
Development of Graphene-Based Polymeric Nanocomposites: A Brief Overview


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
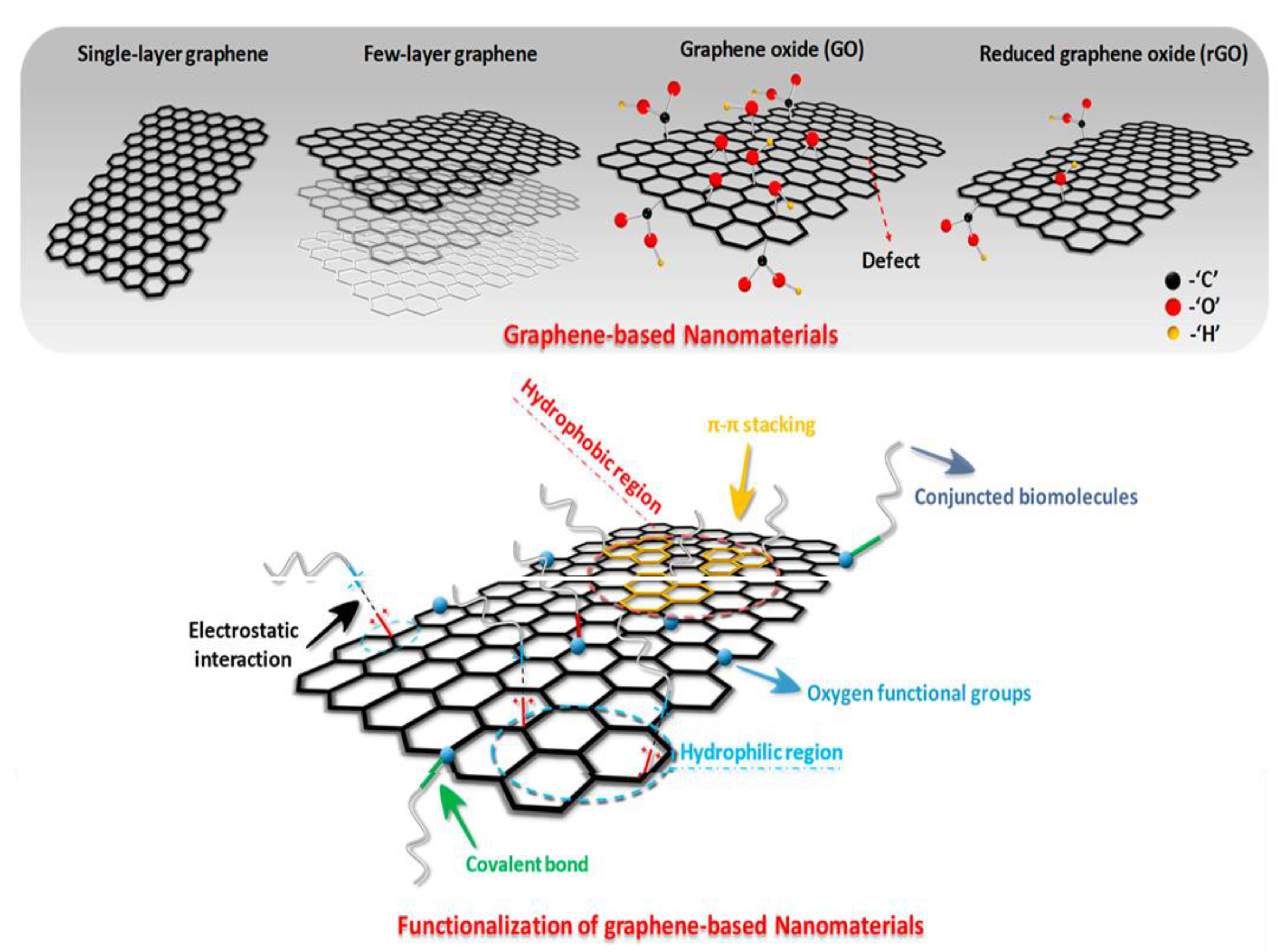
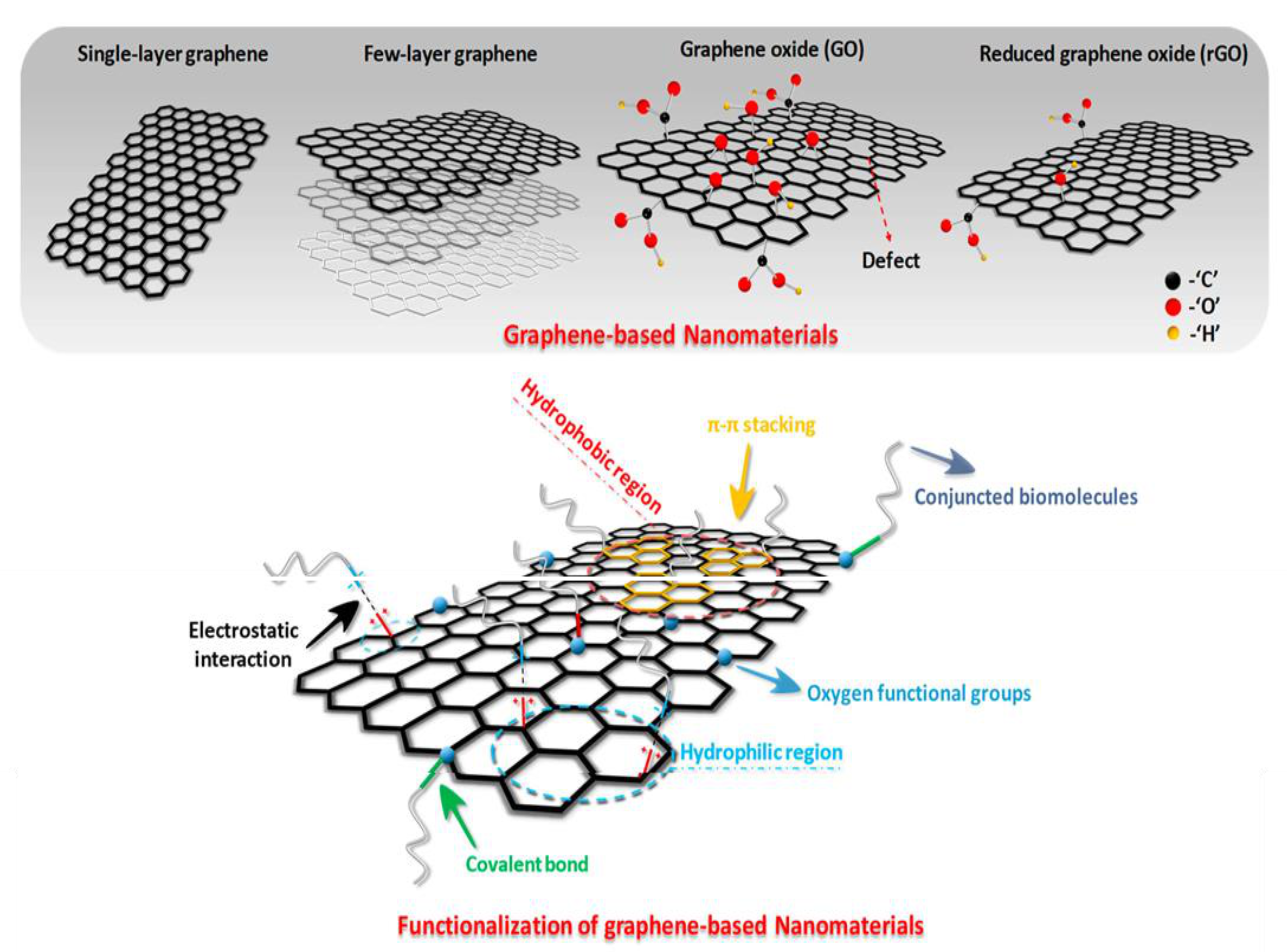
2. Graphene and Its Derivatives: Synthesis and Characteristics
2.1. Properties and Synthesis Methods of Graphene
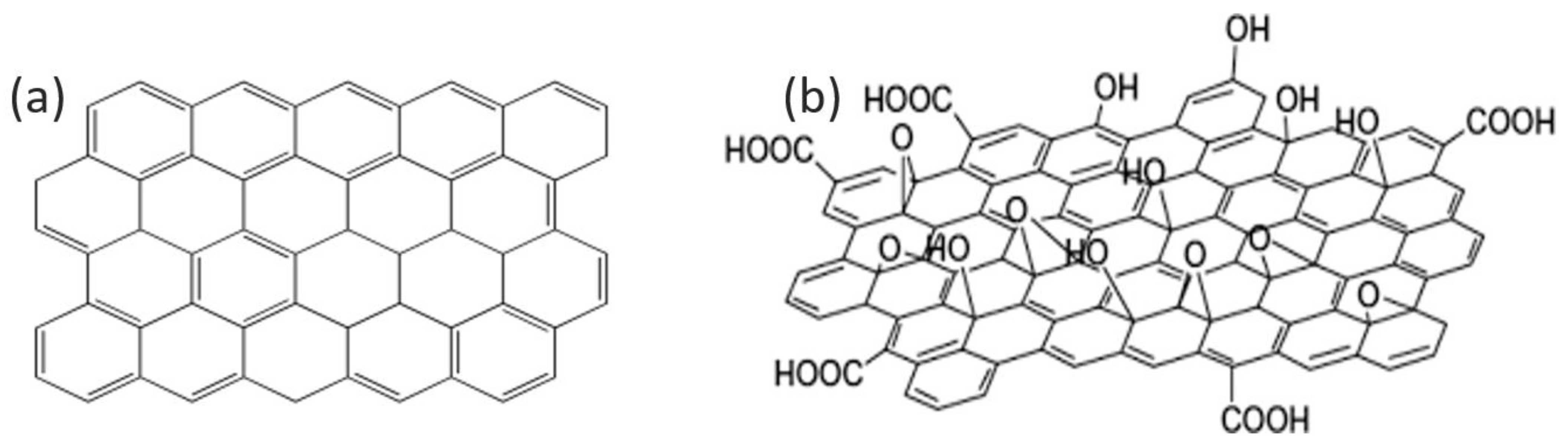
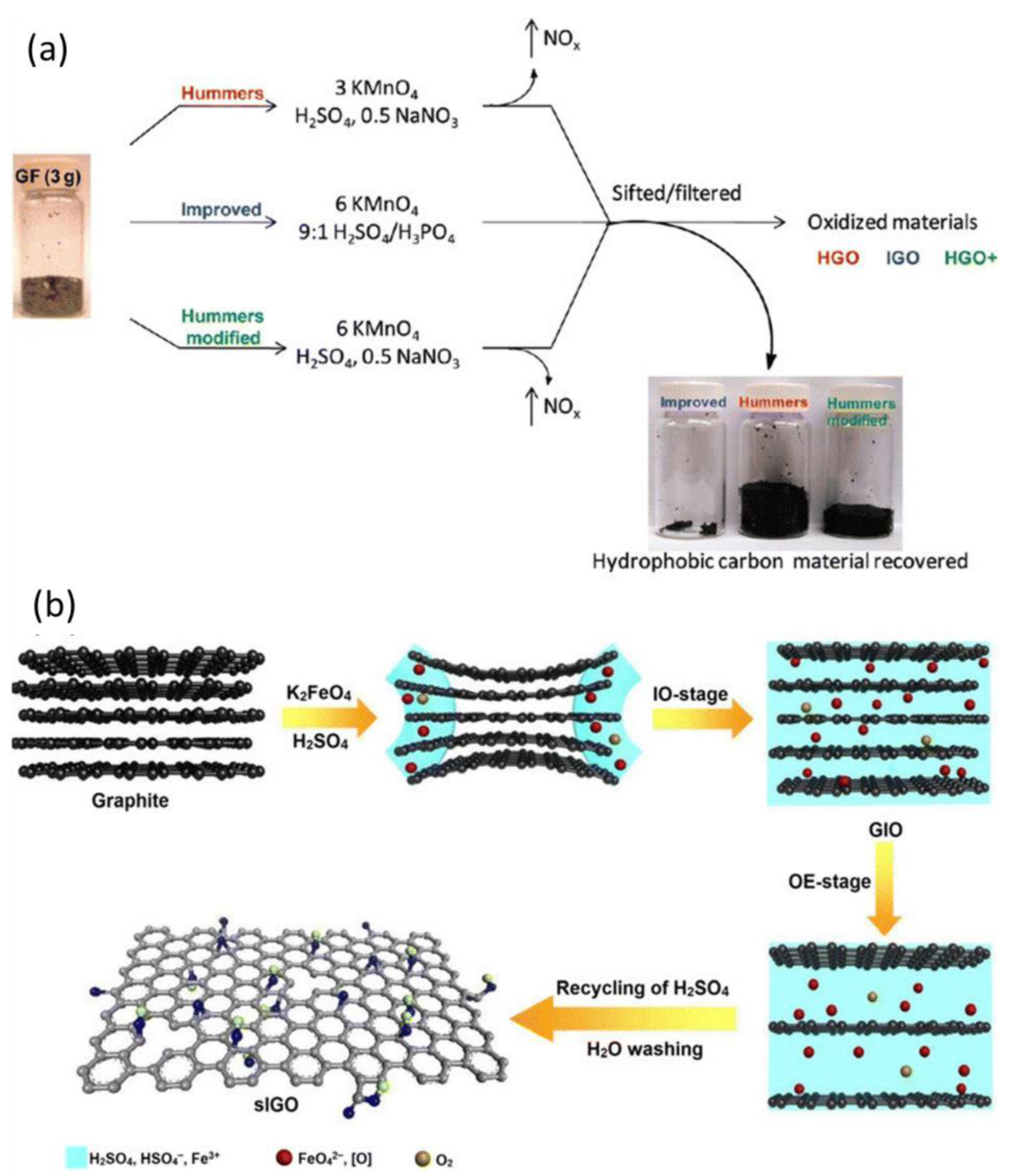
2.2. Characteristics and Synthesis Methods of Graphene Oxide
2.3. Synthesis and Characteristics of Reduced Graphene Oxide
3. Functionalization Procedures for the Development of Graphene/Polymer Nanocomposites
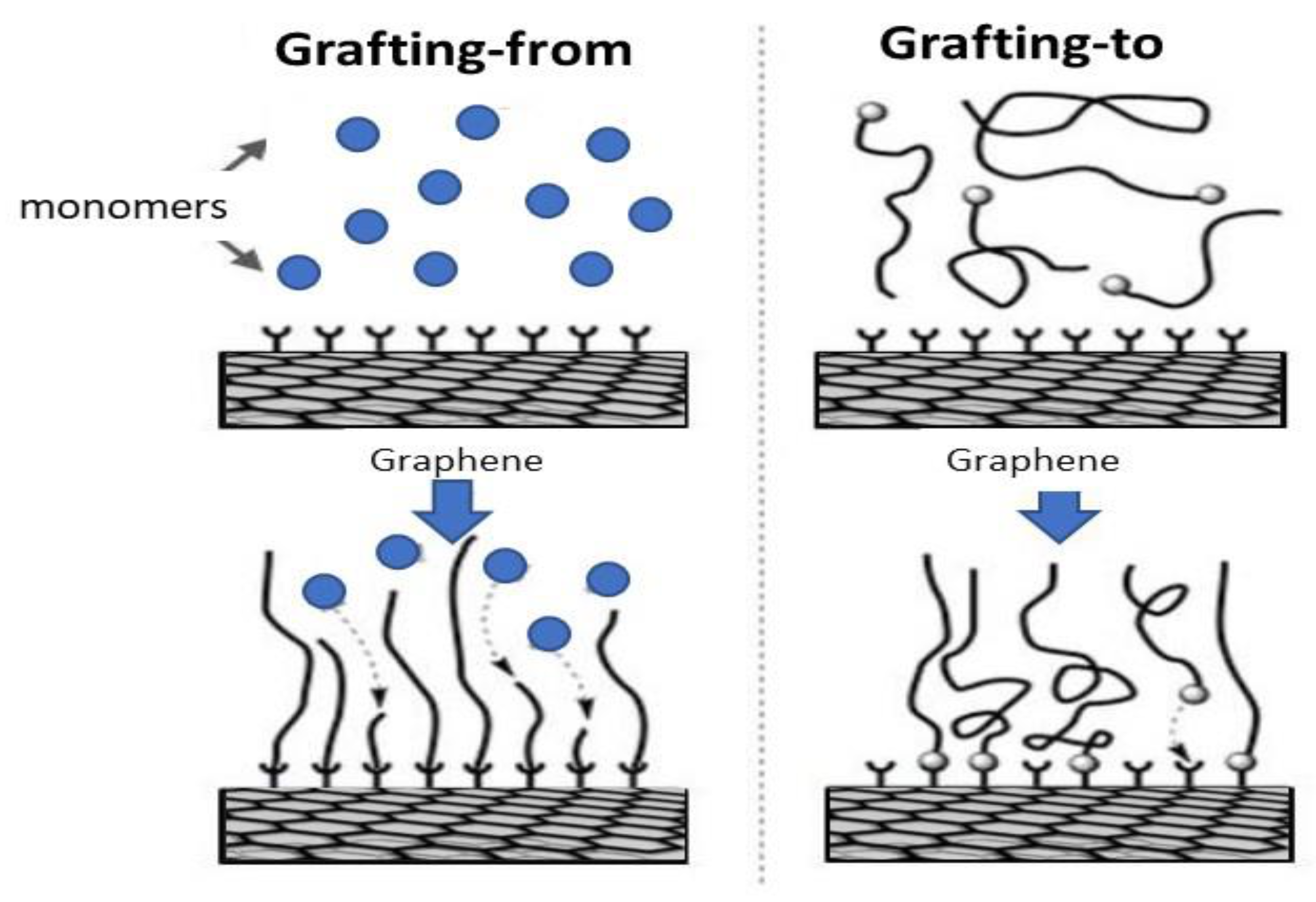
3.1. Covalent Functionalization with Polymers
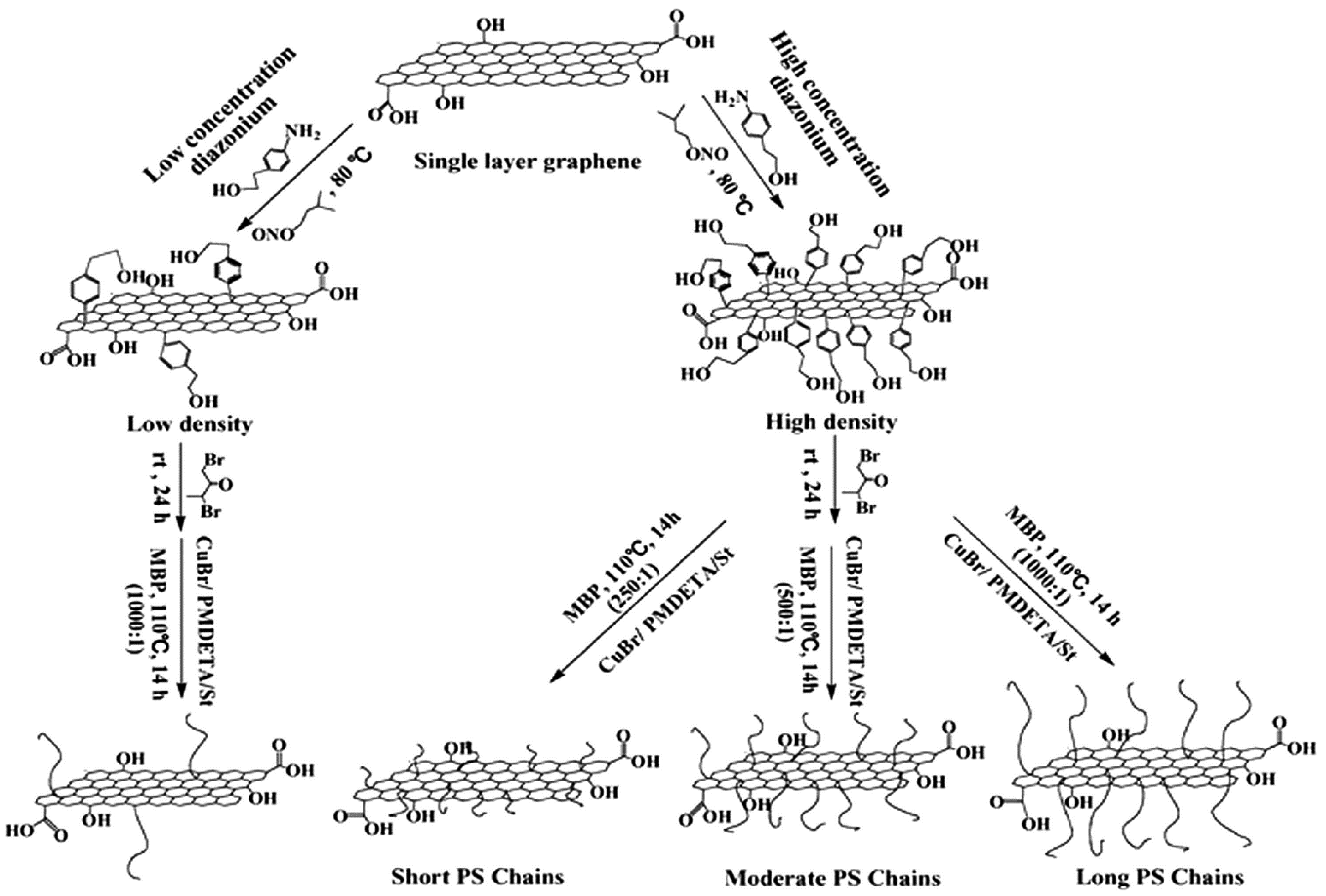
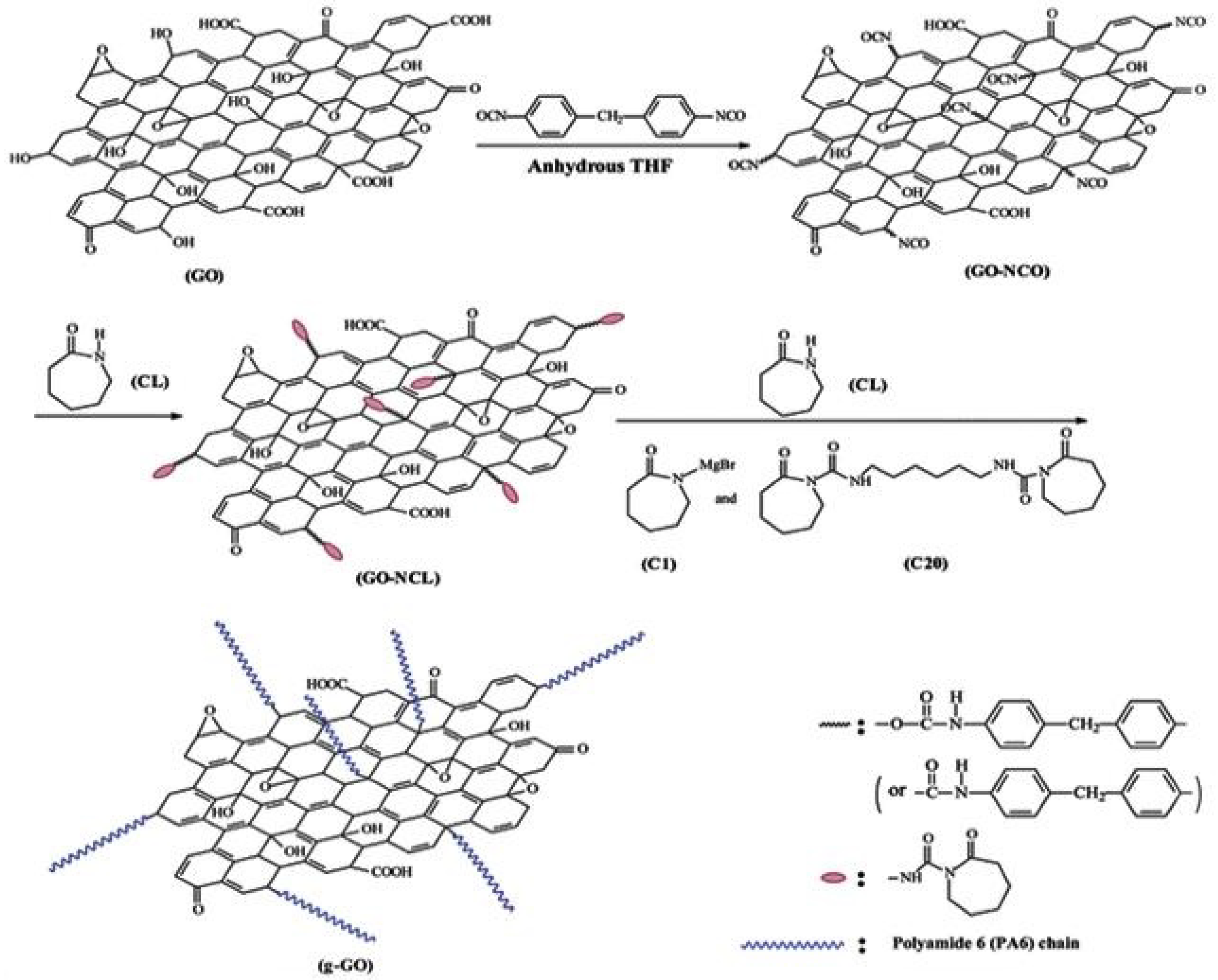
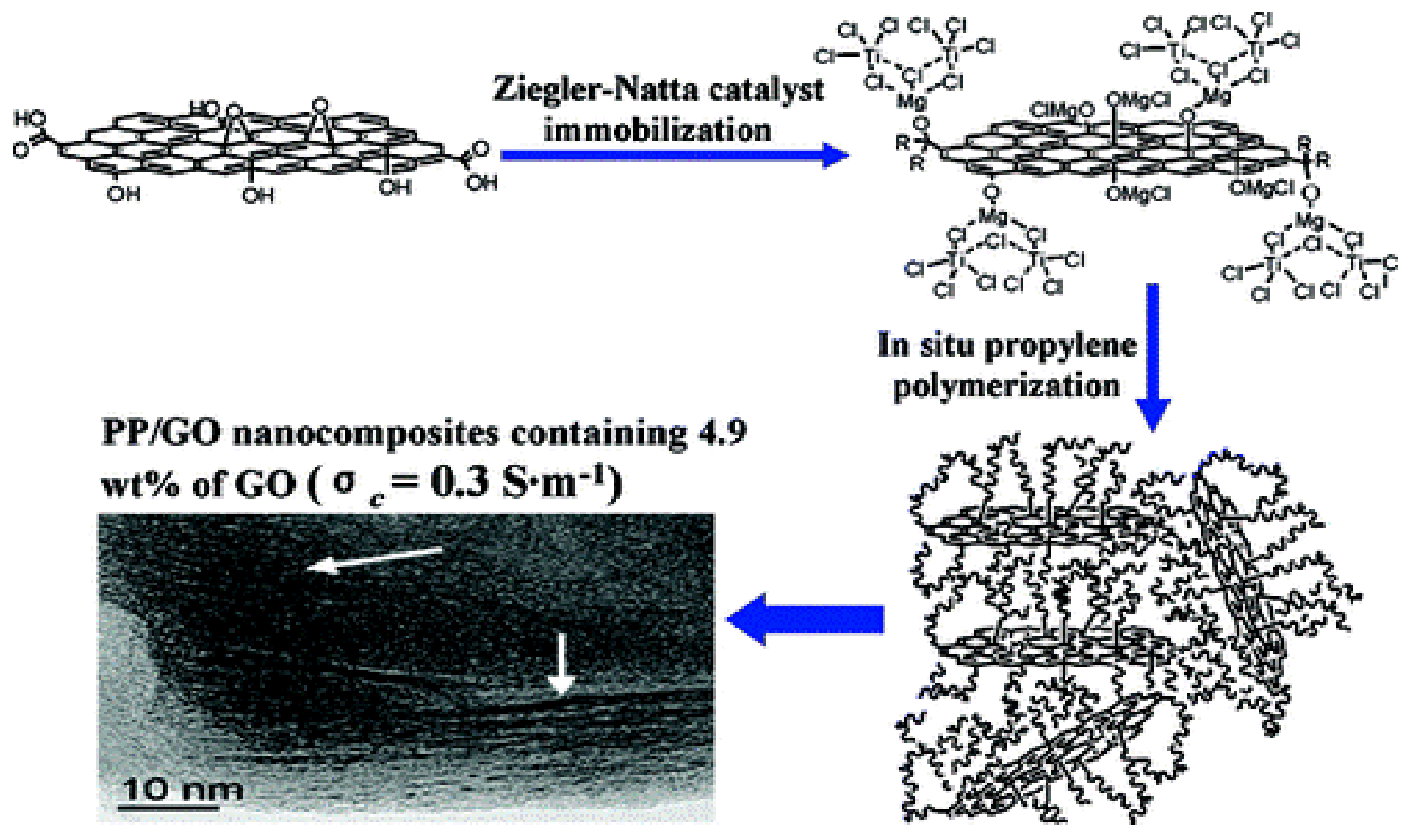
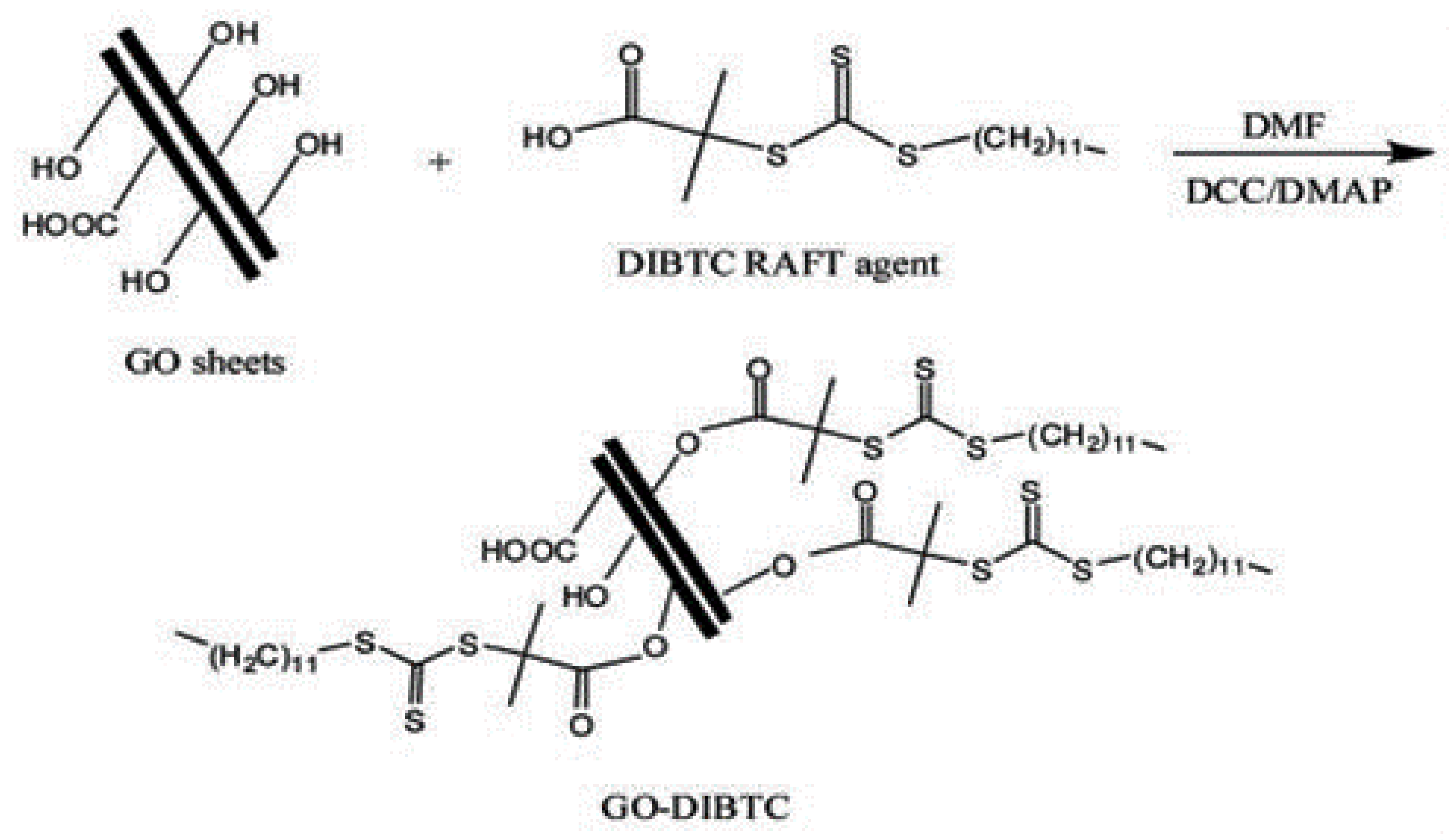
3.1.1. Grafting-from Methods
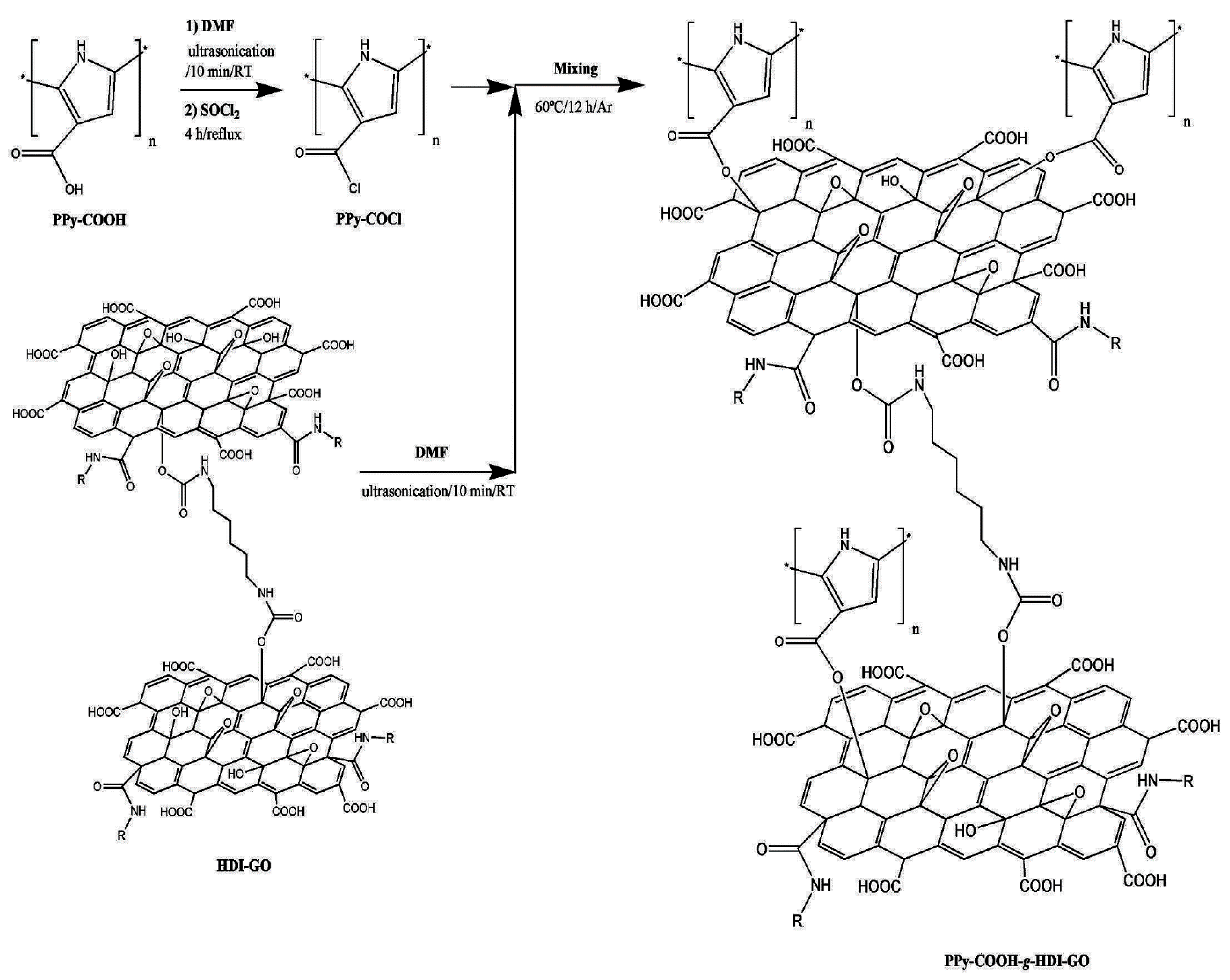
3.1.2. Grafting-to Methods
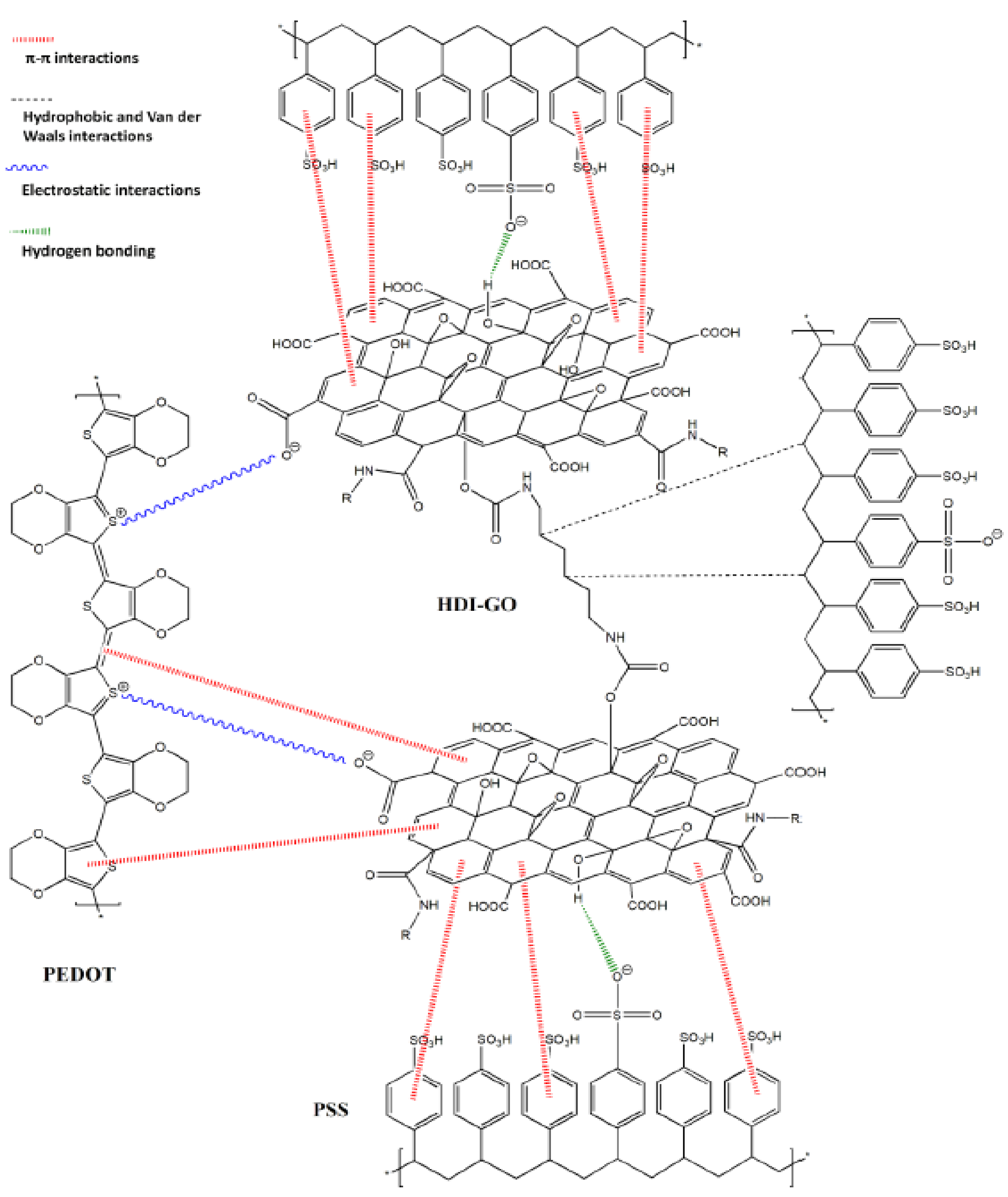
3.2. Non-Covalent Functionalization with Polymers
4. Applications of Graphene/Polymer Nanocomposites in the Field of Energy
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Edwards, D.C. Polymer-filler interactions in rubber reinforcement. J. Mater. Sci. 1990, 25, 4175–4185. [Google Scholar] [CrossRef]
- Harito, C.; Bavykin, D.V.; Yuliarto, B.; Dipojono, H.K.; Walsh, F.C. Polymer Nanocomposites Having a High Filler Content: Synthesis, Structures, Properties, and Applications. Nanoscale 2019, 11, 4653–4682. [Google Scholar] [CrossRef] [PubMed]
- Diez-Pascual, A.M.; Gomez-Fatou, M.A.; Ania, F.; Flores, A. Nanoindentation in polymer nanocomposites. Prog. Mater. Sci. 2015, 67, 1–94. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, M.A.; Rafiee, J.; Wang, Z.; Song, H.; Yu, Z.Z.; Koratkar, N. Enhanced Mechanical Properties of Nanocomposites at Low Graphene Content. ACS Nano 2009, 3, 3884–3890. [Google Scholar] [CrossRef]
- Diez-Pascual, A.M.; Naffakh, M.; Marco, C.; Ellis, G.; Gomez-Fatou, M.A. High-performance nanocomposites based on polyetherketones. Prog. Mater. Sci. 2012, 57, 1106–1190. [Google Scholar] [CrossRef]
- Stoller, M.D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R.S. Graphene-based Ultracapacitors. Nano. Lett. 2008, 8, 3498–3502. [Google Scholar] [CrossRef]
