
Influence of TPU/EVA Phase Morphology Evolution on Supercritical Carbon Dioxide Extrusion Foaming


Abstract
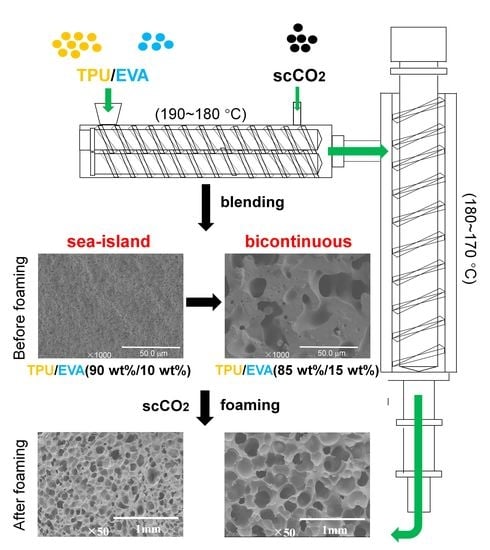
1. Introduction
2. Experimental
2.1. Materials
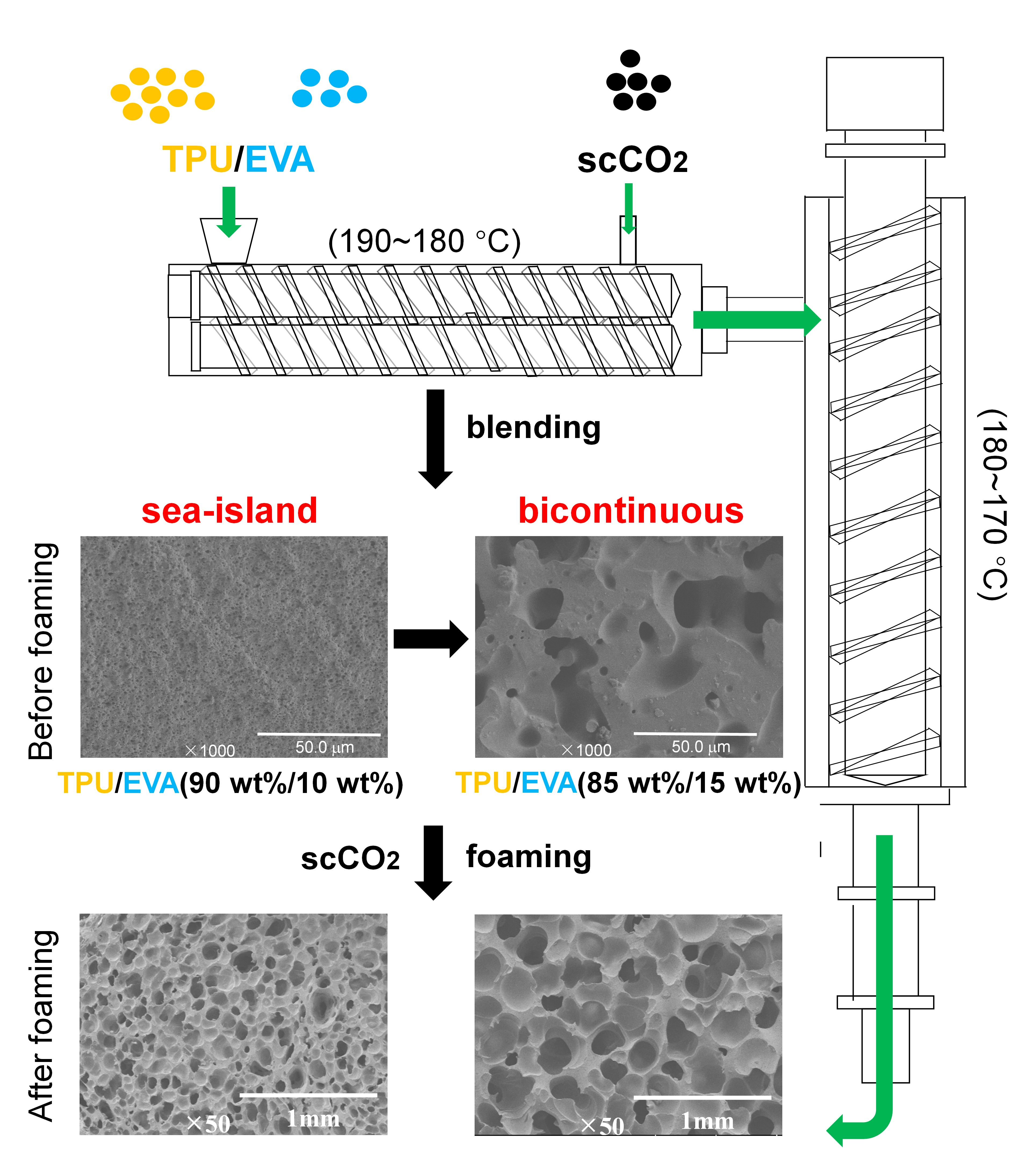
2.2. Sample Preparation
2.3. Characterization
2.3.1. Morphological Study
2.3.2. Rheological Properties
2.3.3. Differential Scanning Calorimetry (DSC)
2.3.4. X-ray Diffraction (XRD)
2.3.5. Polarizing Microscope (POM)
2.3.6. Foaming Characteristics
3. Results and Discussion
3.1. Morphology
3.2. Rheological Properties of TPU/EVA Blends
3.3. Crystallization Properties of TPU/EVA Blends
3.4. XRD Analysis of TPU/EVA Blend
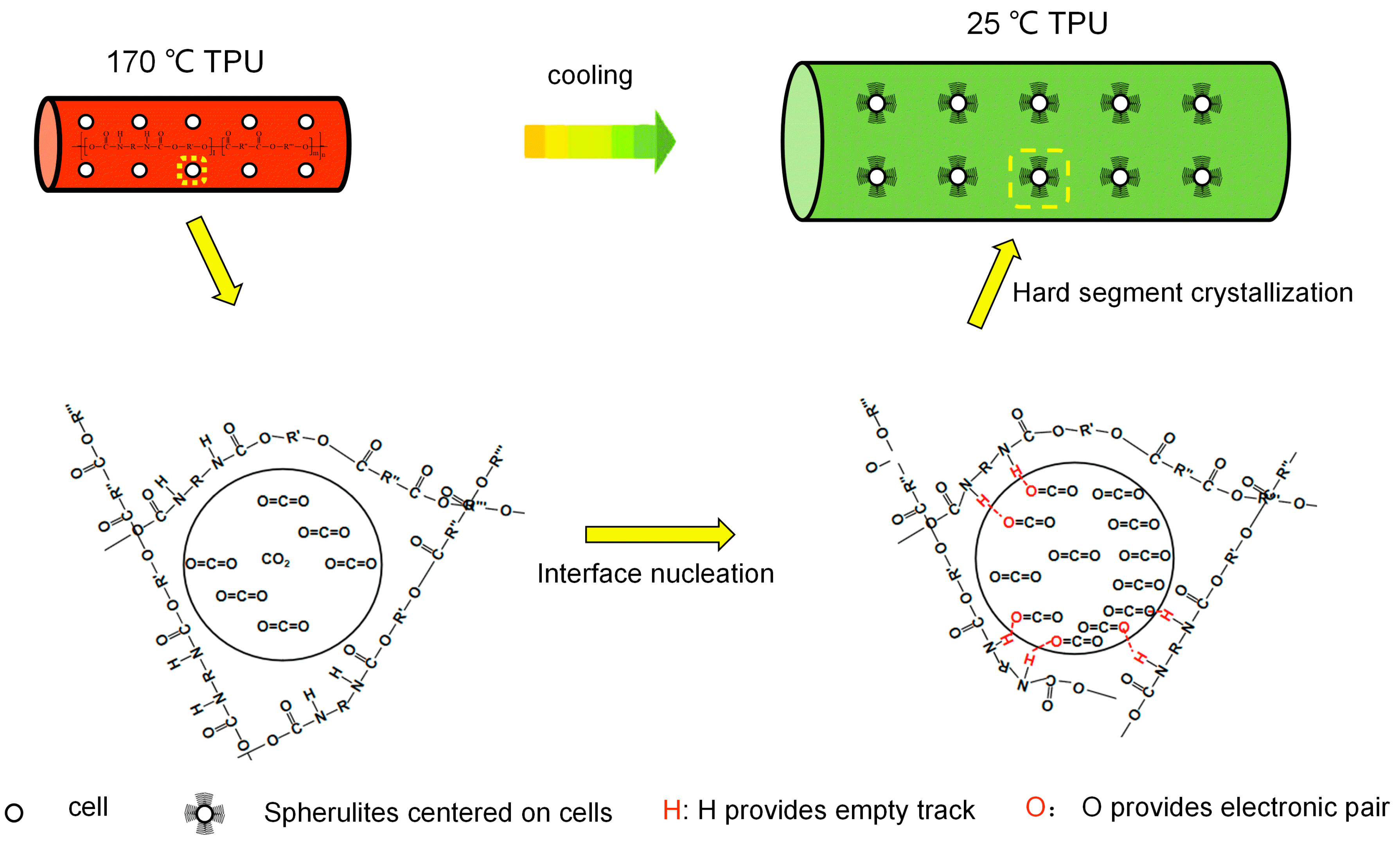
3.5. Crystal Morphology of TPU/EVA Blend
3.6. Crystallization Kinetics of TPU/EVA Blend
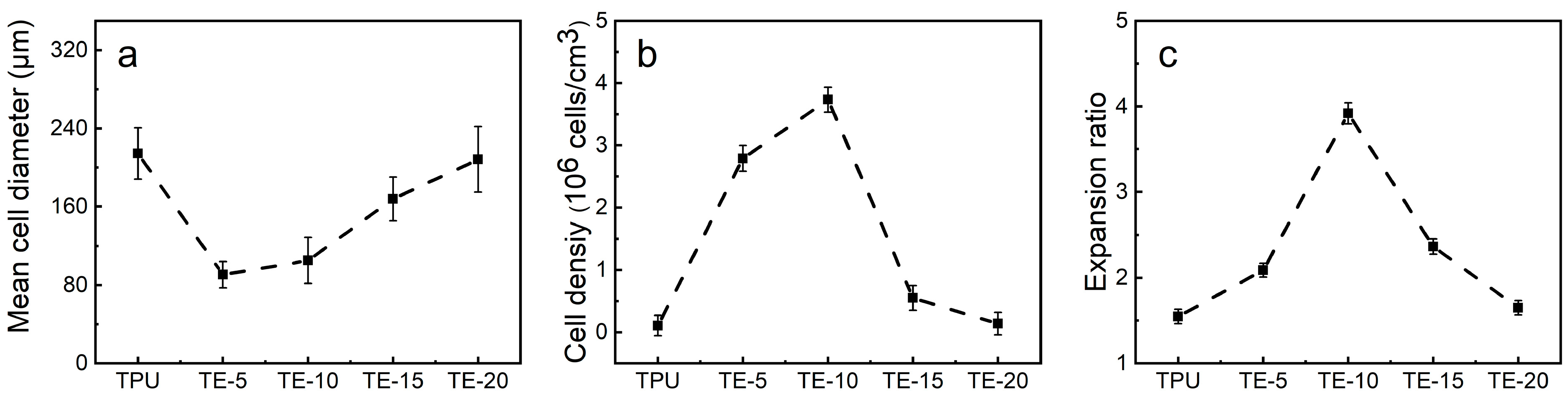
3.7. Foaming Performance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lan, B.; Li, P.; Yang, Q.; Gong, P. Dynamic self generation of hydrogen bonding and relaxation of polymer chain segment in stabilizing thermoplastic polyurethane microcellular foams. Mater. Today Commun. 2020, 24, 101056. [Google Scholar] [CrossRef]
- Ge, C.; Wang, S.; Zhai, W. Influence of cell type and skin-core structure on the tensile elasticity of the microcellular thermoplastic polyurethane foam. J. Cell. Plast. 2019, 56, 207–226. [Google Scholar] [CrossRef]
- Hu, G.; Feng, F. Effect of nanoparticles orientation on morphology of polymeric nanocomposite foams: Preparation of foamed nanocomposite fibers by supercritical carbon dioxide. Polym. Technol. Mater. 2020, 59, 1407–1416. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.Q.; Zhu, Z.B. Internal structure and crystallinity investigation of segmented thermoplastic polyurethane elastomer degradation in supercritical methanol. Polym. Degrad. Stab. 2017, 140, 17–24. [Google Scholar] [CrossRef]
