
Free Volume and Permeability of Mixed Matrix Membranes Made from a Terbutil-M-terphenyl Polyamide and a Porous Polymer Network

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of 3-Carboxy-phenylboronic Acid Results
2.2. 5-Terbuthyl-m-terphenyl-3,3″-dicarboxylic Acid, tBTmDA
2.3. Synthesis of Diacid Chloride
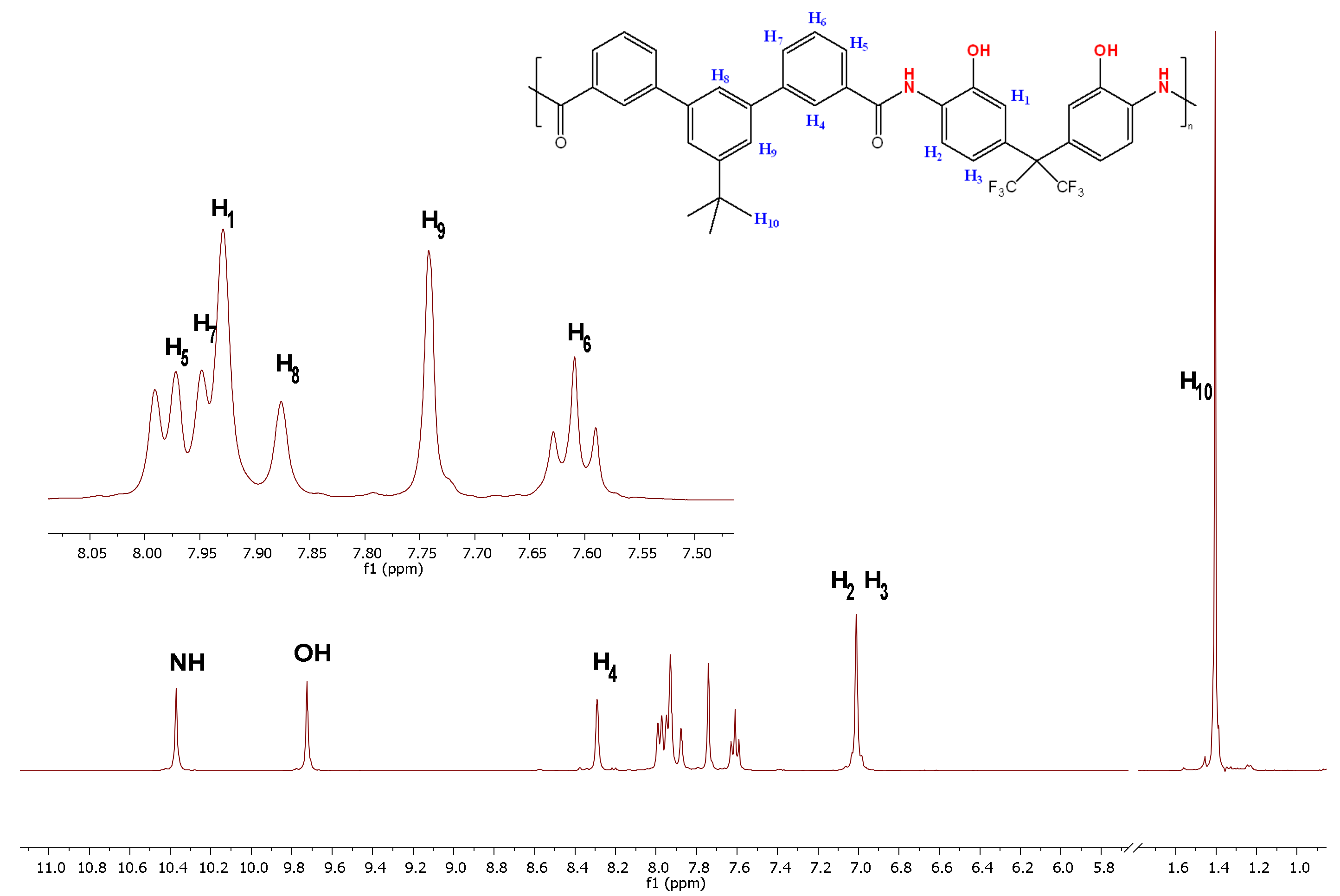
2.4. Polymer Synthesis
2.5. Polymer Characterization
2.6. Polymer Matrix Films
2.7. Preparation of Mixed Matrix Membranes
2.8. Thermal Rearrangement Mixed Matrix Membranes (TR-MMMs)
3. Membrane Characterization
3.1. Thermogravimetric Analysis
3.2. Differential Scanning Calorimetry
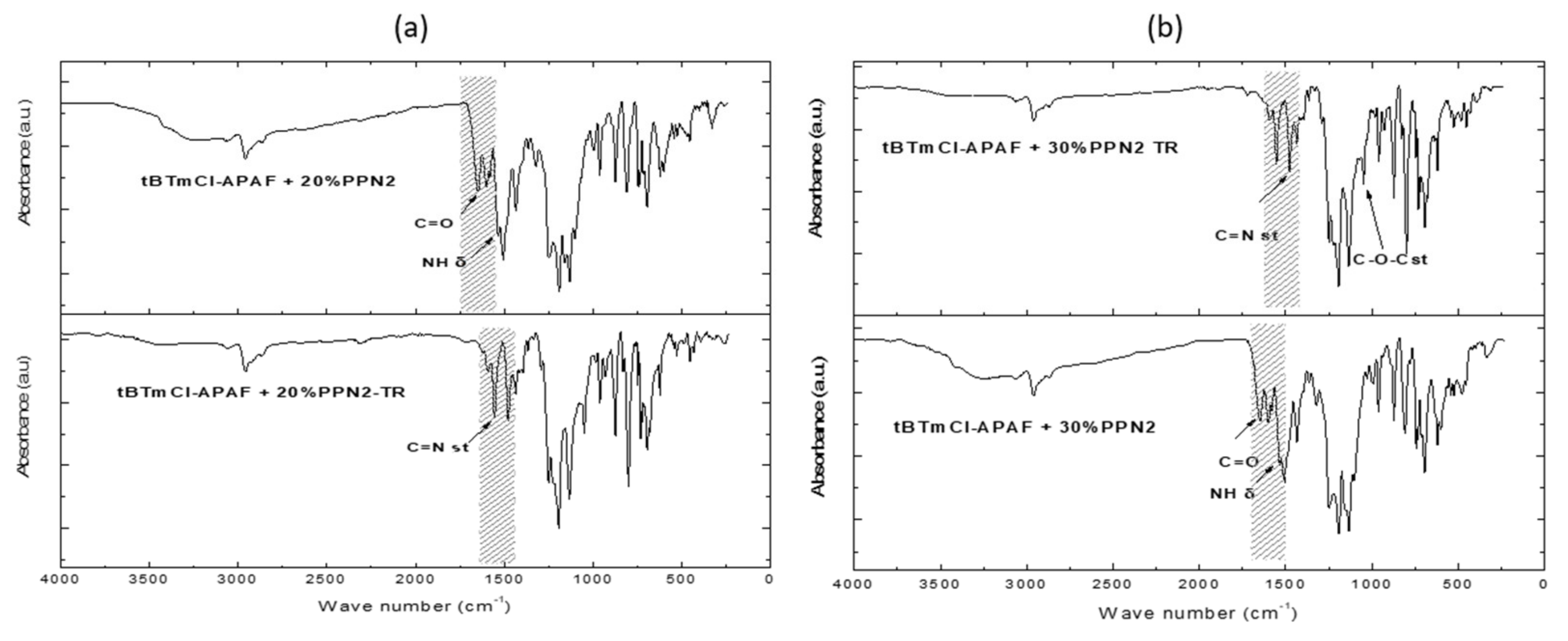
3.3. Fourier Transform Infrared Spectroscopy (ATR-FTIR)
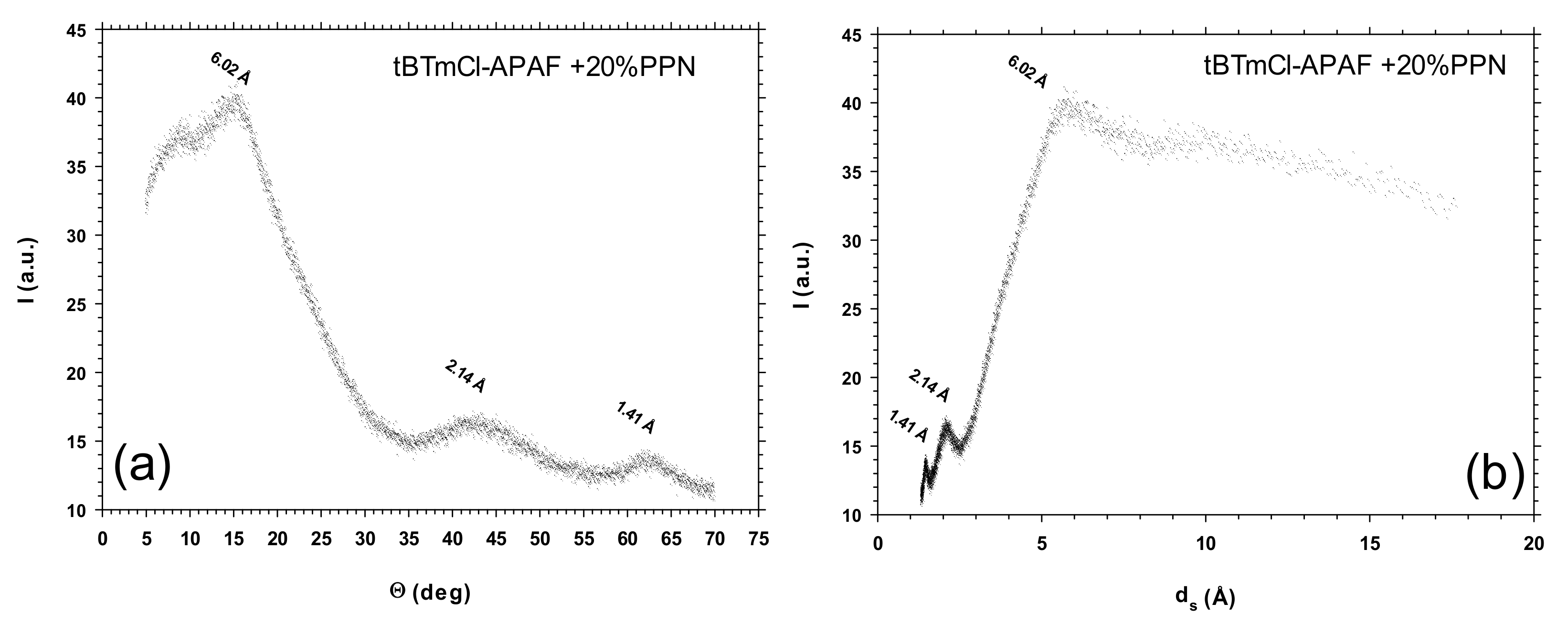
3.4. Wide-Angle X-ray Scattering
3.5. Mechanicals Properties
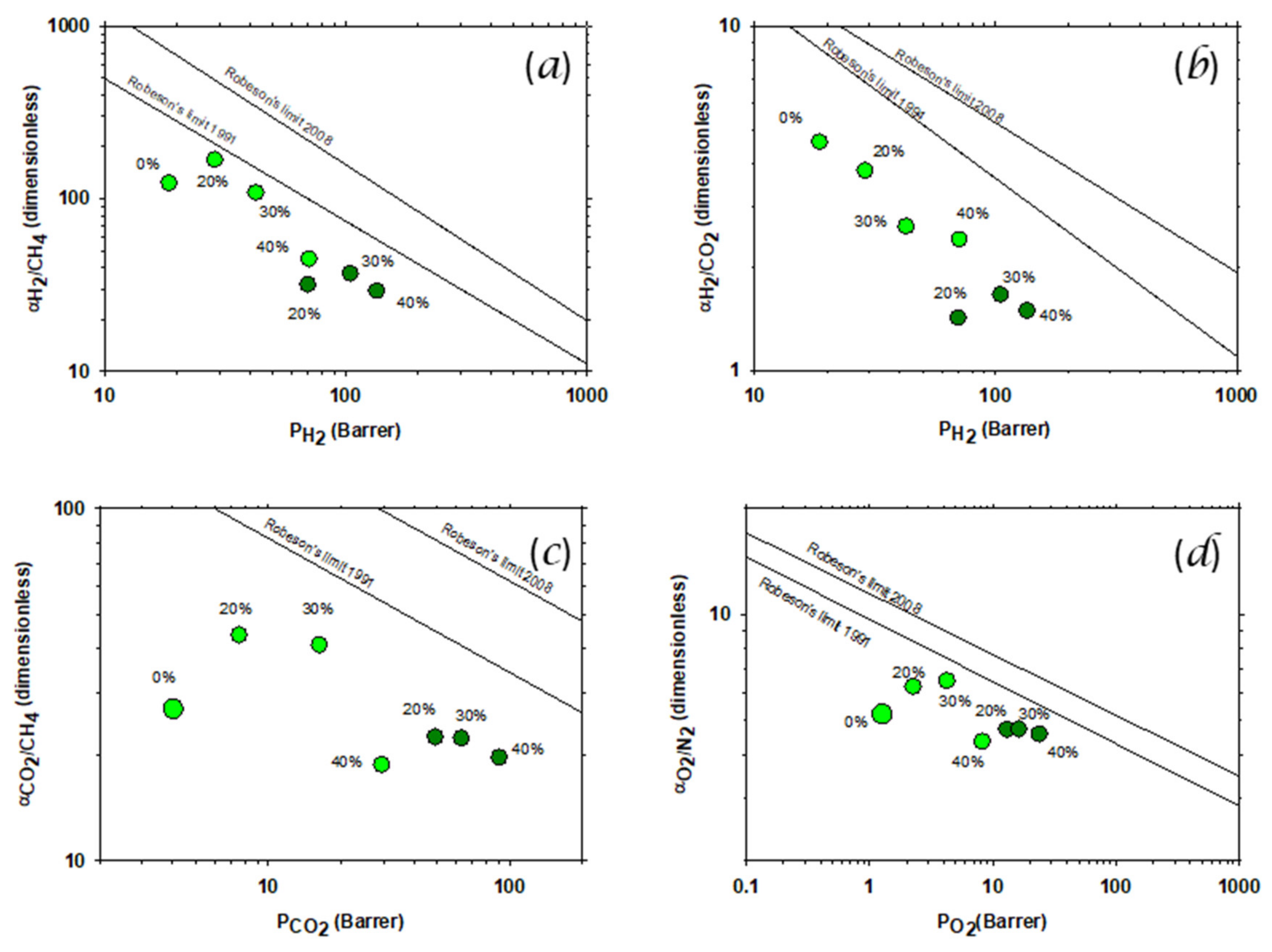
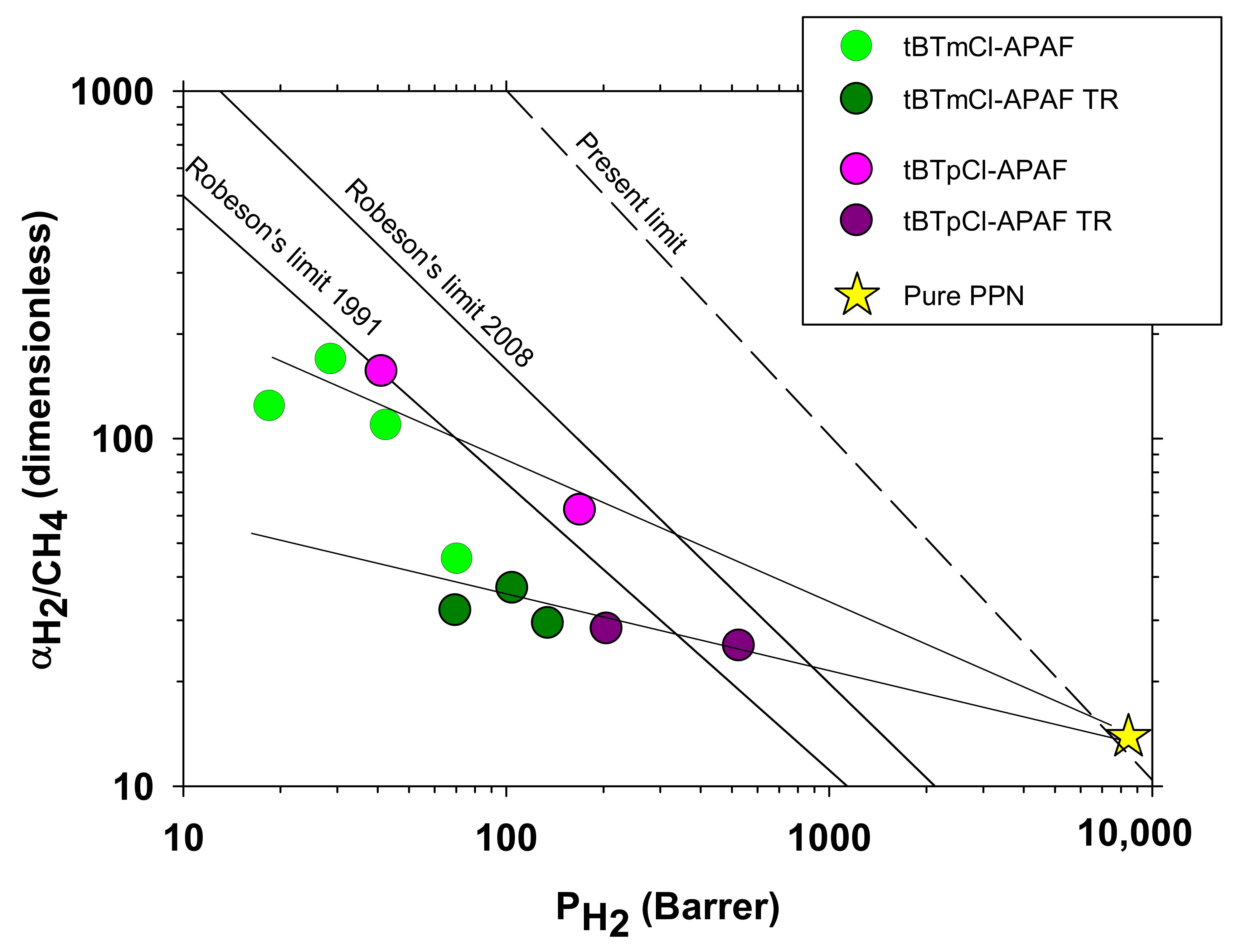
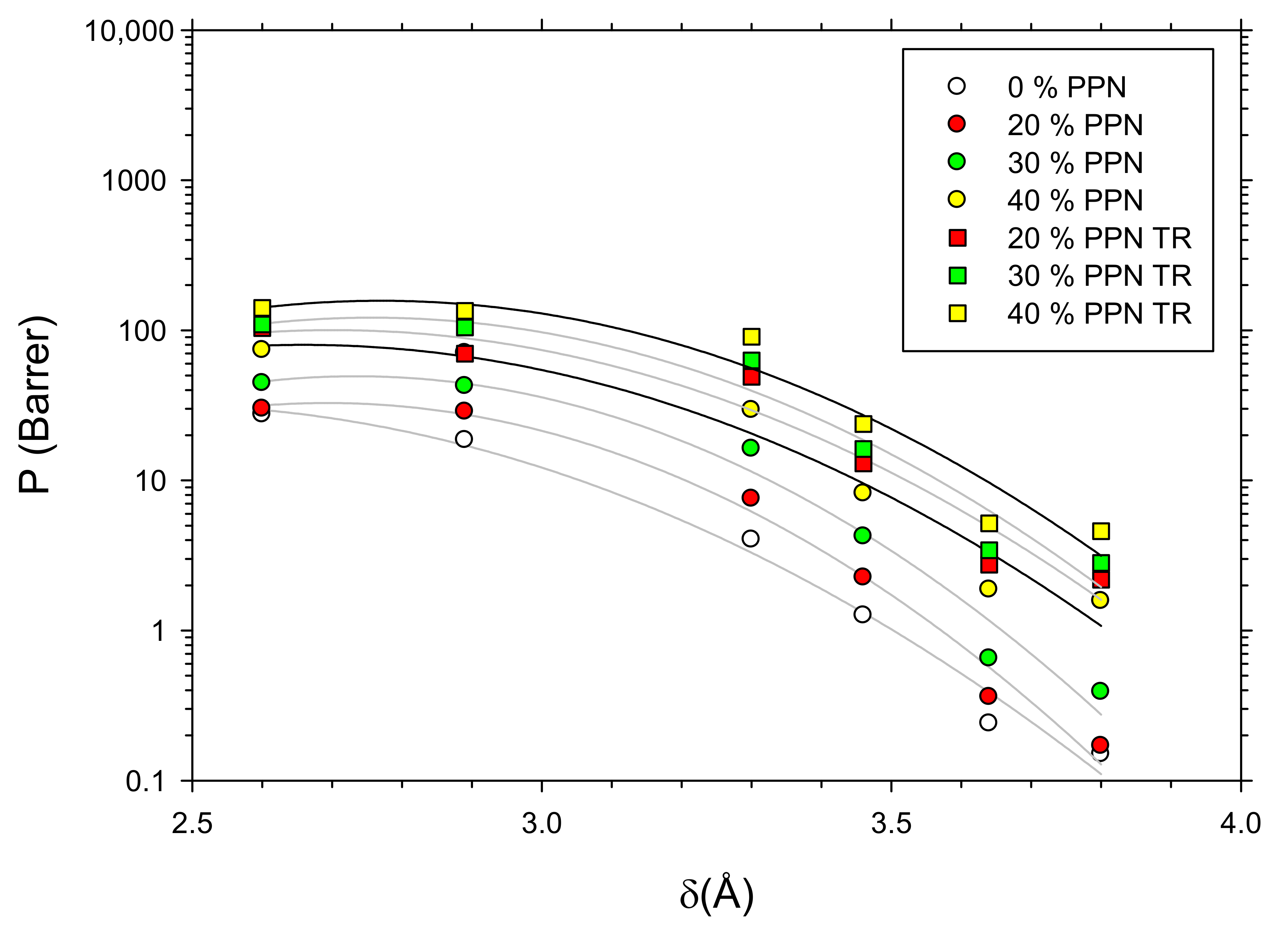
3.6. Gas Transport: Permeability and Selectivity
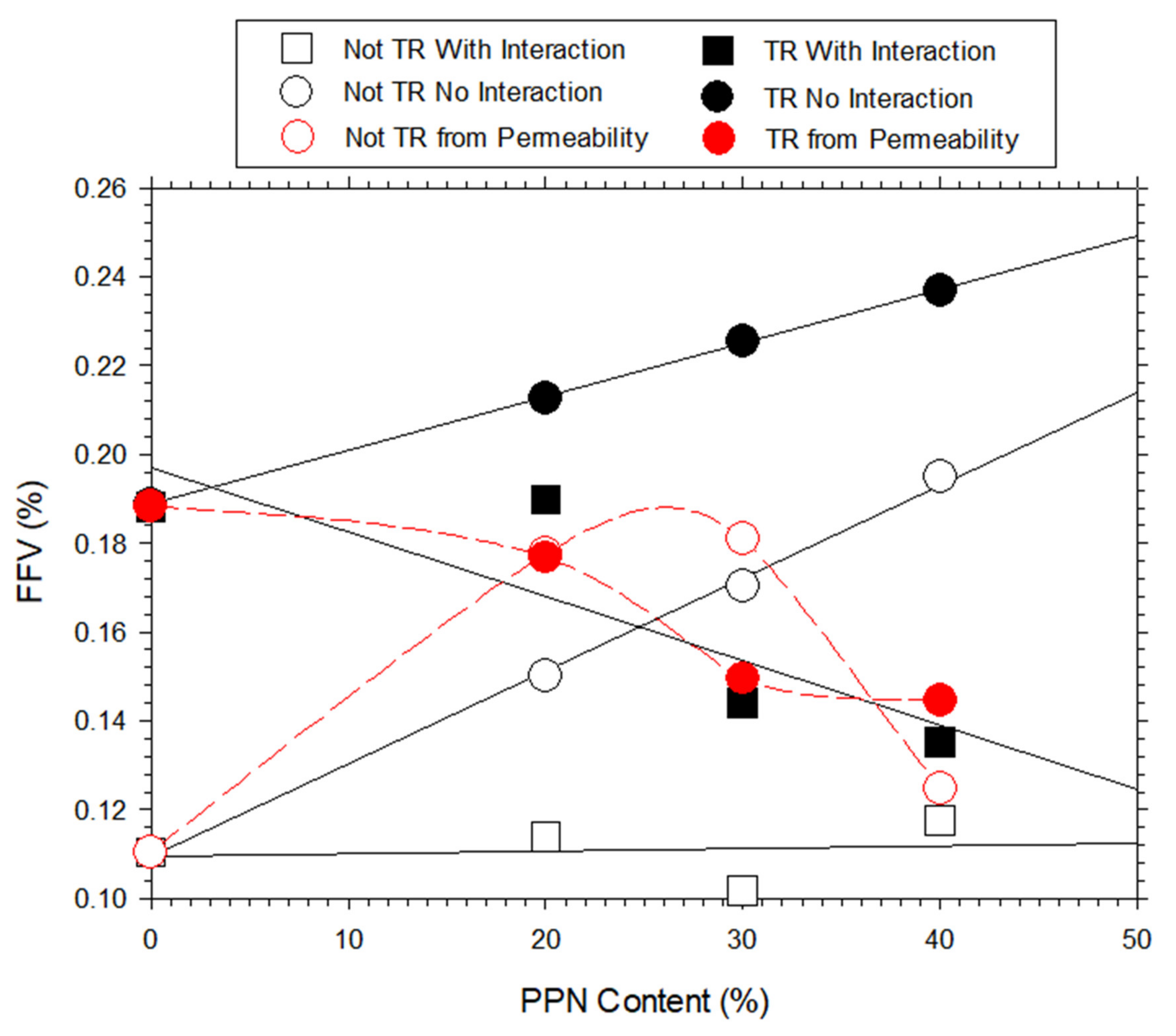
3.7. Density and Free Volume
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Du, Z.; Liu, C.; Zhai, J.; Guo, X.; Xiong, Y.; Su, W.; He, G. A Review of Hydrogen Purification Technologies for Fuel Cell Vehicles. Catalysts 2021, 11, 393. [Google Scholar] [CrossRef]
- Muñoz, R.; Meier, L.; Diaz, I.; Jeison, D. A review on the state-of-the-art of physical/chemical and biological technologies for biogas upgrading. Rev. Environ. Sci. Bio/Technol. 2015, 14, 727–759. [Google Scholar] [CrossRef] [Green Version]
- Angelidaki, I.; Treu, L.; Tsapekos, P.; Luo, G.; Campanaro, S.; Wenzel, H.; Kougias, P.G. Biogas upgrading and utilization: Current status and perspectives. Biotechnol. Adv. 2018, 36, 452–466. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.-G.; Hu, T.-L.; Bu, X.-H. Metal-Organic Framework Materials for the Separation and Purification of Light Hydrocarbons. Adv. Mater. 2020, 32, 1806445. [Google Scholar] [CrossRef] [PubMed]
