
Liquid-Crystal Ordering and Microphase Separation in the Lamellar Phase of Rod-Coil-Rod Triblock Copolymers. Molecular Theory and Computer Simulations


 , ,
, , 
Abstract
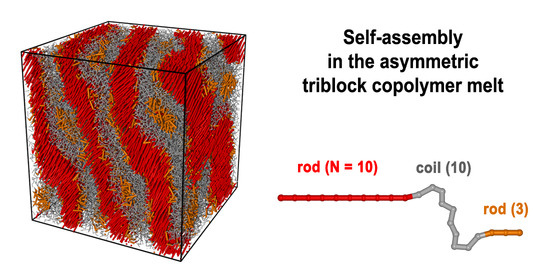
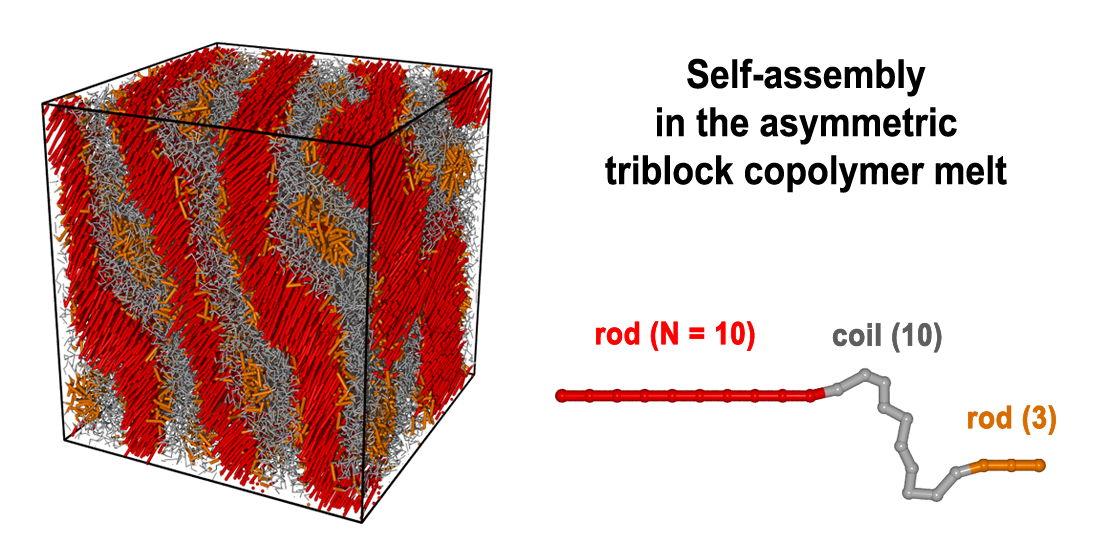
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular-Statistical Theory of Triblock Copolymers
2.1. General Density Functional Theory
2.2. Free Energy of the Lamellar Phase
3. Correlation Functions in Rod-Coil-Rod Triblock Copolymers
3.1. Ornstein-Zernike Equations
3.2. Density-Density Correlation Functions of Rod-Coil-Rod Triblock Copolymers
4. Results and Discussion
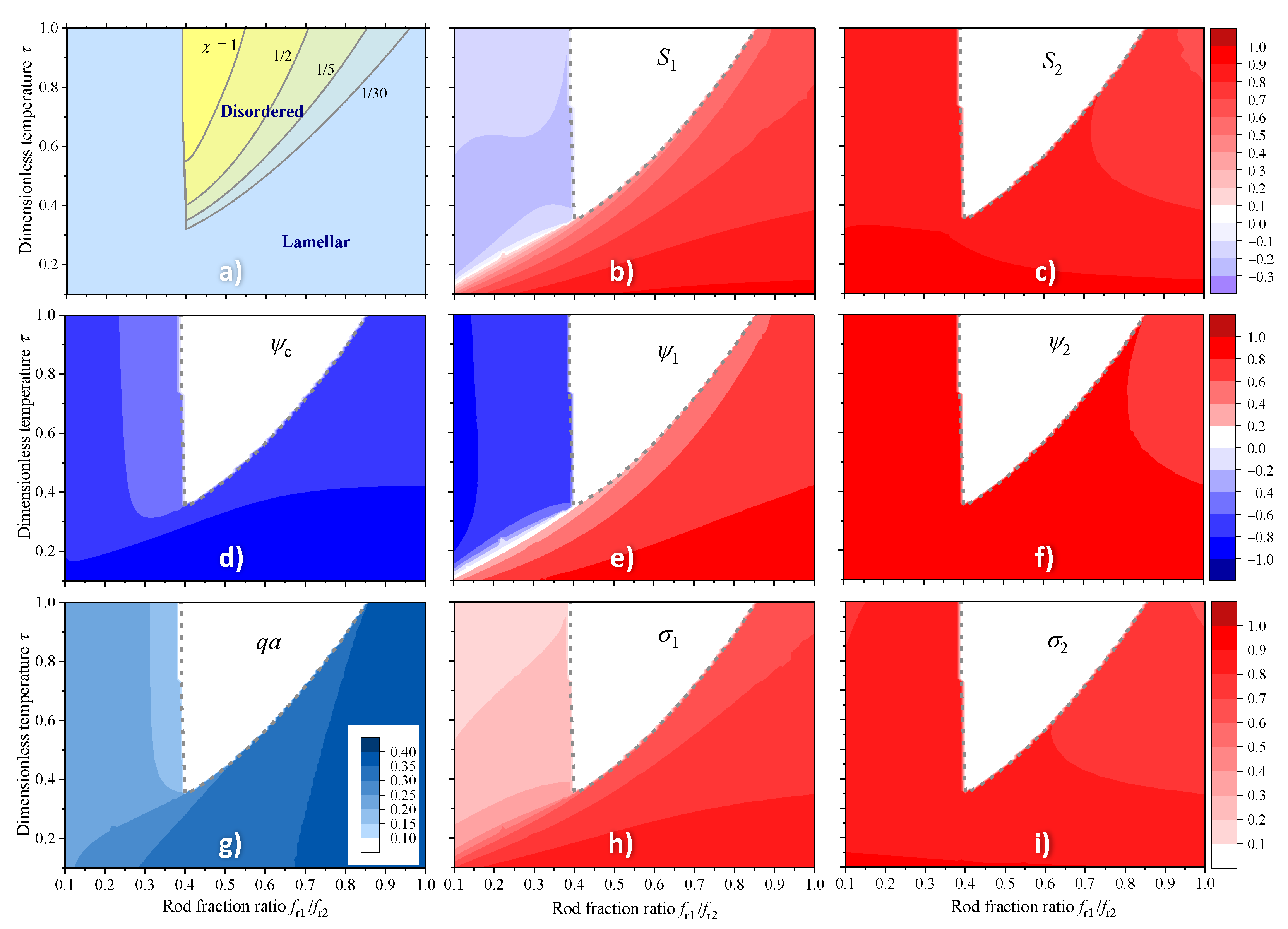
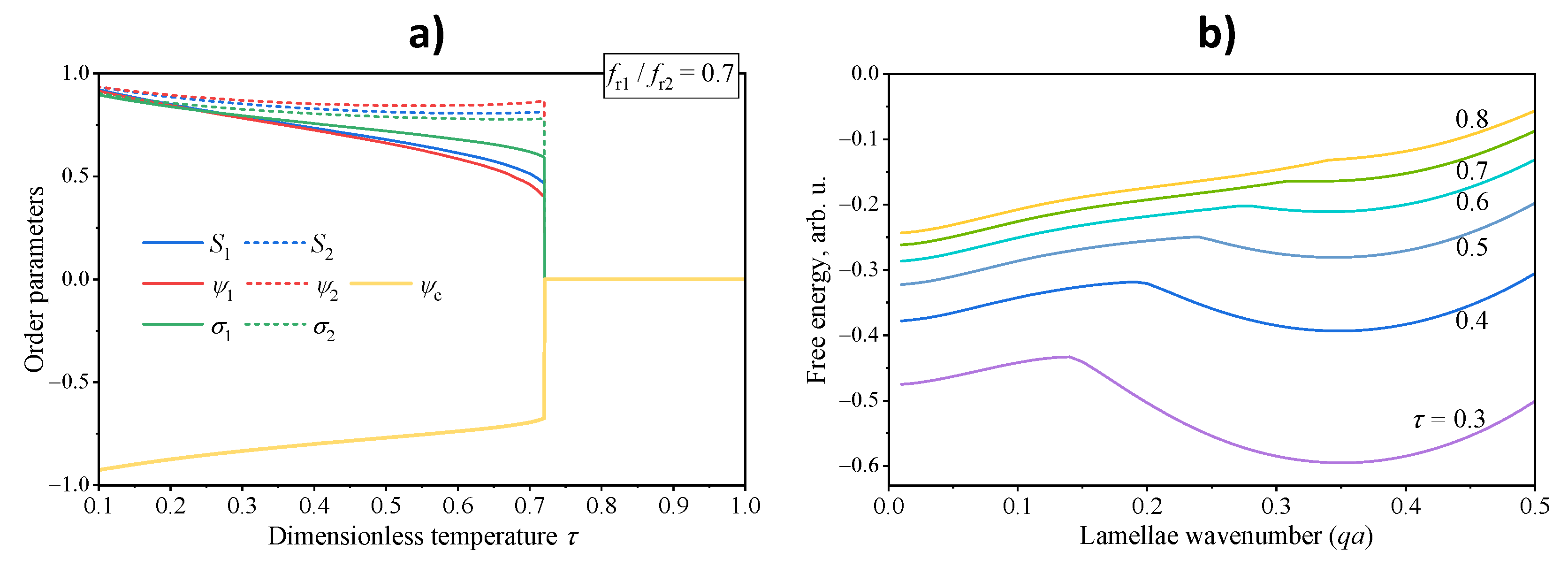
4.1. Phase Diagrams and Transitions
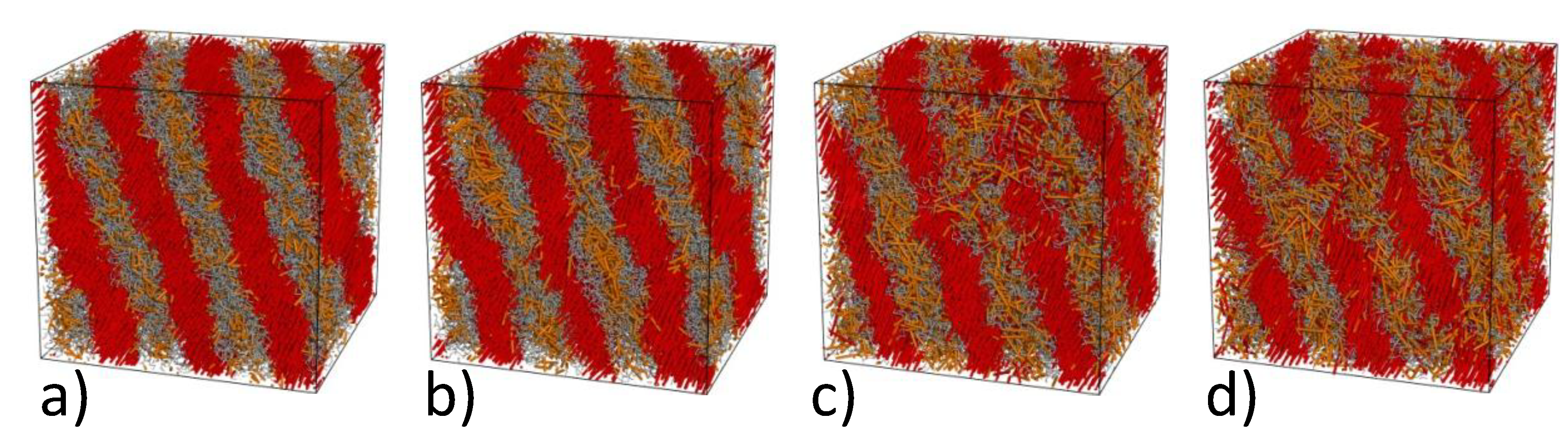
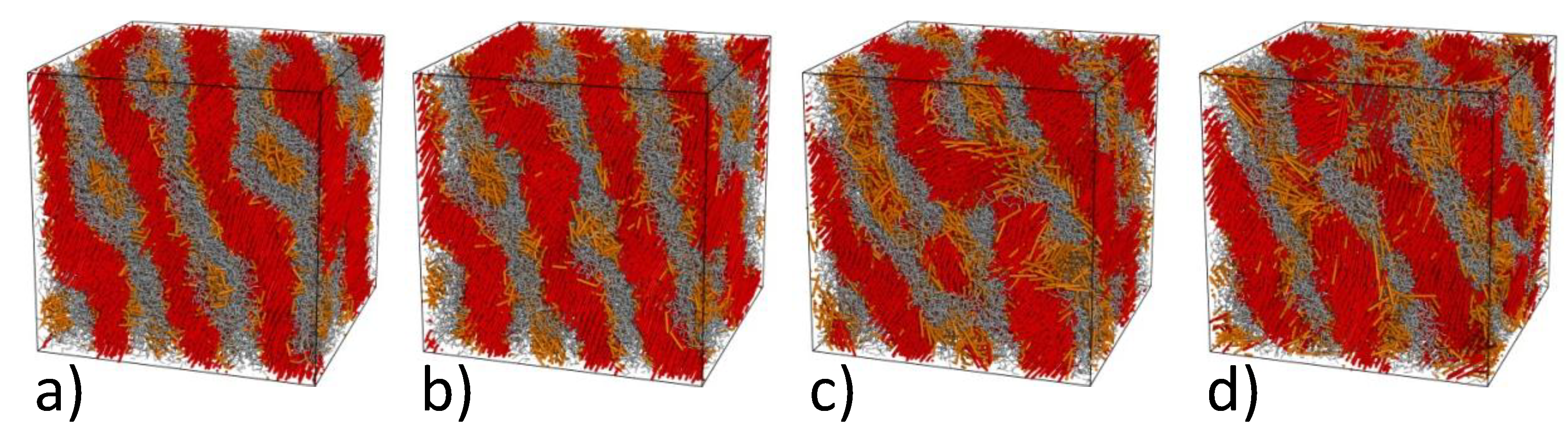
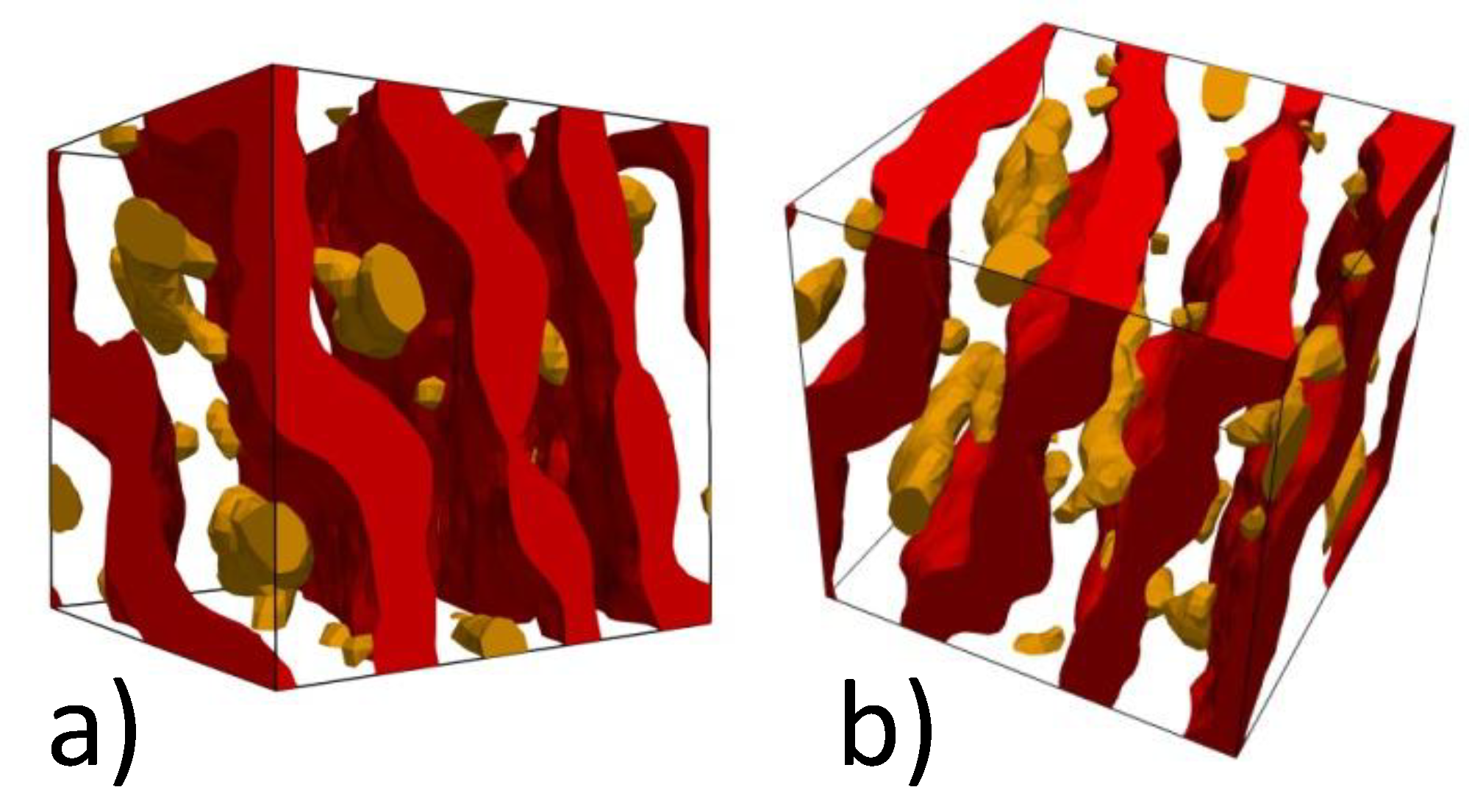
4.2. Computer Simulations of Lamellar Phase
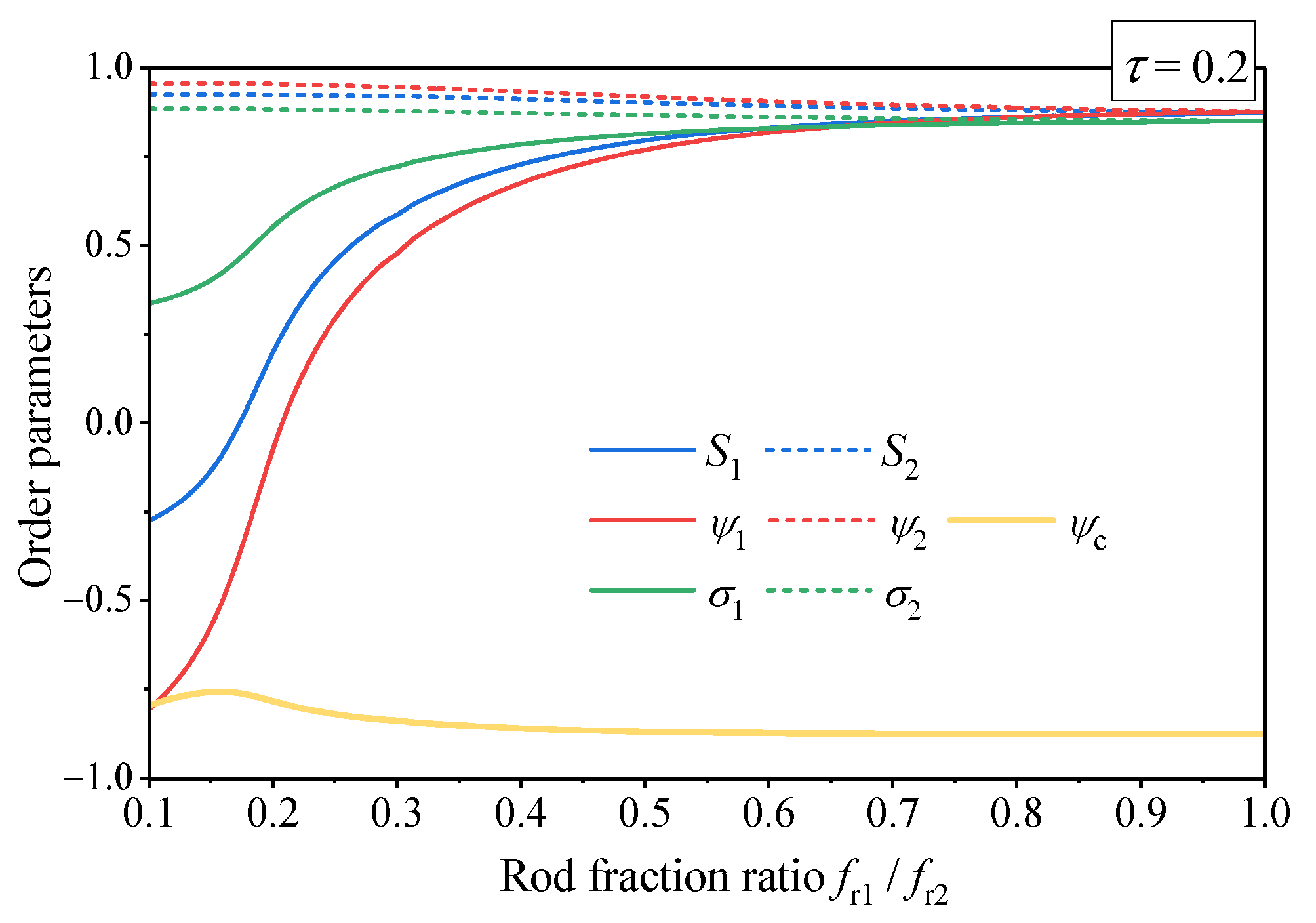
4.3. The Effect of Polymer Chain Asymmetry on Lamellar Ordering
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Expansion of the Mean-Field Potentials
Appendix B. Derivation of the Ornstein-Zernike Equations for Rod-Coil-Rod Triblock Copolymers
Appendix C. Solution of the Ornstein-Zernike Equations
Appendix D. Density-Density Correlation Functions of Rod-Coil-Rod Polymer Chains
Appendix D.1. Rod-Coil Total Correlation Function
Appendix D.2. Total Correlation Function of Segments of the Same Rod
Appendix D.3. Total Correlation Function between Segments of the Two Different Rods
References
- Xiao, L.L.; Zhou, X.; Yue, K.; Guo, Z.H. Synthesis and Self-Assembly of Conjugated Block Copolymers. Polymers 2020, 13, 110. [Google Scholar] [CrossRef]
- Yassar, A.; Miozzo, L.; Gironda, R.; Horowitz, G. Rod-coil and all-conjugated block copolymers for photovoltaic applications. Prog. Polym. Sci. 2013, 38, 791–844. [Google Scholar] [CrossRef]
