Huang, J.; Zheng, C.; Wu, C. Expression of cotton PLATZ1 in transgenic Arabidopsis reduces sensitivity to osmotic and salt stress for germination and seedling establishment associated with modification of the abscisic acid, gibberellin, and ethylene signalling pathways. BMC Plant Biol. 2018, 18, 218. [Google Scholar] [CrossRef]
- Liu, S.; Yang, R.; Liu, M.; Zhang, S.; Yan, K.; Yang, G.; Huang, J.; Zheng, C.; Wu, C. PLATZ2 negatively regulates salt tolerance in Arabidopsis seedlings by directly suppressing the expression of the CBL4/SOS3 and CBL10/SCaBP8 genes. J. Exp. Bot. 2020, 71, 5589–5602. [Google Scholar] [CrossRef]
- Naish, M.; Alonge, M.; Włodzimierz, P.; Tock, A.J.; Abramson, B.W.; Schmücker, A.; Mandáková, T.; Jamge, B.; Lambing, C.; Kuo, P.; et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 2021, 374, eabi7489. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; Chen, H.; Zhang, Z.; Wu, J.; Wang, X. A near-complete genome assembly of Brassica rapa provides new insights into the evolution of centromeres. Plant Biotechnol. J. 2023, 21, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qi, Y.; Wang, L.; Wang, L.; Yan, X.; Dang, Z.; Li, W.; Zhao, W.; Pei, X.; Li, X.; et al. Genomic Comparison and Population Diversity Analysis Provide Insights into the Domestication and Improvement of Flax. iScience 2020, 23, 100967. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, H.; Wang, F.; Li, J.; Wang, Y.; Zhao, Q.; Wang, Y.; Wang, X.; Lei, X.; Sun, R.; et al. Telomere to telomere flax (Linum usitatissimum L.) genome assembly unlocks insights beyond fatty acid metabolism pathways. Hortic. Res. 2025, 12, uhaf127. [Google Scholar] [CrossRef]
- Huis, R.; Hawkins, S.; Neutelings, G. Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol. 2010, 10, 71. [Google Scholar] [CrossRef]
- Chytilová, M.; Mudroňová, D.; Nemcová, R.; Gancarčíková, S.; Buleca, V.; Koščová, J.; Tkáčiková, L. Anti-inflammatory and immunoregulatory effects of flax-seed oil and Lactobacillus plantarum—Biocenol LP96 in gnotobiotic pigs challenged with enterotoxigenic Escherichia coli. Res. Vet. Sci. 2013, 95, 103–109. [Google Scholar] [CrossRef]
- Heller, K.; Sheng, Q.C.; Guan, F.; Alexopoulou, E.; Hua, L.S.; Wu, G.W.; Jankauskiene, Z.; Fu, W.Y. A comparative study between Europe and China in crop management of two types of flax: Linseed and fibre flax. Ind. Crop Prod. 2015, 68, 24–31. [Google Scholar] [CrossRef]
- Santos, H.O.; Price, J.C.; Bueno, A.A. Beyond Fish Oil Supplementation: The Effects of Alternative Plant Sources of Omega-3 Polyunsaturated Fatty Acids upon Lipid Indexes and Cardiometabolic Biomarkers-An Overview. Nutrients 2020, 12, 3159. [Google Scholar] [CrossRef]
- Wang, N.; Qi, F.; Wang, F.; Lin, Y.; Xiaoyang, C.; Peng, Z.; Zhang, B.; Qi, X.; Deyholos, M.K.; Zhang, J. Evaluation of Differentially Expressed Genes in Leaves vs. Roots Subjected to Drought Stress in Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2023, 24, 12019. [Google Scholar] [CrossRef]
- Wang, N.; Lin, Y.; Qi, F.; Xiaoyang, C.; Peng, Z.; Yu, Y.; Liu, Y.; Zhang, J.; Qi, X.; Deyholos, M.K.; et al. Comprehensive Analysis of Differentially Expressed Genes and Epigenetic Modification-Related Expression Variation Induced by Saline Stress at Seedling Stage in Fiber and Oil Flax, Linum usitatissimum L. Plants 2022, 11, 2053. Plants 2022, 11, 2053. [Google Scholar] [CrossRef]
- Garcia-Hernandez, M.; Berardini, T.Z.; Chen, G.; Crist, D.; Doyle, A.; Huala, E.; Knee, E.; Lambrecht, M.; Miller, N.; Mueller, L.A.; et al. TAIR: A resource for integrated Arabidopsis data. Funct. Integr. Genom. 2002, 2, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Khaja, R.; MacDonald, J.R.; Zhang, J.; Scherer, S.W. Methods for identifying and mapping recent segmental and gene duplications in eukaryotic genomes. Methods Mol. Biol. 2006, 338, 9–20. [Google Scholar] [CrossRef]
- Melnikova, N.V.; Dmitriev, A.A.; Belenikin, M.S.; Koroban, N.V.; Speranskaya, A.S.; Krinitsina, A.A.; Krasnov, G.S.; Lakunina, V.A.; Snezhkina, A.V.; Sadritdinova, A.F.; et al. Identification, Expression Analysis, and Target Prediction of Flax Genotroph MicroRNAs Under Normal and Nutrient Stress Conditions. Front. Plant Sci. 2016, 7, 399. [Google Scholar] [CrossRef]
