1. Introduction
Poly(ε-caprolactone) (PCL) is a biocompatible and biodegradable industrial polymer with production output amounting to tens of thousands of tons every year. The melting temperature of PCL is low (60 °C), and crystallization proceeds relatively rapidly (at its minimum, the half-time of crystallization is in the order of 0.1 s). The range of current and possible applications of PCL includes rapid prototyping, biomedical products such as sutures and dental splints, controllable drug-release systems, and others [
1,
2,
3]. The properties of PCL can be further tuned to better suit the relevant application by modifying its chemical structure. One option in this direction is creating cross-links between polymer chains of PCL with a suitable cross-linking agent. Such cross-linked polymers based on PCL were shown to have greater mechanical strength and possess a two-way shape memory effect which is not exhibited by the PCL itself [
4,
5,
6,
7].
Crosslinking influences the crystallization behavior of the polymer. Under non-isothermal conditions, the crystallization temperature of PCL changes due to cross-linking [
8,
9]. Optimizing the processing of such modified polymers requires knowledge of their crystallization behavior under different conditions. Notably, the high cooling rates realizable in modern production techniques emphasize the need for assessing the nucleation and crystal growth rate in a wide temperature range, including the deeply supercooled state. At deep supercooling homogeneous crystal nucleation becomes the dominant nucleation process, following the predictions of classical nucleation theory (CNT) [
10].
The development of fast scanning calorimetry (FSC) [
11] has opened the doors to investigate the nucleation and crystallization process of fast crystallizing polymers at low temperatures and deep supercooling conditions [
12,
13,
14,
15]. These investigations have provided experimental data on homogeneous nucleation kinetics in polymers and demonstrated that CNT is applicable for describing homogeneous crystal nucleation in polymers. Therefore, combining FSC and CNT should better allow understanding homogeneous nucleation in cross-linked PCL as well. It may be possible to clarify if cross-links are incorporated in the crystal lattice or how cross-links influence the nucleation kinetics. In the present paper, we will use standard approaches of CNT in treating crystallization to perform a theoretical analysis of the obtained experimental data.
However, employing the basic ideas and methods of CNT in polymer crystallization, some specific features must be appropriately accounted for in the theoretical treatment. In particular, non-crystallizable units in the polymer molecule can prevent chains from correct packing. For example, introducing D-isomer units into poly(
l-lactic acid) (PLLA) chains reduces polymer crystallinity and slows down both nucleation and crystallization [
16]. Crosslinking may act as another sterical hindrance for the chain packing. The spatial density of cross-links introduces another length scale that can affect nucleation and crystallization processes. Comparing nucleation and crystallization kinetics of differently cross-linked polymers to a non-cross-linked sample, a more detailed and correct understanding of structure formation in polymer melts can be, consequently, achieved.
Furthermore, cross-linking should restrict molecular mobility to a greater extent than the inclusion of non-crystallizing units in linear polymer chains. The viscosity of the polymer increases with increasing cross-linking [
17]. The cross-linking of PCL has a strong effect on the rheological properties of the polymer, and already at a relatively low cross-link density, the behavior of the polymer changes from viscous to elastic [
8]. These variations affect crystal nucleation and growth via variations of the kinetic coefficients in the expressions for the nucleation and growth rates. On the other hand, they may also result in a more pronounced effect of elastic stresses in the work of critical cluster formation, and the driving force for crystal growth resulting in an additional impact on structure formation processes in the polymers analyzed [
18], in particular, a decrease in both nucleation and growth rates. Further, at temperatures below the glass transition range, the polymer may not reach the metastable equilibrium state in cooling prior to crystal nucleation. This factor may lead to an additional decrease in the nucleation rate in cooling and support nucleation in heating [
19]. In parallel with the change in viscosity, an increase in the glass transition temperature and a slowing down of relaxation processes are expected. It is worthwhile to note that, both glass transition temperature and enthalpy relaxation kinetics of PLLA are not affected by D-isomer’s introduction [
20], while the nucleation kinetics and crystal growth show a strong dependency, thus providing a good comparison.
Recently, we have reported [
9] the effect of cross-linking density in poly(ε-caprolactone) (PCL) on the non-isothermal crystallization rate of this polymer. The samples of PCL at different degrees of cross-linking were melted and then cooled at different cooling rates; the crystallinity of the sample after this treatment was determined by measuring the fusion enthalpy at subsequent heating. It was found that cross-linking slows down crystallization. The cooling rate at which half of the maximum possible crystallinity of the sample is reached inversely correlates with the cross-link density. Cross-linking modifies the chemical structure of the polymer but keeps most of the monomeric units unchanged. With this, it is an interesting model system to study the influence of the cross-link induced changes in viscosity [
21] on crystal nucleation and growth kinetics.
In the present work, we extend the previous study [
9] by analyzing crystallization at isothermal conditions down to and below the glass transition temperature. Particularly, we investigate the effect of cross-link density on crystal nucleation and crystallization rates of PCL, as well as the glass transition temperature and relaxation kinetics. In advancing the theoretical analysis, we will analyze which factors employing and going beyond standard CNT are the basic ones determining the effect of cross-linking on crystallization in the polymer analyzed.
2. Materials and Methods
Samples of cross-linked PCL were prepared by heating commercial PCL (Aldrich, St. Louis, MO, USA, average
Mn = 45,000 g·mol
−1, density ρ = 1.142 g·cm
−3) with different amounts of radical initiator benzoyl peroxide (BPO) (Aldrich, St. Louis, MO, USA, 75% water stabilized), as described previously [
4]. The spatial density of cross-links for each sample was determined using an equilibrium swelling method [
9],
Table 1.
Fast scanning calorimetry experiments were performed using a Flash DSC 1 (Mettler Toledo, Greifensee, Switzerland) with a UFS1 sensor (Mettler Toledo, Greifensee, Switzerland). In every experiment, 10–50 ng of the specimen was placed on the chip sensor, heated up to 150 °C, and cooled down to melt the sample and achieve better thermal contact with the chip sensor before performing measurements.
Tammann’s two-stage nucleation and growth approach, with a non-isothermal growth stage [
12,
22,
23], was employed to investigate the kinetics of crystal nucleation and crystallization of neat and cross-linked PCL. The temperature program includes melting with consecutive fast cooling to an annealing temperature, allowing crystal nucleation and, in some cases, also crystallization. The chosen cooling rate prevents the formation of crystal nuclei before reaching the annealing temperature. The duration and the temperature of the annealing step are varied. After annealing, the sample is cooled at 5000 K/s to −80 °C and then heated to 90 °C (analysis scan). See
Figure 1 for details of the temperature program.
