Bimetallic Aluminum 5,6-Dihydro-7,7-dimethyl quinolin-8-olates as Pro-Initiators for the ROP of ε-CL; Probing the Nuclearity of the Active Initiator


 ,
, 
Abstract
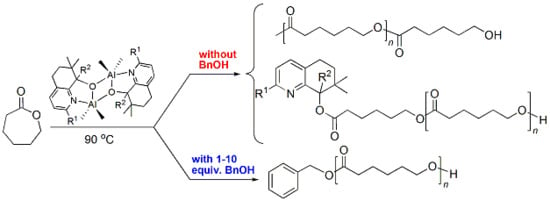
:
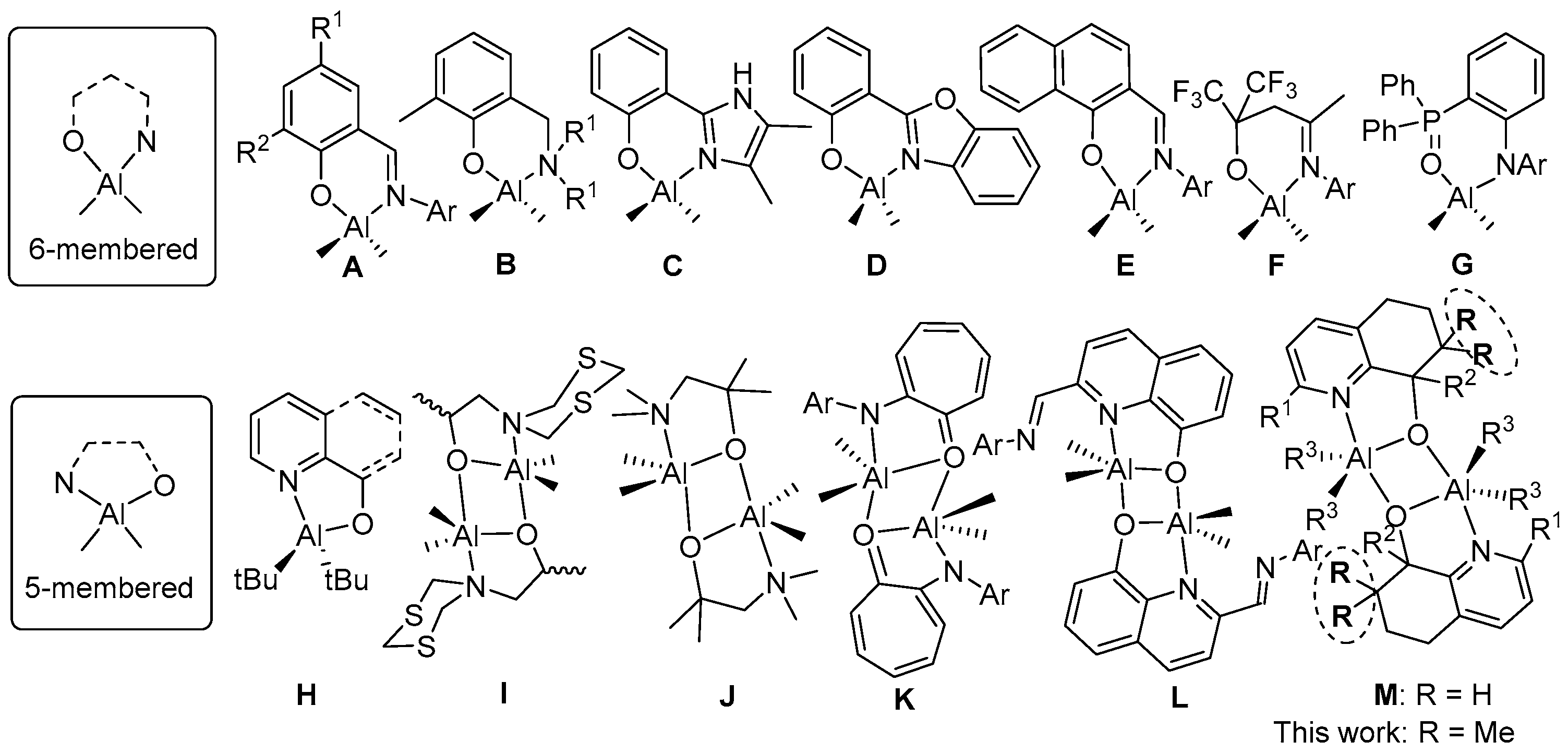
1. Introduction
2. Materials and Methods
