
Population Structure and Genetic Diversity of Colletotrichum gloeosporioides on Citrus in China


Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Fungal Strains
2.2. DNA Extraction, PCR Amplification, and Sequencing
2.3. Data Analysis
3. Results
3.1. GAPDH Sequence Haplotype Diversity
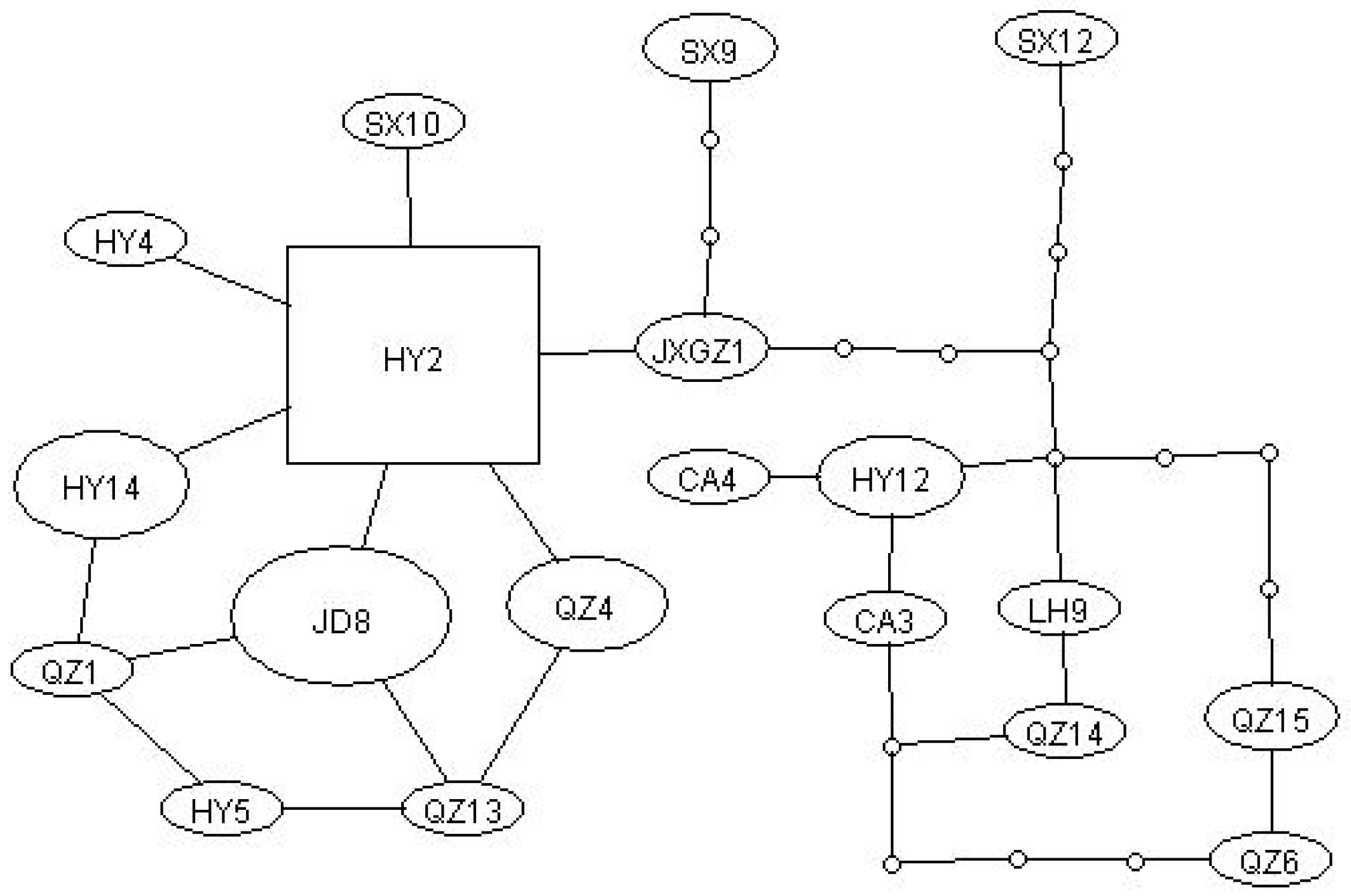
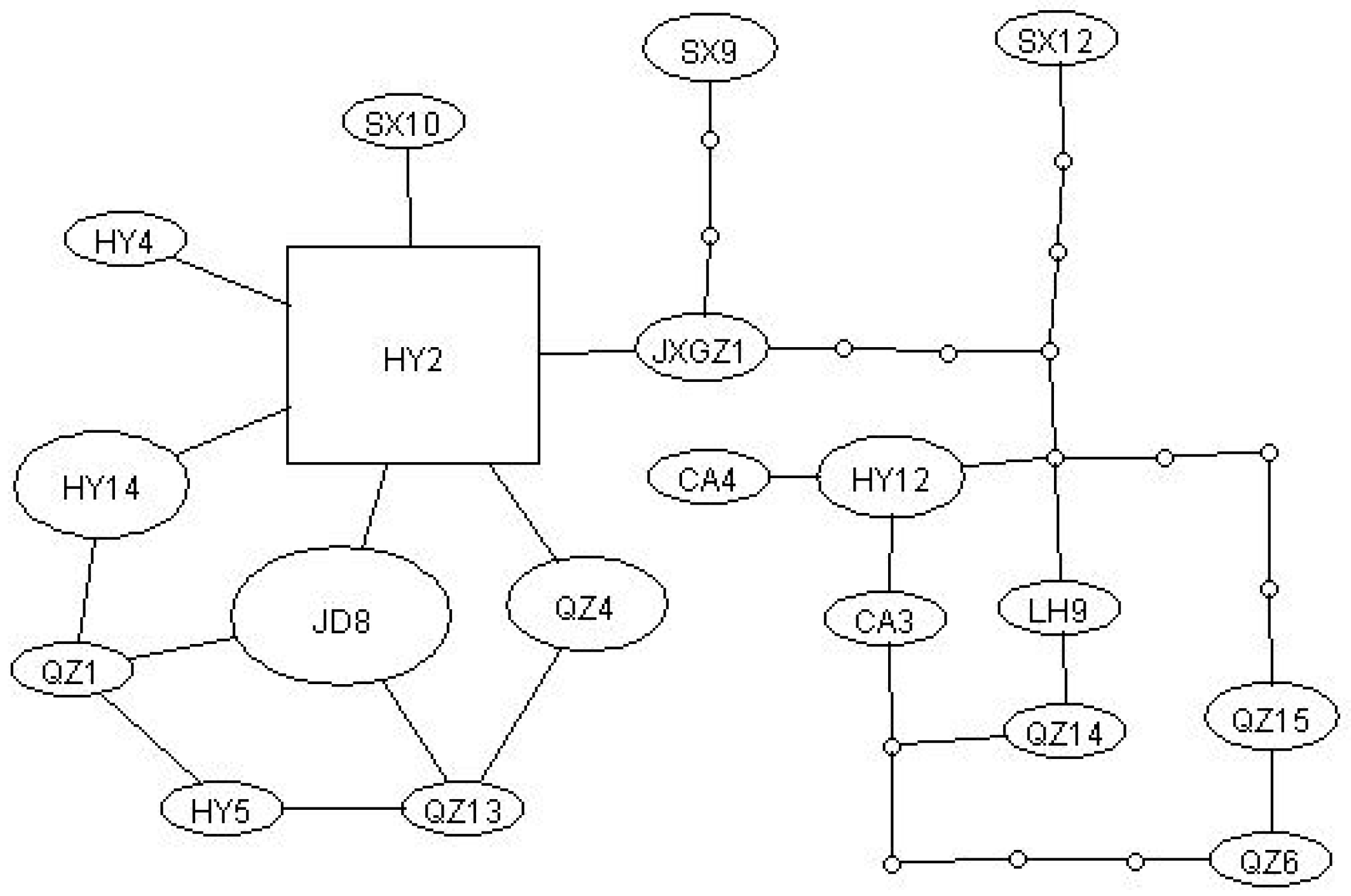
3.2. Haplotype Network Analysis
3.3. Genetic Differentiation Analysis
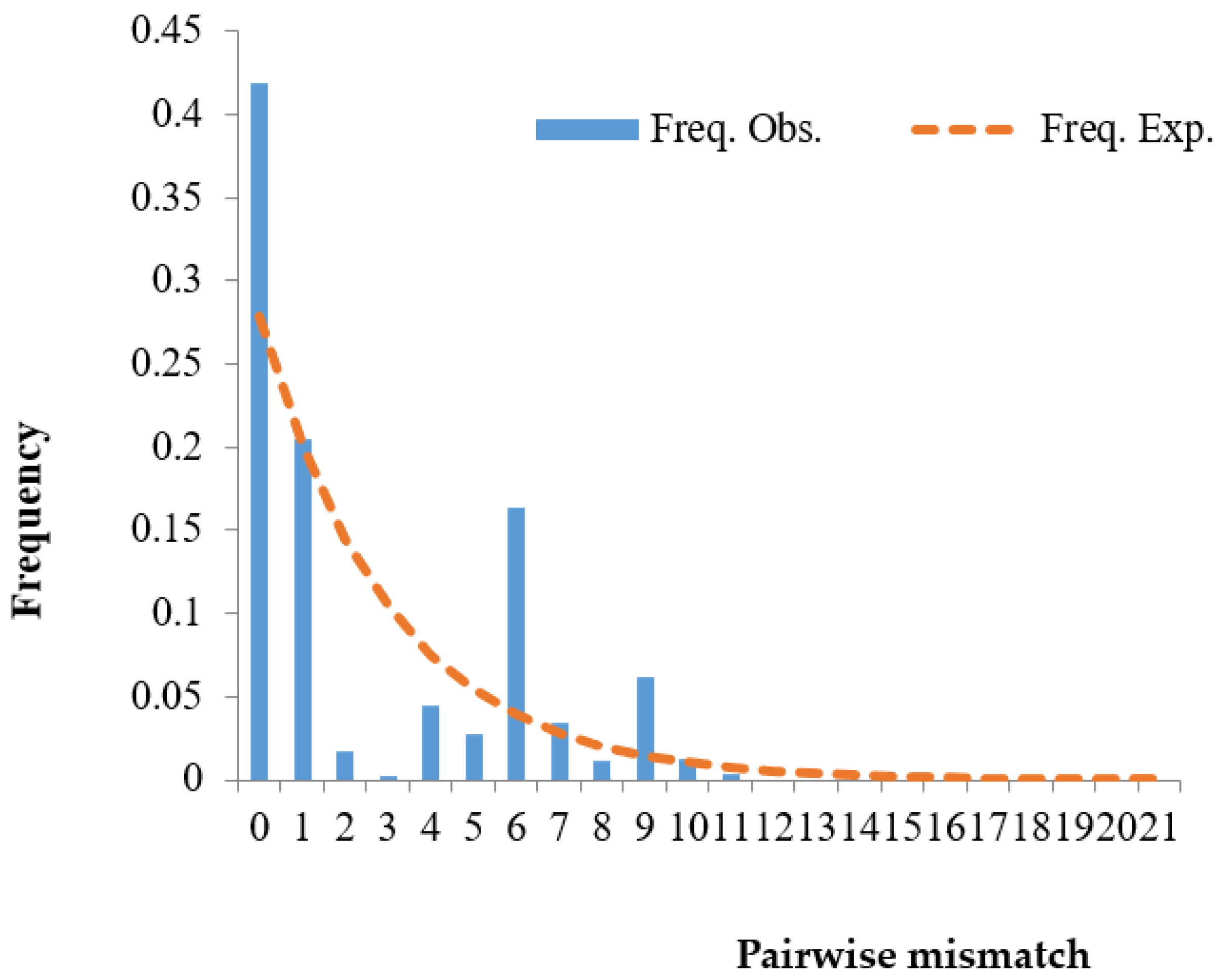
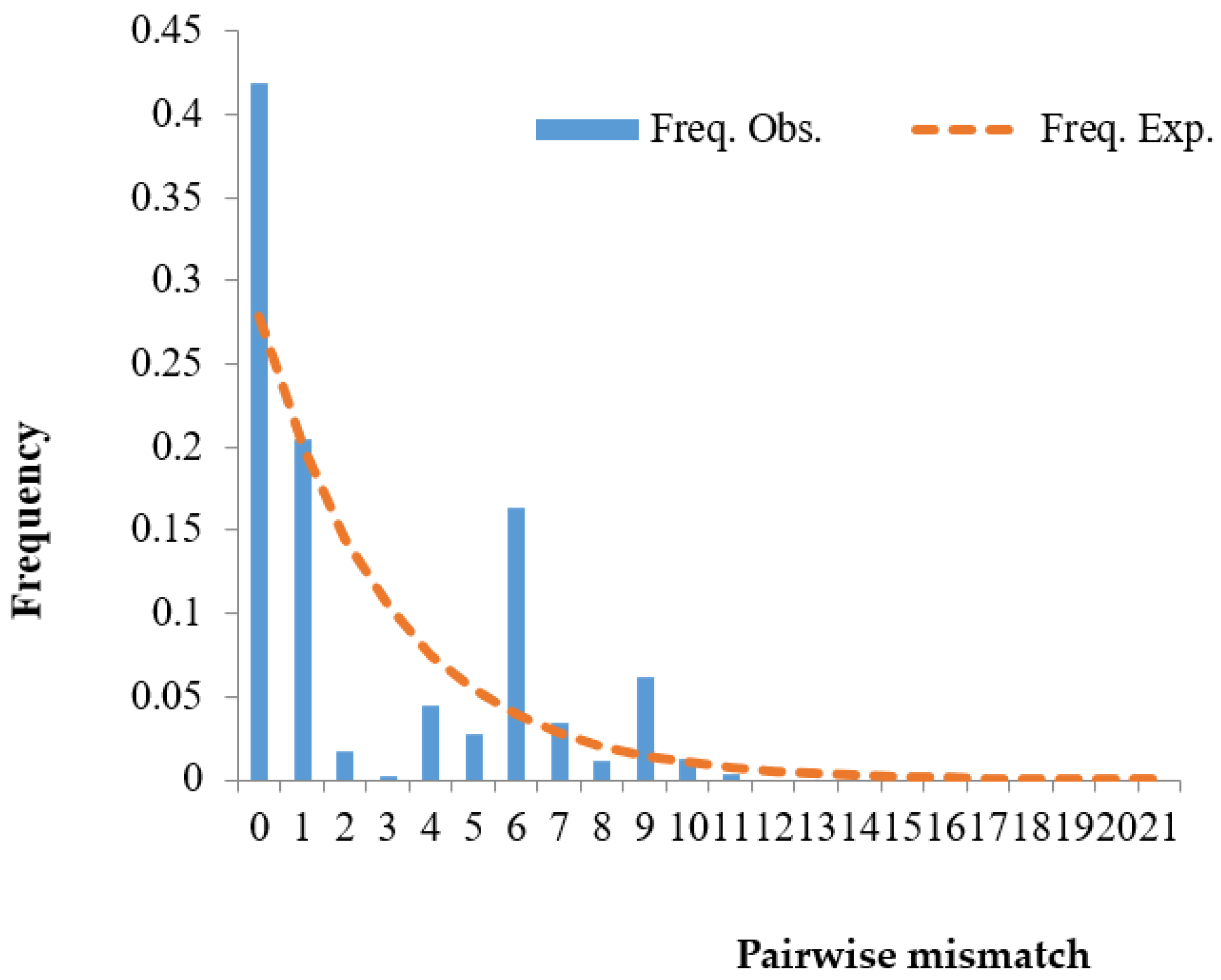
3.4. Population Expansion Analysis
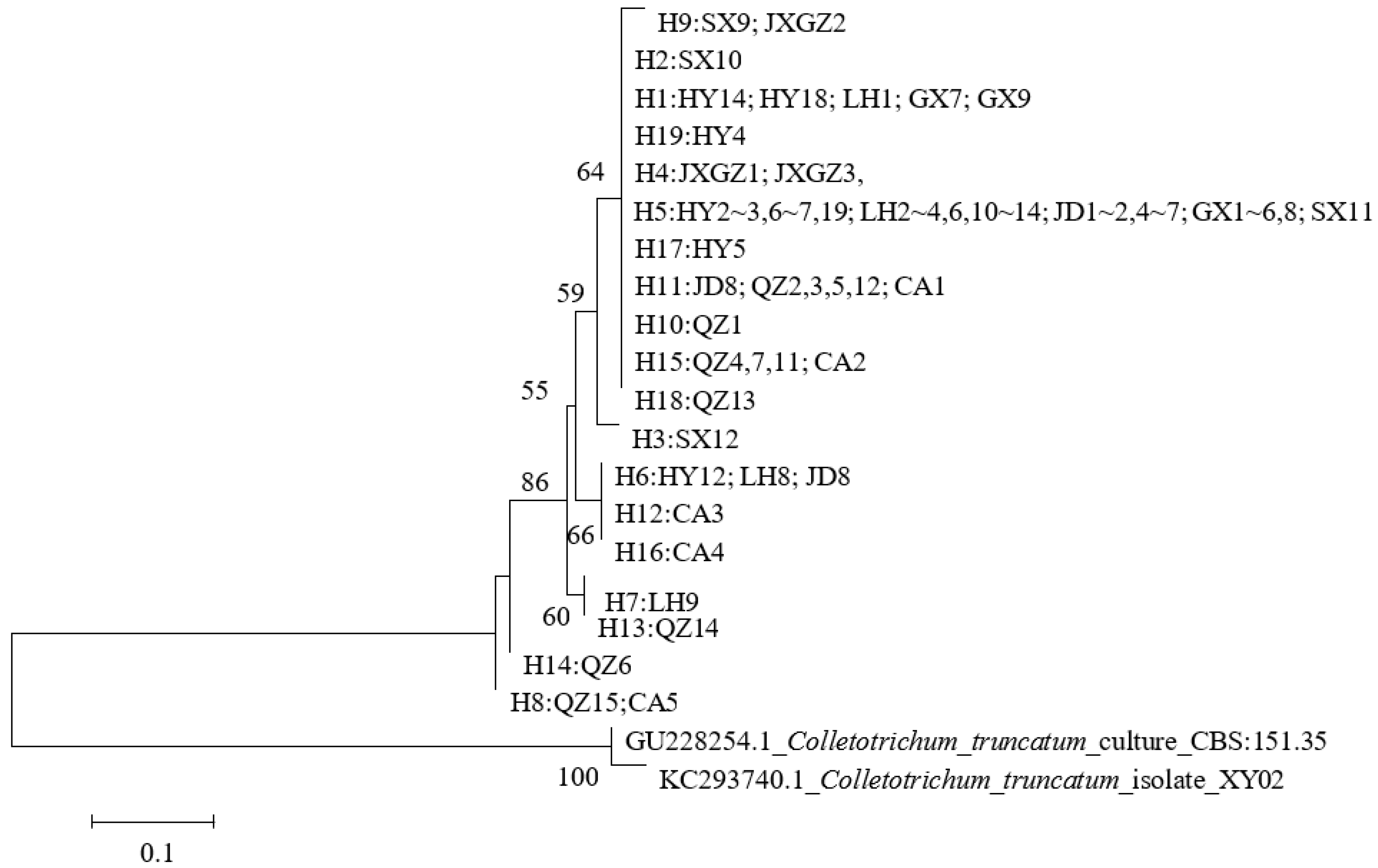
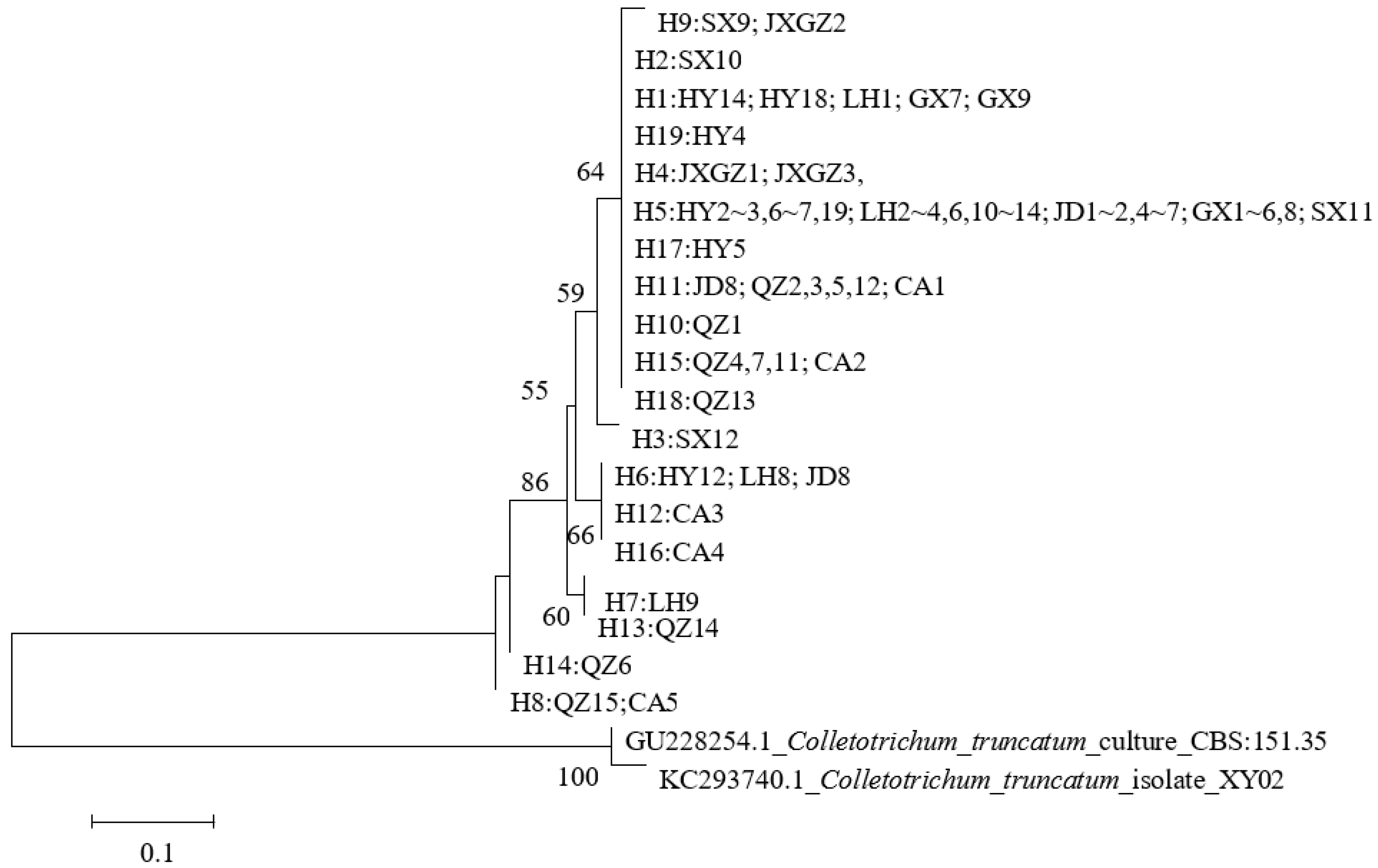
3.5. Phylogenetic Relationship Analysis Based on GAPDH Sequences
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, G.A.; Terol, J.; Ibanez, V.; López-García, A.; Pérez-Román, E.; Borredá, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R. Genomics of the origin and evolution of Citrus. Nature 2018, 554, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uysal, A.; Kurt, Ş.; Guarnaccia, V. Distribution and characterization of Colletotrichum species associated with Citrus anthracnose in eastern Mediterranean region of Turkey. Eur. J. Plant Pathol. 2022, 163, 125–141. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Pinnaka, A.K.; Shenoy, B.D. Resolving the Colletotrichum siamense species complex using ApMat marker. Fungal Divers. 2015, 71, 247–264. [Google Scholar] [CrossRef]