Depending on the annealing conditions, the thermal effects of enthalpy relaxation at the glass transition, cold crystallization (the growth stage in Tammann’s scheme), and melting occur. Because of the decreasing crystallization rates with increasing cross-link density [
9], for each sample, a specific heating rate of the analysis scan was chosen. The selected heating rates were so fast that no, or only minor, crystallization occurs for samples not containing homogeneous nuclei. At the same time, the heating rate must be slow enough that for samples containing significant amounts of homogeneous nuclei, crystallization is easily detectable. This way, for all samples containing homogeneously formed crystal nuclei, cold crystallization was observed, independent of the cross-link density dependent crystallization rate.
The optimal heating rate for the analysis scans for the cross-linked PCL samples must be chosen depending on the crystallization rate of the particular polymer. Too slow heating will promote crystallization of the polymer during the analysis scan originating from a few heterogeneous nuclei. At the same time, too fast heating causes a broadening of the cold crystallization peak or even disappearance of cold crystallization and makes an independent evaluation of cold-crystallization and melting enthalpies impossible. As shown previously [
9], the crystallization rate of the cross-linked PCL slows down with an increasing degree of cross-linking. Thus, the optimal heating rate depends on the cross-link density. The chosen heating rates were 5000 K/s for pure PCL, 3000 K/s for cross-linked PCL with 3% BPO, 1000 K/s for cross-linked PCL with 5% BPO, and 500 K/s for cross-linked PCL with 10% BPO.
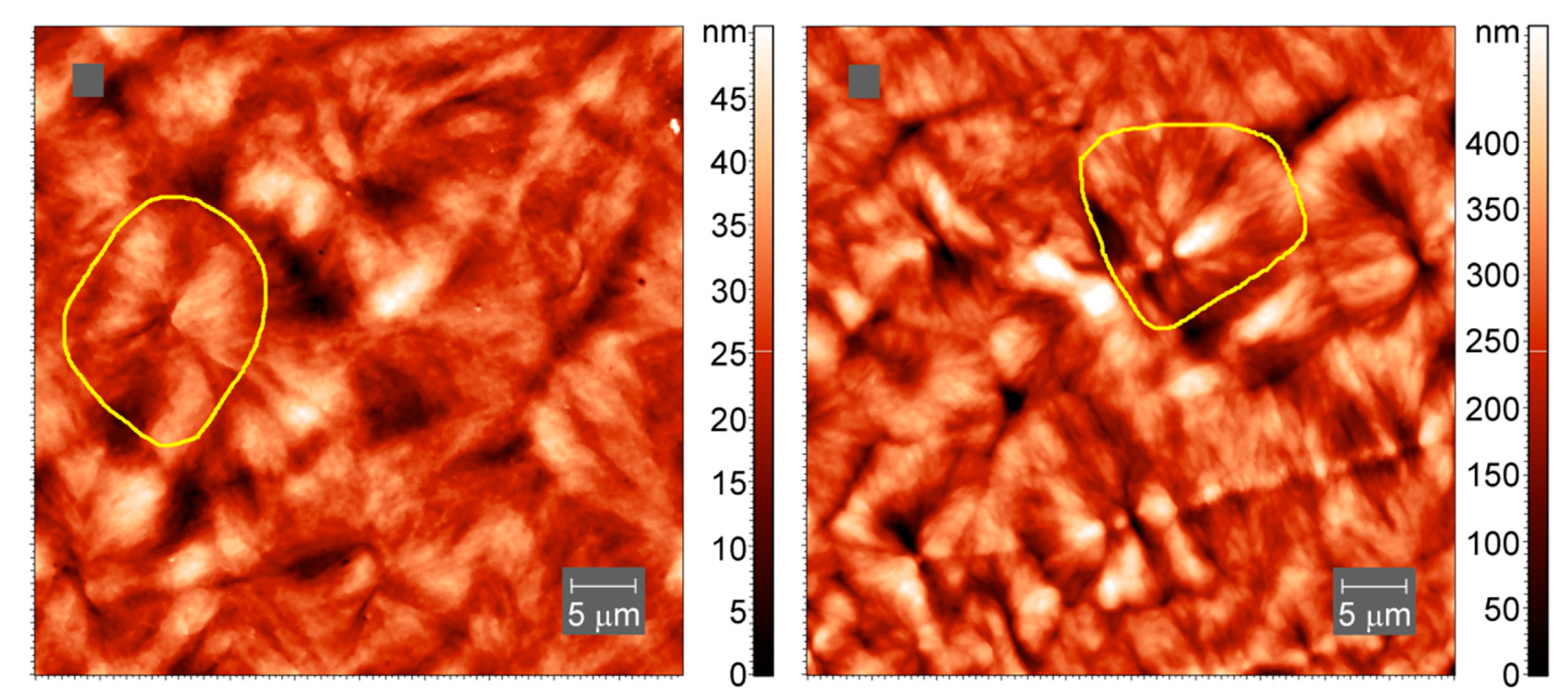
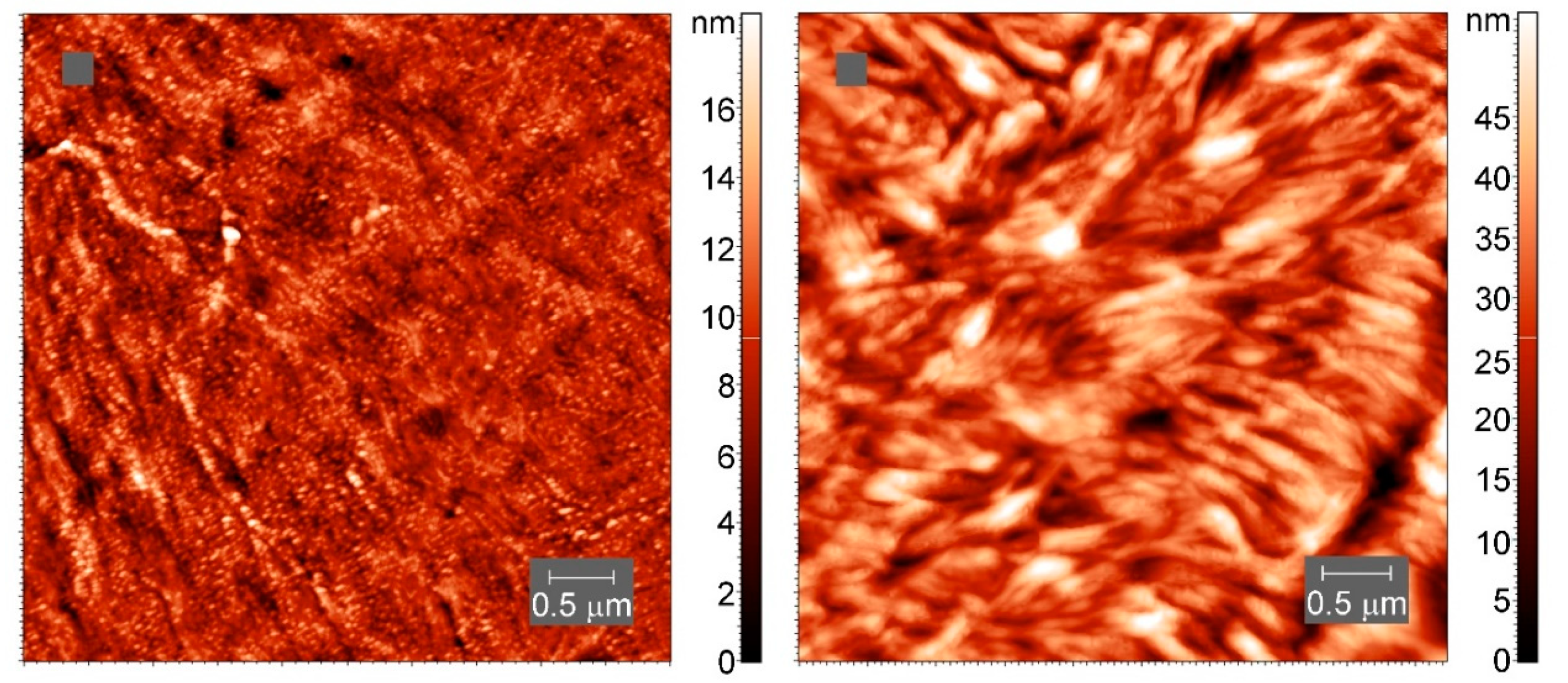
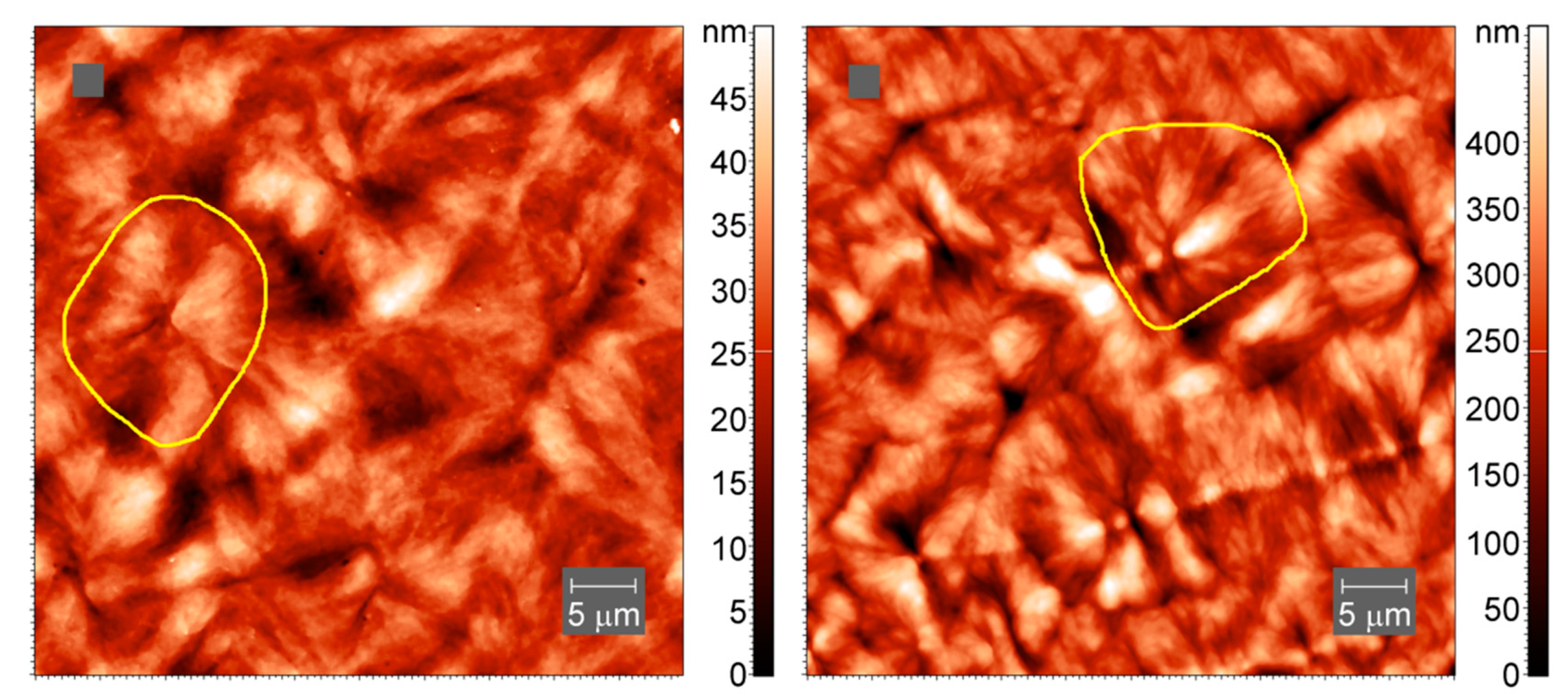
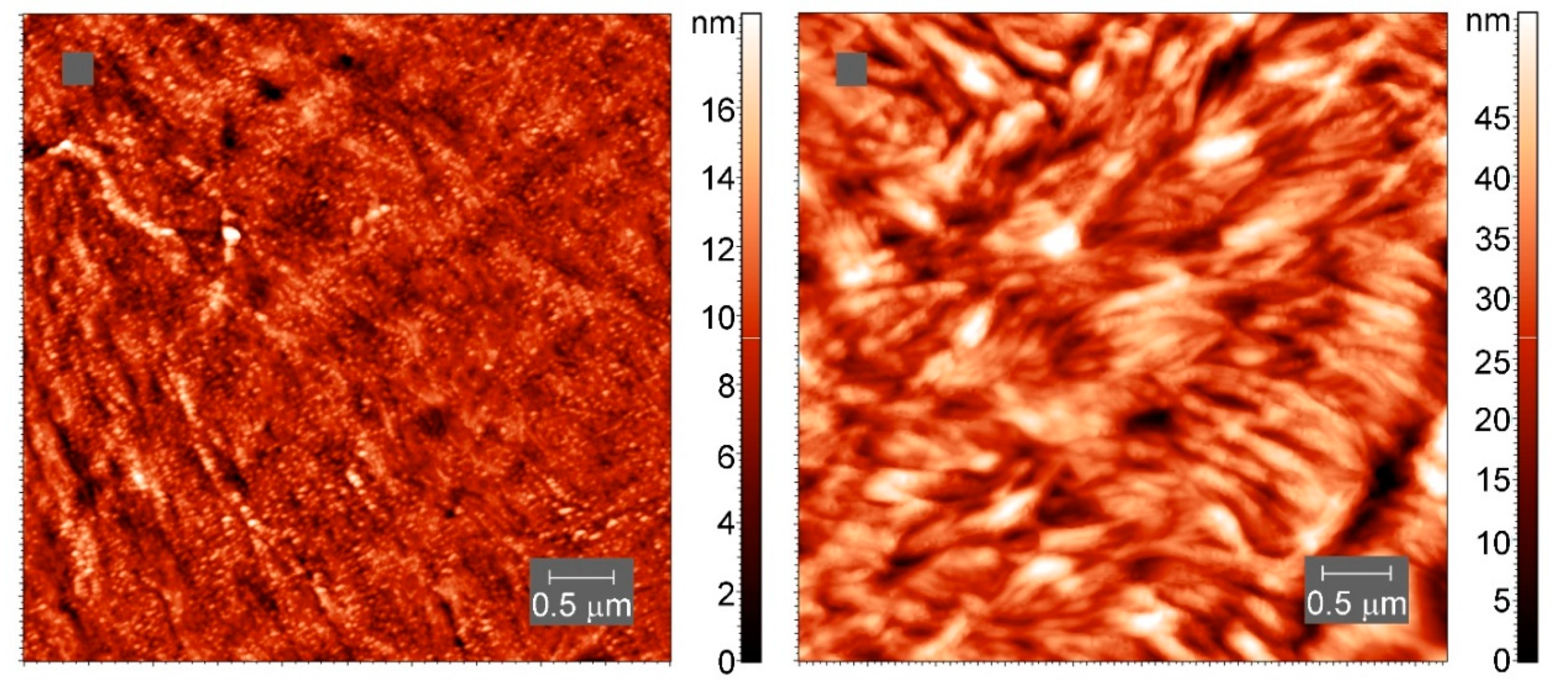