- Wang, H.; Cui, L.-F.; Yang, Y.; Casalongue, H.S.; Robinson, J.T.; Liang, Y.; Cui, Y.; Dai, H. Mn3O4-Graphene Hybrid as a High-Capacity Anode Material for Lithium Ion Batteries. J. Am. Chem. Soc. 2010, 132, 13978–13980. [Google Scholar] [CrossRef] [Green Version]
- Díez-Pascual, A.M.; Luceño Sánchez, J.A.; Peña Capilla, R.; García Díaz, P. Recent Developments in Graphene/Polymer Nanocomposites for Application in Polymer Solar Cells. Polymers 2018, 10, 217. [Google Scholar] [CrossRef] [Green Version]
- Kou, R.; Shao, Y.; Wang, D.; Engelhard, M.H.; Kwak, J.H.; Wang, J.; Viswanathan, V.V.; Wang, C.; Lin, Y.; Wang, Y.; et al. Enhanced activity and stability of Pt catalysts on functionalized graphene sheets for electrocatalytic oxygen reduction. Electrochem. Commun. 2009, 11, 954–957. [Google Scholar] [CrossRef]
- Luceño-Sánchez, J.A.; Charas, A.; Díez-Pascual, A.M. Effect of HDI-Modified GO on the Thermoelectric Performance of Poly(3,4-ethylenedioxythiophene):Poly(Styrenesulfonate) Nanocomposite Films. Polymers 2021, 13, 1503. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Díez-Pascual, A.M.; Lázaro, E.; Vera, S.; Gómez-Fatou, M.A. Chemical sensors based on polymer composites with carbon nanotubes and graphene: The role of the polymer. J. Mater. Chem. A 2014, 2, 14289–14328. [Google Scholar] [CrossRef]
- Díez-Pascual, A.M.; Díez-Vicente, A.L. Poly(propylene fumarate)/Polyethylene Glycol-Modified Graphene Oxide Nanocomposites for Tissue Engineering. ACS Appl. Mater. Interfaces 2016, 8, 17902–17914. [Google Scholar] [CrossRef]
- Chang, H.; Wu, H. Graphene-Based Nanomaterials: Synthesis, Properties, and Optical and Optoelectronic Applications. Adv. Funct. Mater. 2013, 23, 1984–1997. [Google Scholar] [CrossRef]
- Weiss, N.O.; Zhou, H.; Liao, L.; Liu, Y.; Jiang, S.; Huang, Y.; Duan, X. Graphene: An Emerging Electronic Material. Adv. Mater. 2012, 24, 5782–5825. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Nair, R.R.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.R.; Geim, A.K. Fine structure constant defines visual transparency of graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Cai, W.; Colombo, L.; Ruoff, R.S. Evolution of Graphene Growth on Ni and Cu by Carbon Isotope Labeling. Nano Lett. 2009, 9, 4268–4272. [Google Scholar] [CrossRef] [Green Version]
- Charrier, A.; Coati, A.; Argunova, T.; Thibaudau, F.; Garreau, Y.; Pinchaux, R.; Forbeaux, I.; Debever, J.-M.; Sauvage-Simkin, M.; Themlin, J.-M. Solid-state decomposition of silicon carbide for growing ultra-thin heteroepitaxial graphite films. J. Appl. Phys. 2002, 92, 2479–2484. [Google Scholar] [CrossRef]
