Synthesis of Well-Defined Gold Nanoparticles Using Pluronic: The Role of Radicals and Surfactants in Nanoparticles Formation


Abstract
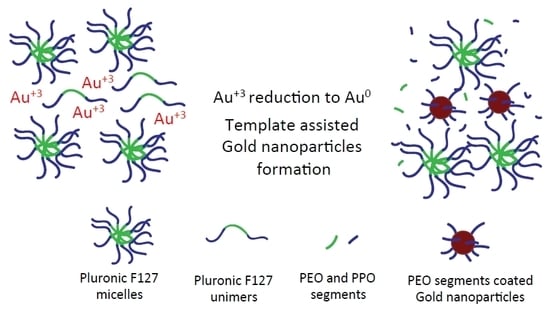
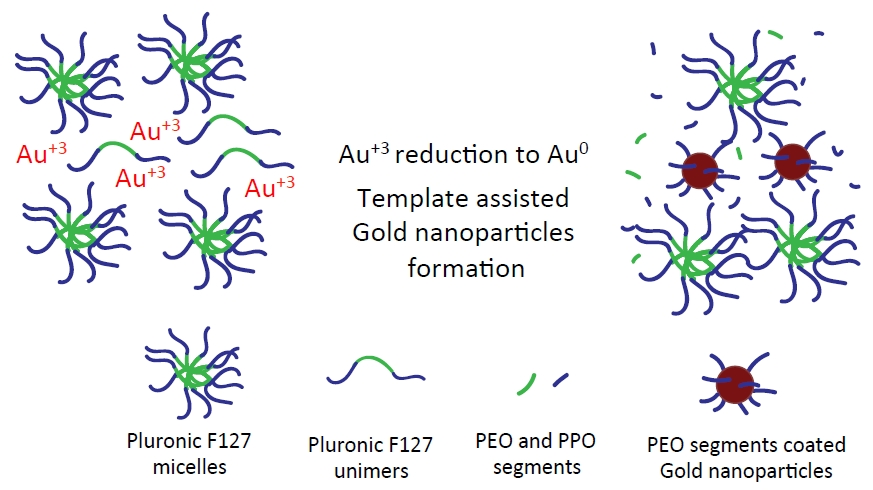
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of GNP
2.3. GNP Purification
2.4. UV–Vis Spectroscopy
2.5. Physico-Chemical Analysis
2.5.1. DLS Analysis
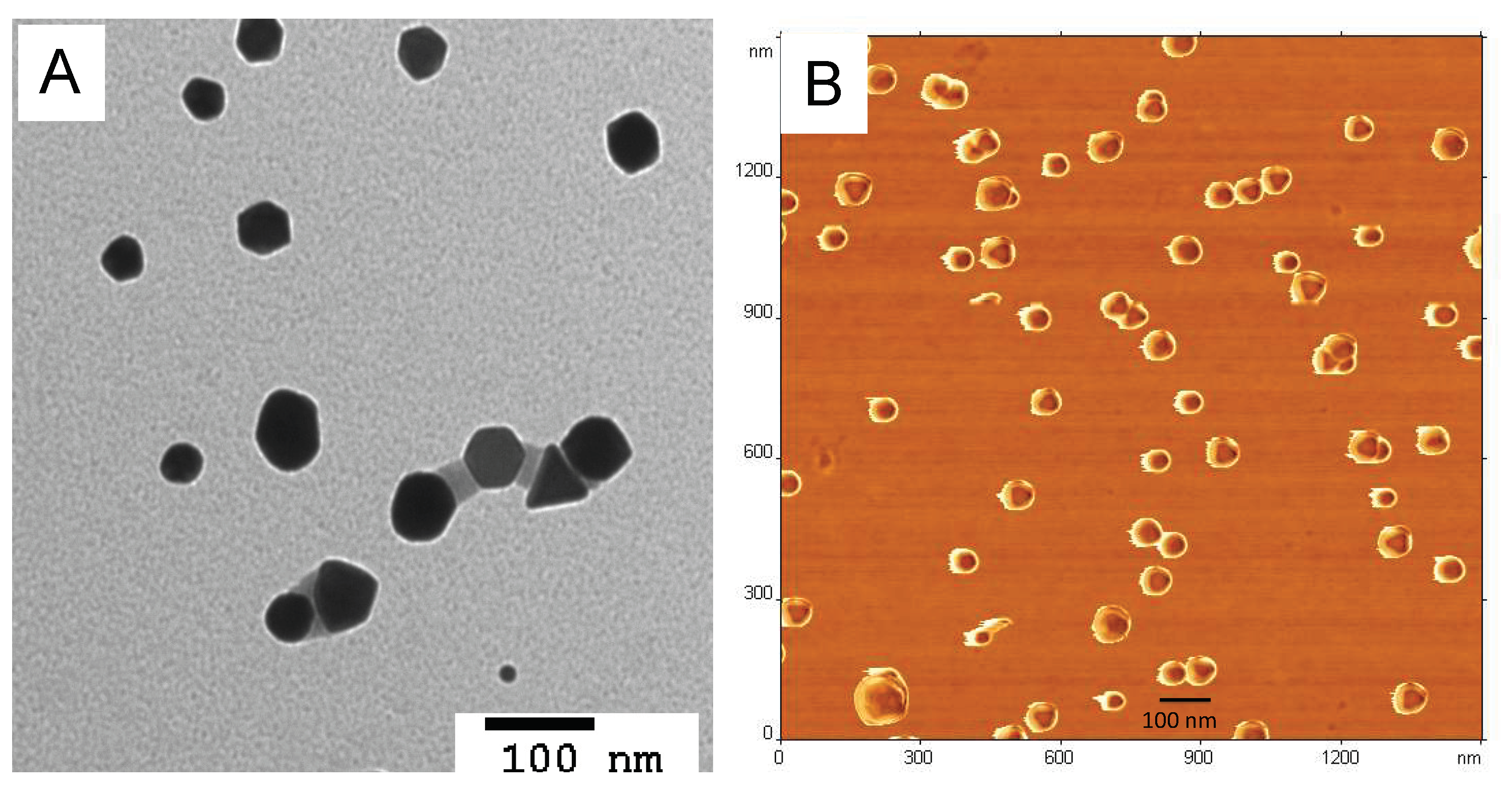
2.5.2. TEM Analysis
2.5.3. AFM Analysis
2.5.4. Surface Analysis of GNP and Reaction Products
2.5.5. FT-IR
2.5.6. NMR
2.5.7. GPC
2.5.8. TGA
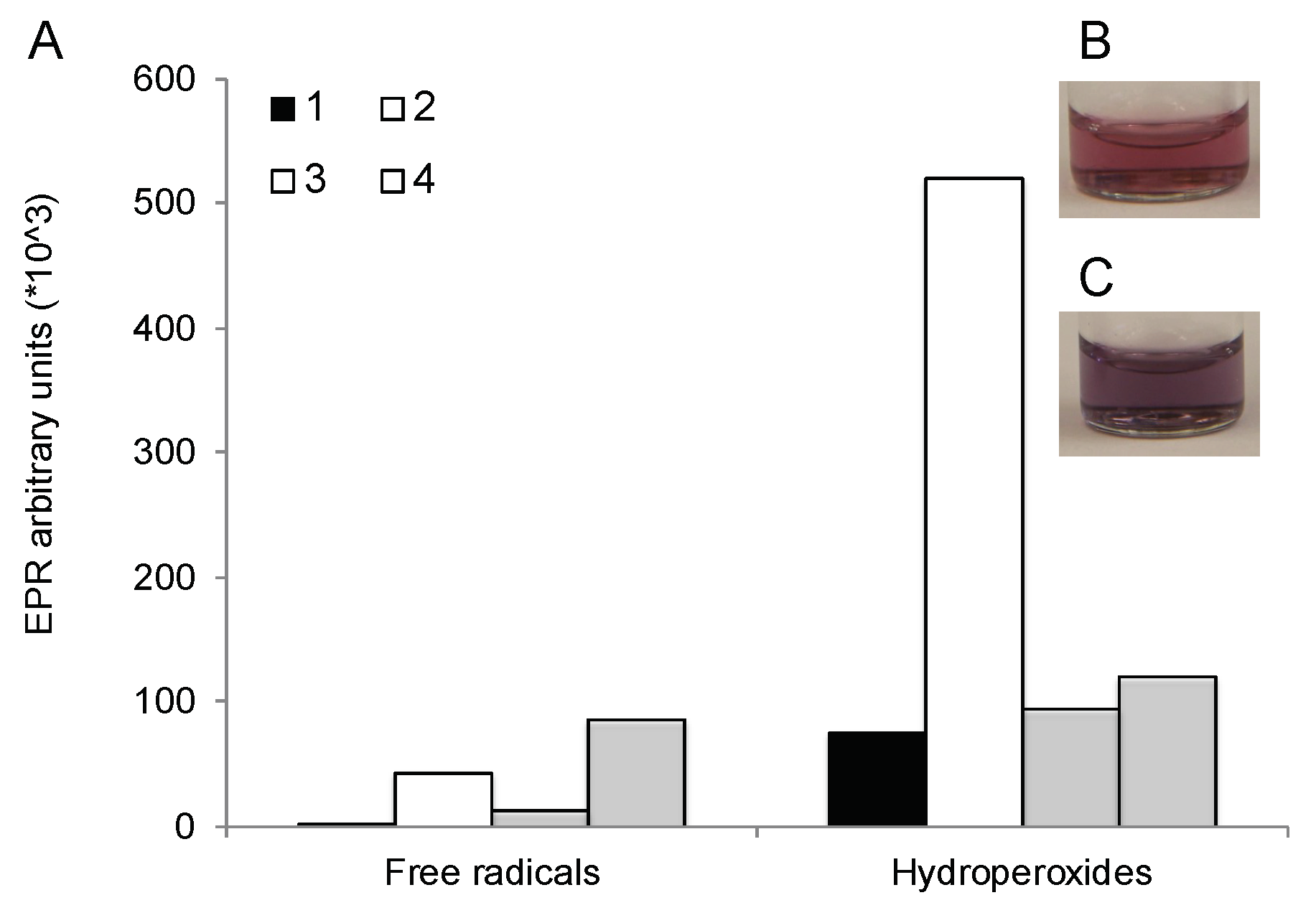
2.5.9. Free Radical and Hydroperoxide Assays
2.5.10. In Vitro Stability of Purified GNP
3. Results
3.1. Preliminary Considerations and Choice of a Lead Copolymer
3.2. Effect of the Temperature on Au3+ Reduction and GNP Formation
3.3. Effect of HAuCl4 Concentration on the GNP Formation and Characteristics
3.4. Effect of F127 Concentration on the GNP Formation and Characteristics
3.5. Relationship between Polymer Aggregation and GNP Formation
3.6. Effect of pH on the GNP Formation and Characteristics
3.7. Effect of NaCl on the GNP Formation
3.8. Chemical Conversion of the Block Copolymer During the Reaction
3.9. Batch to Batch Variation in the Formation of GNP
3.10. Effect of Lyophilization of the Copolymer Solutions on GNP Formation