Atomic force microscopy (AFM) images of the samples after crystallization at slow cooling were collected in tapping mode in air using an atomic force microscope Titanium (LLC “NT-MDT”, Moscow, Russia). The instrument is equipped with a multiprobe revolution cartridge of CNG cantilevers (LLC “NT-MDT”, Moscow, Russia) with force constants ranging from 3 to 10 N m−1 and resonance frequencies from 120 to 150 kHz. The scan frequency was 1 Hz. The crystalline samples were prepared by melting the polymers between glass slides and allowing the melt to slowly cool in air, after which the samples were cut with a microtome. All images were obtained at 25 °C.
3. Results of the Experiments
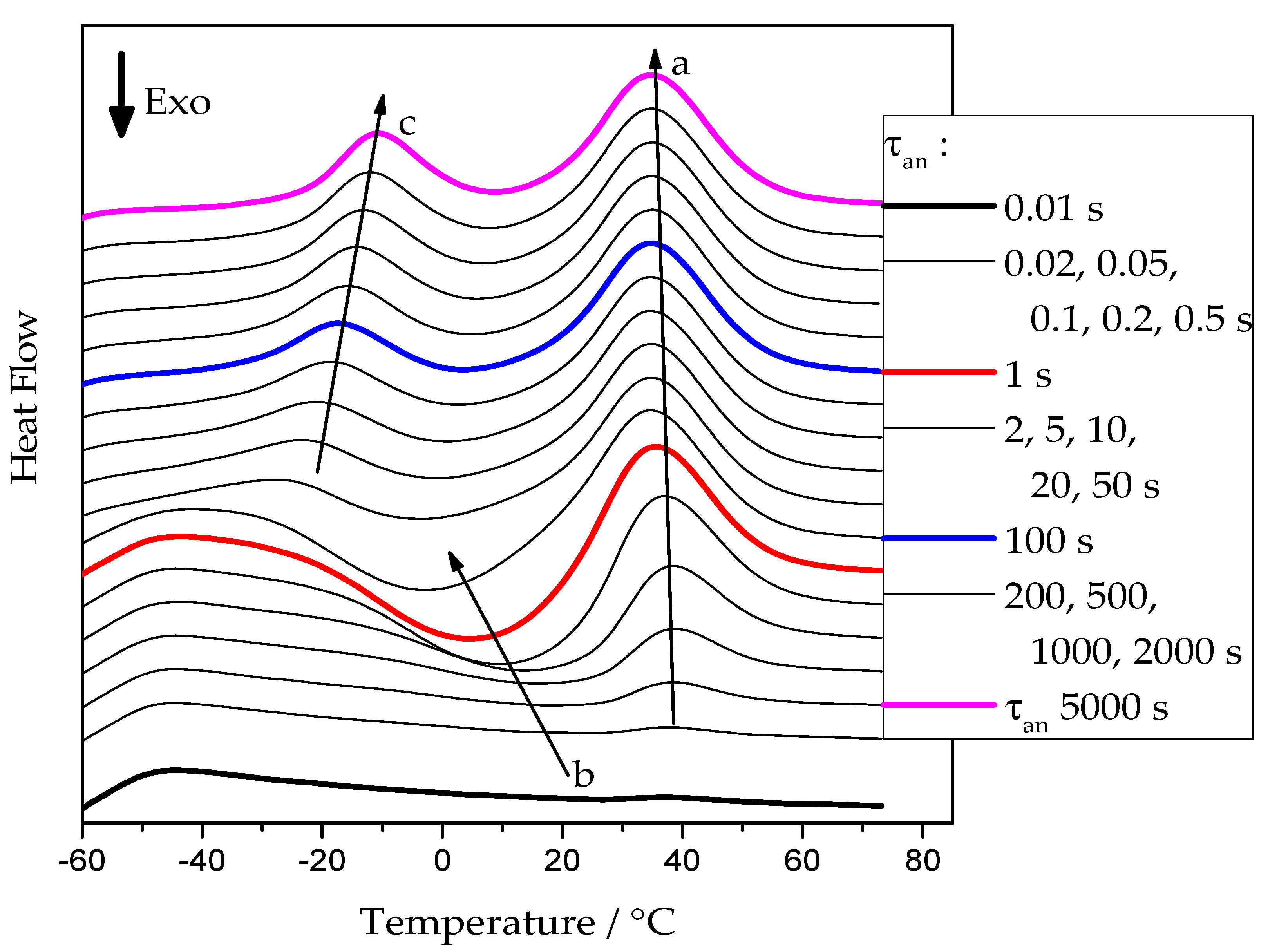
Figure 2 presents examples of heating scans recorded for the neat PCL sample after annealing at −50 °C. Similar curves were obtained for annealing temperatures from −65 °C up to 20 °C for all cross-linked PCL samples.
