
Intrinsically Disordered Synthetic Polymers in Biomedical Applications


 ,
,  and
and
Abstract
1. Introduction
2. Intrinsically Disordered Synthetic Polymers
2.1. Biomedical Applications
2.2. Synthesis of Intrinsically Disordered Polymers for Biomedical Applications
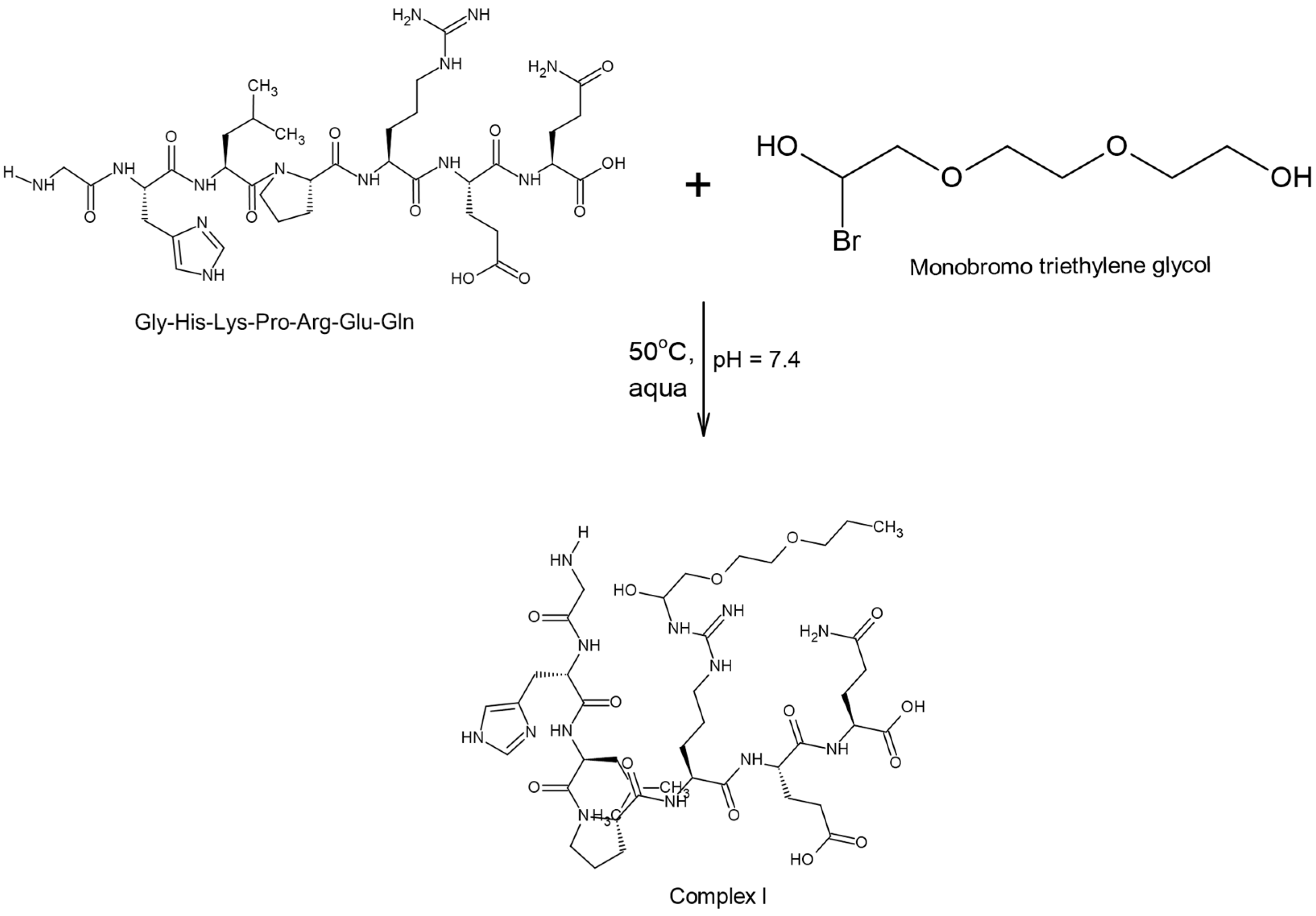
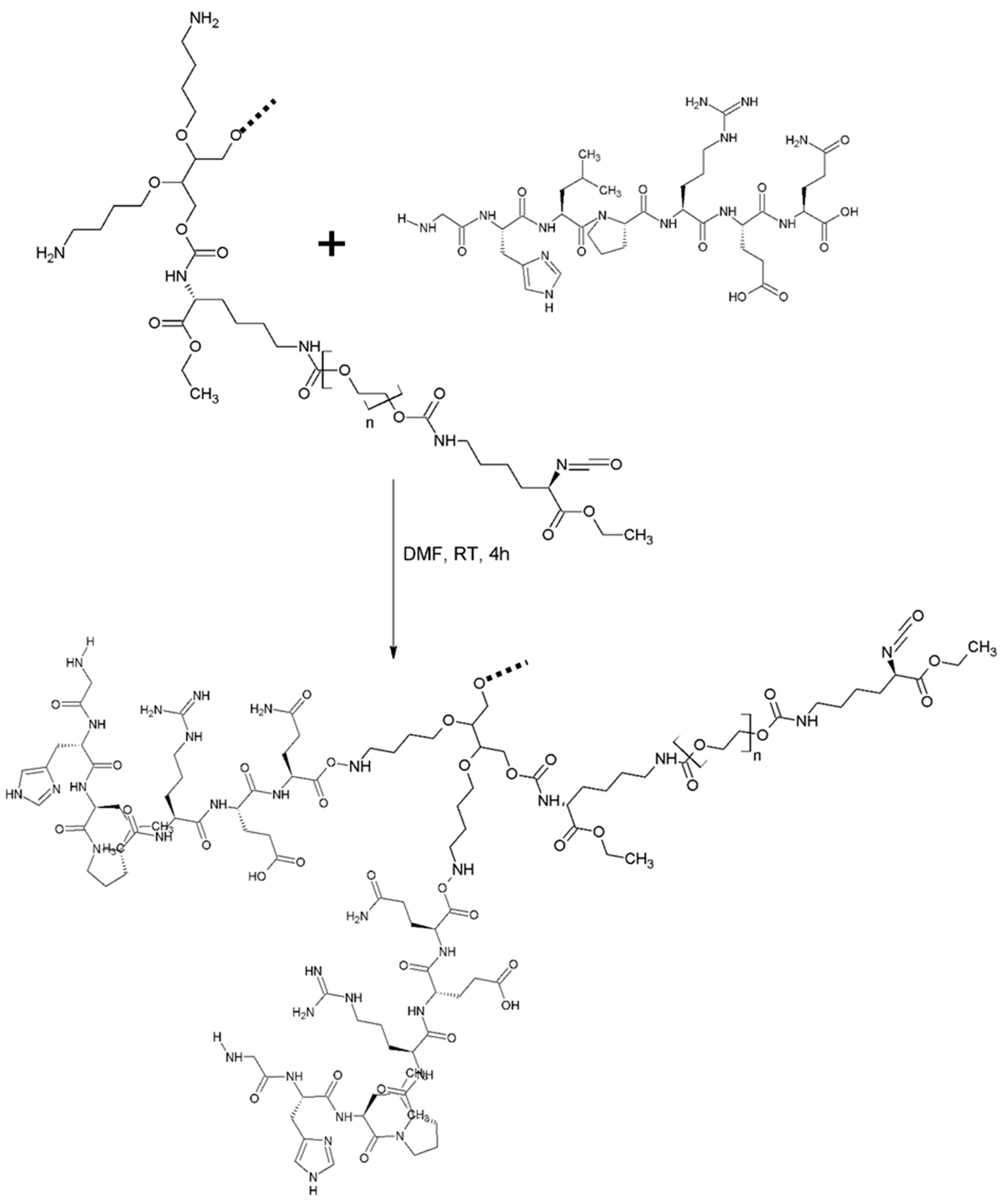
2.2.1. Modification of the Chain Extender with a Structure-Breaking Peptide Oligomer
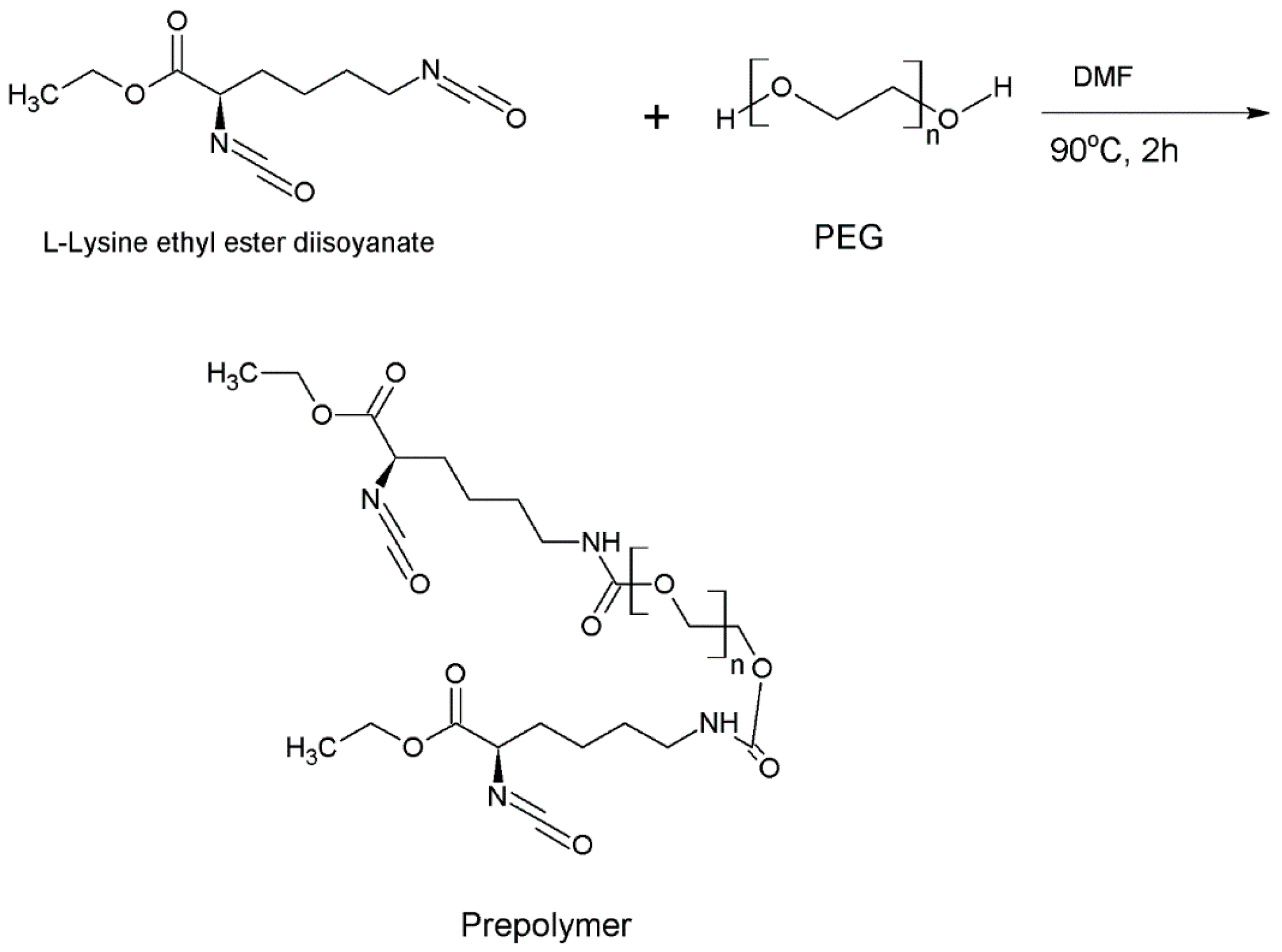
2.2.2. Synthesis of the Prepolymer and Synthesis of Intrinsically Disordered Polymer
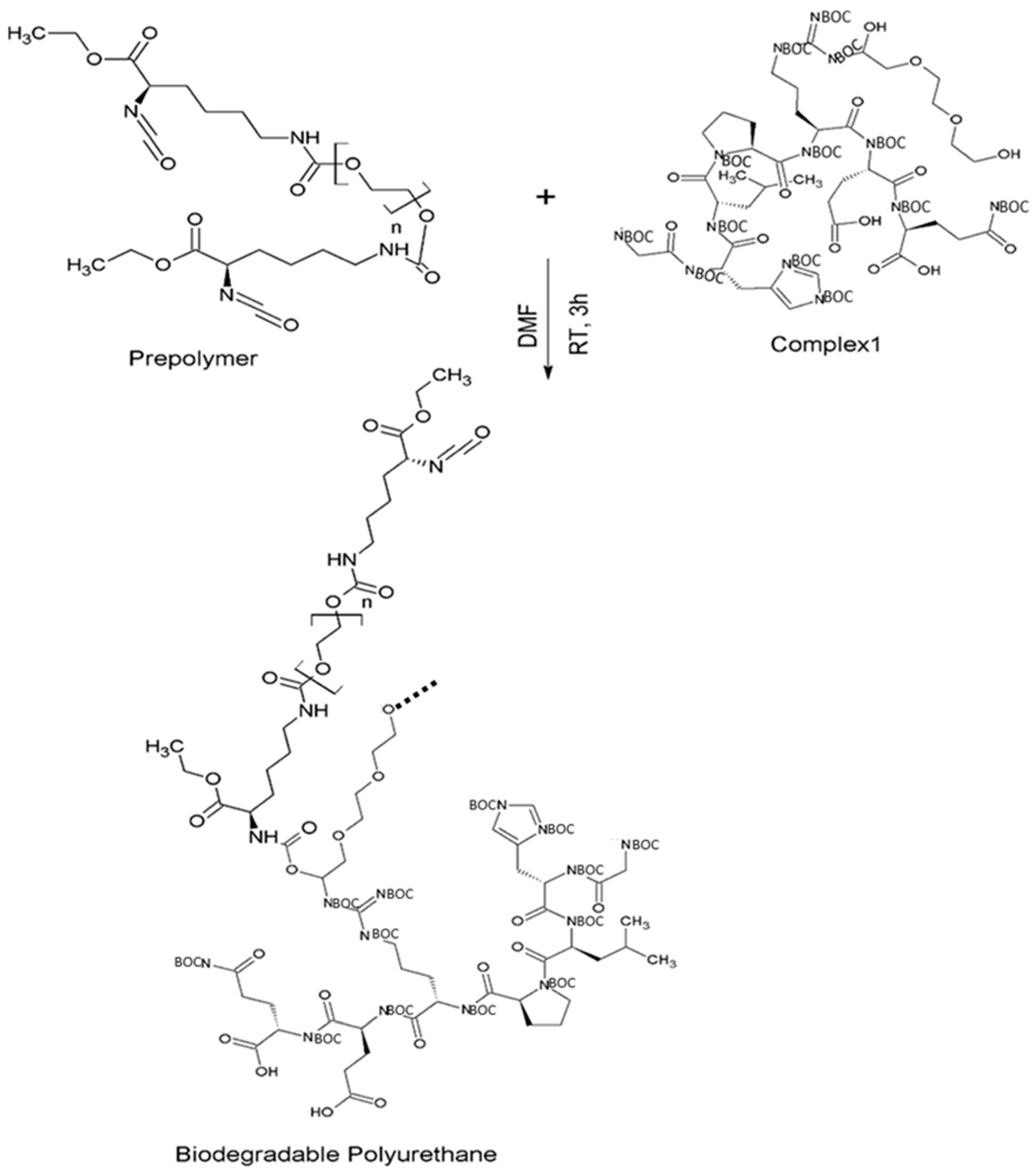
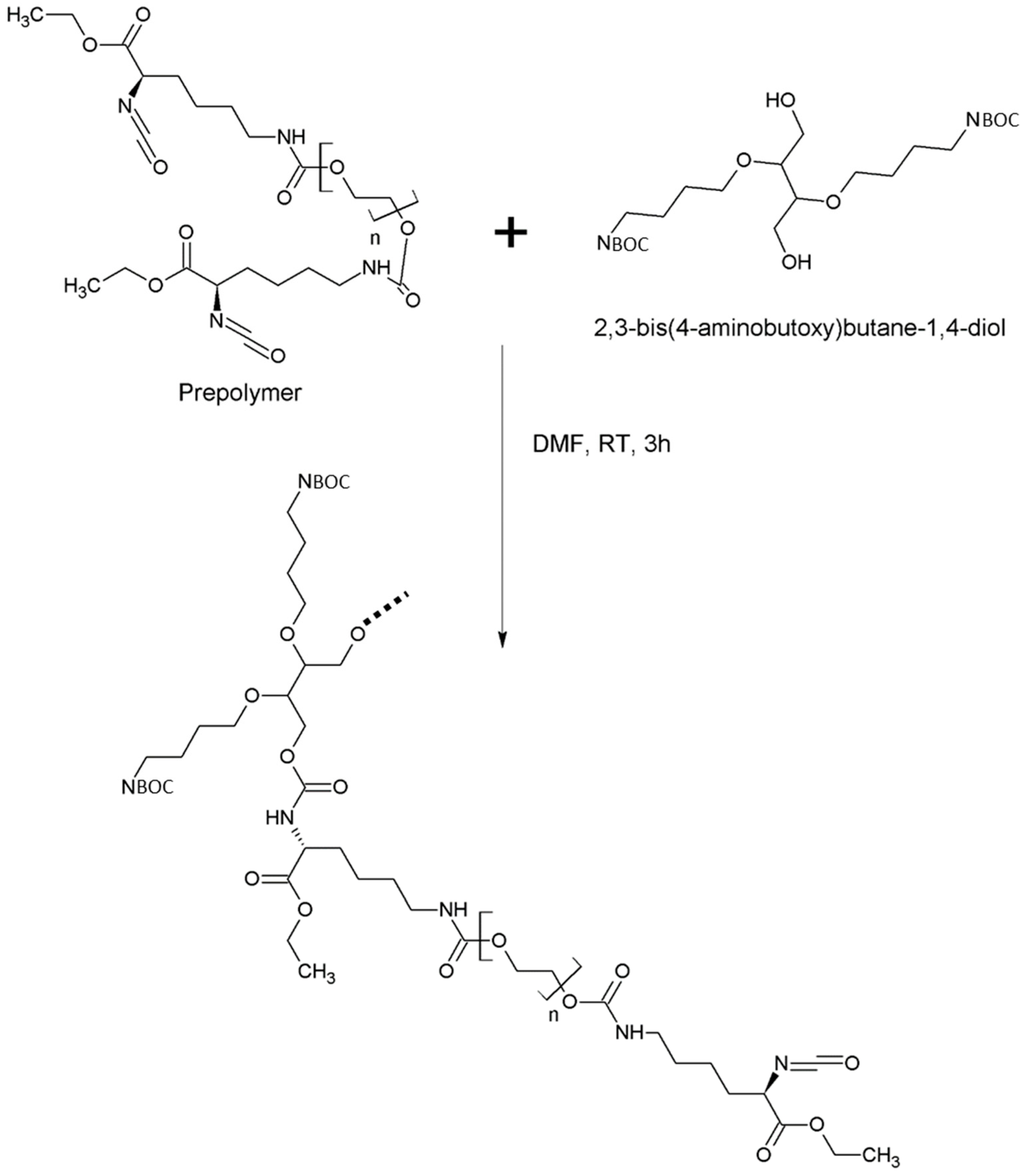
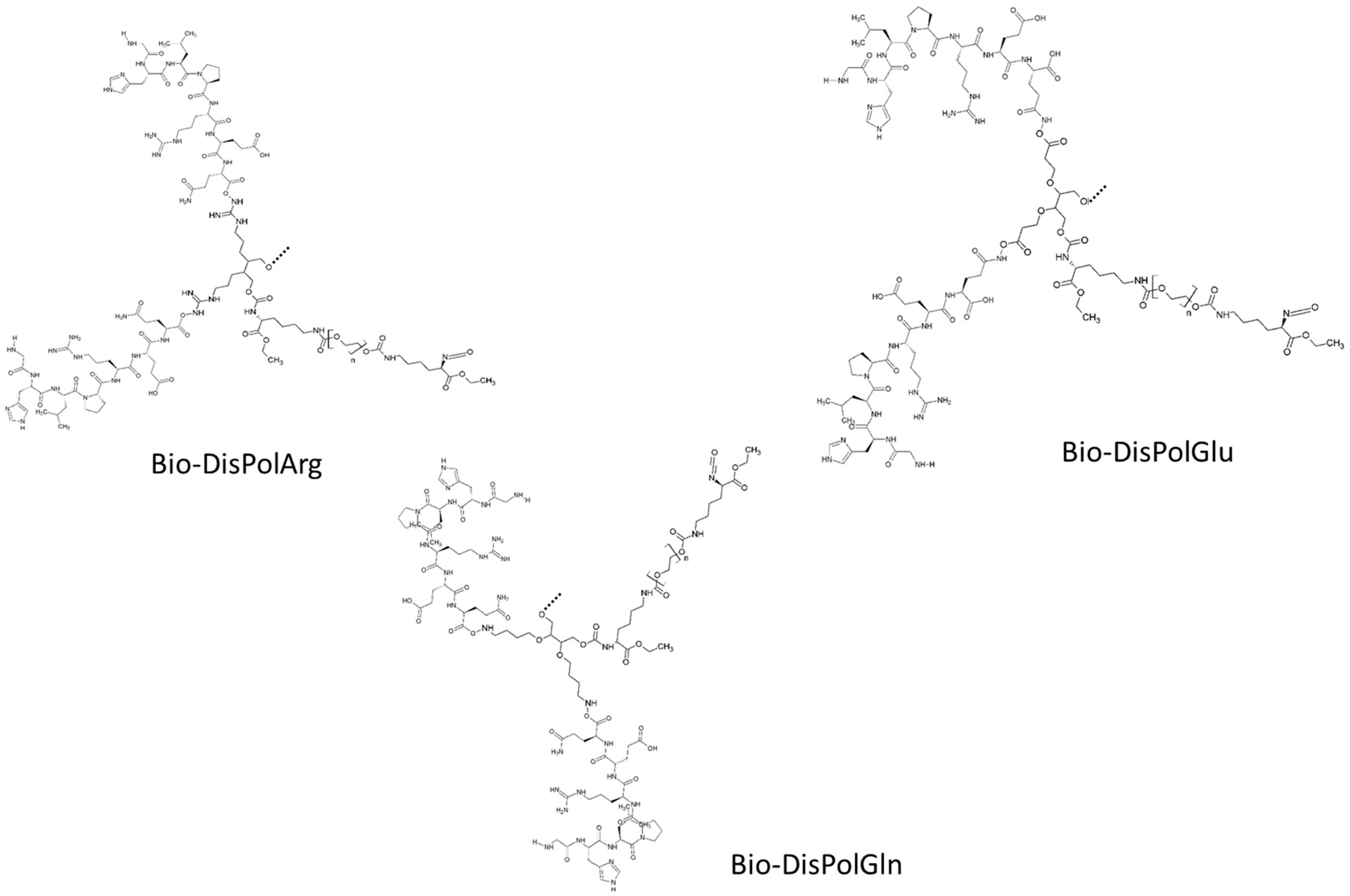
2.2.3. Synthesis of Intrinsically Disordered Polymers
2.3. Characterization and Production of New Class of Bio-Mimicked Intrinsically Disordered Polymers
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Quiroz, F.G.; Chilkoti, A. Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat. Mater. 2015, 14, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for Drug Delivery Systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Pillai, O.; Panchagnula, R. Polymers in drug delivery. Curr. Opin. Chem. Biol. 2001, 5, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.K.; Kim, S.W. Recent advances in polymeric drug delivery systems. Biomater. Res. 2020, 24, 12. [Google Scholar] [CrossRef]
- Silva, A.C.Q.; Silvestre, A.J.D.; Vilela, C.; Freire, C.S.R. Natural Polymers-Based Materials: A Contribution to a Greener Future. Molecules 2021, 27, 94. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef]
- Siepmann, J.; Siegel, R.A.; Rathbone, M.J. (Eds.) Fundamentals and Applications of Controlled Release Drug Delivery; Springer: Boston, MA, USA, 2012; ISBN 978-1-4614-0880-2. [Google Scholar]
- Park, H.; Otte, A.; Park, K. Evolution of drug delivery systems: From 1950 to 2020 and beyond. J. Control. Release 2022, 342, 53–65. [Google Scholar] [CrossRef]
- Mezhuev, Y.O.; Varankin, A.V.; Luss, A.L.; Dyatlov, V.A.; Tsatsakis, A.M.; Shtilman, M.I.; Korshak, Y.V. Immobilization of dopamine on the copolymer of N-vinyl-2-pyrrolidone and allyl glycidyl ether and synthesis of new hydrogels. Polym. Int. 2020, 69, 1275–1282. [Google Scholar] [CrossRef]
- Nair, P.R. Delivering Combination Chemotherapies and Targeting Oncogenic Pathways via Polymeric Drug Delivery Systems. Polymers 2019, 11, 630. [Google Scholar] [CrossRef]
- Shevchenko, K.G.; Garkushina, I.S.; Canfarotta, F.; Piletsky, S.A.; Barlev, N.A. Nano-molecularly imprinted polymers (nanoMIPs) as a novel approach to targeted drug delivery in nanomedicine. RSC Adv. 2022, 12, 3957–3968. [Google Scholar] [CrossRef]
- Rawal, S.U.; Patel, B.M.; Patel, M.M. New Drug Delivery Systems Developed for Brain Targeting. Drugs 2022, 82, 749–792. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.K.; Gauba, A.; Shrivastava, N.; Tripathi, R.M.; Jain, S.K.; Pandey, A.K. Polymeric micelles and cancer therapy: An ingenious multimodal tumor-targeted drug delivery system. Drug Deliv. Transl. Res. 2023, 13, 135–163. [Google Scholar] [CrossRef] [PubMed]
- Marschütz, M.K.; Bernkop-Schnürch, A. Oral peptide drug delivery: Polymer–inhibitor conjugates protecting insulin from enzymatic degradation in vitro. Biomaterials 2000, 21, 1499–1507. [Google Scholar] [CrossRef]