- Aroon, M.A.; Ismail, A.F.; Matsuura, T.; Montazer-Rahmati, M.M. Performance studies of mixed matrix membranes for gas separation: A review. Sep. Purif. Technol. 2010, 75, 229–242. [Google Scholar] [CrossRef]
- Robeson, L.M. The upper bound revisited. J. Membr. Sci. 2008, 320, 390–400. [Google Scholar] [CrossRef]
- Robeson, L.M.; Burgoyne, W.F.; Langsam, M.; Savoca, A.C.; Tien, C.F. High performance polymers for membrane separation. Polymer 1994, 35, 4970–4978. [Google Scholar] [CrossRef]
- Robeson, L.M. Correlation of separation factor versus permeability for polymeric membranes. J. Membr. Sci. 1991, 62, 165–185. [Google Scholar] [CrossRef]
- Etxeberria-Benavides, M.; David, O.; Johnson, T.; Łozińska, M.M.; Orsi, A.; Wright, P.A.; Mastel, S.; Hillenbrand, R.; Kapteijn, F.; Gascon, J. High performance mixed matrix membranes (MMMs) composed of ZIF-94 filler and 6FDA-DAM polymer. J. Membr. Sci. 2018, 550, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Park, H.B.; Jung, C.H.; Lee, Y.M.; Hill, A.J.; Pas, S.J.; Mudie, S.T.; Van Wagner, E.; Freeman, B.D.; Cookson, D.J. Polymers with Cavities Tuned for Fast Selective Transport of Small Molecules and Ions. Science 2007, 318, 254. [Google Scholar] [CrossRef] [PubMed]
- Rezakazemi, M.; Ebadi Amooghin, A.; Montazer-Rahmati, M.M.; Ismail, A.F.; Matsuura, T. State-of-the-art membrane based CO2 separation using mixed matrix membranes (MMMs): An overview on current status and future directions. Prog. Polym. Sci. 2014, 39, 817–861. [Google Scholar] [CrossRef]