At short annealing times, no significant thermal effects are present. With increasing annealing time, a cold-crystallization peak develops, arrow
b, indicating the presence of an increasing number of crystal nuclei that can grow to crystals in the temperature range of sufficiently high growth rate [
12]. At higher temperatures, the corresponding melting endotherm appears. The position of this endotherm, see arrow
a, is essentially independent of the annealing conditions. It is determined by the time available for a melting-recrystallization process on heating [
24]. For annealing times above 5 s, a low-temperature endothermic peak develops. It corresponds to the melting of small crystals already grown at the annealing temperature. After this initial melting, the PCL re-crystallizes and re-melts continuously until the final melting occurs, arrow
a.
The values of the cold crystallization enthalpy and overall latent heat (sum of cold-crystallization and all melting enthalpies) were determined as illustrated in
Figure 3. Cold crystallization enthalpy corresponds to the area of peak 1, integrated with a linear baseline between points
a and
b. The overall latent heat is the integral, employing a linear baseline between points
a and
c.
It can be seen from
Figure 2 that, with a longer annealing time, an endothermal peak appears between −30 and 0 °C, indicated by the arrow
c. This peak corresponds to the melting of tiny polymer crystals which have grown during the extended annealing. However, the peak may not represent the entire melting endotherm, as the developed melt immediately re-crystallizes to form more stable crystals. The latter effect is exothermal and, thus, partially compensates for the low-temperature melting effect. The re-crystallized crystals finally melt during the high-temperature endotherm. This is illustrated in
Figure 3b; the red curve represents the low-temperature melting of the tiny polymer crystals, the blue curve represents the exotherm of recrystallization or cold-crystallization, and the magenta curve corresponds to the melting endotherm of the re-crystallized polymer. The black curve corresponds to the overall effect, which is the sum of the heat flows of the individual processes. The shaded area corresponds to the overall latent heat, which is not affected by the melting-recrystallization process since only the sum of the different heat flows is measured.
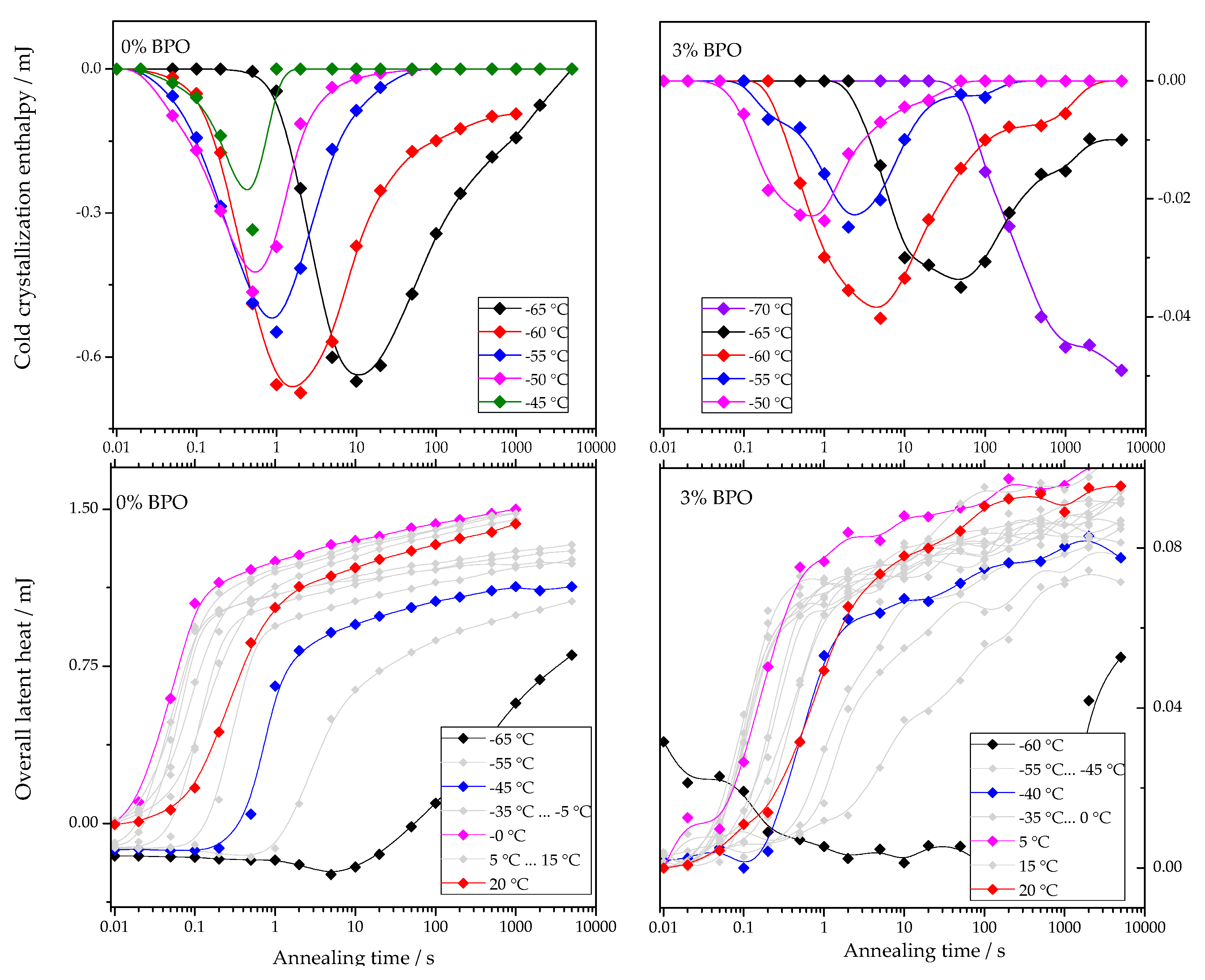
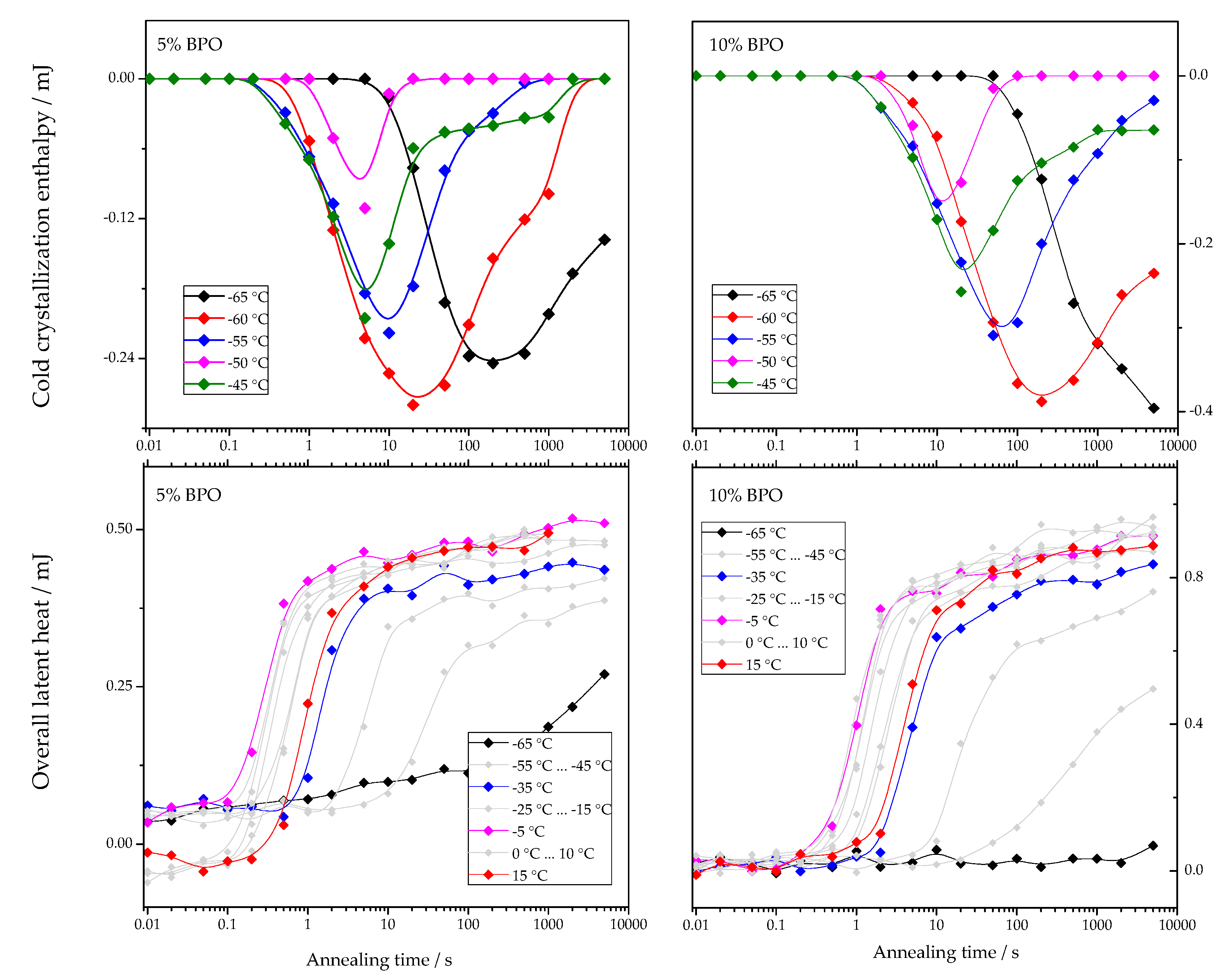
The dependencies of cold-crystallization enthalpy and overall latent heat on annealing time for different annealing temperatures are shown for all studied PCL samples in
Figure 4.
As shown in