- Liu, C.L.; Lin, C.H.; Kuo, C.C.; Lin, S.T.; Chen, W.C. Conjugated rod–coil block copolymers: Synthesis, morphology, photophysical properties, and stimuli-responsive applications. Prog. Polym. Sci. 2011, 36, 603–637. [Google Scholar] [CrossRef]
- Segalman, R.A.; McCulloch, B.; Kirmayer, S.; Urban, J.J. Block Copolymers for Organic Optoelectronics. Macromolecules 2009, 42, 9205–9216. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X.F.; Wei, H.B.; Wan, X.H. Tunable assembly of amphiphilic rod-coil block copolymers in solution. Chem. Soc. Rev. 2013, 42, 9127. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.D.; Alcazar, D.; Krikorian, V.; Toney, M.F.; Thomas, E.L.; Segalman, R.A. Crystalline Structure in Thin Films of DEH-PPV Homopolymer and PPV-b-PI Rod-Coil Block Copolymers. Macromolecules 2007, 41, 58–66. [Google Scholar] [CrossRef]
- Lee, S.; Cheng, L.C.; Yager, K.G.; Mumtaz, M.; Aissou, K.; Ross, C.A. In Situ Study of ABC Triblock Terpolymer Self-Assembly under Solvent Vapor Annealing. Macromolecules 2019, 52, 1853–1863. [Google Scholar] [CrossRef]
- Verheyen, L.; Timmermans, B.; Koeckelberghs, G. Influence of the Sequence in Conjugated Triblock Copolymers on Their Aggregation Behavior. Macromolecules 2018, 51, 6421–6429. [Google Scholar] [CrossRef]
- Honeker, C.C.; Thomas, E.L. Impact of Morphological Orientation in Determining Mechanical Properties in Triblock Copolymer Systems. Chem. Mater. 1996, 8, 1702–1714. [Google Scholar] [CrossRef]
- Erukhimovich, I.Y. Weak segregation theory and non-conventional morphologies in the ternary ABC triblock copolymers. Eur. Phys. J. E 2005, 18, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Mayes, A.M.; de la Cruz, M.O. Microphase separation in multiblock copolymer melts. J. Chem. Phys. 1989, 91, 7228–7235. [Google Scholar] [CrossRef]
- Noolandi, J.; Shi, A.C.; Linse, P. Theory of Phase Behavior of Poly(oxyethylene)-Poly(oxypropylene)-Poly(oxyethylene) Triblock Copolymers in Aqueous Solutions. Macromolecules 1996, 29, 5907–5919. [Google Scholar] [CrossRef]