- Berger, C.; Song, Z.; Li, T.; Li, X.; Ogbazghi, A.Y.; Feng, R.; Dai, Z.; Marchenkov, A.N.; Conrad, E.H.; First, P.N.; et al. Ultrathin Epitaxial Graphite: 2D Electron Gas Properties and a Route toward Graphene-based Nanoelectronics. J. Phys. Chem. B 2004, 108, 19912–19916. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-Y.; Lu, A.-Y.; Xu, Y.; Chen, F.-R.; Khlobystov, A.N.; Li, L.-J. High-Quality Thin Graphene Films from Fast Electrochemical Exfoliation. ACS Nano 2011, 5, 2332–2339. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Zheng, Q.; Kim, J.K. Synthesis, Structure and Properties of Graphene and Graphene Oxide. In Graphene for Transparent Conductors. Synthesis, Properties and Applications; Springer: New York, NY, USA, 2015; pp. 29–94. ISBN 978-1493927685. [Google Scholar]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Lin, F.; Tong, X.; Wang, Y.; Bao, J.; Wang, Z.M. Graphene oxide liquid crystals: Synthesis, phase transition, rheological property, and applications in optoelectronics and display. Nano Res. Lett. 2015, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yao, B.; Li, C.; Shi, G. An improved Hummers method for eco-friendly synthesis of graphene oxide. Carbon 2013, 64, 225–229. [Google Scholar] [CrossRef]
- Peng, L.; Xu, Z.; Liu, Z.; Wei, Y.; Sun, H.; Li, Z.; Zhao, X.; Gao, C. An iron-based green approach to 1-h production of single-layer graphene oxide. Nat Commun. 2015, 6, 5716. [Google Scholar] [CrossRef] [Green Version]
- Ciesielski, A.; Samorì, P. Graphene via sonication assisted liquid-phase exfoliation. Chem. Soc. Rev. 2014, 43, 381–398. [Google Scholar] [CrossRef]
- Yang, H.; Li, H.; Zhai, J.; Yu, H. Simple Synthesis of Graphene Oxide Using Ultrasonic Cleaner from Expanded Graphite. Ind. Eng. Chem. 2014, 53, 17878–17883. [Google Scholar] [CrossRef]
- Tene, T.; Tubon Usca, G.; Guevara, M.; Molina, R.; Veltri, F.; Arias, M.; Caputi, L.S.; Vacacela Gomez, C. Toward Large-Scale Production of Oxidized Graphene. Nanomaterials 2020, 10, 279. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Li, C.; Shi, G. Functional Composite Materials based on Chemically Converted Graphene. Adv. Mater. 2011, 23, 1089–1115. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Chen, H.