
Adhesion of Bis-Salphen-Based Coordination Polymers to Graphene: Insights from Free Energy Perturbation Study


Abstract
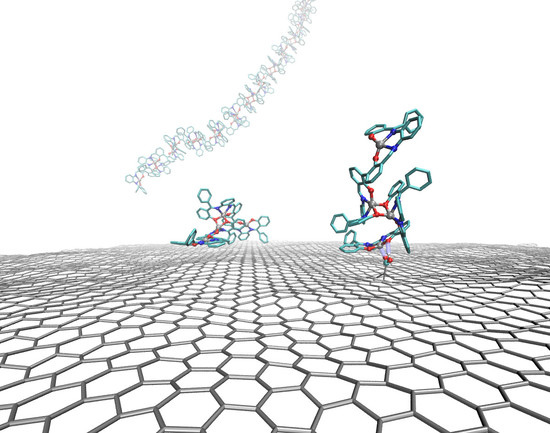
1. Introduction
2. Materials and Methods
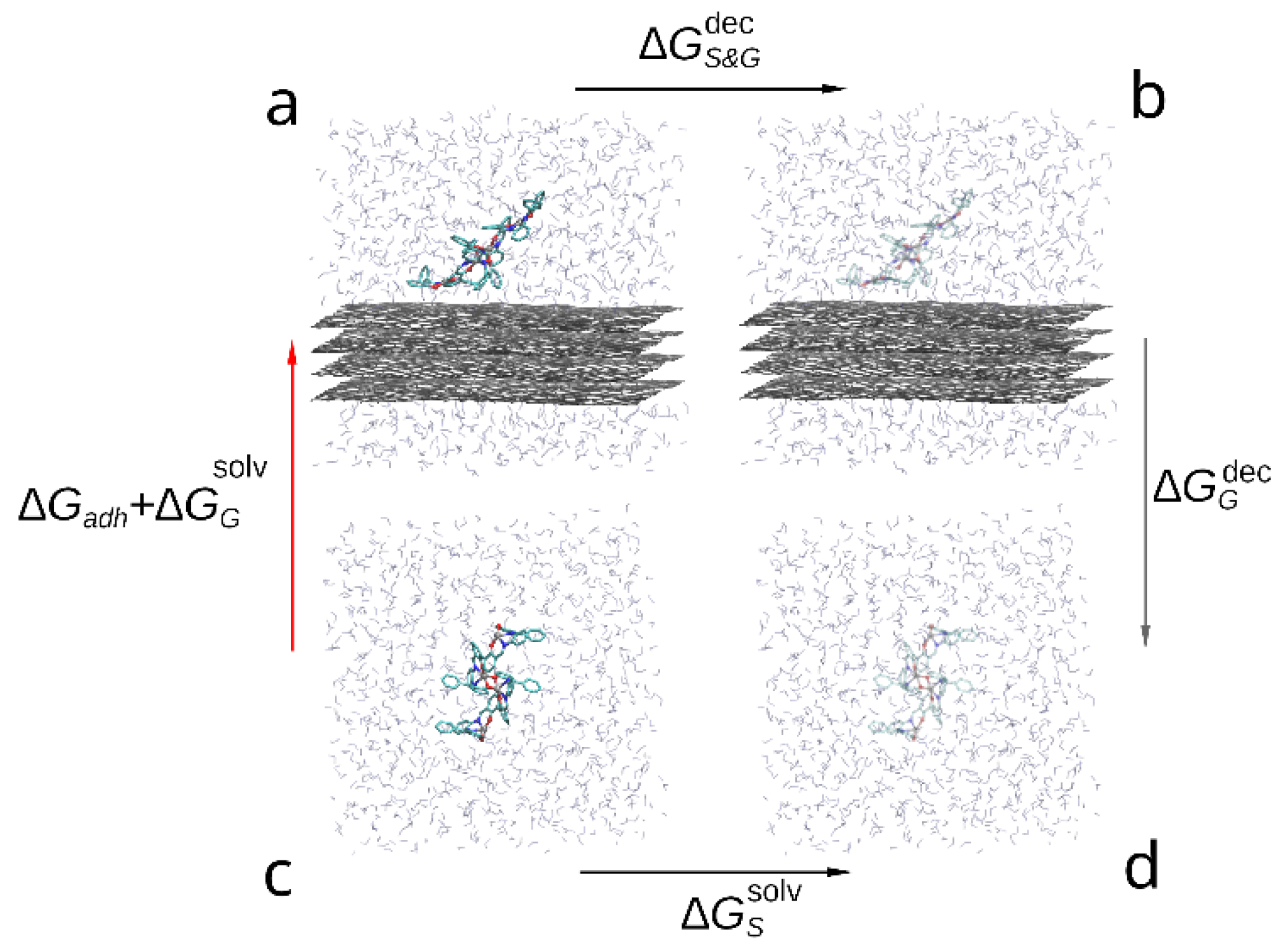
2.1. Ab Initio Energy of Adhesion
2.2. Molecular Dynamic Simulations
3. Results
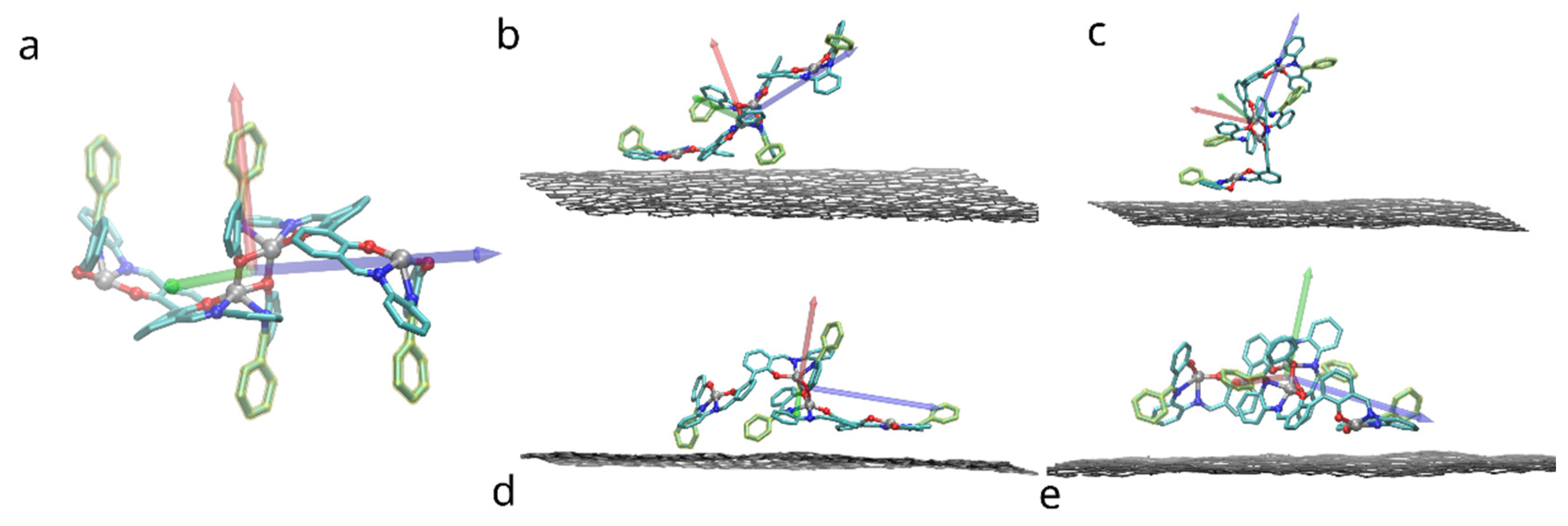
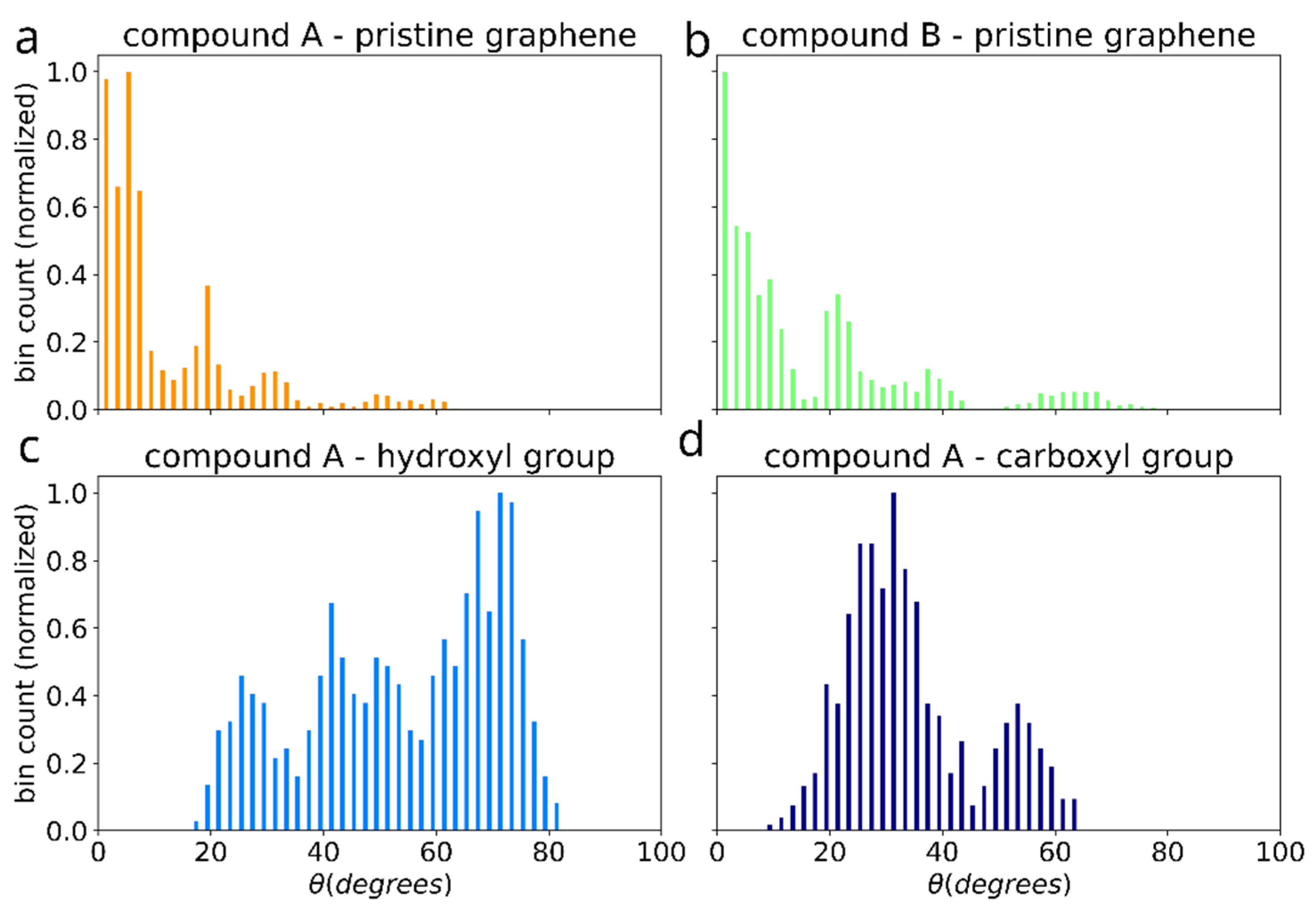
3.1. Binding of a Single Salphen Complex to Functionalized Graphene
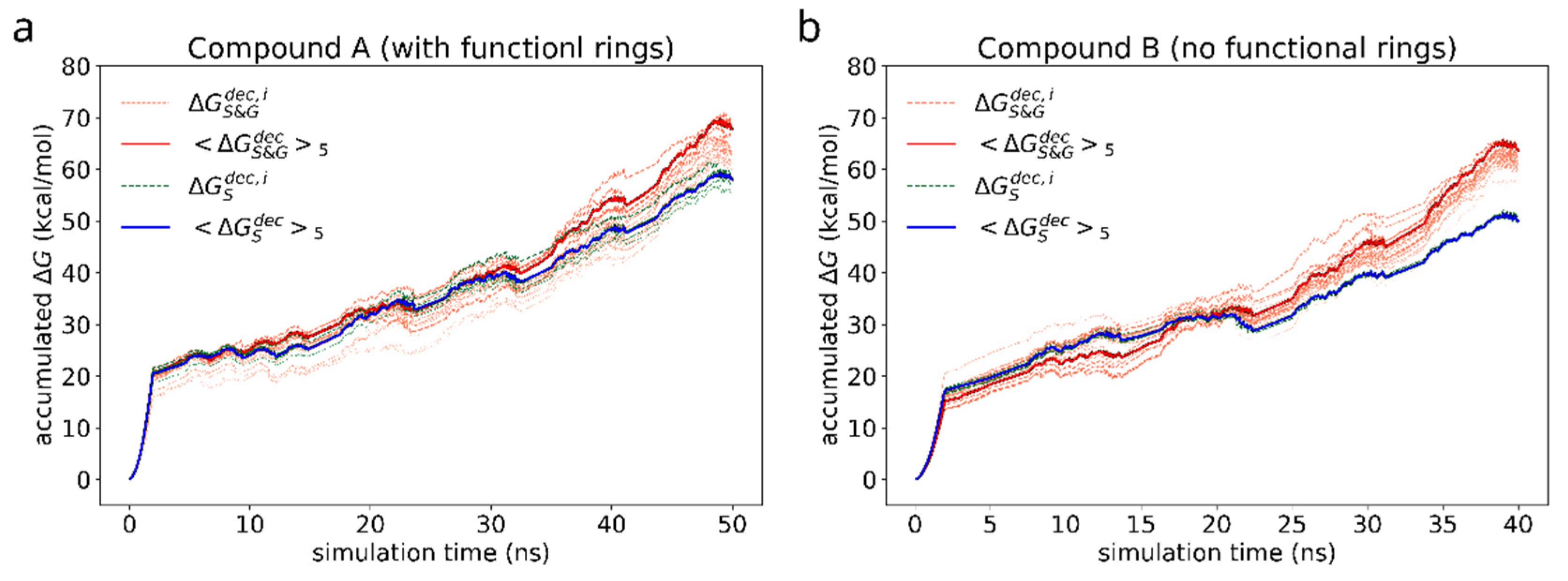
3.2. Free Energy of Adhesion of a Bis-Saplhen Dimer
3.2.1. The Effect of Compound Functionalization
3.2.2. The Effect of Graphene Functionalization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Wang, S.; Peng, J.; Tan, Y.; Li, C.; Boey, F.Y.C.; Long, Y. Liquid Thermo-Responsive Smart Window Derived from Hydrogel. Joule 2020, 4, 2458–2474. [Google Scholar] [CrossRef]
- Kim, S.J.; Choi, K.; Lee, B.; Kim, Y.; Hong, B.H. Materials for Flexible, Stretchable Electronics: Graphene and 2D Materials. Annu. Rev. Mater. Res. 2015, 45, 63–84. [Google Scholar] [CrossRef]
- Xu, Z.; Buehler, M.J. Nanoengineering Heat Transfer Performance at Carbon Nanotube Interfaces. ACS Nano 2009, 3, 2767–2775. [Google Scholar] [CrossRef] [PubMed]
- Musland, L.; Flage-Larsen, E. Thermoelectric Effect in Superlattices; Applicability of Coherent and Incoherent Transport Models. Comput. Mater. Sci. 2018, 153, 88–96. [Google Scholar] [CrossRef]
- Coropceanu, I.; Boles, M.A.; Talapin, D.V. Systematic Mapping of Binary Nanocrystal Superlattices: The Role of Topology in Phase Selection. J. Am. Chem. Soc. 2019; Just Accepted Manuscript. [Google Scholar] [CrossRef]