- Weir, B.S.; Johnston, P.R.; Damm, U. The Colletotrichum gloeosporioides species complex. Stud. Mycol. 2012, 73, 115–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Chen, G.Q.; Hou, X.; Fu, Y.S.; Cai, L.; Hyde, K.D.; Li, H.Y. Colletotrichum species associated with cultivated citrus in China. Fungal Divers. 2013, 61, 61–74. [Google Scholar] [CrossRef]
- Zhan, J.; McDonald, B.A. Experimental measures of pathogen competition and relative fitness. Annu. Rev. Phytopathol. 2013, 51, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, G.; Huang, F.; Zhang, J.; Hyde, K.; Li, H. Phyllosticta species associated with citrus diseases in China. Fungal Divers. 2011, 52, 209–224. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, E.; Van de Peer, Y. zt: A sofware tool for simple and partial mantel tests. J. Stat. Softw. 2002, 7, 1–12. [Google Scholar] [CrossRef]
- He, L.I.; Zhu, D.X.; Jian-Ping, X.U.; Zhou, G.Y.; Mo, H.U.; Tian, F. Population genetic structure of Colletotrichum gloeosporioides causing anthracnose of Camellia oleifera in China. Acta Phytopathol. Sin. 2014, 44, 620–628. [Google Scholar]
- Pu, Z.X.; Lu, L.M.; Huang, Z.H.; Hu, X.R.; Du, D.C.; Chen, G.Q. Genetic diversity of Colletotrichum gloeosporioides from Zhejiang by ISSR molecular markers. J. Fujian Agric. For. Univ. (Nat. Sci. Ed.) 2017, 6, 607–611. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Strain | Site (Code) | Longitude, Latitude | Host (Code) | Genbank Accession of GAPDH | Hyplotype (Hn) | Collection Year |
|---|---|---|---|---|---|---|
| HY2 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus sinensis Osbeck (QC) | MT449239 | H5 | 2018 |