- Rahimi, M.; Charmi, G.; Matyjaszewski, K.; Banquy, X.; Pietrasik, J. Recent developments in natural and synthetic polymeric drug delivery systems used for the treatment of osteoarthritis. Acta Biomater. 2021, 123, 31–50. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, A.N.; Torchilin, V.P. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Deliv. Rev. 2004, 56, 1273–1289. [Google Scholar] [CrossRef]
- Hwang, D.; Ramsey, J.D.; Kabanov, A.V. Polymeric micelles for the delivery of poorly soluble drugs: From nanoformulation to clinical approval. Adv. Drug Deliv. Rev. 2020, 156, 80–118. [Google Scholar] [CrossRef]
- Kumari, P.; Ghosh, B.; Biswas, S. Nanocarriers for cancer-targeted drug delivery. J. Drug Target. 2016, 24, 179–191. [Google Scholar] [CrossRef]
- Fournier, E.; Passirani, C.; Montero-Menei, C.N.; Benoit, J.P. Biocompatibility of implantable synthetic polymeric drug carriers: Focus on brain biocompatibility. Biomaterials 2003, 24, 3311–3331. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, S.; Ge, L.; Wu, W.; Jiang, X. Translatable High Drug Loading Drug Delivery Systems Based on Biocompatible Polymer Nanocarriers. Biomacromolecules 2018, 19, 1732–1745. [Google Scholar] [CrossRef]
- Kohane, D.S.; Langer, R. Biocompatibility and drug delivery systems. Chem. Sci. 2010, 1, 441–446. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly (Ethylene Glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef]
- Liu, J.; Liew, S.S.; Wang, J.; Pu, K. Bioinspired and Biomimetic Delivery Platforms for Cancer Vaccines. Adv. Mater. 2022, 34, 2103790. [Google Scholar] [CrossRef] [PubMed]
- Artyukhov, A.A.; Nechaeva, A.M.; Shtilman, M.I.; Chistyakov, E.M.; Svistunova, A.Y.; Bagrov, D.V.; Kuskov, A.N.; Docea, A.O.; Tsatsakis, A.M.; Gurevich, L.; et al. Nanoaggregates of Biphilic Carboxyl-Containing Copolymers as Carriers for Ionically Bound Doxorubicin. Materials 2022, 15, 7136. [Google Scholar] [CrossRef]
- Hauet, T.; Eugene, M. A new approach in organ preservation: Potential role of new polymers. Kidney Int. 2008, 74, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Advanced Polymers for Three-Dimensional (3D) Organ Bioprinting. Micromachines 2019, 10, 814. [Google Scholar] [CrossRef]
- Thomson, R.C.; Wake, M.C.; Yaszemski, M.J.; Mikos, A.G. Biodegradable Polymer Scaffolds to Regenerate Organs. In Biopolymers II; Peppas, N.A., Langer, R.S., Eds.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1995; Volume 122, pp. 245–274. ISBN 978-3-540-58788-0. [Google Scholar]
- Siren, E.M.J.; Luo, H.D.; Tam, F.; Montgomery, A.; Enns, W.; Moon, H.; Sim, L.; Rey, K.; Guan, Q.; Wang, J.-J.; et al. Prevention of vascular-allograft rejection by protecting the endothelial glycocalyx with immunosuppressive polymers. Nat. Biomed. Eng. 2021, 5, 1202–1216. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Liu, X.; Kumar, S.; Gochman, G.; Ji, Y.; Liao, Y.-P.; Chang, C.H.; Situ, W.; Lu, J.; et al. Use of Polymeric Nanoparticle Platform Targeting the Liver to Induce Treg-Mediated Antigen-Specific Immune Tolerance in a Pulmonary Allergen Sensitization Model. ACS Nano 2019, 13, 4778–4794. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. Organ engineering—Combining stem cells, biomaterials, and bioreactors to produce bioengineered organs for transplantation. Bioessays 2013, 35, 163–172. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Zamboulis, A.; Koumentakou, I.; Michailidou, G.; Noordam, M.J.; Bikiaris, D.N. Biocompatible Synthetic Polymers for Tissue Engineering Purposes. Biomacromolecules 2022, 23, 1841–1863. [Google Scholar] [CrossRef]