Figure 4, the cold-crystallization enthalpy values have an extremum, while the overall latent heat follows a sigmoidal curve. The initial growth of the cold-crystallization enthalpy is a result of the development of crystal nuclei during annealing. At the same time, the values of the overall latent heat remain close to zero until cold-crystallization reaches the maximum since the formation, and, consequently, the “disordering” of the nuclei does not yield any measurable heat effect.
Further annealing leads to crystal growth during annealing even at the lowest annealing temperatures. Thus, the increasing volume occupied by these crystals does not contribute to the cold-crystallization on heating anymore, and the absolute value of the cold-crystallization enthalpy decreases. At the same time, the overall latent heat starts to deviate from zero and grows continuously. Parameterizing the time dependencies of the cold-crystallization enthalpy and the overall latent heat allows comparing nucleation and crystallization rates between the samples with different cross-link densities. Therefore, the time dependencies of the overall latent heat (
) were fitted with Equation (1), following the procedure described in [
12]:
The first term of this equation corresponds to the standard Johnson-Mehl-Avrami-Kolmogorov (JMAK) description of overall crystallization. Here,
is the enthalpy of melting at the final stage of primary crystallization,
is the halftime of crystallization, and
is the Avrami exponent of crystallization. The second term describes secondary crystallization, and its treatment is based on the assumption of a linear increase in melting enthalpy with the logarithm of time.
Figure 5 shows an example of the fit of the overall latent heat of the pure PCL sample after annealing at –45 and –60 °C with Equation (1). The corresponding fitting parameters are: at –45 °C
= (3.1 ± 0.1) × 10
−4 J,
= 5.3 ± 0.3 s,
= 2.5 ± 0.4,
A2 = (1.0 ± 0.2) × 10
−5 J; at –60 °C
= (4.4 ± 0.2) × 10
−4 J,
= 119 ± 13 s,
= 1.0 ± 0.1,
A2 = 0 J.
The parametrization of the cold crystallization time-dependencies was carried out as follows. For each cross-link density of PCL, a set of cold-crystallization curves was obtained. Among each set, the curve with the highest absolute cold-crystallization enthalpy value was chosen. The data points after the extremum of the cold-crystallization curve were discarded, and the remaining data points were fitted with the JMAK-equation:
where
is the maximum of the cold crystallization enthalpy of the considered polymer,
is the halftime of nucleation,
is the Avrami exponent for the nucleation process. For the rest of the curves
and
were kept fixed, and only nucleation halftime was allowed to fit. Again, the points after the extremum of the curve were discarded since they are strongly disturbed by the growth of crystals.