-M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Cote, L.J.; Cruz-Silva, R.; Huang, J. Flash reduction and patterning of graphene oxide and its polymer composite. J. Am. Chem. Soc. 2009, 131, 11027–11032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, L.; Wei, S.; He, Y.; Xia, H.; Chen, Q.; Sun, H.-B.; Xiao, F.-S. Direct imprinting of microcircuits on graphene oxides film by femtosecond laser reduction. Nano Today 2010, 5, 15–20. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.; Kohlhaas, K.M.; Zimmey, E.J.; Stach, E.A.; Piner, R.D.; Nquyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef]
- Periasamy, M.; Thirumalaikumar, M. Methods of enhancement of reactivity and selectivity of sodium borohydride for applications in organic chemistry. J. Organomet. Chem. 2000, 609, 137–151. [Google Scholar] [CrossRef]
- Bei, H.; Yang, Y.; Zhang, Q.; Tian, Y.; Luo, X.; Yang, M.; Zhao, X. Graphene-Based Nanocomposites for Neural Tissue Engineering. Molecules 2019, 24, 658. [Google Scholar] [CrossRef] [Green Version]
- Diez-Pascual, A.M. Chemical Functionalization of Carbon Nanotubes with Polymers: A Brief Overview. Macromol 2021, 1, 64–83. [Google Scholar] [CrossRef]
- Liu, J.; Ye, Y.; Xue, Y.; Xie, X.; Mai, Y. Recent advances in covalent functionalization of carbon nanomaterials with polymers: Strategies and perspectives. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Functional polymers by atom transfer radical polymerization. Prog. Polym. Sci. 2001, 26, 337–377. [Google Scholar] [CrossRef]
- Fang, M.; Wang, K.; Lu, H.; Yang, Y.; Nutt, S.J. Covalent polymer functionalization of Graphene nanosheets and mechancial properties of composites. J. Mater. Chem. 2009, 19, 7098–7105. [Google Scholar] [CrossRef]
- Gonçalves, G.; Marques Paula, A.A.P.; Barros-Timmons, A.; Bdkin, I.; Singh, M.K.; Emami, N.; Grácio, J. Graphene oxide modified with PMMA via ATRP as a reinforcement filler. J. Mater. Chem. 2010, 20, 9927–9934. [Google Scholar] [CrossRef] [Green Version]
- Roghani-Mamaqani, H.; Khezri, K. A grafting from approach to graft polystyrene chains at the surface of graphene nanolayers by RAFT polymerization: Various graft densities from hydroxyl groups. Appl. Surf. Sci. 2016, 360, 373–382. [Google Scholar] [CrossRef]
- Etmimi, H.M.; Tonge, M.P.; Sanderson, R.D. Synthesis and characterization of polystyrene-graphite nanocomposites via surface RAFT-mediated miniemulsion polymerization. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 1621–1632. [Google Scholar] [CrossRef]