- Leibler, L. Theory of Microphase Separation in Block Copolymers. Macromolecules 1980, 13, 1602–1617. [Google Scholar] [CrossRef]
- Aliev, M.A.; Kuzminyh, N.Y.; Ugolkova, E.A. Phase separation in polydisperse rod–coil block copolymers. Phys. A Stat. Mech. Appl. 2013, 392, 6214–6231. [Google Scholar] [CrossRef]
- Matsen, M.W.; Barrett, C. Liquid-crystalline behavior of rod-coil diblock copolymers. J. Chem. Phys. 1998, 109, 4108–4118. [Google Scholar] [CrossRef]
- Müller, M.; Schick, M. Ordered Phases in Rod-Coil Diblock Copolymers. Macromolecules 1996, 29, 8900–8903. [Google Scholar] [CrossRef]
- Pryamitsyn, V.; Ganesan, V. Self-assembly of rod-coil block copolymers. J. Chem. Phys. 2004, 120, 5824–5838. [Google Scholar] [CrossRef]
- Chen, J.Z.; Zhang, C.X.; Sun, Z.Y.; Zheng, Y.S.; An, L.J. A novel self-consistent-field lattice model for block copolymers. J. Chem. Phys. 2006, 124, 104907. [Google Scholar] [CrossRef]
- Shah, M.; Pryamitsyn, V.; Ganesan, V. A Model for Self-Assembly in Side Chain Liquid Crystalline Block Copolymers. Macromolecules 2007, 41, 218–229. [Google Scholar] [CrossRef]
- Gao, J.; Song, W.; Tang, P.; Yang, Y. Self-assembly of semiflexible block copolymers: 2D numerical implementation of self-consistent field theory. Soft Matter 2011, 7, 5208. [Google Scholar] [CrossRef]
- Gao, J.; Tang, P.; Yang, Y. Non-lamellae structures of coil-semiflexible diblock copolymers. Soft Matter 2013, 9, 69–81. [Google Scholar] [CrossRef]
- Kriksin, Y.A.; Khalatur, P.G. Parallel Algorithm for 3D SCF Simulation of Copolymers with Flexible and Rigid Blocks. Macromol. Theory Simul. 2012, 21, 382–399. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, J.; Sun, Z.; Shi, T.; An, L.; Jia, Y. Self-assembly of linear ABC coil-coil-rod triblock copolymers. Polymer 2010, 51, 3315–3319. [Google Scholar] [CrossRef]