3.11. Formation of Reacting Oxygen Species during the Reaction
3.12. Mechanism of HAuCl4·3H2O Reduction by Pluronic
- The decomposition of the hydroperoxides present in Pluronic leads to Pluronic chains degradation and formation of lower molecular mass alcohols. This process results in the release of a hydroxonium ion that contributes to acidification of the media and formation of the superoxide O2−• and hydroxyl radical HO• or hydrogen peroxide that can participate in further redox reactions. Here we present the oxidation and decomposition of the PPO block because it is known to degrade faster than the PEO [20]. However, similar processes can also proceed at the PEO block and PEG homopolymer.

 2HO• → H2O2
2HO• → H2O2 - The reactive oxygen species formed in (1), (2), and (3) promote the reduction of Au3+. Since tetracloroauric ion [AuCl4]− undergoes acid and alkalli hydrolysis and gradually transforms into tetrahydroxoaurate ion [Au(OH)4]− at basic conditions [21] the reduction reactions proceed through different mechanisms depending on pH. Most likely these reactions are complex and involve multiple elementary steps and intermediates. Their formal description is further complicated by the coexistence of several forms of aquachlorohydroxo complexes of gold(III) that can participate in the reducton with varying reactivity. Therefore, we present only the simplified schemes of the overall reactions with either tetracloroauric or tetrahydroxoaurate ions at acidic or alkaline conditions, respectively. We also would like to point out that the reactivities of the superoxide radical O2−• and hyrogen peroxide can depend on pH. The superoxide radical at low pH exists mainly in the protonated form of the hydroperoxyl radical HO2• (pKa ~4.8), while the hyrogen peroxide (pKa ~11.6) at alkaline conditions can fom the hydroperoxide anion HO2−.In acidic conditionsO2−• + H3O+ → HO2• + H2O[AuCl4]− + 3HO2• + 3H2O → Au0 + 3O2 + 3H3O+ + 4Cl−[AuCl4]− + 3O2−• → Au0 + 3O2 + 4Cl−2[AuCl4]− + 3H2O2 + 6H2O → 2Au0 + 3O2 + 6H3O+ + 8Cl−In alkaline conditionsAu(OH)4− + 3O2−• → Au0 + 3O2 + 4OH−H2O2 + OH− → H2O + HO2−2Au(OH)4− + 3HO2− → 2Au0 + 3O2 + 5OH− + 3H2OReactions (5), (6), (7), (8), and (10) are reversible, but we assume that their equilibria are shifted to the right due to the rapid consumption of the molecular oxygen in subsequent reactions with Pluronic. The latter may be accelerated by the complexation of [AuCl4]− and related gold complexes with the block copolymer chains, as in this case the polymer oxidation may proceed either concurrently or immediately after the reduction of the gold ions.