- Hossieny, N.J.; Barzegari, M.R.; Nofar, M.; Mahmood, S.H.; Park, C.B. Crystallization of hard segment domains with the presence of butane for microcellular thermoplastic polyurethane foams. Polymer 2013, 55, 651–662. [Google Scholar] [CrossRef]
- Dai, C.; Zhang, C.; Huang, W.; Chang, K.-C.; Lee, L.J. Thermoplastic polyurethane microcellular fibers via supercritical carbon dioxide based extrusion foaming. Polym. Eng. Sci. 2013, 53, 2360–2369. [Google Scholar] [CrossRef]
- Kropp, D.; Michaeli, W.; Herrmann, T.; Schröder, O. Foam Extrusion of Thermoplastic Elastomers Using CO2 as Blowing Agent. J. Cell. Plast. 1998, 34, 304–311. [Google Scholar] [CrossRef]
- Rizvi, R.; Naguib, H. Porosity and composition dependence on electrical and piezoresistive properties of thermoplastic polyurethane nanocomposites. J. Mater. Res. 2013, 28, 2415–2425. [Google Scholar] [CrossRef]
- Sahnoune, A. Foaming of Thermoplastic Elastomers with Water. J. Cell. Plast. 2001, 37, 149–159. [Google Scholar] [CrossRef]
- Nalawade, S.P.; Picchioni, F.; Marsman, J.H.; Janssen, L. The FT-IR studies of the interactions of CO2 and polymers having different chain groups. J. Supercrit. Fluids 2006, 36, 236–244. [Google Scholar] [CrossRef]
- Huang, A.; Peng, X.; Turng, L.-S. In-situ fibrillated polytetrafluoroethylene (PTFE) in thermoplastic polyurethane (TPU) via melt blending: Effect on rheological behavior, mechanical properties, and microcellular foamability. Polymer 2018, 134, 263–274. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, K.; Hu, S.; Liu, Q.; Zhao, X.; Liu, Y. Improved cell morphology and reduced shrinkage ratio of ETPU beads by reactive blending. Polym. Test. 2017, 63, 38–46. [Google Scholar] [CrossRef]