- Wang, A.; Ye, J.; Humphrey, M.G.; Zhang, C. Graphene and Carbon-Nanotube Nanohybrids Covalently Functionalized by Porphyrins and Phthalocyanines for Optoelectronic Properties. Adv. Mater. 2018, 30, 1705704. [Google Scholar] [CrossRef]
- Mustonen, K.; Susi, T.; Kaskela, A.; Laiho, P.; Tian, Y.; Nasibulin, A.G.; Kauppinen, E.I. Influence of the Diameter of Single-Walled Carbon Nanotube Bundles on the Optoelectronic Performance of Dry-Deposited Thin Films. Beilstein J. Nanotechnol. 2012, 3, 692–702. [Google Scholar] [CrossRef]
- Escárcega-Bobadilla, M.V.; Zelada-Guillén, G.A.; Pyrlin, S.V.; Wegrzyn, M.; Ramos, M.M.D.; Giménez, E.; Stewart, A.; Maier, G.; Kleij, A.W. Nanorings and Rods Interconnected by Self-Assembly Mimicking an Artificial Network of Neurons. Nat Commun 2013, 4, 2648. [Google Scholar] [CrossRef]
- Hong, S.W.; Jeong, W.; Ko, H.; Kessler, M.R.; Tsukruk, V.V.; Lin, Z. Directed Self-Assembly of Gradient Concentric Carbon Nanotube Rings. Adv. Funct. Mater. 2008, 18, 2114–2122. [Google Scholar] [CrossRef]
- Basu, S.; Patra, P.; Sarkar, J. Dewetting Assisted Self-Assembly of Carbon Nanotube into Circular Nanorings. Chem Eng Sci 2022, 261, 117961. [Google Scholar] [CrossRef]
- Koner, K.; Karak, S.; Kandambeth, S.; Karak, S.; Thomas, N.; Leanza, L.; Perego, C.; Pesce, L.; Capelli, R.; Moun, M.; et al. Porous Covalent Organic Nanotubes and Their Assembly in Loops and Toroids. Nat. Chem. 2022, 14, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Kato, Y.; Higashiharaguchi, S.; Aratsu, K.; Isobe, A.; Saito, T.; Prabhu, D.D.; Kitamoto, Y.; Hollamby, M.J.; Smith, A.J.; et al. Self-Assembled Poly-Catenanes from Supramolecular Toroidal Building Blocks. 