2.1. General Considerations and Materials
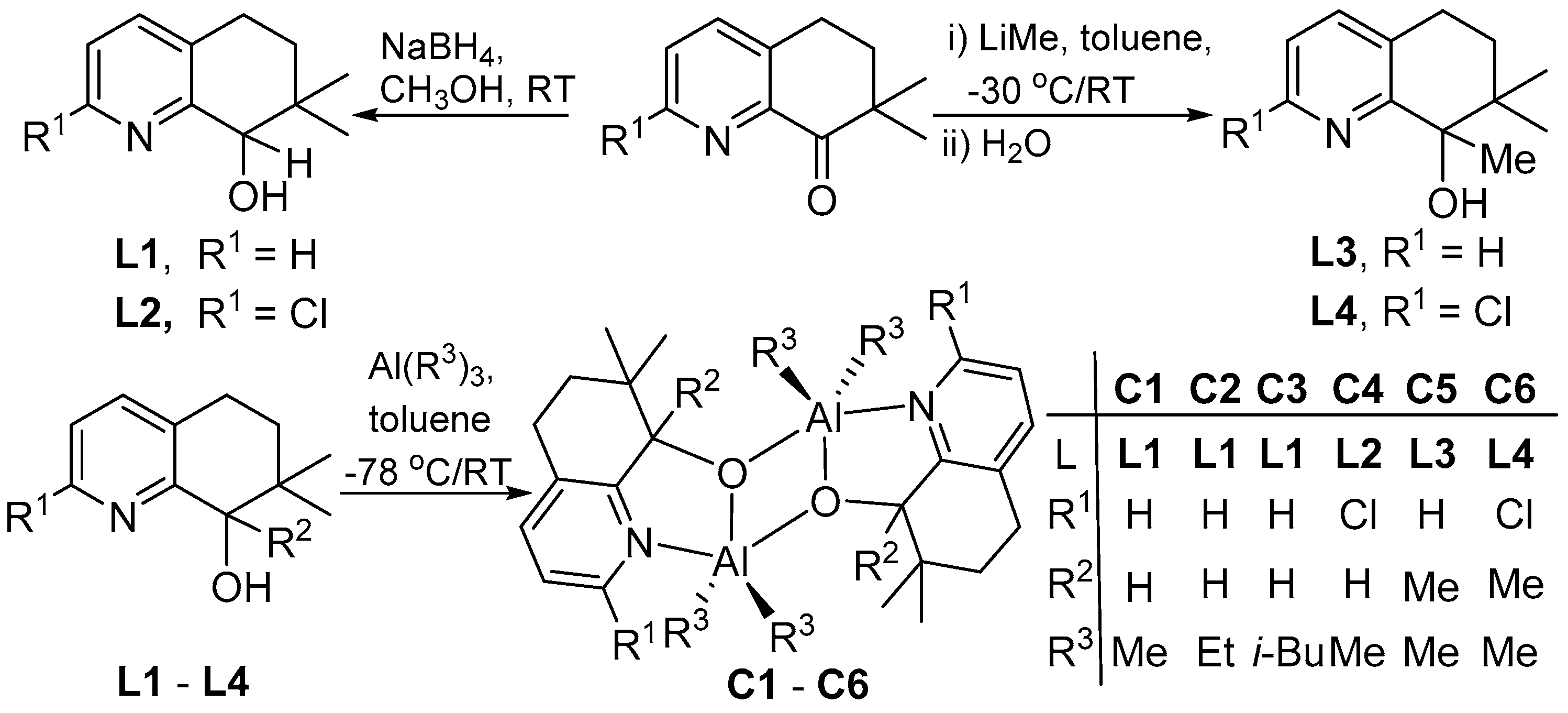
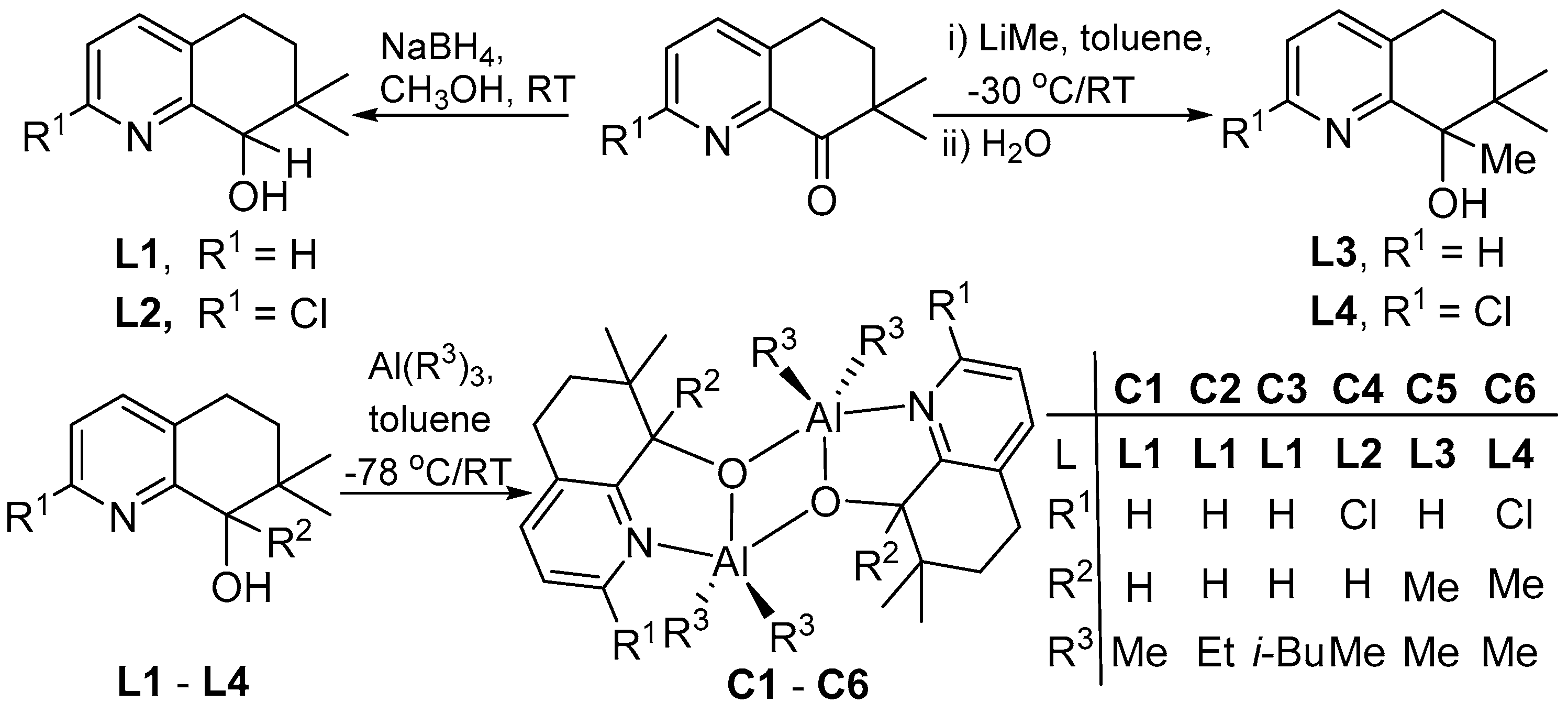