| HY3 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus sinensis Osbeck (QC) | MT449240 | H5 | 2018 |
| HY4 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus unchiu Hort. ex Tanaka (WG) | MT449241 | H19 | 2018 |
| HY5 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus unchiu Hort. ex Tanaka (WG) | MT449242 | H17 | 2018 |
| HY6 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449243 | H5 | 2018 |
| HY7 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449244 | H5 | 2018 |
| HY12 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus unchiu Hort. ex Tanaka (WG) | MT449249 | H6 | 2018 |
| HY14 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus unchiu Hort. ex Tanaka (WG) | MT449251 | H1 | 2018 |
| HY18 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449255 | H1 | 2018 |
| HY19 | Huangyan, Zhejiang (HY) | 121.17, 28.64 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449256 | H5 | 2018 |
| LH1 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus unchiu Hort. ex Tanaka (WG) | MT449257 | H1 | 2018 |
| LH2 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus unchiu Hort. ex Tanaka (WG) | MT449258 | H5 | 2018 |
| LH3 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449259 | H5 | 2018 |
| LH4 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449260 | H5 | 2018 |
| LH6 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449262 | H5 | 2018 |
| LH8 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449264 | H6 | 2018 |
| LH9 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus unchiu Hort. ex Tanaka (WG) | MT449265 | H7 | 2018 |