- Chakraborty, G.; Gupta, A.; Pugazhenthi, G.; Katiyar, V. Facile dispersion of exfoliated graphene/PLA nanocomposites via in situ polycondensation with a melt extrusion process and its rheological studies. J. Appl. Polym. Sci. 2018, 135, 46476. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Guan, W. In situ ring-opening polymerization of poly(l-lactide)-graft-graphene oxide and its effect on the crystallization kinetics and morphology of biodegradable poly(l-lactide) at low loadings. RSC Adv. 2014, 4, 9463–9470. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, X.; Li, H.; Yan, C. Facile preparation route for graphene oxide reinforced polyamide 6 composites via in situ anionic ring-opening polymerization. J. Mater. Chem. 2012, 22, 24081–24091. [Google Scholar] [CrossRef]
- Huang, Y.; Qin, Y.; Zhou, Y.; Niu, H.; Yu, Z.; Dong, J. Polypropylene/Graphene Oxide Nanocomposites Prepared by In Situ Ziegler−Natta Polymerization. Chem. Mater. 2010, 22, 4096–4102. [Google Scholar] [CrossRef]
- Bahrami, H.; Ramazani, S.A.A.; Kheradmand, A.; Shafiee, M.; Baniasadi, H. Investigation of Thermomechanical Properties of UHMWPE/Graphene Oxide Nanocomposites Prepared by In situ Ziegler–Natta Polymerization. Adv. Polym. Technol. 2015, 34. [Google Scholar] [CrossRef]
- Luceño-Sánchez, J.A.; Maties, G.; Gonzalez-Arellano, C.; Diez-Pascual, A.M. Synthesis and Characterization of Graphene Oxide Derivatives via Functionalization Reaction with Hexamethylene Diisocyanate. Nanomaterials 2018, 8, 870. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zhen, W. Preparation and characterization of amidated graphene oxide and its effect on the performance of poly(lactic acid). Iranian Polym. J. 2018, 27, 239–252. [Google Scholar] [CrossRef]
- Barrera-Andrade, J.M.; Rojas-García, E.; García-Valdés, J.; Valenzuela, M.A.; Albiter, E. Incorporation of amide functional groups to graphene oxide during the photocatalytic degradation of free cyanide. Mater. Lett. 2020, 280, 128538. [Google Scholar] [CrossRef]
- Sturala, J.; Luxa, J.; Pumera, M.; Sofer, Z. Chemistry of Graphene Derivatives: Synthesis, Applications, and Perspectives. Chemistry 2018, 24, 5992–6006. [Google Scholar] [CrossRef]
- Yu, D.; Yang, Y.; Durstock, M.; Baek, J.-B.; Dai, L. Soluble P3HT-Grafted Graphene for Efficient Bilayer Heterojunction Photovoltaic Devices. ACS Nano 2010, 4, 5633–5640. [Google Scholar] [CrossRef] [Green Version]
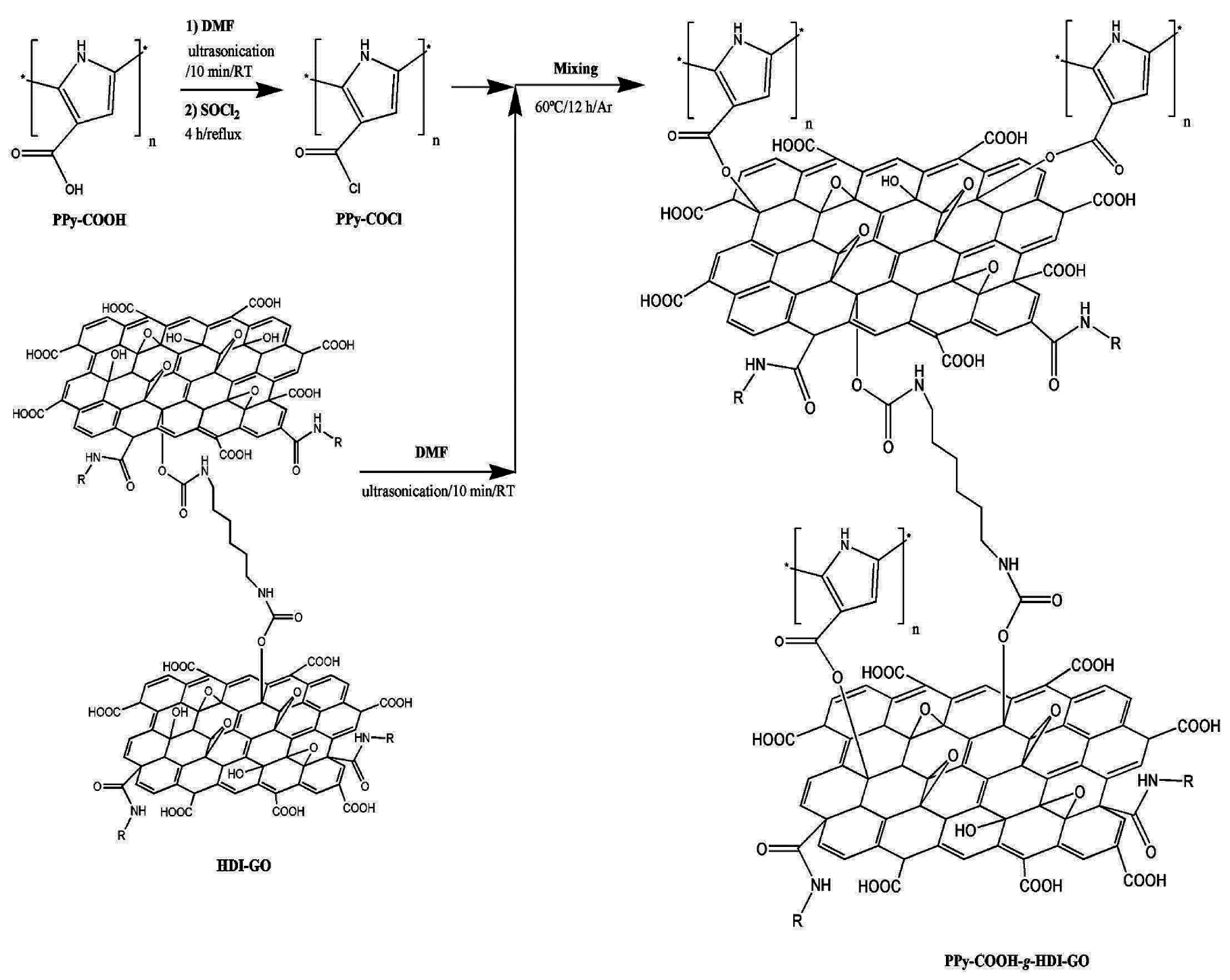