- Düchs, D.; Sullivan, D.E. Entropy-induced smectic phases in rod coil copolymers. J. Phys. Condens. Matter 2002, 14, 12189–12202. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, R.C.; Sullivan, D.E.; Chen, J.Z.Y. Smectic phases in rod-coil diblock copolymers. J. Phys. Condens. Matter 2007, 19, 376107. [Google Scholar] [CrossRef]
- Tang, J.; Jiang, Y.; Zhang, X.; Yan, D.; Chen, J.Z.Y. Phase Diagram of Rod-Coil Diblock Copolymer Melts. Macromolecules 2015, 48, 9060–9070. [Google Scholar] [CrossRef]
- Song, W.; Tang, P.; Qiu, F.; Yang, Y.; Shi, A.C. Phase behavior of semiflexible-coil diblock copolymers: A hybrid numerical SCFT approach. Soft Matter 2011, 7, 929–938. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, J.Z.Y. Influence of Chain Rigidity on the Phase Behavior of Wormlike Diblock Copolymers. Phys. Rev. Lett. 2013, 110, 138305. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, Y.; Chen, J.Z.Y. Phase transitions in semiflexible–rod diblock copolymers: A self-consistent field theory. Soft Matter 2014, 10, 8932–8944. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, P.; Shi, A.C. Liquid crystalline bilayers self-assembled from rod–coil diblock copolymers. Soft Matter 2017, 13, 4607–4615. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Y.; Chen, J.Z.Y. Complex liquid-crystal nanostructures in semiflexible ABC linear triblock copolymers: A self-consistent field theory. J. Chem. Phys. 2016, 145, 184902. [Google Scholar] [CrossRef]
- Song, W.; Tang, P.; Zhang, H.; Yang, Y.; Shi, A.C. New Numerical Implementation of Self-Consistent Field Theory for Semiflexible Polymers. Macromolecules 2009, 42, 6300–6309. [Google Scholar] [CrossRef]
- Han, X.-G.; Liang, N.; Zhang, H. Self-assembly in rod/coil block copolymers: Degenerate behavior under nonconfinement. Condens. Matter Phys. 2020, 23, 33603. [Google Scholar] [CrossRef]
- Zhang, Q.; Lin, J.; Wang, L.; Xu, Z. Theoretical modeling and simulations of self-assembly of copolymers in solution. Prog. Polym. Sci. 2017, 75, 1–30. [Google Scholar] [CrossRef]