| LH10 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449266 | H5 | 2018 |
| LH11 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449267 | H5 | 2018 |
| LH12 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449268 | H5 | 2018 |
| LH13 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus reticulata Blanco cv. Succosa (BDZ) | MT449269 | H5 | 2018 |
| LH14 | Linhai, Zhejiang (LH) | 121.29, 28.76 | Citrus unchiu Hort. ex Tanaka (WG) | KC293710 | H5 | 2018 |
| JD1 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449270 | H5 | 2018 |
| JD2 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449271 | H5 | 2018 |
| JD3 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449272 | H6 | 2018 |
| JD4 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449273 | H5 | 2018 |
| JD5 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449274 | H5 | 2018 |
| JD6 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449275 | H5 | 2018 |
| JD7 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449276 | H5 | 2018 |
| JD8 | Jiande, Zhejiang (JD) | 119.56, 29.54 | Citrus unchiu Hort. ex Tanaka (WG) | MT449277 | H11 | 2018 |
| GX1 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449278 | H5 | 2018 |
| GX2 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449279 | H5 | 2018 |
| GX3 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449280 | H5 | 2018 |
| GX4 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449281 | H5 | 2018 |
| GX5 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449282 | H5 | 2018 |
| GX6 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449283 | H5 | 2018 |
| GX7 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449284 | H1 | 2018 |
| GX8 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449285 | H5 | 2018 |
| GX9 | Mengshan, Guangxi (GX) | 110.53, 24.2 | Mashui tangerine (MSJ) | MT449286 | H1 | 2018 |
| SX9 | Chenggu, Shanxi (SX) | 109.03, 34.31 | Citrus unchiu Hort. ex Tanaka (WG) | KC293717 | H9 | 2012 |
| SX10 | Chenggu, Shanxi (SX) | 109.03, 34.31 | Citrus unchiu Hort. ex Tanaka (WG) | KC293718 | H2 | 2012 |
| SX11 | Chenggu, Shanxi (SX) | 109.03, 34.31 | Citrus unchiu Hort. ex Tanaka (WG) | KC293719 | H5 | 2011 |
| SX12 | Chenggu, Shanxi (SX) | 109.03, 34.31 | Citrus unchiu Hort. ex Tanaka (WG) | KC293720 | H3 | 2011 |
| QZ1 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449295 | H10 | 2019 |
| QZ2 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449296 | H11 | 2019 |
| QZ3 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449297 | H11 | 2019 |
| QZ4 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449298 | H15 | 2019 |
| QZ5 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449299 | H11 | 2019 |
| QZ6 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449300 | H14 | 2019 |
| QZ7 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus unchiu Hort. ex Tanaka (WG) | MT449301 | H15 | 2019 |
| QZ11 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus paradise Macf (PTY) | MT449305 | H15 | 2019 |
| QZ12 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus paradise Macf (PTY) | MT449306 | H11 | 2019 |