400 Nat. 2020, 583, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Pyrlin, S.V.; Hine, N.D.M.; Kleij, A.W.; Ramos, M.M.D. Self-Assembly of Bis-Salphen Compounds: From Semiflexible Chains to Webs of Nanorings. Soft Matter 2018, 14, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Nirmalraj, P.N.; Lyons, P.E.; De, S.; Coleman, J.N.; Boland, J.J. Electrical Connectivity in Single-Walled Carbon Nanotube Networks. Nano. Lett. 2009, 9, 3890–3895. [Google Scholar] [CrossRef] [PubMed]
- Buldum, A.; Lu, J.P. Contact Resistance between Carbon Nanotubes. Phys. Rev. B 2001, 63, 161403. [Google Scholar] [CrossRef]
- Alberti, S.A.N.; Schneider, J.; Müller-Plathe, F. Mobility of Polymer Melts in a Regular Array of Carbon Nanotubes. J. Chem. Theory Comput. 2021, 18, 3295. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B.O. A Review of Methods for the Calculation of Solution Free Energies and the Modelling of Systems in Solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella Sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Virnau, P.; Muller, M. Calculation of Free Energy through Successive Umbrella Sampling. J. Chem. Phys. 2004, 120, 10925. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef]
- Park, S.; Schulten, K. Calculating Potentials of Mean Force from Steered Molecular Dynamics Simulations. J. Chem. Phys. 2004, 120, 5946–5961. [Google Scholar] [CrossRef]
- Park, S.; Khalili-Araghi, F.; Tajkhorshid, E.; Schulten, K. Free Energy Calculation from Steered Molecular Dynamics Simulations Using Jarzynski’s Equality. J. Chem. Phys. 2003, 119, 3559. [Google Scholar] [CrossRef]
- Comer, J.; Gumbart, J.C.; Hénin, J.; Lelievre, T.; Pohorille, A.; Chipot, C. The Adaptive Biasing Force Method: Everything You Always Wanted to Know but Were Afraid to Ask. J. Phys. Chem. B 2015, 119, 1129–1151. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the Opls Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Pohorille, A.; Jarzynski, C.; Chipot, C. Good Practices in Free-Energy Calculations. J. Phys. Chem. B 2010, 114, 10235–10253. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Roux, B.; Chipot, C. Standard Binding Free Energies from Computer Simulations: What Is the Best Strategy? J. Chem. Theory Comput. 2013, 9, 794–802. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient Estimation of Free Energy Differences from Monte Carlo Data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Kim, I.; Allen, T.W. Bennett’s Acceptance Ratio and Histogram Analysis Methods Enhanced by Umbrella Sampling along a Reaction Coordinate in Configurational Space. J. Chem. Phys. 2012, 136, 164103. [Google Scholar] [CrossRef]
- Abrams, J.B.; Rosso, L.; Tuckerman, M.E. Efficient and Precise Solvation Free Energies via Alchemical Adiabatic Molecular Dynamics. J. Chem. Phys. 2006, 125, 074115. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Williams-Noonan, B.J.; Yuriev, E.; Chalmers, D.K. Free Energy Methods in Drug Design: Prospects of “Alchemical Perturbation” in Medicinal Chemistry. J. Med. Chem. 2018, 61, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.K.; Datta, A. Disentangling the Liquid Phase Exfoliation of Two-Dimensional Materials: An “in Silico” Perspective. Phys. Chem. Chem. Phys. 2020, 22, 22157–22179. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Salgado, S.; Maheshwari, V.; Liu, J. DNA Adsorbed on Graphene and Graphene Oxide: Fundamental Interactions, Desorption and Applications. Curr. Opin. Colloid Interface Sci. 2016, 26, 41–49. [Google Scholar] [CrossRef]
- Ghaderi, N.; Peressi, M. First-Principle Study of Hydroxyl Functional Groups on Pristine, Defected Graphene, and Graphene Epoxide. J. Phys. Chem. C 2010, 114, 21625–21630. [Google Scholar] [CrossRef]