- Liu, F.; Sun, T.; Tang, P.; Zhang, H.; Qiu, F. Understanding chain folding morphology of semicrystalline polymers based on a rod–coil multiblock model. Soft Matter 2017, 13, 8250–8263. [Google Scholar] [CrossRef]
- Wilson, M.R.; Thomas, A.B.; Dennison, M.; Masters, A.J. Computer simulations and theory of polymer tethered nanorods: The role of flexible chains in influencing mesophase stability. Soft Matter 2009, 5, 363–368. [Google Scholar] [CrossRef]
- Lintuvuori, J.S.; Wilson, M.R. A coarse-grained simulation study of mesophase formation in a series of rod-coil multiblock copolymers. Phys. Chem. Chem. Phys. 2009, 11, 2116. [Google Scholar] [CrossRef]
- Singh, Y. Molecular theory of liquid crystals: Application to the nematic phase. Phys. Rev. A 1984, 30, 583–593. [Google Scholar] [CrossRef]
- Sluckin, T.J.; Shukla, P. Molecular field theory of nematics: Density functional approach. I. Bulk effects. J. Phys. A Math. Gen. 1983, 16, 1539–1553. [Google Scholar] [CrossRef]
- Gorkunov, M.V.; Osipov, M.A.; Lagerwall, J.P.F.; Giesselmann, F. Order-disorder molecular model of the smectic-A-smectic-Cphase transition in materials with conventional and anomalously weak layer contraction. Phys. Rev. E 2007, 76, 051706. [Google Scholar] [CrossRef]
- Longa, L.; Stelzer, J.; Dunmur, D. Density functional approach to study the elastic constants of biaxial nematic liquid crystals. J. Chem. Phys. 1998, 109, 1555–1566. [Google Scholar] [CrossRef]
- Osipov, M.A. Molecular theories of liquid crystals. In Handbook of Liquid Crystals; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2014; pp. 40–71. [Google Scholar] [CrossRef]
- Osipov, M.A.; Gorkunov, M.V. Molecular theory of liquid-crystal ordering in rod-coil diblock copolymers. Phys. Rev. E 2019, 100, 042701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osipov, M.A.; Gorkunov, M.V.; Antonov, A.A. Density Functional Approach to the Molecular Theory of Rod-Coil Diblock Copolymers. Polym. Sci. Ser. A 2020, 62, 562–577. [Google Scholar] [CrossRef]