- Li, F.Z.; Ouyang, L.; Shi, P.; Wang, L.; Liu, Y.J. Preparation and Study of EVA/Poplar Wood Powders Composite Foaming Material. Adv. Mater. Res. 2014, 1053, 276–282. [Google Scholar] [CrossRef]
- Ma, J.; Duan, Z.; Xue, C.; Deng, F. Morphology and mechanical properties of EVA/OMMT nanocomposite foams. J. Thermoplast. Compos. Mater. 2012, 26, 555–569. [Google Scholar] [CrossRef]
- Ji, Z.; Ma, J.; Qin, X.; Wu, Y.; Xu, R.; Ma, Z.; Xue, C.; Qin, J.; Shao, L. Improved dimensional stability of styrene butadiene rubber/ethylene vinyl acetate composite foams with skeleton support structure based on alternately cross-linking process. Polymer 2018, 157, 103–110. [Google Scholar] [CrossRef]
- Jacobs, M.A.; Kemmere, M.F.; Keurentjes, J.T. Foam processing of poly(ethylene-co-vinyl acetate) rubber using supercritical carbon dioxide. Polymer 2004, 45, 7539–7547. [Google Scholar] [CrossRef]
- Maiti, M.; Jasra, R.V.; Kusum, S.K.; Chaki, T.K. Microcellular Foam from Ethylene Vinyl Acetate/Polybutadiene Rubber (EVA/BR) Based Thermoplastic Elastomers for Footwear Applications. Ind. Eng. Chem. Res. 2012, 51, 10607–10612. [Google Scholar] [CrossRef]
- Rodriguez-Perez, M.; Simoes, R.; Roman-Lorza, S.; Alvarez-Lainez, M.; Montoya-Mesa, C.; Constantino, C.; de Saja, J. Foaming of EVA/starch blends: Characterization of the structure, physical properties, and biodegradability. Polym. Eng. Sci. 2011, 52, 62–70. [Google Scholar] [CrossRef]
- Dutta, J.; Naskar, K. Investigation of morphology, mechanical, dynamic mechanical and thermal behaviour of blends based on ethylene vinyl acetate (EVA) and thermoplastic polyurethane (TPU). RSC Adv. 2014, 4, 60831–60841. [Google Scholar] [CrossRef]
- Ma, J.; Shao, L.; Xue, C.; Deng, F.; Duan, Z. Compatibilization and properties of ethylene vinyl acetate copolymer (EVA) and thermoplastic polyurethane (TPU) blend based foam. Polym. Bull. 2014, 71, 2219–2234. [Google Scholar] [CrossRef]