- Mehandzhiyski, A.Y.; Morita, M.; Oya, Y.; Kato, N.; Mori, K.; Koyanagi, J. Effect of Electrostatic Interactions on the Interfacial Energy between Thermoplastic Polymers and Graphene Oxide: A Molecular Dynamics Study. Polymers 2022, 14, 2579. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA Method for Ab Initio Order- N Materials Simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Junquera, J.; Paz, Ó.; Sánchez-Portal, D.; Artacho, E. Numerical Atomic Orbitals for Linear-Scaling Calculations. Phys. Rev. B 2001, 64, 235111. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Sordo, J.A.; Chin, S.; Sordo, T.L. On the Counterpoise Correction for the Basis Set Superposition Error in Large Systems. Theor. Chim. Acta 1988, 74, 101–110. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Luty, B.A.; Davis, M.E.; Tironi, I.G.; van Gunsteren, W.F. A Comparison of Particle-Particle, Particle-Mesh and Ewald Methods for Calculating Electrostatic Interactions in Periodic Molecular Systems. Mol. Simul. 1994, 14, 11–20. [Google Scholar] [CrossRef]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. Tools: Advances in RESP and ESP Charge Derivation and Force Field Library Building. Phys. Chem. Chem. Phys. 2010, 12, 7821. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02. J. Comput. Chem. 2004, 24, 1748–1757. [Google Scholar]
- Stote, R.H.; Karplus, M. Zinc Binding in Proteins and Solution: A Simple but Accurate Nonbonded Representation. Proteins Struct. Funct. Genet. 1995, 23, 12–31. [Google Scholar] [CrossRef]
- Beutler, T.C.; Mark, A.E.; van Schaik, R.C.; Gerber, P.R.; van Gunsteren, W.F. Avoiding Singularities and Numerical Instabilities in Free Energy Calculations Based on Molecular Simulations. Chem. Phys. Lett. 1994, 222, 529–539. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Skiniotis, G. Development of OPLS-AA/M Parameters for Simulations of G Protein-Coupled Receptors and Other Membrane Proteins. J. Chem. Theory Comput. 2022, 18, 4482–4489. [Google Scholar] [CrossRef] [PubMed]