| QZ13 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus paradise Macf (PTY) | MT449307 | H18 | 2019 |
| QZ14 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus paradise Macf (PTY) | MT449308 | H13 | 2019 |
| QZ15 | Quzhou, Zhejiang (QZ) | 118.88, 28.98 | Citrus paradise Macf (PTY) | MT449309 | H8 | 2019 |
| CA1 | Chun’an, Zhejiang (CA) | 118.56, 29.4 | Citrus unchiu Hort. ex Tanaka (WG) | MT449310 | H11 | 2019 |
| CA2 | Chun’an, Zhejiang (CA) | 118.56, 29.4 | Citrus unchiu Hort. ex Tanaka (WG) | MT449311 | H15 | 2019 |
| CA3 | Chun’an, Zhejiang (CA) | 118.56, 29.4 | Citrus unchiu Hort. ex Tanaka (WG) | MT449312 | H12 | 2019 |
| CA4 | Chun’an, Zhejiang (CA) | 118.56, 29.4 | Citrus unchiu Hort. ex Tanaka (WG) | MT449313 | H16 | 2019 |
| CA5 | Chun’an, Zhejiang (CA) | 118.56, 29.4 | Citrus unchiu Hort. ex Tanaka (WG) | MT449314 | H8 | 2019 |
| JXGZ1 | Ganzhou, Jiangxi (JXGZ) | 114.9, 25.8 | Citrus sinensis Osbeck (QC) | KC293706 | H4 | 2011 |
| JXGZ2 | Ganzhou, Jiangxi (JXGZ) | 114.9, 25.8 | Citrus sinensis Osbeck (QC) | KC293707 | H9 | 2011 |
| JXGZ3 | Ganzhou, Jiangxi (JXGZ) | 114.9, 25.8 | Citrus unchiu Hort. ex Tanaka (WG) | KC293708 | H4 | 2011 |
| Source | df | SS | MS | Est. Var. | % | Stat | Value | Prob (P) |
|---|---|---|---|---|---|---|---|---|
| geographic populations | ||||||||
| Among Pops | 7 | 6.415 | 0.916 | 0.077 | 19% | PhiPT | 0.190 | 0.001 |
| Within Pops | 55 | 17.997 | 0.327 | 0.327 | 81% | |||
| Total | 62 | 24.413 | 0.404 | 100% | ||||
| host populations | ||||||||