- Banerjee, R.; Ray, S.S. Foamability and Special Applications of Microcellular Thermoplastic Polymers: A Review on Recent Advances and Future Direction. Macromol. Mater. Eng. 2020, 305, 2000366. [Google Scholar] [CrossRef]
- Chen, X.-H.; Yu, P.; Kostromin, S.; Bronnikov, S. Minor-phase particles evolution in a polyethylene/ethylene-propylene copolymer (80/20) blend across mixing: Breakup and coalescence. J. Appl. Polym. Sci. 2013, 130, 3421–3431. [Google Scholar] [CrossRef]
- Arefmanesh, A.; Advani, S. Diffusion-induced growth of a gas bubble in a viscoelastic fluid. Rheol. Acta 1991, 30, 274–283. [Google Scholar] [CrossRef]
- Yang, C.; Zhe, X.; Zhang, M.; Wang, M.; Wu, G. Radiation effects on the foaming of atactic polypropylene with supercritical carbon dioxide. Radiat. Phys. Chem. 2017, 131, 35–40. [Google Scholar] [CrossRef]
- Peón, J.; Vega, J.; Del Amo, B.; Martínez-Salazar, J. Phase morphology and melt viscoelastic properties in blends of ethylene/vinyl acetate copolymer and metallocene-catalysed linear polyethylene. Polymer 2003, 44, 2911–2918. [Google Scholar] [CrossRef]
- Yang, C.-G.; Wang, M.-H.; Zhang, M.-X.; Li, X.-H.; Wang, H.-L.; Xing, Z.; Ye, L.-F.; Wu, G.-Z. Supercritical CO2 Foaming of Radiation Cross-Linked Isotactic Polypropylene in the Presence of TAIC. Molecules 2016, 21, 1660. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, W.; Zhou, C. Phase Behavior and its Viscoelastic Responses of Poly(methyl methacrylate) and Poly(styrene-co-maleic anhydride) Blend Systems. Polym. Bull. 2006, 56, 455–466. [Google Scholar] [CrossRef]
- Nofar, M.; Ameli, A.; Park, C.B. Development of polylactide bead foams with double crystal melting peaks. Polymer 2015, 69, 83–94. [Google Scholar] [CrossRef]