- Carpenter, C.M.; Frank, D.N.; Williamson, K.; Arbet, J.; Wagner, B.D.; Kechris, K.; Kroehl, M.E. tidyMicro: A pipeline for microbiome data analysis and visualization using the tidyverse in R. BMC Bioinform. 2021, 22, 41. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Lu, J.; Xiaoyang, C.; Li, J.; Wu, H.; Wang, Y.; Di, P.; Deyholos, M.K.; Zhang, J. Whole-Genome Identification of the Flax Fatty Acid Desaturase Gene Family and Functional Analysis of the LuFAD2.1 Gene Under Cold Stress Conditions. Plant Cell Environ. 2025, 48, 2221–2239. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, T.; Wang, Z.; Zhang, F.; Li, N.; Jiang, W. Genome-Wide Identification and Expression Analysis of the PLATZ Transcription Factor in Tomato. Plants 2023, 12, 2632. [Google Scholar] [CrossRef]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Wang, L.; Liu, H.; Jiang, N.; He, W.; Yan, H.; Wu, M.; Xiang, Y. Whole-genome identification and multiple abiotic stresses expression pattern profiling analysis of PLATZ transcription factor family members in Pecan (Carya illinoensis). Int. J. Biol. Macromol. 2023, 248, 125959. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Li, J.; Tang, H.; Paterson, A.H. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell 2014, 26, 2792–2802. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.W.; Donoghue, P. Whole-Genome Duplication and Plant Macroevolution. Trends Plant Sci. 2018, 23, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef]
- Dhatterwal, P.; Mehrotra, S.; Miller, A.J.; Mehrotra, R. Promoter profiling of Arabidopsis amino acid transporters: Clues for improving crops. Plant Mol. Biol. 2021, 107, 451–475. [Google Scholar] [CrossRef]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.P. ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity. New Phytol. 2014, 202, 35–49. [Google Scholar] [CrossRef]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and Their Regulatory Roles in Plant-Environment Interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nagpal, P.; Villarino, G.; Trinidad, B.; Bird, L.; Huang, Y.; Reed, J.W. miR167 limits anther growth to potentiate anther dehiscence. Development 2019, 146, dev.174375. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Di, D.; Wu, L.; Yang, J.; Lu, Y.; Shi, W. MicroRNAs Are Involved in Regulating Plant Development and Stress Response through Fine-Tuning of TIR1/AFB-Dependent Auxin Signaling. Int. J. Mol. Sci. 2022, 23, 510. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, J.; Wang, W.; Xu, L.A. ARF family identification in Tamarix chinensis reveals the salt responsive expression of TcARF6 targeted by miR167. PeerJ 2020, 8, e8829. [Google Scholar] [CrossRef]
- Gu, F.; Ren, Y.; Manzoor, M.A.; Wang, T.; Huang, R.; Chen, N.; Song, C.; Zhang, Y. Plant AT-rich protein and zinc-binding protein (PLATZ) family in Dendrobium huoshanense: Identification, evolution and expression analysis. BMC Plant Biol. 2024, 24, 1276. [Google Scholar] [CrossRef]
- Chen, H.; Ma, X.; Lv, G.; Wang, Z.; Wang, L.; Yan, B.; Shang, W.; Wang, X.; Ma, Z.; Zheng, W. Genome-Wide Analysis of the PLATZ Gene Family in Oryza Genus: Evolution, Expression During Inflorescence Development and Stress Responses. Agronomy 2025, 15, 117. [Google Scholar] [CrossRef]
- Cai, K.; Song, X.; Yue, W.; Liu, L.; Ge, F.; Wang, J. Identification and Functional Characterization of Abiotic Stress Tolerance-Related PLATZ Transcription Factor Family in Barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2024, 25, 10191. [Google Scholar] [CrossRef]
- Liu, M.; Wang, C.; Ji, Z.; Lu, J.; Zhang, L.; Li, C.; Huang, J.; Yang, G.; Yan, K.; Zhang, S.; et al. Regulation of drought tolerance in Arabidopsis involves the PLATZ4-mediated transcriptional repression of plasma membrane aquaporin PIP2;8. Plant J. 2023, 115, 434–451. [Google Scholar] [CrossRef]
- Zhao, J.; Zheng, L.; Wei, J.; Wang, Y.; Chen, J.; Zhou, Y.; Chen, M.; Wang, F.; Ma, Y.; Xu, Z. The soybean PLATZ transcription factor GmPLATZ17 suppresses drought tolerance by interfering with stress-associated gene regulation of GmDREB5. Crop. J. 2022, 10, 1014–1025. [Google Scholar] [CrossRef]
- Zhang, K.; Lan, Y.; Wu, M.; Wang, L.; Liu, H.; Xiang, Y. PhePLATZ1, a PLATZ transcription factor in moso bamboo (Phyllostachys edulis), improves drought resistance of transgenic Arabidopsis thaliana. Plant Physiol. Biochem. 2022, 186, 121–134. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).