2.2. Syntheses of 2-R1-7,7-Me2,8-R2C9H6N-8-OH (L)
2.3. Syntheses of [{2-R1-7,7-Me2-8-R2C9H6N-8-O}AlR32]2 (C)
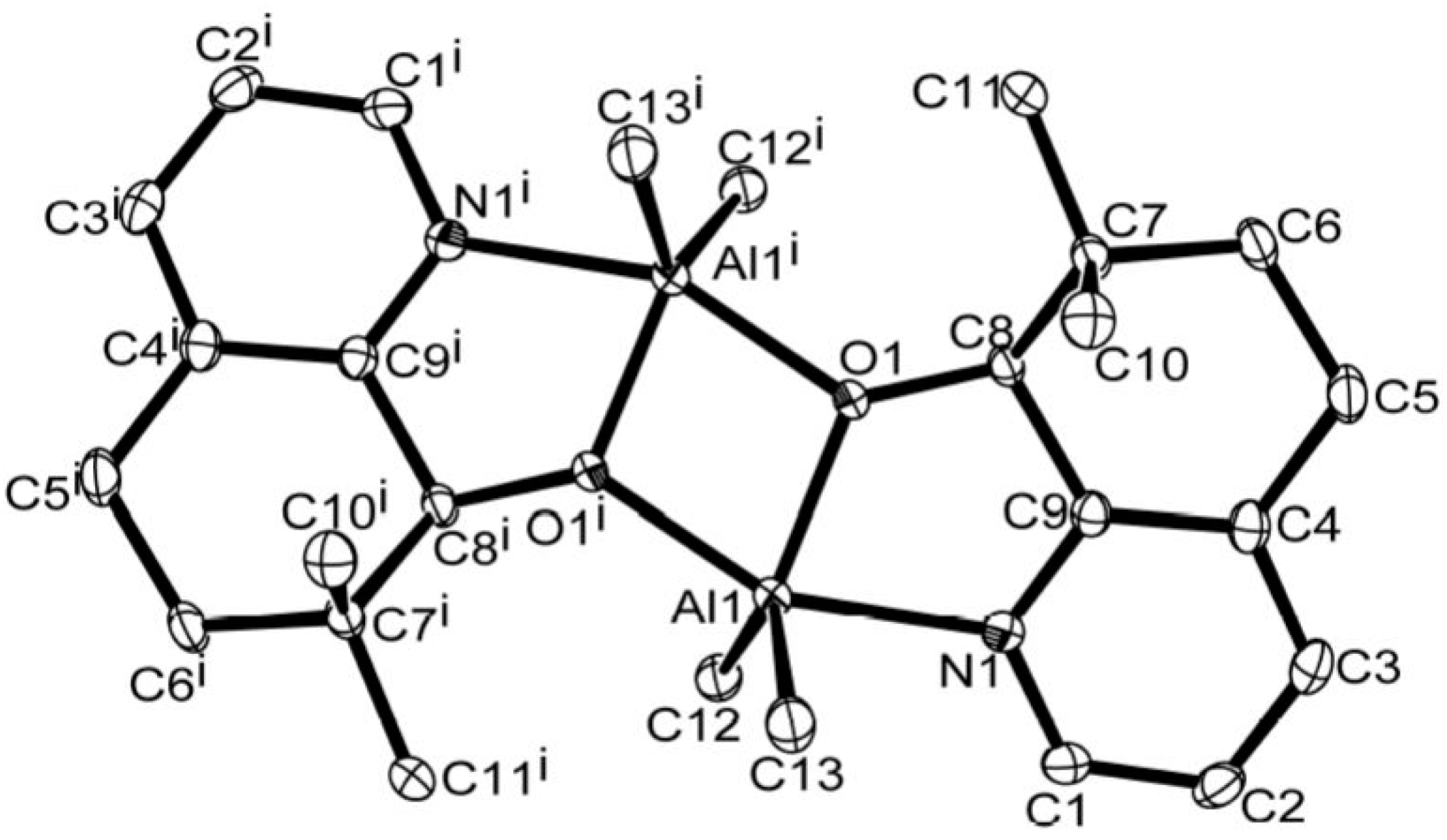