- Li, J.; Li, Z.; Ye, L.; Zhao, X.; Coates, P.; Caton-Rose, F.; Martyn, M. Structure evolution and orientation mechanism of long-chain-branched poly (lactic acid) in the process of solid die drawing. Eur. Polym. J. 2017, 90, 54–65. [Google Scholar] [CrossRef]
- Schick, C.; Androsch, R.; Schmelzer, J.W.P. Homogeneous crystal nucleation in polymers. J. Phys. Condens. Matter 2017, 29, 453002. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, X.; Liu, G.; Xing, Q.; Cavallo, D.; Jiang, Q.; Müller, A.J.; Wang, D. Interfacial nucleation in iPP/PB-1 blends promotes the formation of polybutene-1 trigonal crystals. Polymer 2018, 138, 396–406. [Google Scholar] [CrossRef]
- Herc, A.S.; Bojda, J.; Nowacka, M.; Lewiński, P.; Maniukiewicz, W.; Piorkowska, E.; Kowalewska, A. Crystallization, structure and properties of polylactide/ladder poly(silsesquioxane) blends. Polymer 2020, 201, 122563. [Google Scholar] [CrossRef]
- Haidet, B.B.; Hughes, E.T.; Mukherjee, K. Nucleation control and interface structure of rocksalt PbSe on (001) zincblende III-V surfaces. Phys. Rev. Mater. 2020, 4, 033402. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Wang, K. Interfacial crystallization behavior of poly(ether-ether-ketone) on polyimide-modified CCF300 carbon fibers. Polym. Compos. 2020, 41, 2433–2445. [Google Scholar] [CrossRef]
- Farrow, G.; Preston, D. Measurement of crystallinity in drawn polyethylene terephthalate fibres by X-ray diffraction. Br. J. Appl. Phys. 1960, 11, 353–358. [Google Scholar] [CrossRef]
- Hindeleh, A.; Johnson, D. Crystallinity and crystallite size measurement in polyamide and polyester fibres. Polymer 1978, 19, 27–32. [Google Scholar] [CrossRef]
- Hong, S.; Zhang, X.; Zhang, R.; Wang, L.; Zhao, J.; Han, C.C. Liquid–Liquid Phase Separation and Crystallization in Thin Films of a Polyolefin Blend. Macromolecules 2008, 41, 2311–2314. [Google Scholar] [CrossRef]