- Osipov, M.A.; Gorkunov, M.V.; Berezkin, A.V.; Antonov, A.A.; Kudryavtsev, Y.V. Molecular theory of the tilting transition and computer simulations of the tilted lamellar phase of rod–coil diblock copolymers. J. Chem. Phys. 2020, 152, 184906. [Google Scholar] [CrossRef]
- Reenders, M.; ten Brinke, G. Compositional and Orientational Ordering in Rod-Coil Diblock Copolymer Melts. Macromolecules 2002, 35, 3266–3280. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Wu, F.; Lin, J.; Cai, C.; Wang, L.; Chen, J.; Gao, L. Programmable Morphology Evolution of Rod-Coil-Rod Block Copolymer Assemblies Induced by Variation of Chain Ordering. Langmuir 2021, 37, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Droghetti, H.; Pagonabarraga, I.; Carbone, P.; Asinari, P.; Marchisio, D. Dissipative particle dynamics simulations of tri-block co-polymer and water: Phase diagram validation and microstructure identification. J. Chem. Phys. 2018, 149, 184903. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Song, S.; Long, X.P.; Zhang, C.Y.; Chen, Y.M. Dissipative Particle Dynamics Simulation on Self-Assembly Behavior of Rod-Coil-Rod Triblock Copolymer in Solutions. Macromol. Theory Simul. 2014, 23, 490–499. [Google Scholar] [CrossRef]
- Omar, A.K.; Hanson, B.; Haws, R.T.; Hu, Z.; Bout, D.A.V.; Rossky, P.J.; Ganesan, V. Aggregation Behavior of Rod-Coil-Rod Triblock Copolymers in a Coil-Selective Solvent. J. Phys. Chem. B 2014, 119, 330–337. [Google Scholar] [CrossRef]
- Prhashanna, A.; Khan, S.A.; Chen, S.B. Micelle morphology and chain conformation of triblock copolymers under shear: LA-DPD study. Colloids Surf. Physicochem. Eng. 2016, 506, 457–466. [Google Scholar] [CrossRef]