- Galizia, M.; Chi, W.S.; Smith, Z.P.; Merkel, T.C.; Baker, R.W.; Freeman, B.D. 50th Anniversary Perspective: Polymers and Mixed Matrix Membranes for Gas and Vapor Separation: A Review and Prospective Opportunities. Macromolecules 2017, 50, 7809–7843. [Google Scholar] [CrossRef]
- Lau, C.H.; Mulet, X.; Konstas, K.; Doherty, C.M.; Sani, M.-A.; Separovic, F.; Hill, M.R.; Wood, C.D. Hypercrosslinked Additives for Ageless Gas-Separation Membranes. Angew. Chem. Int. Ed. 2016, 55, 1998–2001. [Google Scholar] [CrossRef]
- Thapa, S.; Meng, L.; Hettiarachchi, E.; Bader, Y.K.; Dickie, D.A.; Rubasinghege, G.; Ivanov, S.A.; Vreeland, E.C.; Qin, Y. Charge-Separated and Lewis Paired Metal-Organic Framework for Anion Exchange and CO2 Chemical Fixation. Chem. A Eur. J. 2020, 26, 13788–13791. [Google Scholar] [CrossRef] [PubMed]
- Seoane, B.; Coronas, J.; Gascon, I.; Benavides, M.E.; Karvan, O.; Caro, J.; Kapteijn, F.; Gascon, J. Metal-organic framework based mixed matrix membranes: A solution for highly efficient CO2 capture? Chem. Soc. Rev. 2015, 44, 2421–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liang, F.; Bux, H.; Yang, W.; Caro, J. Zeolitic imidazolate framework ZIF-7 based molecular sieve membrane for hydrogen separation. J. Membr. Sci. 2010, 354, 48–54. [Google Scholar] [CrossRef]
- Smith, S.J.D.; Hou, R.; Lau, C.H.; Konstas, K.; Kitchin, M.; Dong, G.; Lee, J.; Lee, W.H.; Seong, J.G.; Lee, Y.M.; et al. Highly permeable Thermally Rearranged Mixed Matrix Membranes (TR-MMM). J. Membr. Sci. 2019, 585, 260–270. [Google Scholar] [CrossRef]
- Aguilar-Lugo, C.; Suárez-García, F.; Hernández, A.; Miguel, J.A.; Lozano, Á.E.; de la Campa, J.G.; Álvarez, C. New Materials for Gas Separation Applications: Mixed Matrix Membranes Made from Linear Polyimides and Porous Polymer Networks Having Lactam Groups. Ind. Eng. Chem. Res. 2019, 58, 9585–9595. [Google Scholar] [CrossRef]