- Luo, J.; Liang, Y.; Yang, J.; Niu, H.; Dong, J.-Y.; Han, C.C. Effects of liquid–liquid phase separation on crystallization kinetics and morphology of isotactic polypropylene/poly (ethylene-co-octene) in-reactor alloy. Polymer 2012, 53, 2465–2475. [Google Scholar] [CrossRef]
- Shi, W.; Chen, F.; Zhang, Y.; Han, C.C. Viscoelastic Phase Separation and Interface Assisted Crystallization in a Highly Immiscible iPP/PMMA Blend. ACS Macro Lett. 2012, 1, 1086–1089. [Google Scholar] [CrossRef]
- Park, J.; Seo, Y.; Cho, S.; Whang, D.; Kim, K.; Chang, T. Formation of 1,1,12,12-tetramethyl[1.1]silaferrocenophane and poly(ferrocenylsilane) in the reaction of ferrous chloride with the dilithium salt of dicyclopentadienyldimethylsilanes. J. Organomet. Chem. 1995, 489, 23–25. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Vincent, M.F.; Eckert, C.A. Infrared cell for supercritical fluid–polymer interactions. Rev. Sci. Instrum. 1996, 67, 1586–1589. [Google Scholar] [CrossRef]
- Macknight, W.J.; Yang, M. Property-structure relationships in poly-urethanes: Infrared studies. J. Polym. Sci. Polym. Symp. 2007, 42, 817–832. [Google Scholar] [CrossRef]
- Pan, H.; Cao, Z.; Chen, Y.; Wang, X.; Jia, S.; Yang, H.; Zhang, H.; Dong, L. Effect of molecular stereoregularity on the transcrystallinization properties of poly(l-lactide)/basalt fiber composites. Int. J. Biol. Macromol. 2019, 137, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.; Sekar, M.; Thanikachalam, V.; Manikandan, G. Thermal decomposition and kinetic analyses of sulfonamide Schiff’s bases in oxygen atmosphere—A comparative study. Chem. Data Collect. 2017, 9, 229–243. [Google Scholar] [CrossRef]
- Seo, Y.; Kang, T.; Hong, S.M.; Choi, H.J. Nonisothermal crystallization behaviors of a polyolefin terpolymer and its foam. Polymer 2007, 48, 3844–3849. [Google Scholar] [CrossRef]