- Sultana, N. Scaffolds for Tissue Engineering. In Biodegradable Polymer-Based Scaffolds for Bone Tissue Engineering; SpringerBriefs in Applied Sciences and Technology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–17. ISBN 978-3-642-34801-3. [Google Scholar]
- Ma, P.X.; Langer, R. Fabrication of Biodegradable Polymer Foams for Cell Transplantation and Tissue Engineering. In Tissue Engineering; Humana Press: Totowa, NJ, USA, 1998; Volume 18, pp. 47–56. ISBN 978-0-89603-516-4. [Google Scholar]
- Olmos, D.; González-Benito, J. Polymeric Materials with Antibacterial Activity: A Review. Polymers 2021, 13, 613. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, T.; Gauthaman, K.; Hammad, A.H.; Navare, K.J.; Alshahrie, A.A.; Bencherif, S.A.; Tamayol, A.; Memic, A. Oxygen-Releasing Antibacterial Nanofibrous Scaffolds for Tissue Engineering Applications. Polymers 2020, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, L. Imminent antimicrobial bioink deploying cellulose, alginate, EPS and synthetic polymers for 3D bioprinting of tissue constructs. Carbohydr. Polym. 2021, 260, 117774. [Google Scholar] [CrossRef]
- Dragostin, I.; Dragostin, O.; Pelin, A.-M.; Grigore, C.; Zamfir, C.L. The importance of polymers for encapsulation process and for enhanced cellular functions. J. Macromol. Sci. Part A 2017, 54, 489–493. [Google Scholar] [CrossRef]
- Gentile, F.T.; Doherty, E.J.; Rein, D.H.; Shoichet, M.S.; Winn, S.R. Polymer science for macroencapsulation of cells for central nervous system transplantation. React. Polym. 1995, 25, 207–227. [Google Scholar] [CrossRef]
- Sabbagh, F.; Muhamad, I.I.; Niazmand, R.; Dikshit, P.K.; Kim, B.S. Recent progress in polymeric non-invasive insulin delivery. Int. J. Biol. Macromol. 2022, 203, 222–243. [Google Scholar] [CrossRef]
- Sheridan, M.; Shea, L.; Peters, M.; Mooney, D. Bioabsorbable polymer scaffolds for tissue engineering capable of sustained growth factor delivery. J. Control. Release 2000, 64, 91–102. [Google Scholar] [CrossRef]
- Macko, M.; Szczepański, Z.; Mikołajewski, D.; Nowak, J.; Mikołajewska, E.; Furtak, J.; Listopadzki, S. Design and manufacture of artificial organs made of polymers. MATEC Web Conf. 2019, 254, 06006. [Google Scholar] [CrossRef]
- Ishihara, K. Bioinspired phospholipid polymer biomaterials for making high performance artificial organs. Sci. Technol. Adv. Mater. 2000, 1, 131–138. [Google Scholar] [CrossRef]
- Nguyen, B.T.D.; Thi, H.Y.N.; Thi, B.P.N.; Kang, D.-K.; Kim, J.F. The Roles of Membrane Technology in Artificial Organs: Current Challenges and Perspectives. Membranes 2021, 11, 239. [Google Scholar] [CrossRef]
- Jurak, M.; Wiącek, A.E.; Ładniak, A.; Przykaza, K.; Szafran, K. What affects the biocompatibility of polymers? Adv. Colloid Interface Sci. 2021, 294, 102451. [Google Scholar] [CrossRef] [PubMed]
- Holmes, T.C. Novel peptide-based biomaterial scaffolds for tissue engineering. Trends Biotechnol. 2002, 20, 16–21. [Google Scholar] [CrossRef]