- Doherty, B.; Zhong, X.; Gathiaka, S.; Li, B.; Acevedo, O. Revisiting OPLS Force Field Parameters for Ionic Liquid Simulations. J. Chem. Theory Comput. 2017, 13, 6131–6135. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Aljuhani, A.; Ahmed, H.E.A.; Ihmaid, S.K.; Omar, A.M.; Althagfan, S.S.; Alahmadi, Y.M.; Ahmad, I.; Patel, H.; Ahmed, S.; Almikhlafi, M.A.; et al. In Vitro and Computational Investigations of Novel Synthetic Carboxamide-Linked Pyridopyrrolopyrimidines with Potent Activity as SARS-CoV-2-M Pro Inhibitors. RSC Adv. 2022, 12, 26895–26907. [Google Scholar] [CrossRef]
- Monti, M.; Stener, M.; Aschi, M. A Computational Approach for Modeling Electronic Circular Dichroism of Solvated Chromophores. J. Comput. Chem. 2022, 15, 2023–2036. [Google Scholar] [CrossRef]
- Ding, K.; Xu, C.; Peng, Z.; Long, X.; Shi, J.; Li, Z.; Zhang, Y.; Lai, J.; Chen, L.; Cai, Y.-P.; et al. Tuning the Solvent Alkyl Chain to Tailor Electrolyte Solvation for Stable Li-Metal Batteries. ACS Appl. Mater. Interfaces 2022, 14, 44470–44478. [Google Scholar] [CrossRef]
- Jewett, A.I.; Stelter, D.; Lambert, J.; Saladi, S.M.; Roscioni, O.M.; Ricci, M.; Autin, L.; Maritan, M.; Bashusqeh, S.M.; Keyes, T.; et al. Moltemplate: A Tool for Coarse-Grained Modeling of Complex Biological Matter and Soft Condensed Matter Physics. J. Mol. Biol. 2021, 433, 166841. [Google Scholar] [CrossRef]