- Lau, C.H.; Konstas, K.; Doherty, C.M.; Kanehashi, S.; Ozcelik, B.; Kentish, S.E.; Hill, A.J.; Hill, M.R. Tailoring Physical Aging in Super Glassy Polymers with Functionalized Porous Aromatic Frameworks for CO2 Capture. Chem. Mater. 2015, 27, 4756–4762. [Google Scholar] [CrossRef]
- Cheng, X.Q.; Konstas, K.; Doherty, C.M.; Wood, C.D.; Mulet, X.; Xie, Z.; Ng, D.; Hill, M.R.; Shao, L.; Lau, C.H. Hyper-Cross-Linked Additives that Impede Aging and Enhance Permeability in Thin Polyacetylene Films for Organic Solvent Nanofiltration. ACS Appl. Mater. Interfaces 2017, 9, 14401–14408. [Google Scholar] [CrossRef]
- Smith, S.J.D.; Lau, C.H.; Mardel, J.I.; Kitchin, M.; Konstas, K.; Ladewig, B.P.; Hill, M.R. Physical aging in glassy mixed matrix membranes; tuning particle interaction for mechanically robust nanocomposite films. J. Mater. Chem. A 2016, 4, 10627–10634. [Google Scholar] [CrossRef]
- Kim, S.; Hou, J.; Wang, Y.; Ou, R.; Simon, G.P.; Seong, J.G.; Lee, Y.M.; Wang, H. Highly permeable thermally rearranged polymer composite membranes with a graphene oxide scaffold for gas separation. J. Mater. Chem. A 2018, 6, 7668–7674. [Google Scholar] [CrossRef]
- Aguilar-Lugo, C.; Lee, W.H.; Miguel, J.A.; de la Campa, J.G.; Prádanos, P.; Bae, J.Y.; Lee, Y.M.; Álvarez, C.; Lozano, Á.E. Highly Permeable Mixed Matrix Membranes of Thermally Rearranged Polymers and Porous Polymer Networks for Gas Separations. ACS Appl. Polym. Mater. 2021, 3, 5224–5235. [Google Scholar] [CrossRef]
- Hou, R.; Ghanem, B.S.; Smith, S.J.D.; Doherty, C.M.; Setter, C.; Wang, H.; Pinnau, I.; Hill, M.R. Highly permeable and selective mixed-matrix membranes for hydrogen separation containing PAF-1. J. Mater. Chem. A 2020, 8, 14713–14720. [Google Scholar] [CrossRef]