- Hong, Y.; Lin, Z.; Yang, Y.; Jiang, T.; Shang, J.; Luo, Z. Biocompatible Conductive Hydrogels: Applications in the Field of Biomedicine. Int. J. Mol. Sci. 2022, 23, 4578. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J. Controlled biodegradability of polymers—A key to drug delivery systems. Biomaterials 1984, 5, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.; Garbuzenko, O.B.; Ber, E.; Chandna, P.; Khandare, J.J.; Pozharov, V.P.; Minko, T. Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging? J. Control. Release 2008, 130, 107–114. [Google Scholar] [CrossRef]
- Gamucci, O.; Bertero, A.; Gagliardi, M.; Bardi, G. Biomedical Nanoparticles: Overview of Their Surface Immune-Compatibility. Coatings 2014, 4, 139–159. [Google Scholar] [CrossRef]
- Ishihara, K. Blood-Compatible Surfaces with Phosphorylcholine-Based Polymers for Cardiovascular Medical Devices. Langmuir 2019, 35, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- DeFife, K.M.; Grako, K.; Cruz-Aranda, G.; Price, S.; Chantung, R.; Macpherson, K.; Khoshabeh, R.; Gopalan, S.; Turnell, W.G. Poly(ester amide) Co-polymers Promote Blood and Tissue Compatibility. J. Biomater. Sci. Polym. Ed. 2009, 20, 1495–1511. [Google Scholar] [CrossRef]
- Jaganathan, S.K.; Balaji, A.; Vellayappan, M.V.; Subramanian, A.P.; John, A.A.; Asokan, M.K.; Supriyanto, E. Review: Radiation-induced surface modification of polymers for biomaterial application. J. Mater. Sci. 2015, 50, 2007–2018. [Google Scholar] [CrossRef]
- Hu, Y.; Winn, S.R.; Krajbich, I.; Hollinger, J.O. Porous polymer scaffolds surface-modified with arginine-glycine-aspartic acid enhance bone cell attachment and differentiationin vitro. J. Biomed. Mater. Res. 2003, 64A, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Huyer, L.D.; Mandla, S.; Wang, Y.; Campbell, S.B.; Yee, B.; Euler, C.; Lai, B.F.; Bannerman, A.D.; Lin, D.S.Y.; Montgomery, M.; et al. Macrophage Immunomodulation Through New Polymers that Recapitulate Functional Effects of Itaconate as a Power House of Innate Immunity. Adv. Funct. Mater. 2021, 31, 2003341. [Google Scholar] [CrossRef]
- Vishwakarma, A.; Bhise, N.S.; Evangelista, M.B.; Rouwkema, J.; Dokmeci, M.R.; Ghaemmaghami, A.M.; Vrana, N.E.; Khademhosseini, A. Engineering Immunomodulatory Biomaterials to Tune the Inflammatory Response. Trends Biotechnol. 2016, 34, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Tzianabos, A.O. Polysaccharide Immunomodulators as Therapeutic Agents: Structural Aspects and Biologic Function. Clin. Microbiol. Rev. 2000, 13, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Dane, K.Y.; Nembrini, C.; Tomei, A.A.; Eby, J.K.; O’Neil, C.P.; Velluto, D.; Swartz, M.A.; Inverardi, L.; Hubbell, J.A. Nano-sized drug-loaded micelles deliver payload to lymph node immune cells and prolong allograft survival. J. Control. Release 2011, 156, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Guelcher, S.A. Biodegradable Polyurethanes: Synthesis and Applications in Regenerative Medicine. Tissue Eng. Part B: Rev. 2008, 14, 3–17. [Google Scholar] [CrossRef]
- Tatai, L.; Moore, T.G.; Adhikari, R.; Malherbe, F.; Jayasekara, R.; Griffiths, I.; Gunatillake, P.A. Thermoplastic biodegradable polyurethanes: The effect of chain extender structure on properties and in-vitro degradation. Biomaterials 2007, 28, 5407–5417. [Google Scholar] [CrossRef]