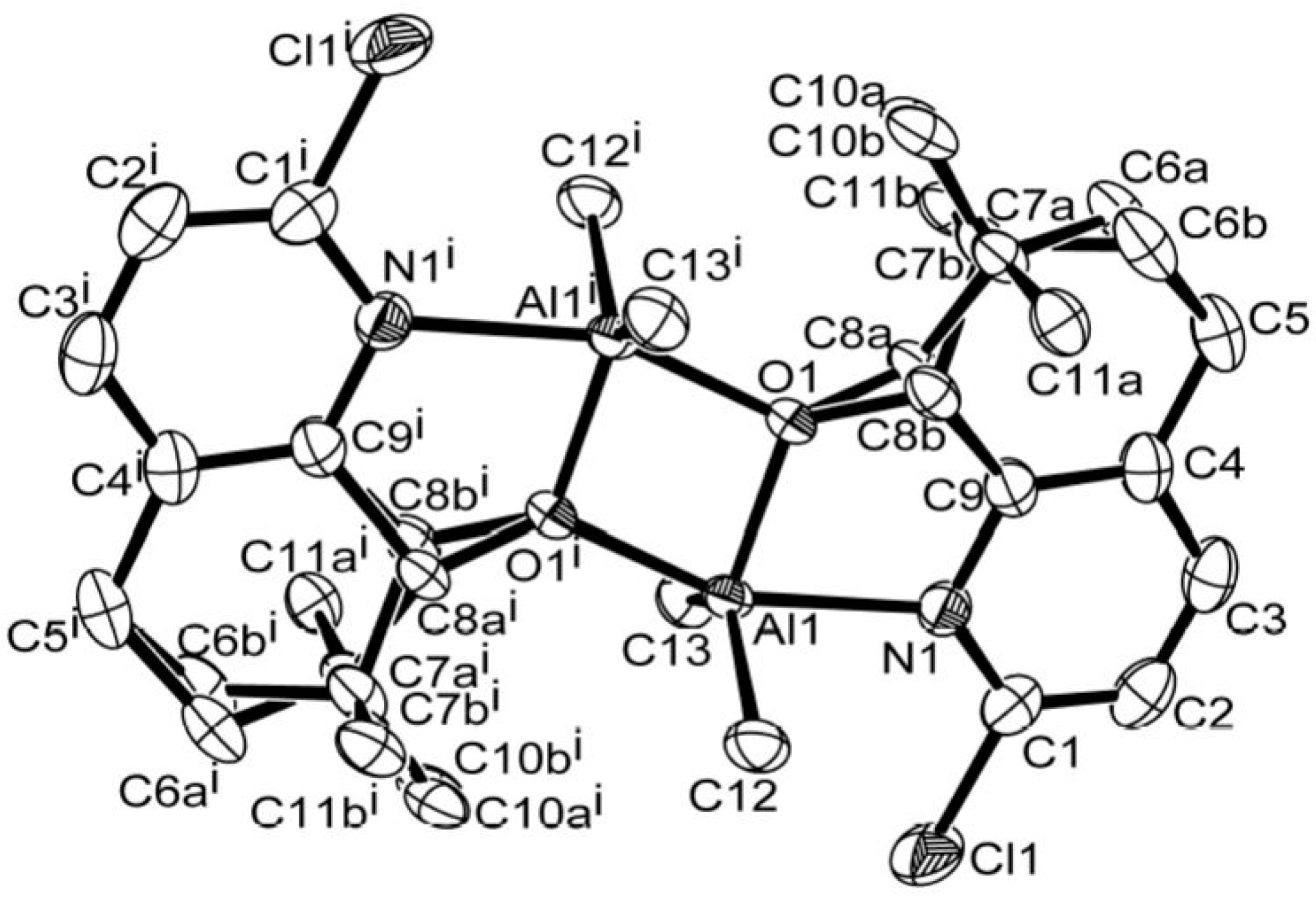
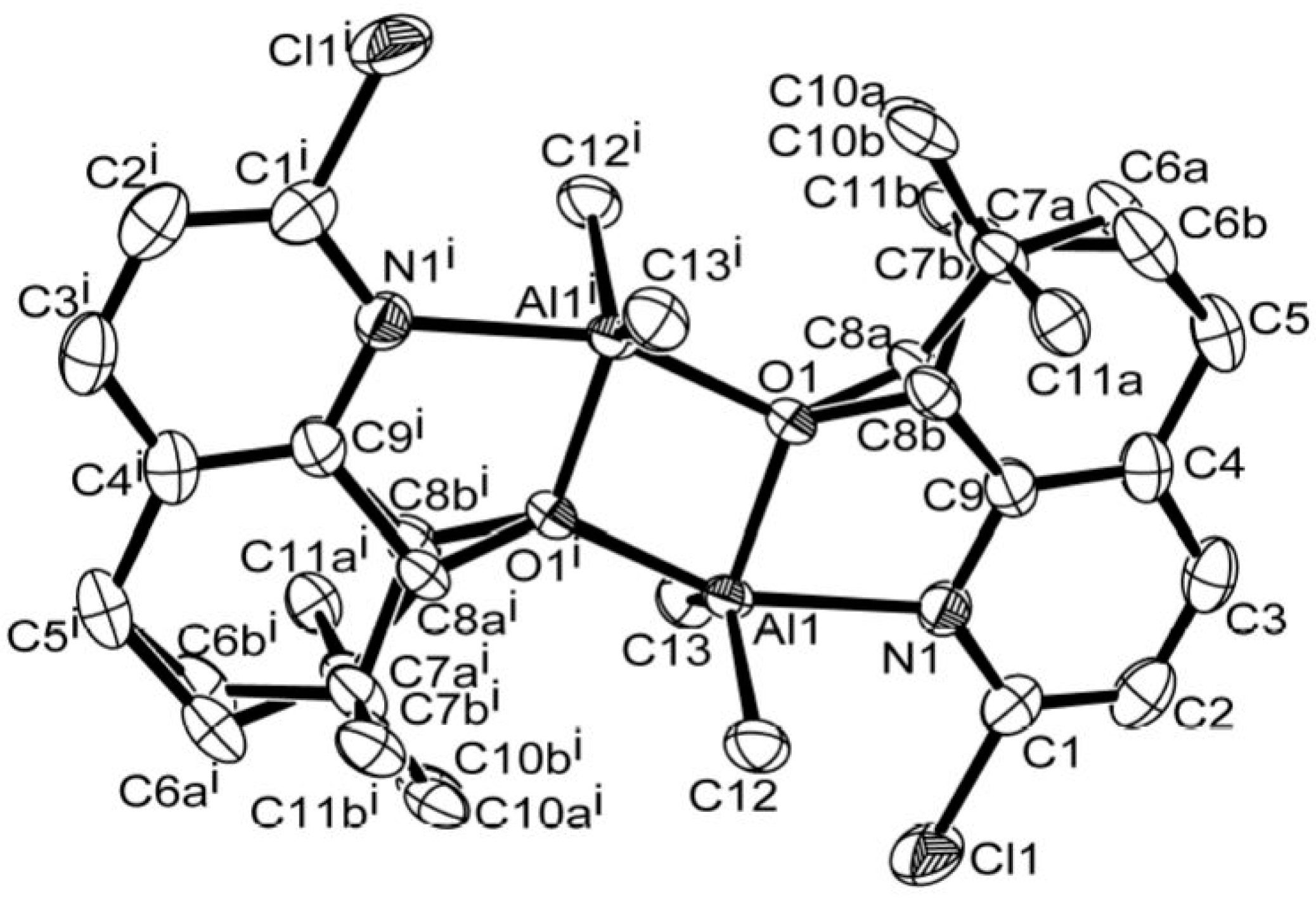
2.4. X-ray Crystallographic Studies
2.5. General Procedure for ε-Caprolactone Polymerization