- Finally, the molecular oxygen formed in the reduction reactions (5), (6), (7), (8), and (10) propagates hydroxyperoxidation of either PPO or PEO chains in Pluronic.




4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed]
- Nash, M.A.; Lai, J.J.; Hoffman, A.S.; Yager, P.; Stayton, P.S. Smart Diblock Copolymers as Templates for Magnetic-Core Gold-Shell Nanoparticle Synthesis. Nano Lett. 2010, 10, 85–91. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oishi, M.; Hayashi, H.; Uno, T.; Ishii, T.; Iijima, M.; Nagasaki, Y. One-pot synthesis of pH-responsive PEGylated nanogels containing gold nanoparticles by autoreduction of chloroaurate ions within nanoreactors. Macromol. Chem. Phys. 2007, 208, 1176–1182. [Google Scholar] [CrossRef]
- Sun, X.P.; Jiang, X.; Dong, S.J.; Wang, E.K. One-step synthesis and size control of dendrimer-protected gold nanoparticles: A heat-treatment-based strategy. Macromol. Rapid. Commun. 2003, 24, 1024–1028. [Google Scholar] [CrossRef]
- Toroz, D.; Corni, S. Peptide synthesis of gold nanoparticles: The early steps of gold reduction investigated by density functional theory. Nano Lett. 2011, 11, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Longenberger, L.; Mills, G. Formation of Metal Particles in Aqueous Solutions by Reactions of Metal Complexes with Polymers. J. Phys. Chem. 1995, 99, 475–478. [Google Scholar] [CrossRef]
- Sakai, T.; Alexandridis, P. Spontaneous formation of gold nanoparticles in poly (ethylene oxide)-poly (propylene oxide) solutions: Solvent quality and polymer structure effects. Langmuir 2005, 21, 8019–8025. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Alexandridis, P. Size- and shape-controlled synthesis of colloidal gold through autoreduction of the auric cation by poly (ethylene oxide)-poly (propylene oxide) block copolymers in aqueous solutions at ambient conditions. Nanotechnology 2005, 16, S344–S353. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Alexandridis, P. Mechanism of gold metal ion reduction, nanoparticle growth and size control in aqueous amphiphilic block copolymer solutions at ambient conditions. J. Phys. Chem. B 2005, 109, 7766–7777. [Google Scholar] [CrossRef]
- Shou, Q.; Guo, C.; Yang, L.; Jia, L.; Liu, C.; Liu, H. Effect of pH on the single-step synthesis of gold nanoparticles using PEO-PPO-PEO triblock copolymers in aqueous media. J. Colloid Interface Sci. 2011, 363, 481–489. [Google Scholar] [CrossRef]
- Chen, S.; Guo, C.; Hu, G.H.; Wang, J.; Ma, J.H.; Liang, X.F.; Zheng, L.; Liu, H.Z. Effect of hydrophobicity inside PEO-PPO-PEO block copolymer micelles on the stabilization of gold nanoparticles: Experiments. Langmuir 2006, 22, 9704–9711. [Google Scholar] [CrossRef] [PubMed]
- Khullar, P.; Singh, V.; Mahal, A.; Kaur, H.; Banipal, T.S.; Kaur, G.; Bakshi, M.S. Tuning the Shape and Size of Gold Nanoparticles with Triblock Polymer Micelle Structure Transitions and Environments. J. Phys. Chem. C 2011, 115, 10442–10454. [Google Scholar] [CrossRef]