- Holian, B.L.; Voter, A.F.; Ravelo, R. Thermostatted Molecular Dynamics: How to Avoid the Toda Demon Hidden in Nose-Hoover Dynamics. Phys. Rev. E 1995, 52, 2338. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Stoll, E. Molecular-Dynamics Study of a Three-Dimensional One-Component Model for Distortive Phase Transitions. Phys. Rev. B 1978, 17, 1302. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Mobley, D.L.; Case, D.A. Nonlinear Scaling Schemes for Lennard-Jones Interactions in Free Energy Calculations. J. Chem. Phys. 2007, 127, 214108. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Pande, V.S. Comparison of Efficiency and Bias of Free Energies Computed by Exponential Averaging, the Bennett Acceptance Ratio, and Thermodynamic Integration. J. Chem. Phys. 2005, 122, 144107. [Google Scholar] [CrossRef] [PubMed]
- Klimovich, P.V.; Shirts, M.R.; Mobley, D.L. Guidelines for the Analysis of Free Energy Calculations. J. Comput. Aided Mol. Des. 2015, 29, 397–411. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Kharisov, B.I.; Kharissova, O.V.; Dimas, A.V. The Dispersion, Solubilization and Stabilization in “Solution” of Single-Walled Carbon Nanotubes. RSC Adv. 2016, 6, 68760–68787. [Google Scholar] [CrossRef]
- Cao, X.Z.; Merlitz, H.; Wu, C.X.; Ungar, G.; Sommer, J.U. A Theoretical Study of Dispersion-to-Aggregation of Nanoparticles in Adsorbing Polymers Using Molecular Dynamics Simulations. Nanoscale 2016, 8, 6964–6968. [Google Scholar] [CrossRef]
- Elkashef, M.; Wang, K.; Abou-Zeid, M.N. Acid-Treated Carbon Nanotubes and Their Effects on Mortar Strength. Front. Struct. Civ. Eng. 2015, 10, 180–188. [Google Scholar] [CrossRef]
- Liang, S.; Li, G.; Tian, R. Multi-Walled Carbon Nanotubes Functionalized with a Ultrahigh Fraction of Carboxyl and Hydroxyl Groups by Ultrasound-Assisted Oxidation. J. Mater. Sci. 2016, 51, 3513–3524. [Google Scholar] [CrossRef]