- Lopez-Iglesias, B.; Suárez-García, F.; Aguilar-Lugo, C.; González Ortega, A.; Bartolomé, C.; Martínez-Ilarduya, J.M.; de la Campa, J.G.; Lozano, Á.E.; Álvarez, C. Microporous Polymer Networks for Carbon Capture Applications. ACS Appl. Mater. Interfaces 2018, 10, 26195–26205. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Torres-Cuevas, E.S.; Palacio, L.; Prádanos, P.; Freeman, B.D.; Lozano, Á.E.; Hernández, A.; Comesaña-Gándara, B. Gas Permeability, Fractional Free Volume and Molecular Kinetic Diameters: The Effect of Thermal Rearrangement on ortho-hydroxy Polyamide Membranes Loaded with a Porous Polymer Network. Membranes 2022, 12, 200. [Google Scholar] [CrossRef] [PubMed]
- Klotz, E.J.F.; Claridge, T.D.W.; Anderson, H.L. Homo- and Hetero-[3]Rotaxanes with Two π-Systems Clasped in a Single Macrocycle. J. Am. Chem. Soc. 2006, 128, 15374–15375. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.P.; Czenkusch, K.; Wi, S.; Gleason, K.L.; Hernández, G.; Doherty, C.M.; Konstas, K.; Bastow, T.J.; Álvarez, C.; Hill, A.J.; et al. Investigation of the chemical and morphological structure of thermally rearranged polymers. Polymer 2014, 55, 6649–6657. [Google Scholar] [CrossRef] [Green Version]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.C.; Hsieh, K.H. Synthesis and characterization of bismaleimides derived from polyurethanes. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 1665–1672. [Google Scholar] [CrossRef]
- Soto, C.; Aguilar Lugo, C.; Rodríguez, S.; Palacio, L.; Lozano, Á.E.; Prádanos, P.; Hernandez, A. Enhancement of CO2/CH4 permselectivity via thermal rearrangement of mixed matrix membranes made from an o-hydroxy polyamide with an optimal load of a porous polymer network. Sep. Purif. Technol. 2020, 247, 116895. [Google Scholar] [CrossRef]