| Among Pops | 4 | 3.403 | 0.851 | 0.047 | 12% | PhiPT | 0.115 | 0.002 |
| Within Pops | 58 | 21.010 | 0.362 | 0.362 | 88% | |||
| Total | 62 | 24.413 | 0.409 | 100% |
| Geographical Populations | HY | LH | JD | GX | SX | QZ | CA | JXGZ | Host Populations | QC | WG | BDZ | MSJ | PTY |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HY | 0.000 | QC | 0.000 | |||||||||||
| LH | 0.052 | 0.000 | WG | 0.047 | 0.000 | |||||||||
| JD | 0.056 | 0.096 | 0.000 | BDZ | 0.048 | 0.029 | 0.000 | |||||||
| GX | 0.039 | 0.052 | 0.028 | 0.000 | MSJ | 0.132 | 0.137 | 0.012 | 0.000 | |||||
| SX | 0.005 | 0.051 | 0.046 | 0.065 | 0.000 | PTY | 0.069 | 0.075 | 0.051 | 0.164 | 0.000 | |||
| QZ | 0.020 | 0.030 | 0.010 | 0.123 | 0.040 | 0.000 | - | - | - | - | - | - | ||
| CA | 0.223 | 0.144 | 0.183 | 0.368 | 0.049 | 0.027 | 0.000 | - | - | - | - | - | - | |
| JXGZ | 0.321 | 0.270 | 0.300 | 0.462 | 0.102 | 0.220 | 0.280 | 0.000 | - | - | - | - | - | - |
| Hn (Number of Strains) | Geographical Origin | Host Origin | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HY (n) | LH (n) | JD (n) | GX (n) | SX (n) | QZ (n) | CA (n) | JXGZ (n) | QC (n) | WG (n) | BDZ (n) | MSJ (n) | PTY (n) | |
| H1 (2) | H1 (1) | H5 (6) | H1 (2) | H2 (1) | H8 (1) | H8 (1) | H4 (2) | H4 (1) | H1 (2) H2 (1) H3 (1) H4 (1) H5 (9) H6 (2) H7 (1) H8 (1) H9 (1) H10 (1) H11 (5) H12 (1) H14 (1) H15 (3) H16 (1) H17 (1) H19 (1) | H1 (1) | H1 (2) | H8 (1) | |
| H5 (5) | H5 (9) | H6 (1) | H5 (7) | H3 (1) | H10 (1) | H11 (1) | H9 (1) | H5 (2) | H5 (10) | H5 (7) | H11 (1) | ||