- Luceño Sánchez, J.A.; Peña Capilla, R.; Díez-Pascual, A.M. Grafting of Polypyrrole-3-carboxylic Acid to the Surface of Hexamethylene Diisocyanate-Functionalized Graphene Oxide. Nanomaterials 2019, 9, 1095. [Google Scholar] [CrossRef] [Green Version]
- Faghani, A.; Donskyi, I.S.; Fardin Gholami, M.; Ziem, B.; Lippitz, A.; Unger, W.E.S.; Böttcher, C.; Rabe, J.P.; Haag, R.; Adeli, M. Controlled Covalent Functionalization of Thermally Reduced Graphene Oxide to Generate Defined Bifunctional 2D Nanomaterials. Angew. Chem. 2017, 129, 2719–2723. [Google Scholar] [CrossRef]
- He, H.; Gao, C. General Approach to Individually Dispersed, Highly Soluble, and Conductive Graphene Nanosheets Functionalized by Nitrene Chemistry. Chem. Mater. 2010, 22, 5054–5064. [Google Scholar] [CrossRef]
- Li, J.; Zhang, G.; Deng, L.; Zhao, S.; Gao, Y.; Jiang, K.; Sun, R.; Wong, C. In situ polymerization of mechanically reinforced, thermally healable Graphene oxide/polyurethane composites based on Diels–Alder chemistry. J. Mater. Chem. A 2014, 2, 20642–20649. [Google Scholar] [CrossRef]
- Sarkar, S.; Bekyarova, E.; Niyogi, S.; Haddon, R.C. Diels−Alder Chemistry of Graphite and Graphene: Graphene as Diene and Dienophile. J. Am. Chem. Soc. 2011, 133, 3324–3327. [Google Scholar] [CrossRef]
- Li, H.; Cheng, F.; Duft, A.M.; Adronov, A. Functionalization of Single-Walled Carbon Nanotubes with Well-Defined Polystyrene by “Click” Coupling. J. Am. Chem. Soc. 2005, 127, 14518–14524. [Google Scholar] [CrossRef]
- Zhang, Y.; He, H.; Gao, C.; Wu, J. Covalent Layer-by-Layer Functionalization of Multiwalled Carbon Nanotubes by Click Chemistry. Langmuir 2009, 25, 5814–5824. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y. Noncovalent π⋅⋅⋅π interaction between graphene and aromatic molecule: Structure, energy, and nature. J. Chem. Phys. 2014, 140, 094302. [Google Scholar] [CrossRef]
- Pérez, E.M.; Martín, N. π–π interactions in carbon nanostructures. Chem. Soc. Rev. 2015, 44, 6425–6433. [Google Scholar] [CrossRef]
- Yu, W.; Sisi, L.; Haiyan, Y.; Jie, L. Progress in the functional modification of graphene/graphene oxide: A review. RSC Adv. 2020, 10, 15328–15345. [Google Scholar] [CrossRef]
- Liu, L.; Yang, W.; Tao, L.; Li, D. Thermosensitive Graphene nanocomposites formed using pyrene-terminal polymers made by RAFT polymerization. J. Appl. Polym. Sci. Part A 2010, 48, 425–433. [Google Scholar] [CrossRef]