- Smith, Z.P.; Sanders, D.F.; Ribeiro, C.P.; Guo, R.; Freeman, B.D.; Paul, D.R.; McGrath, J.E.; Swinnea, S. Gas sorption and characterization of thermally rearranged polyimides based on 3,3′-dihydroxy-4,4′-diamino-biphenyl (HAB) and 2,2′-bis-(3,4-dicarboxyphenyl) hexafluoropropane dianhydride (6FDA). J. Membr. Sci. 2012, 415–416, 558–567. [Google Scholar] [CrossRef]
- Muñoz, D.M.; de la Campa, J.G.; de Abajo, J.; Lozano, A.E. Experimental and Theoretical Study of an Improved Activated Polycondensation Method for Aromatic Polyimides. Macromolecules 2007, 40, 8225–8232. [Google Scholar] [CrossRef]
- Soto, C.; Torres-Cuevas, E.S.; González-Ortega, A.; Palacio, L.; Lozano, Á.E.; Freeman, B.D.; Prádanos, P.; Hernández, A. Gas Separation by Mixed Matrix Membranes with Porous Organic Polymer Inclusions within o-Hydroxypolyamides Containing m-Terphenyl Moieties. Polymers 2021, 13, 931. [Google Scholar] [CrossRef]
- Han, S.H.; Kwon, H.J.; Kim, K.Y.; Seong, J.G.; Park, C.H.; Kim, S.; Doherty, C.M.; Thornton, A.W.; Hill, A.J.; Lozano, Á.E.; et al. Tuning microcavities in thermally rearranged polymer membranes for CO2 capture. Phys. Chem. Chem. Phys. 2012, 14, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.P.; Hernández, G.; Gleason, K.L.; Anand, A.; Doherty, C.M.; Konstas, K.; Alvarez, C.; Hill, A.J.; Lozano, A.E.; Paul, D.R.; et al. Effect of polymer structure on gas transport properties of selected aromatic polyimides, polyamides and TR polymers. J. Membr. Sci. 2015, 493, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Díez, B.; Cuadrado, P.; Marcos-Fernández, Á.; de la Campa, J.G.; Tena, A.; Prádanos, P.; Palacio, L.; Lee, Y.M.; Alvarez, C.; Lozano, Á.E.; et al. Thermally rearranged polybenzoxazoles made from poly(ortho-hydroxyamide)s. Characterization and evaluation as gas separation membranes. React. Funct. Polym. 2018, 127, 38–47. [Google Scholar] [CrossRef]