- Polte, J.; Emmerling, F.; Radtke, M.; Reinholz, U.; Riesemeier, H.; Thunemann, A.F. Real-Time Monitoring of Copolymer Stabilized Growing Gold Nanoparticles. Langmuir 2010, 26, 5889–5894. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Aswal, V.K.; Kohlbrecher, J. Synthesis and Characterization of High Concentration Block Copolymer-Mediated Gold Nanoparticles. Langmuir 2011, 27, 4048–4056. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Bohorquez, M.; Koch, C.; Trygstad, T.; Pandit, N. A study of the temperature-dependent micellization of pluronic F127. J. Colloid Interface Sci. 1999, 216, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Nukolova, N.V.; Oberoi, H.S.; Cohen, S.M.; Kabanov, A.V.; Bronich, T.K. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials 2011, 32, 5417–5426. [Google Scholar] [CrossRef]
- Innocenzi, P.; Malfatti, L.; Piccinini, M.; Marcelli, A. Evaporation-Induced Crystallization of Pluronic F127 Studied in Situ by Time-Resolved Infrared Spectroscopy. J. Phys. Chem. A 2010, 114, 304–308. [Google Scholar] [CrossRef]
- Kumar, V.; Kalonia, D.S. Removal of peroxides in polyethylene glycols by vacuum drying: Implications in the stability of biotech and pharmaceutical formulations. Aaps Pharmscitech 2006, 7, E47. [Google Scholar] [CrossRef]
- Erlandsson, B. Stability-indicating changes in poloxamers: The degradation of ethylene oxide-propylene oxide block copolymers at 25 and 40 °C. Polym. Degrad. Stab. 2002, 87, 571–575. [Google Scholar] [CrossRef]
- Mironov, I.V.; Makotchenko, E.V. The Hydrolysis of AuCl4- and the Stability of Aquachlorohydroxocomplexes of Gold(III) in Aqueous Solution. J. Solut. Chem. 2009, 38, 725–737. [Google Scholar] [CrossRef]
- Abdullin, T.I.; Bondar, O.V.; Shtyrlin, Y.G.; Kahraman, M.; Culha, M. Pluronic Block Copolymer-Mediated Interactions of Organic Compounds with Noble Metal Nanoparticles for SERS Analysis. Langmuir 2010, 26, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.U.; Cha, S.H.; Shin, K.; Jho, J.Y.; Lee, J.C. Preparation of gold nanowires and nanosheets in bulk block copolymer phases under mild conditions. Adv. Mater. 2004, 16, 459–464. [Google Scholar] [CrossRef]
- Paclawski, K.; Fitzner, K. Kinetics of Gold (III) Chloride Complex Reduction Using Sulfur (IV). Metall. Mater. Trans. B 2004, 35, 1071–1085. [Google Scholar] [CrossRef]
- Paclawski, K.; Fitzner, K. Kinetics of Reduction of Gold(III) Complexes Using H2O2. Metall. Mater. Trans. B 2006, 37, 703–714. [Google Scholar] [CrossRef]
- Rahme, K.; Gauffre, F.; Marty, J.D.; Payre, B.; Mingotaud, C. A systematic study of the stabilization in water of gold nanoparticles by poly (ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) triblock copolymers. J. Phys. Chem. C 2007, 111, 7273–7279. [Google Scholar] [CrossRef]
- Straney, P.J.; Andolina, C.M.; Millstone, J.E. Seedless Initiation as an Efficient, Sustainable Route to Anisotropic Gold Nanoparticles. Langmuir 2013, 29, 4396–4403. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.J.; Thompson, L.B.; Chernak, D.J.; Yang, J.A.; Sivapalan, S.T.; Boulos, S.P.; Huang, J.; Alkilany, A.M.; Sisco, P.N. Gold nanorod crystal growth: From seed-mediated synthesis to nanoscale sculpting. Curr. Opin. Colloid Interface Sci. 2011, 16, 128–134. [Google Scholar] [CrossRef]