- D’Arlas, B.F.; Rueda, L.; de la Caba, K.; Mondragon, I.; Eceiza, A. Microdomain composition and properties differences of biodegradable polyurethanes based on MDI and HDI. Polym. Eng. Sci. 2008, 48, 519–529. [Google Scholar] [CrossRef]
- Mahkam, M.; Sharifi-Sanjani, N. Preparation of new biodegradable polyurethanes as a therapeutic agent. Polym. Degrad. Stab. 2003, 80, 199–202. [Google Scholar] [CrossRef]
- Pedersen, D.D.; Kim, S.; Wagner, W.R. Biodegradable polyurethane scaffolds in regenerative medicine: Clinical translation review. J. Biomed. Mater. Res. Part A 2022, 110, 1460–1487. [Google Scholar] [CrossRef]
- Pfohl, P.; Bahl, D.; Rückel, M.; Wagner, M.; Meyer, L.; Bolduan, P.; Battagliarin, G.; Hüffer, T.; Zumstein, M.; Hofmann, T.; et al. Effect of Polymer Properties on the Biodegradation of Polyurethane Microplastics. Environ. Sci. Technol. 2022, 56, 16873–16884. [Google Scholar] [CrossRef]
- Sobczak, M.; Kędra, K. Biomedical Polyurethanes for Anti-Cancer Drug Delivery Systems: A Brief, Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 8181. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness: Linking regulation to protein dynamics. Mol. Biosyst. 2012, 8, 168–177. [Google Scholar] [CrossRef]
- Roberts, S.; Dzuricky, M.; Chilkoti, A. Elastin-like polypeptides as models of intrinsically disordered proteins. FEBS Lett. 2015, 589, 2477–2486. [Google Scholar] [CrossRef]
- Acosta, S.; Ye, Z.; Aparicio, C.; Alonso, M.; Rodríguez-Cabello, J.C. Dual Self-Assembled Nanostructures from Intrinsically Disordered Protein Polymers with LCST Behavior and Antimicrobial Peptides. Biomacromolecules 2020, 21, 4043–4052. [Google Scholar] [CrossRef]
- Chilkoti, A.; Christensen, T.; Mackay, J.A. Stimulus responsive elastin biopolymers: Applications in medicine and biotechnology. Curr. Opin. Chem. Biol. 2006, 10, 652–657. [Google Scholar] [CrossRef]
- Wright, E.R.; Conticello, V.P. Self-assembly of block copolymers derived from elastin-mimetic polypeptide sequences. Adv. Drug Deliv. Rev. 2002, 54, 1057–1073. [Google Scholar] [CrossRef]
- Meyer, D.; Kong, G.A.; Dewhirst, M.W.; Zalutsky, M.R.; Chilkoti, A. Targeting a genetically engineered elastin-like polypeptide to solid tumors by local hyperthermia. Cancer Res. 2001, 61, 1548–1554. [Google Scholar]
- Chilkoti, A.; Dreher, M.R.; Meyer, D.E.; Raucher, D. Targeted drug delivery by thermally responsive polymers. Adv. Drug Deliv. Rev. 2002, 54, 613–630. [Google Scholar] [CrossRef]
- Dreher, M.R.; Raucher, D.; Balu, N.; Colvin, O.M.; Ludeman, S.M.; Chilkoti, A. Evaluation of an elastin-like polypeptide–doxorubicin conjugate for cancer therapy. J. Control. Release 2003, 91, 31–43. [Google Scholar] [CrossRef]
- Furgeson, D.Y.; Dreher, M.R.; Chilkoti, A. Structural optimization of a “smart” doxorubicin–polypeptide conjugate for thermally targeted delivery to solid tumors. J. Control. Release 2006, 110, 362–369. [Google Scholar] [CrossRef]
- Luo, T.; Kiick, K.L. Collagen-like peptides and peptide–polymer conjugates in the design of assembled materials. Eur. Polym. J. 2013, 49, 2998–3009. [Google Scholar] [CrossRef]
- Nettles, D.L.; Chilkoti, A.; Setton, L.A. Applications of elastin-like polypeptides in tissue engineering. Adv. Drug Deliv. Rev. 2010, 62, 1479–1485. [Google Scholar] [CrossRef]
- Ardell, D.H.; Andersen, S.O. Tentative identification of a resilin gene in Drosophila melanogaster. Insect Biochem. Mol. Biol. 2001, 31, 965–970. [Google Scholar] [CrossRef]