An example of the fit for the time dependence of the cold crystallization enthalpy of PCL with 5% BPO after annealing at −45 and −60 °C with Equation (2) is also shown in
Figure 5. The corresponding fitting parameters are: at −45°C
= −(2.4 ± 0.1)·10
−4 J,
= 5.2 ± 0.3 s,
nn = 1.5 ± 0.1; at −60 °C
= −(2.4 ± 0.1)·10
−4 J,
= 2.8 ± 0.1 s,
nn = 1.5 ± 0.1.
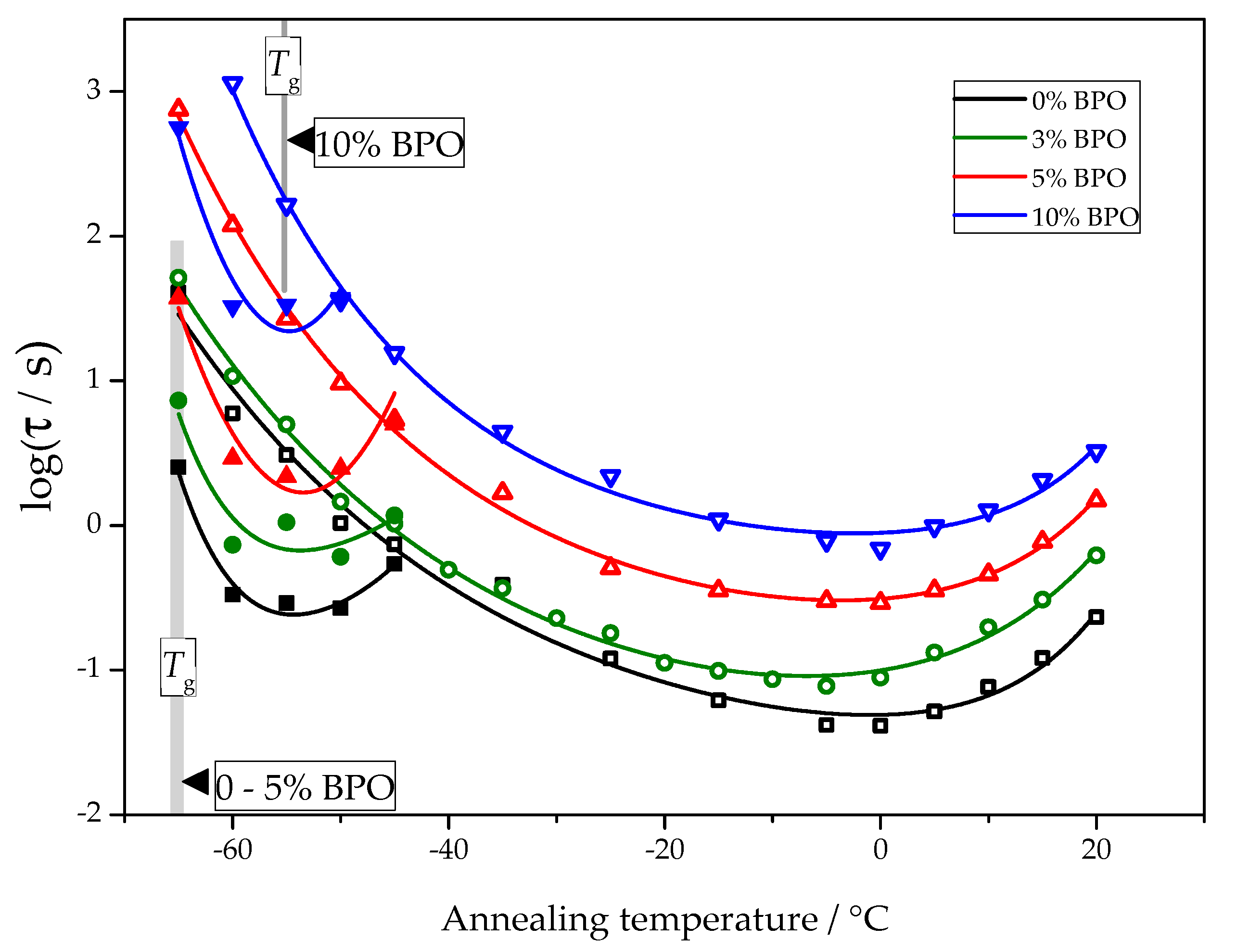
The temperature dependence of nucleation and crystallization halftimes determined using the procedure described above is presented in
Figure 6.
For all samples, the nucleation and crystallization halftime minima appear at about −55 °C and 0 °C, respectively. These minima are equivalent to the maxima of homogeneous nucleation and overall crystallization rates. The independence of the position of the maxima of homogeneous nucleation rates on cross-link density follows directly from the experimental data. The corresponding half-times of cold crystallization are not disturbed by other effects. The position of the maximum of the overall crystallization rate at about 0 °C depends on the interplay between heterogeneous nucleation kinetics and the temperature dependence of the crystal growth rate. With the data shown in
Figure 6, it is impossible to decide if the heterogeneous nucleation rate or growth rate causes the stable position of the minimum crystallization halftime. There are even arguments that the temperature position of the maximum of the overall crystallization rate may be close to the maximum of the crystal growth rate [
25].
Disentangling the influence of heterogenous nucleation kinetics and growth rate requires some general considerations. Starting from the Johnson–Mehl–Avrami–Kolmogorov (JMAK) approach [
18,
26], the temperature of the maximum growth rate becomes accessible [
25]. On heating an amorphous but nuclei containing sample, cold crystallization is observed, see
Figure 2. The cold crystallization peak maximum shifts to higher temperatures with increasing heating rate before the peak eventually vanishes. From the theoretical analysis, it follows that the position of the peak before it vanishes approaches the temperature of the maximum crystal growth rate. The heating rate at which the cold crystallization temperature reaches a plateau depends on the nucleation state of the sample.
Figure 7 shows the heating rate dependence of the cold crystallization peak temperature for three different samples of the cross-linked PCL with 5% BPO. The blue curve was obtained when the sample was heated quickly (1 s isotherm at −80 °C) after rapid cooling at 5000 K/s to −80 °C. Red and black curves were obtained from two distinct samples (with different mass and geometry) after annealing (nucleating) the samples at −60 °C for 20 s (1000 K/s cooling rate). As can be seen from
Figure 7, depending on the sample history, distinct dependencies of the cold crystallization peak temperature on the heating rate are obtained, but the asymptotic temperatures of all curves are nearly the same. The heating rate when the non-nucleated sample reaches the asymptotic cold crystallization peak temperature is much lower than that for the nucleated samples. It indicates that the critical heating rate for preventing nucleation is much lower than the critical heating rate for preventing crystal growth from preexisiting nuclei. In both cases the temperature of maximum growth rate provides the limit for observable crystal growth.