- Ono, T.; Wu, X.; Horiuchi, S.; Furuya, T.; Yoda, S. Two-step foaming process for production of PMMA nanocellular polymer foams via ultra-high pressure and rapid depressurization. J. Supercrit. Fluids 2020, 165, 104963. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TPU (wt%) | EVA (wt%) |
|---|---|---|
| TPU | 100 | 0 |
| TE-5 | 95 | 5 |
| TE-10 | 90 | 10 |
| TE-12 | 88 | 12 |
| TE-13 | 87 | 13 |
| TE-15 | 85 | 15 |
| TE-20 | 80 | 20 |
| EVA | 0 | 100 |
| Sample Designation | Melting Peak Tm (°C) | Melting Enthalpy ∆H (J/g) | Crystallinity (%) |
|---|---|---|---|
| TPU | 159.3 | 2.46 | 1.6 |
| TE-5 | 160.7 | 3.99 | 2.7 |
| TE-10 | 162.6 | 8.57 | 5.8 |
| TE-15 | 159.6 | 6.14 | 4.1 |
| TE-20 | 159.1 | 2.77 | 1.8 |
| Sample | n | |||
|---|---|---|---|---|
| TPU | 1.52 | 2.68 | 0.16 | 0.83 |
| TE-5 | 3.72 | 1.83 | 0.07 | 0.77 |
| TE-10 | 4.20 | 1.67 | 0.08 | 0.78 |
| TE-15 | 2.56 | 1.70 | 0.18 | 0.84 |
| TE-20 | 2.61 | 1.72 | 0.17 | 0.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.-W.; Zhou, T.-T.; Zhang, R.; Hu, S.-F. Influence of TPU/EVA Phase Morphology Evolution on Supercritical Carbon Dioxide Extrusion Foaming. Polymers 2023, 15, 3134. https://doi.org/10.3390/polym15143134
Du J-W, Zhou T-T, Zhang R, Hu S-F. Influence of TPU/EVA Phase Morphology Evolution on Supercritical Carbon Dioxide Extrusion Foaming. Polymers. 2023; 15(14):3134. https://doi.org/10.3390/polym15143134
Chicago/Turabian StyleDu, Jun-Wei, Tian-Tian Zhou, Rong Zhang, and Sheng-Fei Hu. 2023. "Influence of TPU/EVA Phase Morphology Evolution on Supercritical Carbon Dioxide Extrusion Foaming" Polymers 15, no. 14: 3134. https://doi.org/10.3390/polym15143134
APA StyleDu, J.-W., Zhou, T.-T., Zhang, R., & Hu, S.-F. (2023). Influence of TPU/EVA Phase Morphology Evolution on Supercritical Carbon Dioxide Extrusion Foaming. Polymers, 15(14), 3134. https://doi.org/10.3390/polym15143134