| H6 (1) | H6 (1) | H11 (1) | H5 (1) | H11 (4) | H12 (1) | H9 (1) | H6 (1) | H13 (1) | |||||
| H17 (1) | H7 (1) | H9 (1) | H13 (1) | H15 (1) | H15 (1) | ||||||||
| H19 (1) | H14 (1) | H16 (1) | H18 (1) | ||||||||||
| H15 (3) | |||||||||||||
| H18 (1) | |||||||||||||
| Total number of strains | 10 | 12 | 8 | 9 | 4 | 12 | 5 | 3 | 4 | 33 | 12 | 9 | 5 |
| Haploid diversity (h) | 0.680 | 0.417 | 0.406 | 0.346 | 0.750 | 0.792 | 0.800 | 0.444 | 0.625 | 0.876 | 0.292 | 0.346 | 0.800 |
| Unbiased diversity(uh) | 0.756 | 0.455 | 0.464 | 0.389 | 1.000 | 0.864 | 1.000 | 0.667 | 0.833 | 0.903 | 0.318 | 0.389 | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Liang, X.; Kong, J.; Jiao, C.; Li, H.; Gai, Y. Population Structure and Genetic Diversity of Colletotrichum gloeosporioides on Citrus in China. Agronomy 2023, 13, 184. https://doi.org/10.3390/agronomy13010184
Liu B, Liang X, Kong J, Jiao C, Li H, Gai Y. Population Structure and Genetic Diversity of Colletotrichum gloeosporioides on Citrus in China. Agronomy. 2023; 13(1):184. https://doi.org/10.3390/agronomy13010184
Chicago/Turabian StyleLiu, Bei, Xingxing Liang, Jinchao Kong, Chen Jiao, Hongye Li, and Yunpeng Gai. 2023. "Population Structure and Genetic Diversity of Colletotrichum gloeosporioides on Citrus in China" Agronomy 13, no. 1: 184. https://doi.org/10.3390/agronomy13010184
APA StyleLiu, B., Liang, X., Kong, J., Jiao, C., Li, H., & Gai, Y. (2023). Population Structure and Genetic Diversity of Colletotrichum gloeosporioides on Citrus in China. Agronomy, 13(1), 184. https://doi.org/10.3390/agronomy13010184