- Wang, H.; Chung, T.-S.; Paul, D.R. Thickness dependent thermal rearrangement of an ortho-functional polyimide. J. Membr. Sci. 2014, 450, 308–312. [Google Scholar] [CrossRef]
- Lin, H.; Freeman, B.D. Permeation and diffusion. In Springer Handbook of Materials Measurement Methods; Czichos, H., Saito, T., Smith, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 371–387. [Google Scholar] [CrossRef]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry and Use; John Wiley & Sons: New York, NY, USA, 1974. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mw (g/mol) | Mn (g/mol) | Mw/Mn |
|---|---|---|---|
| tBTmCl-APAF | 104 828 | 58 936 | 1.8 |
| Membrane | Before TR Process | After TR Process |
|---|---|---|
| tBTmCl-APAF | 9.93 | 6.29 |
| tBTmCl-APAF + 20% PPN | 6.02 | 6.24 |
| tBTmCl-APAF + 30% PPN | 7.11 | 6.10 |
| tBTmCl-APAF + 40% PPN | 10.92 | 6.23 |
| Membrane | Maximum Stress (MPa) | Elongation at Break (%) | Young’s Modulus (GPa) |
|---|---|---|---|
| tBTmCl-APAF | 25.7 ± 10.2 | 9.90 ± 1.25 | 1.7 ± 0.6 |
| tBTmCl-APAF + 20% PPN | 36.8 ± 6.4 | 2.02 ± 0.39 | 2.2 ± 0.2 |
| tBTmCl-APAF + 30% PPN | 31.9 ± 10.2 | 1.38 ± 0.39 | 1.7 ± 0.3 |
| tBTmCl-APAF + 30% PPN TR | 30.8 ± 7.5 | 1.96 ± 0.47 | 1.9 ± 0.1 |
| PPN Loading | lnA + aFFV | bFFV | cFFV | |
|---|---|---|---|---|
| Before TR | 0% PPN (r = 0.9886) | 6.5 ± 1.5 | 6.5 ± 1.5 | −1.3 ± 0.3 |
| 20% PPN (r = 0.9760) | 10.6 ± 2.8 | 10.6 ± 2.8 | −2.0 ± 0.4 | |
| 30% PPN (r = 0.9760) | 10.9 ± 4.1 | 10.9 ± 4.1 | −2.0 ± 0.4 | |
| 40% PPN (r = 0.9760) | 7.6 ± 1.8 | 7.6 ± 1.8 | −1.4 ± 0.5 | |
| After TR | 20% PPN (r = 0.9557) | −8.8 ± 2.7 | 10.3 ± 1.9 | −2.0 ± 0.5 |