3. Results and Discussion
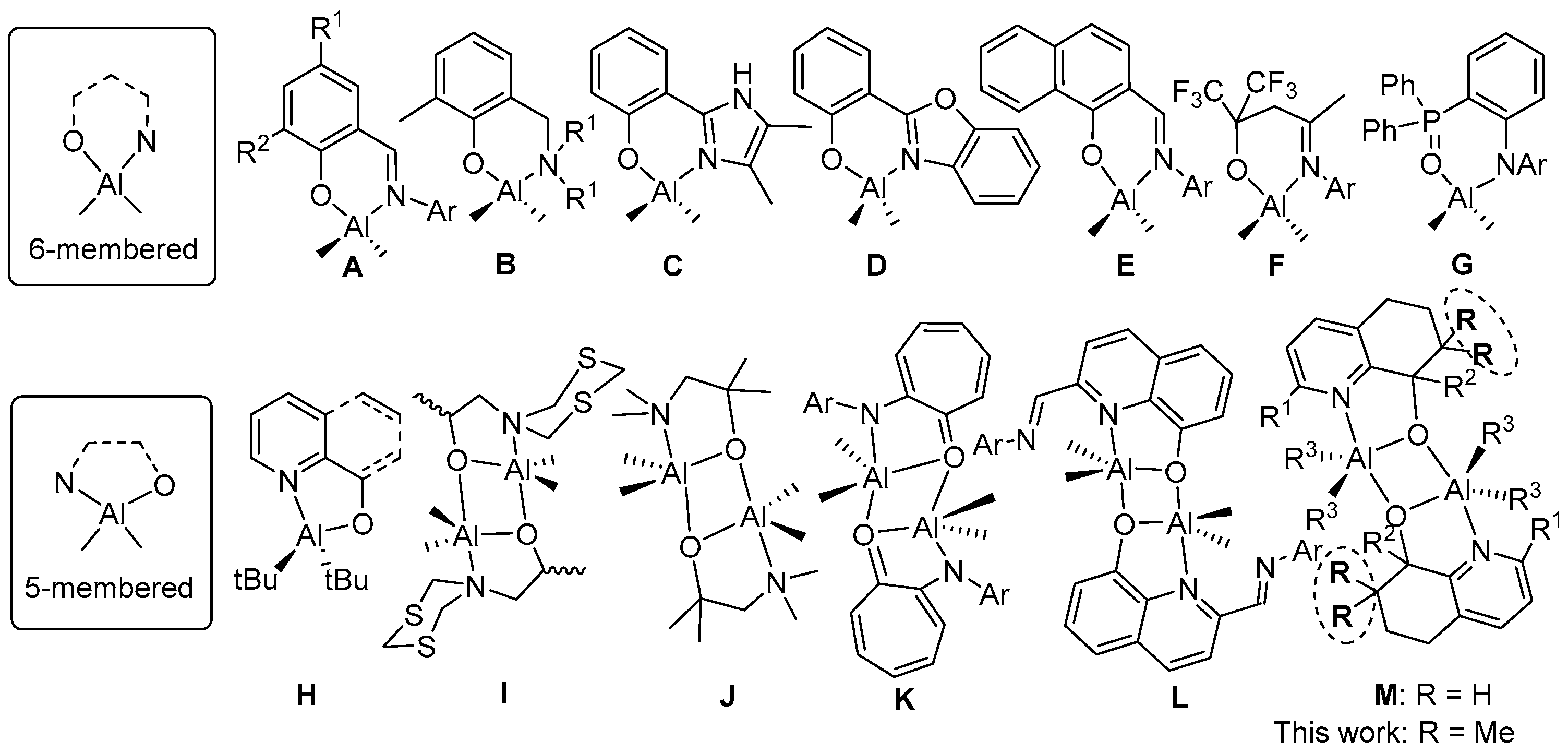
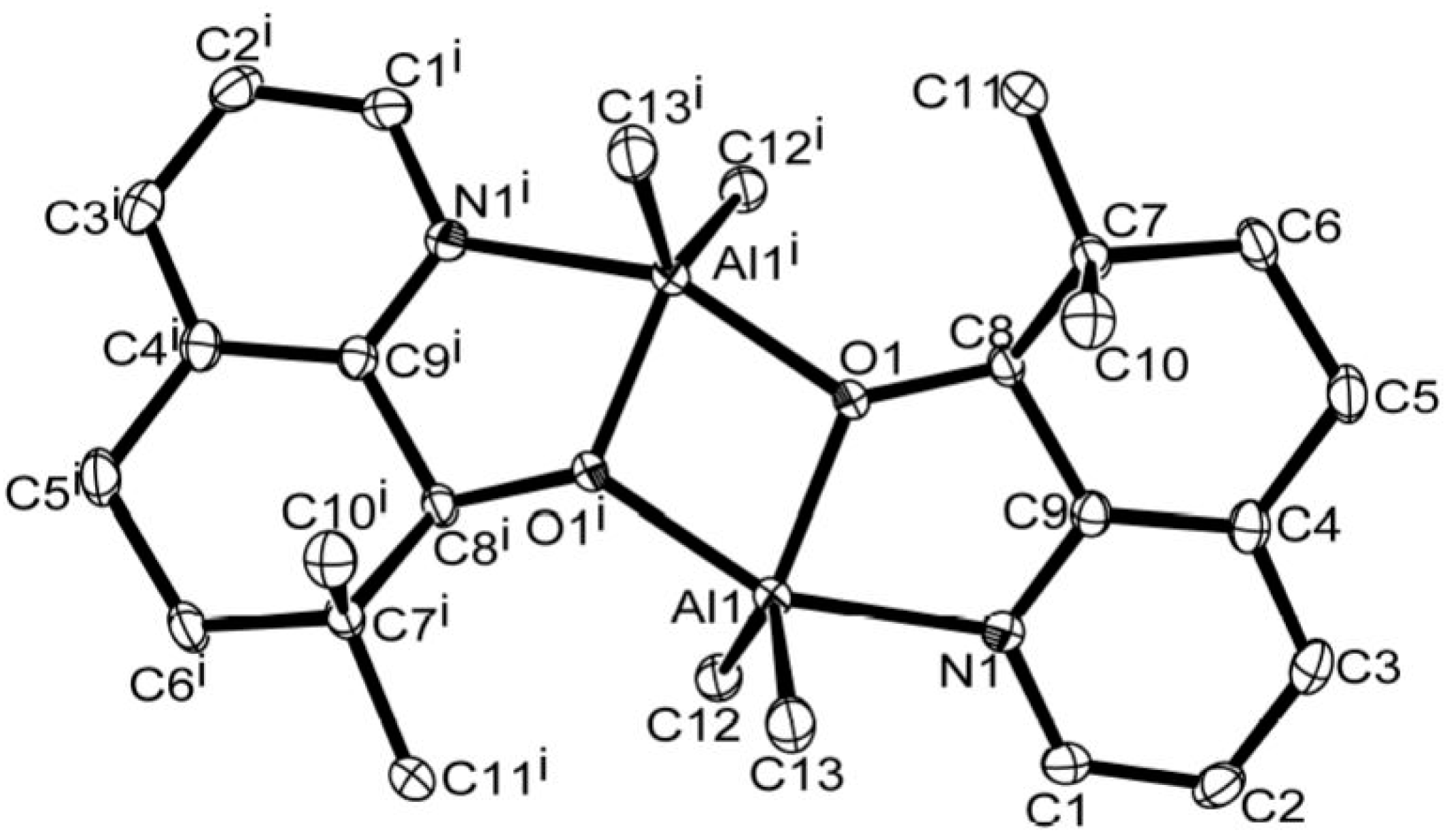