- Yang, C.; Li, Q.; Cai, C.; Lin, J. Nanoparticle-Induced Ellipse-to-Vesicle Morphology Transition of Rod–Coil-Rod Triblock Copolymer Aggregates. Langmuir 2016, 32, 6917–6927. [Google Scholar] [CrossRef]
- González-Pizarro, D.A.; Soto-Figueroa, C.; del Rosario Rodríguez-Hidalgo, M.; Vicente, L. Mesoscopic study of the ternary phase diagram of the PS–PB–PtBMA triblock copolymer: Modification of the phase structure by the composition effect. Soft Matter 2018, 14, 508–520. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, J.; Ma, Z.; Jiang, W. Monte Carlo simulation of the aggregation of rod-flexible triblock copolymers in a thin film. Chem. Phys. 2006, 321, 1–9. [Google Scholar] [CrossRef]
- Su, Y.J.; Huang, J.H. Self-assembly behavior of rod-coil-rod triblock copolymers within a planar slit. Chin. J. Polym. Sci. 2016, 34, 838–849. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, T.; Wang, L.; Lin, S.; Lin, J. Self-assembly of rod-coil-rod triblock copolymers: A route toward hierarchical liquid crystalline structures. Polymer 2016, 103, 64–72. [Google Scholar] [CrossRef]
- Berezkin, A.V.; Kudryavtsev, Y.V.; Osipov, M.A. Tilted Lamellar Phase of the Rod–Coil Diblock Copolymer: Dissipative Particle Dynamics Simulation. Polym. Sci. Ser. A 2020, 62, 430–436. [Google Scholar] [CrossRef]
- Evans, R. Density functionals in the theory of nonuniform fluids. In Fundamentals of Inhomogeneous Fluids; Decker: New York, NY, USA, 1992. [Google Scholar]
- Perera, A.; Patey, G.N.; Weis, J.J. Density functional theory applied to the isotropic-nematic transition in model liquid crystals. J. Chem. Phys. 1988, 89, 6941–6946. [Google Scholar] [CrossRef]
- Osipov, M.A.; Gorkunov, M.V.; Antonov, A.A. Liquid Crystal Ordering in the Hexagonal Phase of Rod-Coil Diblock Copolymers. Polymers 2020, 12, 1262. [Google Scholar] [CrossRef] [PubMed]
- Khokhlov, A.R.; Grosberg, A.Y.; Pande, V.S. Statistical Physics of Macromolecules; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Miller, T.F.; Eleftheriou, M.; Pattnaik, P.; Ndirango, A.; Newns, D.; Martyna, G.J. Symplectic quaternion scheme for biophysical molecular dynamics. J. Chem. Phys. 2002, 116, 8649–8659. [Google Scholar] [CrossRef] [Green Version]