- Goy-Lopez, S.; Taboada, P.; Cambon, A.; Juarez, J.; Alvarez-Lorenzo, C.; Concheiro, A.; Mosquera, V. Modulation of Size and Shape of Au Nanoparticles Using Amino-X-Shaped Poly (ethylene oxide)-Poly (propylene oxide) Block Copolymers. J. Phys. Chem. B 2010, 114, 66–76. [Google Scholar] [CrossRef]
- Genc, R.; Clergeaud, G.; Ortiz, M.; O’Sullivan, C.K. Green Synthesis of Gold Nanoparticles Using Glycerol-Incorporated Nanosized Liposomes. Langmuir 2011, 27, 10894–10900. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, D.W.; Shin, S.I.; Oh, S.G. Preparation of Au Colloids by Polyol Process Using NaHCO3 as a Buffering Agent. Mater. Chem. Phys. 2006, 100, 85–91. [Google Scholar] [CrossRef]
- Steinfeldt, N. In Situ Monitoring of Pt Nanoparticle Formation in Ethylene Glycol Solution by SAXS-Influence of the NaOH to Pt Ratio. Langmuir 2012, 28, 13072–13079. [Google Scholar] [CrossRef]
- Treguer-Delapierre, M.; Majimel, J.; Mornet, S.; Duguet, E.; Ravaine, S. Synthesis of non-spherical gold nanoparticles. Gold Bull. 2008, 41, 195–207. [Google Scholar] [CrossRef]
- Pandit, N.K.; Kisaka, J. Loss of gelation ability of Pluronic(R) F127 in the presence of some salts. Int. J. Pharm. 1996, 145, 129–136. [Google Scholar] [CrossRef]
- Su, Y.L.; Liu, H.Z.; Wang, J.; Chen, J.Y. Study of salt effects on the micellization of PEO-PPO-PEO block copolymer in aqueous solution by FTIR spectroscopy. Langmuir 2002, 18, 865–871. [Google Scholar] [CrossRef]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Carabineiro, S.A.C. Applications of Gold Nanoparticles in Nanomedicine: Recent Advances in Vaccines. Molecules 2017, 22, 857. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HAuCl4 Concentration (mM) | DLS (Reaction Mixture) | DLS (Purified Particles) | Morphology by TEM | ||||
|---|---|---|---|---|---|---|---|
| Deff (nm) | PDI | ζ-Potential (mV) | Deff (nm) | PDI | ζ-Potential (mV) | ||
| 0.05 | 50 ± 1 | 0.53 ± 0.02 | −3.76 ± 0.11 | 77 ± 16 | 0.18 ± 0.04 | −30.23 ± 0.05 | Multi-sized spheres |
| 0.10 | 122 ± 1 | 0.34 ± 0.001 | −3.52 ± 0.03 | 62 ± 1 | 0.25 ± 0.01 | −28.13 ± 1.29 | Multi-sized spheres |
| 0.15 | 216 ± 1 | 0.23 ± 0.01 | −3.18 ± 0.01 | 79 ± 1 | 0.19 ± 0.01 | −28.70 ± 3.41 | Triangles, rods, spheres |
| 0.20 | 231 ± 1 | 0.22 ± 0.02 | −2.45 ± 0.02 | 87 ± 1 | 0.14 ± 0.03 | −27.47± 0.06 | Triangles, plates, spheres |
| 0.25 | 229 ± 2 | 0.23 ± 0.01 | −3.01 ± 0.02 | 72 ± 1 | 0.12 ± 0.02 | −37.73 ± 0.25 | Spheres, triangles |
| F127 Concentration (% w/w) | DLS (Reaction Mixture) | DLS (Purified Particles) | Morphology by TEM | ||||
|---|---|---|---|---|---|---|---|
| Deff (nm) | PDI | ζ-Potential (mV) | Deff (nm) | PDI | ζ-Potential (mV) | ||
| 1.0 | 121 ± 1 | 0.28 ± 0.01 | −12.13 ± 0.30 | 80 ± 1 | 0.29± 0.01 | −25.57 ± 0.40 | Multi-sized spheres, rods, hexagons |