3.1. Synthesis and Characterization of C1–C6
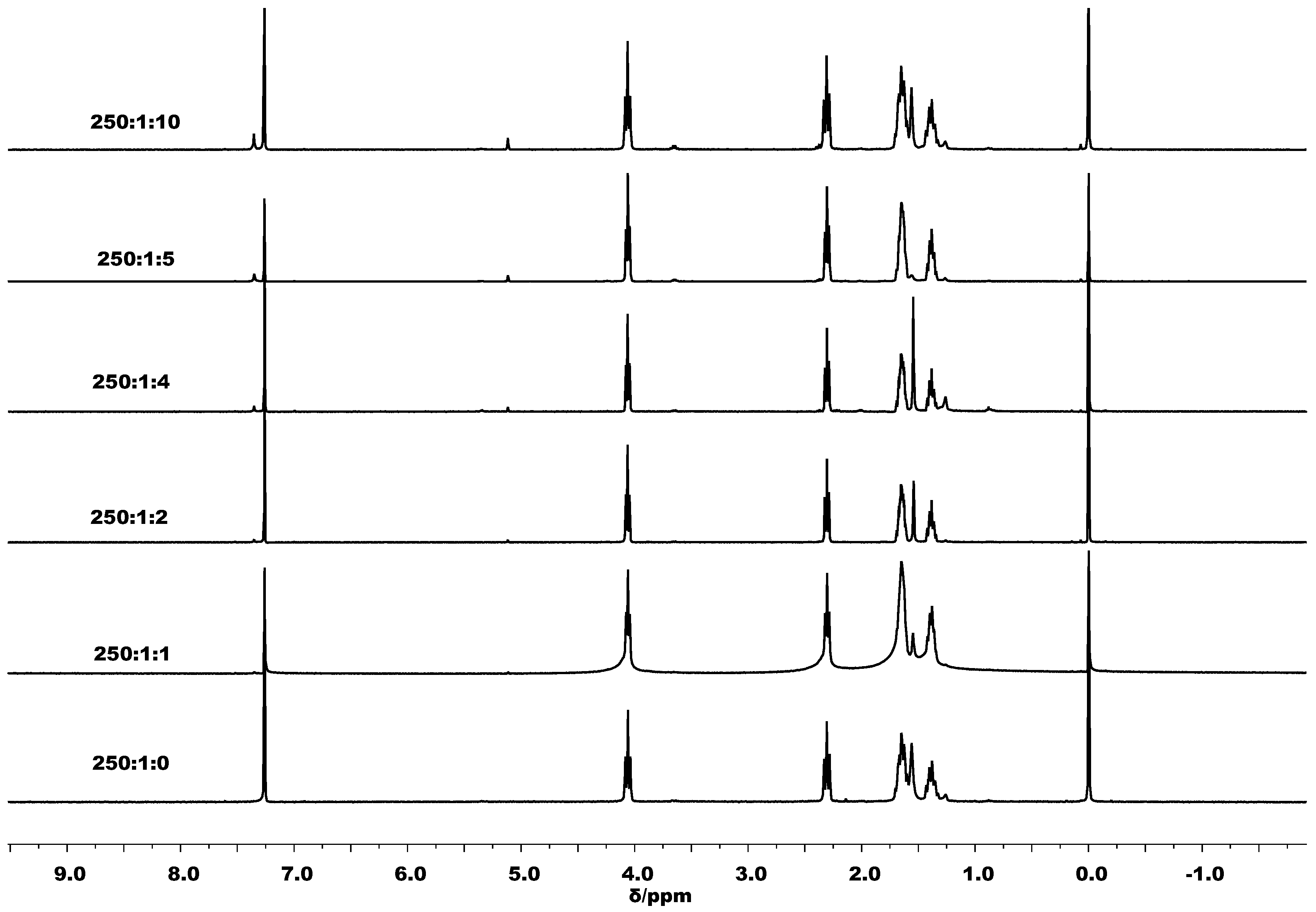
3.2. Ring Opening Polymerization of ε-Caprolactone by C1–C6