- Elvin, C.M.; Carr, A.G.; Huson, M.G.; Maxwell, J.M.; Pearson, R.D.; Vuocolo, T.; Liyou, N.E.; Wong, D.C.C.; Merritt, D.J.; Dixon, N.E. Synthesis and properties of crosslinked recombinant pro-resilin. Nature 2005, 437, 999–1002. [Google Scholar] [CrossRef]
- Balu, R.; Knott, R.; Cowieson, N.P.; Elvin, C.M.; Hill, A.J.; Choudhury, N.R.; Dutta, N.K. Structural ensembles reveal intrinsic disorder for the multi-stimuli responsive bio-mimetic protein Rec1-resilin. Sci. Rep. 2015, 5, 10896. [Google Scholar] [CrossRef]
- Lau, H.K.; Li, L.; Jurusik, A.K.; Sabanayagam, C.R.; Kiick, K.L. Aqueous Liquid–Liquid Phase Separation of Resilin-Like Polypeptide/Polyethylene Glycol Solutions for the Formation of Microstructured Hydrogels. ACS Biomater. Sci. Eng. 2017, 3, 757–766. [Google Scholar] [CrossRef]
- Okesola, B.O.; Lau, H.K.; Derkus, B.; Boccorh, D.K.; Wu, Y.; Wark, A.W.; Kiick, K.L.; Mata, A. Covalent co-assembly between resilin-like polypeptide and peptide amphiphile into hydrogels with controlled nanostructure and improved mechanical properties. Biomater. Sci. 2020, 8, 846–857. [Google Scholar] [CrossRef]
- Coskuner-Weber, O.; Yuce-Erarslan, E.; Uversky, V.N. Paving the Way for Synthetic Intrinsically Disordered Polymers for Soft Robotics. Polymers 2023, 15, 763. [Google Scholar] [CrossRef]
- Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19, 336. [Google Scholar] [CrossRef]
- Coskuner-Weber, O.; Mirzanli, O.; Uversky, V.N. Intrinsically disordered proteins and proteins with intrinsically disordered regions in neurodegenerative diseases. Biophys. Rev. 2022, 14, 679–707. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Monroe, M.B.B. Biostable Segmented Thermoplastic Polyurethane Shape Memory Polymers for Smart Biomedical Applications. ACS Appl. Polym. Mater. 2022, 4, 1956–1965. [Google Scholar] [CrossRef]
- Sikdar, P.; Dip, T.M.; Dhar, A.K.; Bhattacharjee, M.; Hoque, S.; Bin Ali, S. Polyurethane (PU) based multifunctional materials: Emerging paradigm for functional textiles, smart, and biomedical applications. J. Appl. Polym. Sci. 2022, 139, e52832. [Google Scholar] [CrossRef]
- Saad, N.M.; Saridi, M.H.M.; Zubir, S.A. Segmented shape memory polyurethane: Influence of soft segment types and length. Mater. Today Proc. 2022, 66, 2801–2805. [Google Scholar] [CrossRef]
- Bronzeri, L.B.; Gauche, C.; Gudimard, L.; Courtial, E.-J.; Marquette, C.; Felisberti, M.I. Amphiphilic and segmented polyurethanes based on poly(ε-caprolactone)diol and poly(2-ethyl-2-oxazoline)diol: Synthesis, properties, and a preliminary performance study of the 3D printing. Eur. Polym. J. 2021, 151, 110449. [Google Scholar] [CrossRef]
- Veloso-Fernández, A.; Laza, J.M.; Ruiz-Rubio, L.; Martín, A.; Taguado, M.; Benito-Vicente, A.; Martín, C.; Vilas, J.L. Towards a new generation of non-cytotoxic shape memory thermoplastic polyurethanes for biomedical applications. Mater. Today Commun. 2022, 33, 104730. [Google Scholar] [CrossRef]
- Uversky, V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J. Biol. Chem. 2016, 291, 6681–6688. [Google Scholar] [CrossRef]
- Uversky, V.N. The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013, 587, 1891–1901. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef]