| 30% PPN (r = 0.9251) | −10.5 ± 2.7 | 9.1 ± 2.0 | −1.7 ± 0.3 | |
| 40% PPN (r = 0.9251) | −10.1 ± 3.4 | 8.9 ± 1.5 | −1.6 ± 0.4 |
| PPN Loading | FFV from bFFV | FFV from cFFV | |
|---|---|---|---|
| Before TR | 20% PPN (r = 0.9760) | 1.63 | 1.60 |
| 30% PPN (r = 0.9760) | 1.68 | 1.60 | |
| 40% PPN (r = 0.9760) | 1.17 | 1.08 | |
| After TF | 20% PPN (r = 0.9251) | 1.59 | 1.60 |
| 30% PPN (r = 0.9251) | 1.40 | 1.31 | |
| 40% PPN (r = 0.9251) | 1.37 | 1.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto, C.; Carmona, J.; Freeman, B.D.; Palacio, L.; González-Ortega, A.; Prádanos, P.; Lozano, Á.E.; Hernandez, A. Free Volume and Permeability of Mixed Matrix Membranes Made from a Terbutil-M-terphenyl Polyamide and a Porous Polymer Network. Polymers 2022, 14, 3176. https://doi.org/10.3390/polym14153176
Soto C, Carmona J, Freeman BD, Palacio L, González-Ortega A, Prádanos P, Lozano ÁE, Hernandez A. Free Volume and Permeability of Mixed Matrix Membranes Made from a Terbutil-M-terphenyl Polyamide and a Porous Polymer Network. Polymers. 2022; 14(15):3176. https://doi.org/10.3390/polym14153176
Chicago/Turabian StyleSoto, Cenit, Javier Carmona, Benny D. Freeman, Laura Palacio, Alfonso González-Ortega, Pedro Prádanos, Ángel E. Lozano, and Antonio Hernandez. 2022. "Free Volume and Permeability of Mixed Matrix Membranes Made from a Terbutil-M-terphenyl Polyamide and a Porous Polymer Network" Polymers 14, no. 15: 3176. https://doi.org/10.3390/polym14153176
APA StyleSoto, C., Carmona, J., Freeman, B. D., Palacio, L., González-Ortega, A., Prádanos, P., Lozano, Á. E., & Hernandez, A. (2022). Free Volume and Permeability of Mixed Matrix Membranes Made from a Terbutil-M-terphenyl Polyamide and a Porous Polymer Network. Polymers, 14(15), 3176. https://doi.org/10.3390/polym14153176