- Datsyuk, V.; Kalyva, M.; Papagelis, K.; Parthenios, J.; Tasis, D.; Siokou, A.; Kallitsis, I.; Galiotis, C. Chemical Oxidation of Multiwalled Carbon Nanotubes. Carbon N. Y. 2008, 46, 833–840. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functionalization Type | Eadh | Eadh-Eadh (Pristine) |
|---|---|---|
| Pristine | −38.8 | - |
| B-doped | −40.7 | −1.9 |
| N-doped | −41.1 | −2.2 |
| =O | −35.5 | +3.3 |
| -OH | −40.5 | −1.6 |
| -COOH | −44.6 | −5.6 |
| Compound: | Compound A | Compound B |
|---|---|---|
| Solvated (−) | −58.1 ± 5.1 | −50.4 ± 1.2 |
| Adsorbed (−) | −67.7 ± 2.5 | −63.7 ± 0.8 |
| −9.7 ± 5.7 | −13.7 ± 1.5 |
| Functionalization: | Hydroxyl (-OH) | Carboxyl (-COOH) |
|---|---|---|
| Graphen & compound (−) | −5.9 ± 0.1 | −23.0 ± 1.1 |
| ) | −1.2 ± 0.1 | −15.0 ± 1.0 |
| −4.7 ± 0.2 | −8.0 ± 1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyrlin, S.; Lenzi, V.; Silva, A.; Ramos, M.; Marques, L. Adhesion of Bis-Salphen-Based Coordination Polymers to Graphene: Insights from Free Energy Perturbation Study. Polymers 2022, 14, 4525. https://doi.org/10.3390/polym14214525
Pyrlin S, Lenzi V, Silva A, Ramos M, Marques L. Adhesion of Bis-Salphen-Based Coordination Polymers to Graphene: Insights from Free Energy Perturbation Study. Polymers. 2022; 14(21):4525. https://doi.org/10.3390/polym14214525
Chicago/Turabian StylePyrlin, Sergey, Veniero Lenzi, Alexandre Silva, Marta Ramos, and Luís Marques. 2022. "Adhesion of Bis-Salphen-Based Coordination Polymers to Graphene: Insights from Free Energy Perturbation Study" Polymers 14, no. 21: 4525. https://doi.org/10.3390/polym14214525
APA StylePyrlin, S., Lenzi, V., Silva, A., Ramos, M., & Marques, L. (2022). Adhesion of Bis-Salphen-Based Coordination Polymers to Graphene: Insights from Free Energy Perturbation Study. Polymers, 14(21), 4525. https://doi.org/10.3390/polym14214525