- Bu, Z.; Callaway, D.J. Proteins MOVE! Protein dynamics and long-range allostery in cell signaling. Adv. Protein Chem. Struct. Biol. 2011, 83, 163–221. [Google Scholar] [CrossRef]
- Fatafta, H.; Samantray, S.; Sayyed-Ahmad, A.; Coskuner-Weber, O.; Strodel, B. Molecular Simulations of IDPs: From Ensemble Generation to IDP Interactions Leading to Disorder-to-Order Transitions. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2021; Volume 183, pp. 135–185. ISBN 978-0-323-85299-9. [Google Scholar]
- Strodel, B.; Coskuner-Weber, O. Transition Metal Ion Interactions with Disordered Amyloid-β Peptides in the Pathogenesis of Alzheimer’s Disease: Insights from Computational Chemistry Studies. J. Chem. Inf. Model. 2019, 59, 1782–1805. [Google Scholar] [CrossRef] [PubMed]
- Coskuner-Weber, O. Revisiting Cu(II) Bound Amyloid-β40 and Amyloid-β42 Peptides: Varying Coordination Chemistries. J. Turk. Chem. Soc. Sect. A Chem. 2018, 5, 981–1008. [Google Scholar] [CrossRef]
- Malik, U.S.; Niazi, M.B.K.; Jahan, Z.; Zafar, M.I.; Vo, D.-V.N.; Sher, F. Nano-structured dynamic Schiff base cues as robust self-healing polymers for biomedical and tissue engineering applications: A review. Environ. Chem. Lett. 2022, 20, 495–517. [Google Scholar] [CrossRef]
- Tan, R.Y.H.; Lee, C.S.; Pichika, M.R.; Cheng, S.F.; Lam, K.Y. PH Responsive Polyurethane for the Advancement of Biomedical and Drug Delivery. Polymers 2022, 14, 1672. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Qu, X.; Shi, B.; Zheng, Q.; Lin, X.; Chao, S.; Wang, C.; Zhou, J.; Sun, Y.; et al. Ultra-Stretchable and Fast Self-Healing Ionic Hydrogel in Cryogenic Environments for Artificial Nerve Fiber. Adv. Mater. 2022, 34, 2105416. [Google Scholar] [CrossRef]
- Ma, J.; Lee, G.-H.; Kim, J.-H.; Kim, S.-W.; Jo, S.; Kim, C.S. A Transparent Self-Healing Polyurethane–Isophorone-Diisocyanate Elastomer Based on Hydrogen-Bonding Interactions. ACS Appl. Polym. Mater. 2022, 4, 2497–2505. [Google Scholar] [CrossRef]
- Gokaltun, A.A.; Fan, L.; Mazzaferro, L.; Byrne, D.; Yarmush, M.L.; Dai, T.; Asatekin, A.; Usta, O.B. Supramolecular hybrid hydrogels as rapidly on-demand dissoluble, self-healing, and biocompatible burn dressings. Bioact. Mater. 2023, 25, 415–429. [Google Scholar] [CrossRef]
- Adhikari, B.; Stager, M.A.; Krebs, M.D. Cell-instructive biomaterials in tissue engineering and regenerative medicine. J. Biomed. Mater. Res. Part A 2023, 111, 660–681. [Google Scholar] [CrossRef]
- Pourmadadi, M.; Farokh, A.; Rahmani, E.; Eshaghi, M.M.; Aslani, A.; Rahdar, A.; Ferreira, L.F.R. Polyacrylic acid mediated targeted drug delivery nano-systems: A review. J. Drug Deliv. Sci. Technol. 2023, 80, 104169. [Google Scholar] [CrossRef]
- Bami, M.S.; Estabragh, M.A.R.; Khazaeli, P.; Ohadi, M.; Dehghannoudeh, G. pH-responsive drug delivery systems as intelligent carriers for targeted drug therapy: Brief history, properties, synthesis, mechanism and application. J. Drug Deliv. Sci. Technol. 2022, 70, 102987. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, L.; Yang, Q.; Huang, S.; Shi, H.; Long, Q.; Qian, B.; Liu, Z.; Guan, Q.; Liu, M.; et al. Self-healing polyurethane-elastomer with mechanical tunability for multiple biomedical applications in vivo. Nat. Commun. 2021, 12, 4395. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.; Zhang, X. Ultrarobust, tough and highly stretchable self-healing materials based on cartilage-inspired noncovalent assembly nanostructure. Nat. Commun. 2021, 12, 1291. [Google Scholar] [CrossRef] [PubMed]