| 2.0 | 91 ± 1 | 0.43 ± 0.01 | −6.45 ± 0.30 | 54 ± 1 | 0.42 ± 0.03 | −30.20 ± 0.66 | Multi-sized spheres, rods, hexagons, triangles |
| 3.15 | 101 ± 2 | 0.28 ± 0.01 | −5.52 ± 0.02 | 62 ± 1 | 0.29 ± 0.003 | −28.20 ± 0.20 | Spheres, triangles, rods |
| 5.0 | 133 ± 2 | 0.22 ± 0.01 | −4.04 ± 0.20 | 63 ± 1 | 0.19 ± 0.02 | −32.23 ± 1.85 | Spheres, triangles |
| 6.3 | 153 ± 12 | 0.23 ± 0.01 | −3.84 ± 0.06 | 58 ± 1 | 0.17 ± 0.01 | −36.50 ± 0.38 | Spheres, triangles |
| 7.5 | 227 ± 6 | 0.17 ± 0.02 | −3.76 ± 0.20 | 75 ± 1 | 0.15 ± 0.01 | −41.80 ± 0.56 | Spheres |
| 10.0 | 305 ± 3 | 0.11 ± 0.02 | −2.83 ± 0.30 | 86 ± 1 | 0.09 ± 0.01 | −45.30 ± 0.15 | Uniform spheres |
| pH of the F127 Solution a | DLS (Reaction Mixture) | DLS (Purified Particles) | TEM (Purified Particles) | |||||
|---|---|---|---|---|---|---|---|---|
| Deff (nm) | PDI | ζ-Potential (mV) | Deff (nm) | PDI | ζ-Potential (mV) | Deff (nm) | Morpho-Logy | |
| 2.2 (1.24) 2 | 375 ± 3 | 0.11 ± 0.3 | −2.7 ± 0.05 | 114 ± 1 | 0.17 ± 0.02 | −25.8 ± 3.13 | n.d. | Spheres, rods, triangles |
| 4.5 (1.44) 2 | 219 ± 3 | 0.20 ± 0.24 | −3.07 ± 0.13 | 85 ± 1 | 0.16 ± 0.01 | −32.53 ± 0.58 | 77 ± 6 | Spheres |
| 6.7 (3.14) 3 | 160 ±2 | 0.19 ± 0.04 | −3.98 ± 0.18 | 72 ± 1 | 0.12 ± 0.02 | −37.70 ± 0.25 | 54 ± 13 | Spheres |
| 7.5 (5.5) 4 | 115 ±3 | 0.36 ± 0.04 | −4.50 ± 0.66 | 57 ± 1 | 0.26 ± 0.01 | −33.77 ± 0.66 | 23 ± 11 | Spheres |
| 9.0 (6.35) 4 | 56 ±1 | 0.56 ± 0.01 | −4.45 ± 0.21 | 53 ± 1 | 0.36 ± 0.02 | −35.40 ± 1.37 | 12 ± 6 | Spheres |
| 11.5 (7.76) 4 | 65 ± 4 | 0.44 ± 0.02 | −5.20 ± 0.62 | 41 ± 1 | 0.36 ± 0.03 | −32.06 ± 1.45 | 12 ± 4 | Spheres |
| F127 Batch | GNP Synthesized Using Non-Lyophilized F127 | GNP Synthesized Using Lyophilized F127 | ||
|---|---|---|---|---|
| Deff (nm) | PDI | Deff (nm) | PDI | |
| F127_1 | 62 ± 1 | 0.16 ± 0.01 | 47 ± 1 | 0.30 ± 0.004 |
| F127_2 | 157 ± 2 | 0.06 ± 0.02 | 70 ± 1 | 0.07 ± 0.01 |
| F127_3 | Not formed | Not formed | 57 ± 1 | 0.30 ± 0.04 |
| F127_3 (pH 6.5) | 108 ± 2 | 0.17 ± 0.003 | n/a | n/a |
| F127_3 (pH 11.6) | 39 ± 3 | 0.51 ± 0.12 | n/a | n/a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokolsky-Papkov, M.; Kabanov, A. Synthesis of Well-Defined Gold Nanoparticles Using Pluronic: The Role of Radicals and Surfactants in Nanoparticles Formation. Polymers 2019, 11, 1553. https://doi.org/10.3390/polym11101553
Sokolsky-Papkov M, Kabanov A. Synthesis of Well-Defined Gold Nanoparticles Using Pluronic: The Role of Radicals and Surfactants in Nanoparticles Formation. Polymers. 2019; 11(10):1553. https://doi.org/10.3390/polym11101553
Chicago/Turabian StyleSokolsky-Papkov, Marina, and Alexander Kabanov. 2019. "Synthesis of Well-Defined Gold Nanoparticles Using Pluronic: The Role of Radicals and Surfactants in Nanoparticles Formation" Polymers 11, no. 10: 1553. https://doi.org/10.3390/polym11101553
APA StyleSokolsky-Papkov, M., & Kabanov, A. (2019). Synthesis of Well-Defined Gold Nanoparticles Using Pluronic: The Role of Radicals and Surfactants in Nanoparticles Formation. Polymers, 11(10), 1553. https://doi.org/10.3390/polym11101553