The data points from
Figure 7 are fitted by
where
is the cold crystallization peak temperature at heating rate β,
is the asymptotic value of the cold crystallization peak temperature,
is the cold crystallization peak temperature at a heating rate approaching zero,
A is an empirical constant.
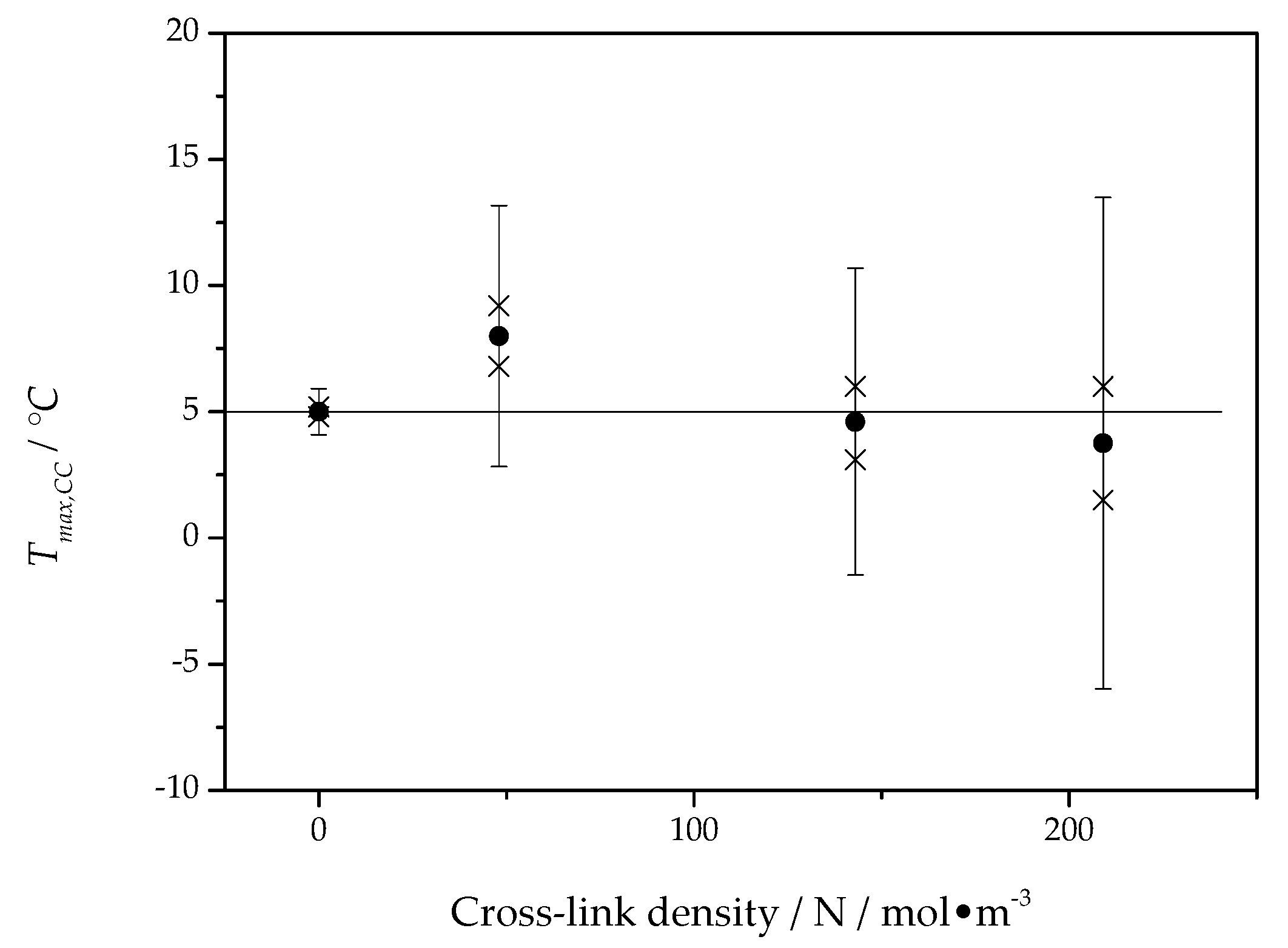
The asymptotic value,
, of the fit function represents a temperature close to the maximum of the crystal growth rate and is presented for all PCL samples as a function of the cross-link density in
Figure 8.
The asymptotic value of the cold crystallization peak temperature is located, independent of cross-link density, at about 5 °C, which is in good agreement with the maximum position of the overall crystallization rate in
Figure 6. This way, the temperature of the maximum of the overall crystallization rate can be interpreted as being caused by the maximum of the crystal growth rate and only marginally influenced by heterogeneous nucleation kinetics. Interestingly, the maximum of the overall crystallization rate in
Figure 6 and the temperature of maximum growth rate from
Figure 8 are independent of the cross-link density.
The classical nucleation theory describes both nucleation and crystal growth rate as dependent on diffusivity [
25]. The diffusion coefficient is commonly linked to viscosity and segmental mobility in polymers [
18]. Therefore, the glass transition temperature of the different PCL samples was determined. Although the heating scans performed during the annealing experiments did show glass transition, the temperature programs had to be optimized to provide a clear comparison between the different PCL samples in this respect. Thus, a separate set of experiments was performed to check the effect of cross-links on the glass transition of PCL. The glass transition temperature at heating rate 1000 K/s is almost the same in neat PCL, cross-linked PCL with 3% and 5% BPO (glass transition midpoints −65.6, −65, and −64.6 °C, respectively), but is notably higher for cross-linked PCL with 10% BPO (glass transition midpoint −54 °C).
Summarizing briefly the experimental data, we come to the following conclusions: With increasing degree of cross-linking, both the nucleation and crystallization half-times increase. In parallel, as shown in
Figure 9, the glass transition range shifts to higher temperatures. In contrast, the temperatures of the maximum nucleation and the overall crystallization rates remain the same independent of the degree of cross-linking. The cold crystallization peak temperature increases generally as a function of heating rate reaching an asymptotic value near to the temperature of the maximum growth rate. A theoretical interpretation of these results is given in terms of classical nucleation theory in the subsequent section. In addition, it is shown there that the average distance between the nearest cross-links is smaller than the estimated lamellae thickness, which indicates the inclusion of cross-links in the crystalline phase of the polymer.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}