- Liang, J.; Huang, Y.; Zhang, L.; Wang, Y. Molecular level dispersion of graphene into poly(vinyl alcohol) and effective reinforcement of their nanocomposites. Adv. Funct. Mater. 2009, 19, 2297–2302. [Google Scholar] [CrossRef]
- Luceño-Sánchez, J.A.; Diez-Pascual, A.M.; Peña Capilla, R.; Garcia-Diaz, P. The Effect of Hexamethylene Diisocyanate-Modified Graphene Oxide as a Nanofiller Material on the Properties of Conductive Polyaniline. Polymers 2019, 11, 1032. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.-H.; Jung, J.-M.; Hwang, I.-T.; Jung, C.-H. Preparation of polyacrylonitrile/Graphene oxide Nanocomposite-derived carbon microstructures by ion beam patterning and post-pyrolisis. Sci. Adv. Mater. 2016, 8, 1714–1718. [Google Scholar] [CrossRef]
- Hsieh, A.G.; Korkut, S.; Punckt, C.; Aksay, I.A. Dispersion Stability of Functionalized Graphene in Aqueous Sodium Dodecyl Sulfate Solutions. Langmuir 2013, 29, 14831–14838. [Google Scholar] [CrossRef]
- Luceño-Sánchez, J.A.; Peña Capilla, R.; Diez-Pascual, A.M. High-Performance PEDOT:PSS/Hexamethylene Diisocyanate- Functionalized Graphene Oxide Nanocomposites: Preparation and Properties. Polymers 2018, 10, 1169. [Google Scholar] [CrossRef] [Green Version]
- Chambers, B.A.; Notarianni, M.; Liu, J.; Motta, N.; Andersson, G.G. Examining the electrical and chemical properties of reduced graphene oxide with varying annealing temperatures in argon atmosphere. Appl. Surf. Sci. 2015, 356, 719–725. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, C.; Yue, B.; Gambhir, S.; Too, C.O.; Wallace, G.G. Electrochemically synthesized polypyrrole/Graphene composite film for lithium batteries. Adv. Energy Mater. 2012, 2, 266–272. [Google Scholar] [CrossRef]
- Guo, C.X.; Wang, M.; Chen, T.; Lou, X.W.; Li, C.M. Graphene based materials: Enhancing solar energy harvesting. Adv. Energy Mater. 2011, 1, 736–741. [Google Scholar] [CrossRef]
- Liang, R.L.; Cao, H.Q.; Qian, D.; Zhang, J.X.; Qu, M.Z. Designed synthesis of SnO2-polyaniline-reduced Graphene oxide nanocomposites as an anode material for lithium-ion battteries. J. Mater. Chem. 2011, 21, 17654–17657. [Google Scholar] [CrossRef]
- Liu, A.; Li, C.; Bai, H.; Shi, G. Electrochemical deposition of polypyrrole/sulfonated Graphene composite films. J. Phys. Chem. C 2010, 114, 22783–22789. [Google Scholar] [CrossRef]
- Zhang, L.L.; Zhao, S.; Tian, X.N.; Zhao, X.S. Layered Graphene Oxide Nanostructures with Sandwiched Conducting Polymers as Supercapacitor Electrodes. Langmuir 2010, 26, 17624–17628. [Google Scholar] [CrossRef]