- Padinjarathil, H.; Mudradi, S.; Balasubramanian, R.; Drago, C.; Dattilo, S.; Kothurkar, N.K.; Ramani, P. Design of an Antibiotic-Releasing Polymer: Physicochemical Characterization and Drug Release Patterns. Membranes 2023, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Segale, L.; Torre, M.L.; Machiste, E.O.; Conte, U. Dissolution behaviour of hydrophilic matrix tablets containing two different polyethylene oxides (PEOs) for the controlled release of a water-soluble drug. Dimensionality study. Biomaterials 2002, 23, 1113–1119. [Google Scholar] [CrossRef]
- Agarwal, S.; Murthy, R. Effect of different polymer concentration on drug release rate and physicochemical properties of mucoadhesive gastroretentive tablets. Indian J. Pharm. Sci. 2015, 77, 705–714. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Li, Z.; Liu, Y.; Xinag, P.; Cui, X.-Y.; Ye, H.; Hu, B.-L.; Lou, L.-P. Physical and chemical characteristics of PM2.5 and its toxicity to human bronchial cells BEAS-2B in the winter and summer. J. Zhejiang Univ. Sci. B 2018, 19, 317–326. [Google Scholar] [CrossRef]
- Funabashi, M.; Ninomiya, F.; Kunioka, M.M. Biodegradability Evaluation of Polymers by ISO 14855-2. Int. J. Mol. Sci. 2009, 10, 3635–3654. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Properties |
|---|---|
| Polymers | The capacity to create components with specified chemical and physical qualities |
| Creation capacity for encapsulating | |
| The ability to design drug delivery systems for specific tissues or cells | |
| The potential to create biocompatible designs | |
| Immune suppression | |
| Supporting the repair of damaged tissues | |
| Potential for growth factor delivery | |
| Possibility of developing biocompatible patterns | |
| Flexibility | |
| Customizability | |
| Immune compatibility | |
| Adjustable surface | |
| IDPs | Self-assembly |
| Highly flexible | |
| The capacity to create for recognition of particular biological targets | |
| Biodegradability and biocompatibility | |
| Anti-inflammatory features | |
| Ability to perform multiple functions | |
| Extreme flexible structure | |
| Binding-folding paradigm | |
| Post-translational modifications | |
| Binding affinity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuce-Erarslan, E.; Domb, A.J.; Kasem, H.; Uversky, V.N.; Coskuner-Weber, O. Intrinsically Disordered Synthetic Polymers in Biomedical Applications. Polymers 2023, 15, 2406. https://doi.org/10.3390/polym15102406
Yuce-Erarslan E, Domb AJ, Kasem H, Uversky VN, Coskuner-Weber O. Intrinsically Disordered Synthetic Polymers in Biomedical Applications. Polymers. 2023; 15(10):2406. https://doi.org/10.3390/polym15102406
Chicago/Turabian StyleYuce-Erarslan, Elif, Abraham (Avi) J. Domb, Haytam Kasem, Vladimir N. Uversky, and Orkid Coskuner-Weber. 2023. "Intrinsically Disordered Synthetic Polymers in Biomedical Applications" Polymers 15, no. 10: 2406. https://doi.org/10.3390/polym15102406
APA StyleYuce-Erarslan, E., Domb, A. J., Kasem, H., Uversky, V. N., & Coskuner-Weber, O. (2023). Intrinsically Disordered Synthetic Polymers in Biomedical Applications. Polymers, 15(10), 2406. https://doi.org/10.3390/polym15102406