- Berezkin, A.V.; Kudryavtsev, Y.V.; Gorkunov, M.V.; Osipov, M.A. Ordering of anisotropic nanoparticles in diblock copolymer lamellae: Simulations with dissipative particle dynamics and a molecular theory. J. Chem. Phys. 2017, 146, 144902. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://lammps.sandia.gov/ (accessed on 30 September 2021).
- Yang, A.J.M.; Fleming, P.D.; Gibbs, J.H. Molecular theory of surface tension. J. Chem. Phys. 1976, 64, 3732–3747. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osipov, M.A.; Gorkunov, M.V.; Antonov, A.A.; Berezkin, A.V.; Kudryavtsev, Y.V. Liquid-Crystal Ordering and Microphase Separation in the Lamellar Phase of Rod-Coil-Rod Triblock Copolymers. Molecular Theory and Computer Simulations. Polymers 2021, 13, 3392. https://doi.org/10.3390/polym13193392
Osipov MA, Gorkunov MV, Antonov AA, Berezkin AV, Kudryavtsev YV. Liquid-Crystal Ordering and Microphase Separation in the Lamellar Phase of Rod-Coil-Rod Triblock Copolymers. Molecular Theory and Computer Simulations. Polymers. 2021; 13(19):3392. https://doi.org/10.3390/polym13193392
Chicago/Turabian StyleOsipov, Mikhail A., Maxim V. Gorkunov, Alexander A. Antonov, Anatoly V. Berezkin, and Yaroslav V. Kudryavtsev. 2021. "Liquid-Crystal Ordering and Microphase Separation in the Lamellar Phase of Rod-Coil-Rod Triblock Copolymers. Molecular Theory and Computer Simulations" Polymers 13, no. 19: 3392. https://doi.org/10.3390/polym13193392
APA StyleOsipov, M. A., Gorkunov, M. V., Antonov, A. A., Berezkin, A. V., & Kudryavtsev, Y. V. (2021). Liquid-Crystal Ordering and Microphase Separation in the Lamellar Phase of Rod-Coil-Rod Triblock Copolymers. Molecular Theory and Computer Simulations. Polymers, 13(19), 3392. https://doi.org/10.3390/polym13193392