- Luceño Sánchez, J.A.; Díez-Pascual, A.M.; Peña Capilla, R. Materials for Photovoltaics: State of Art and Recent Developments. Int. J. Mol. Sci. 2019, 20, 976. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, Y.; Liang, J.; Huang, Y.; Ma, Y.; Wan, X.; Chen, Y. A hybrid material of graphene and poly(3,4 ethyldioxythiophene) with high conductivity, flexibility and transparency. Nano Res. 2009, 2, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Wang, G.; Yang, A.; Tao, X.; Liu, X.; Shen, Y.; Zheng, Z. A transparent, flexible, low temperature and solution processible graphene composite electrode. Adv. Funct. Mater. 2010, 20, 2893–2902. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, W.; Chi, H.; Liu, Y.; Hou, C.L.; Fang, D. Recent development of graphene materials applied in polymer solar cell. Renew. Sustain. Energy Rev. 2015, 43, 973–980. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, Y.; Lee, J.Y.; Ahn, J.-H.; Park, J.H. Flexible and platinum-free dye sensitized solar cells with conducting-polymer coated Graphene counter electrodes. Chemsuschem 2012, 5, 379–382. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Q.; Huang, Y.; Ma, Y.; Yin, S.; Zhang, X.; Sun, W.; Chen, Y. Organic Photovoltaic Devices Based on a Novel Acceptor Material: Graphene. Adv. Mater. 2008, 20, 3924–3930. [Google Scholar] [CrossRef]
- Rafique, S.; Abdullah, S.M.; Shahid, M.M.; Ansari, M.O.; Sulaiman, K. Significantly improved photovoltaic performance in polymer bulk heterojunction solar cells with graphene oxide/PEDOT:PSS double decked hole transport layer. Sci. Rep. 2017, 7, 39555. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díez-Pascual, A.M. Development of Graphene-Based Polymeric Nanocomposites: A Brief Overview. Polymers 2021, 13, 2978. https://doi.org/10.3390/polym13172978
Díez-Pascual AM. Development of Graphene-Based Polymeric Nanocomposites: A Brief Overview. Polymers. 2021; 13(17):2978. https://doi.org/10.3390/polym13172978
Chicago/Turabian StyleDíez-Pascual, Ana M. 2021. "Development of Graphene-Based Polymeric Nanocomposites: A Brief Overview" Polymers 13, no. 17: 2978. https://doi.org/10.3390/polym13172978
APA StyleDíez-Pascual, A. M. (2021). Development of Graphene-Based Polymeric Nanocomposites: A Brief Overview. Polymers, 13(17), 2978. https://doi.org/10.3390/polym13172978