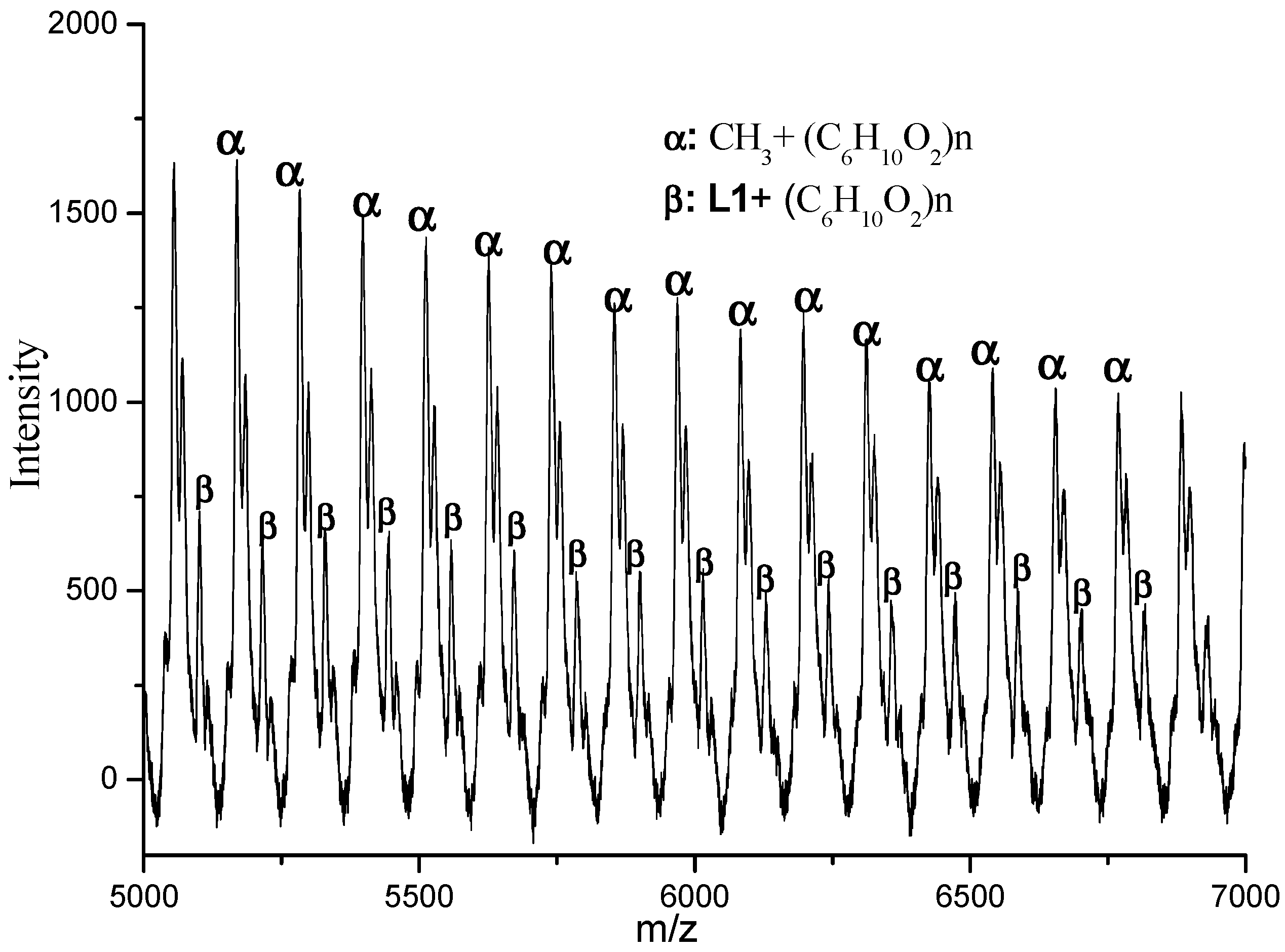
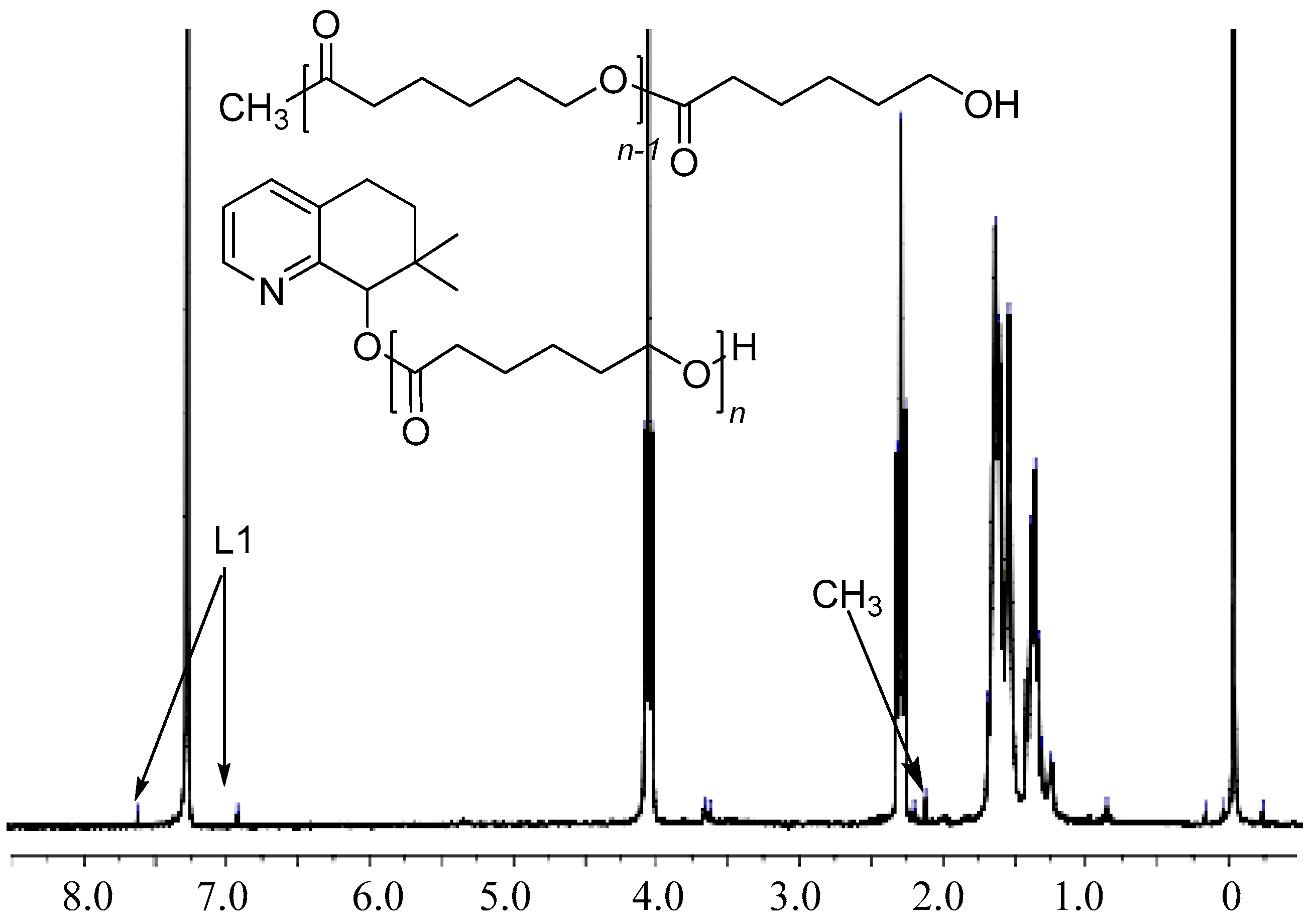
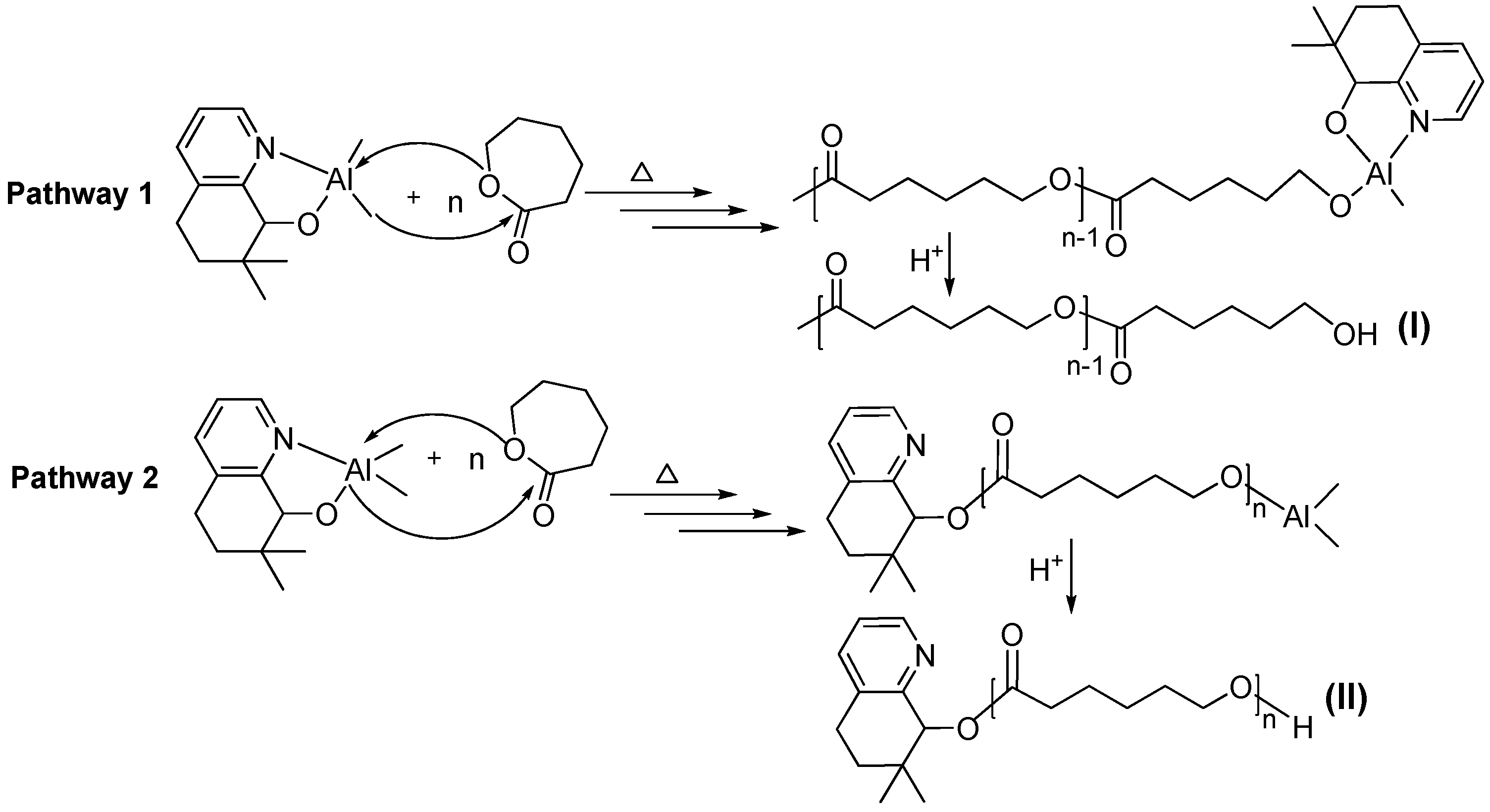
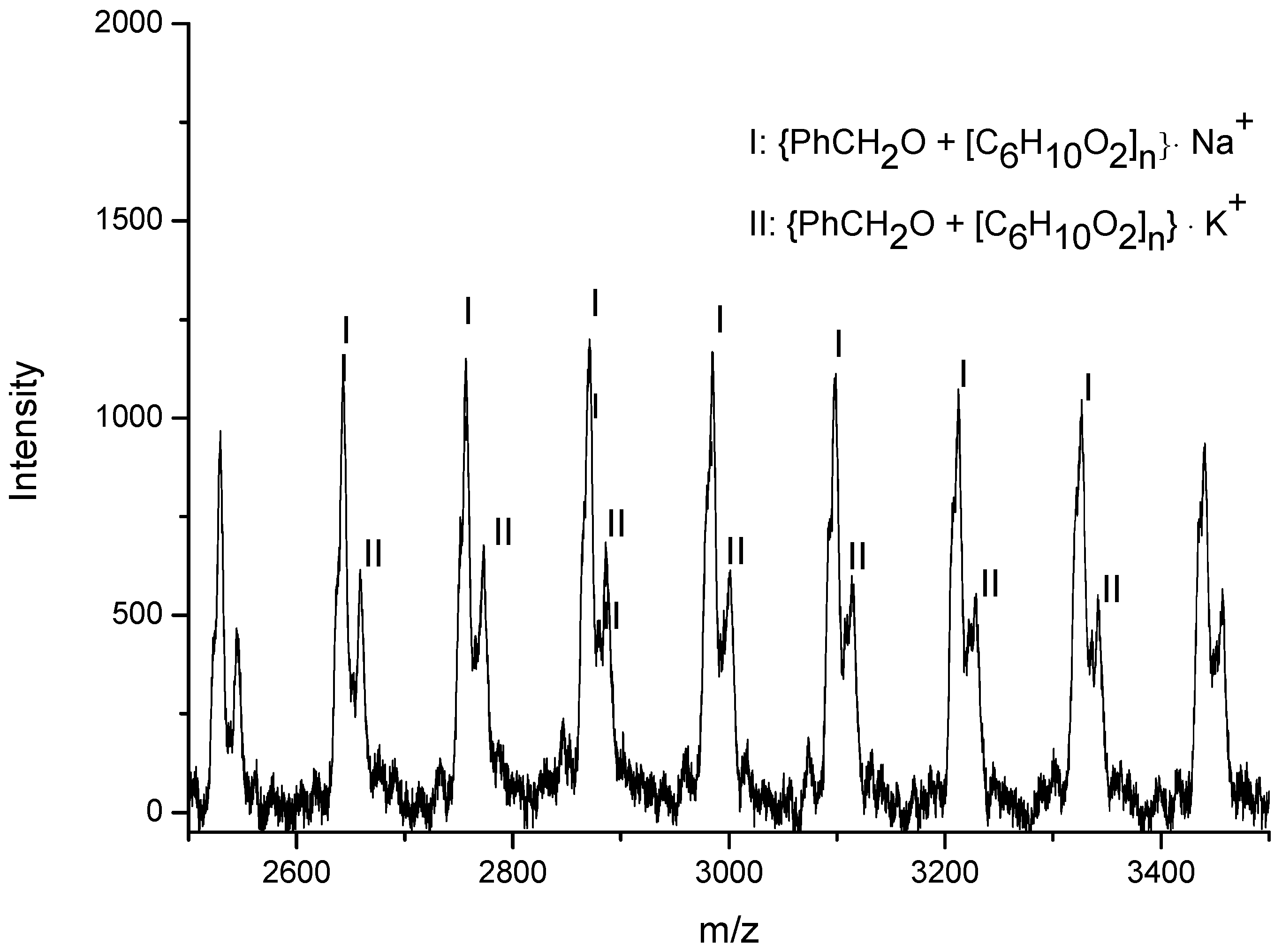
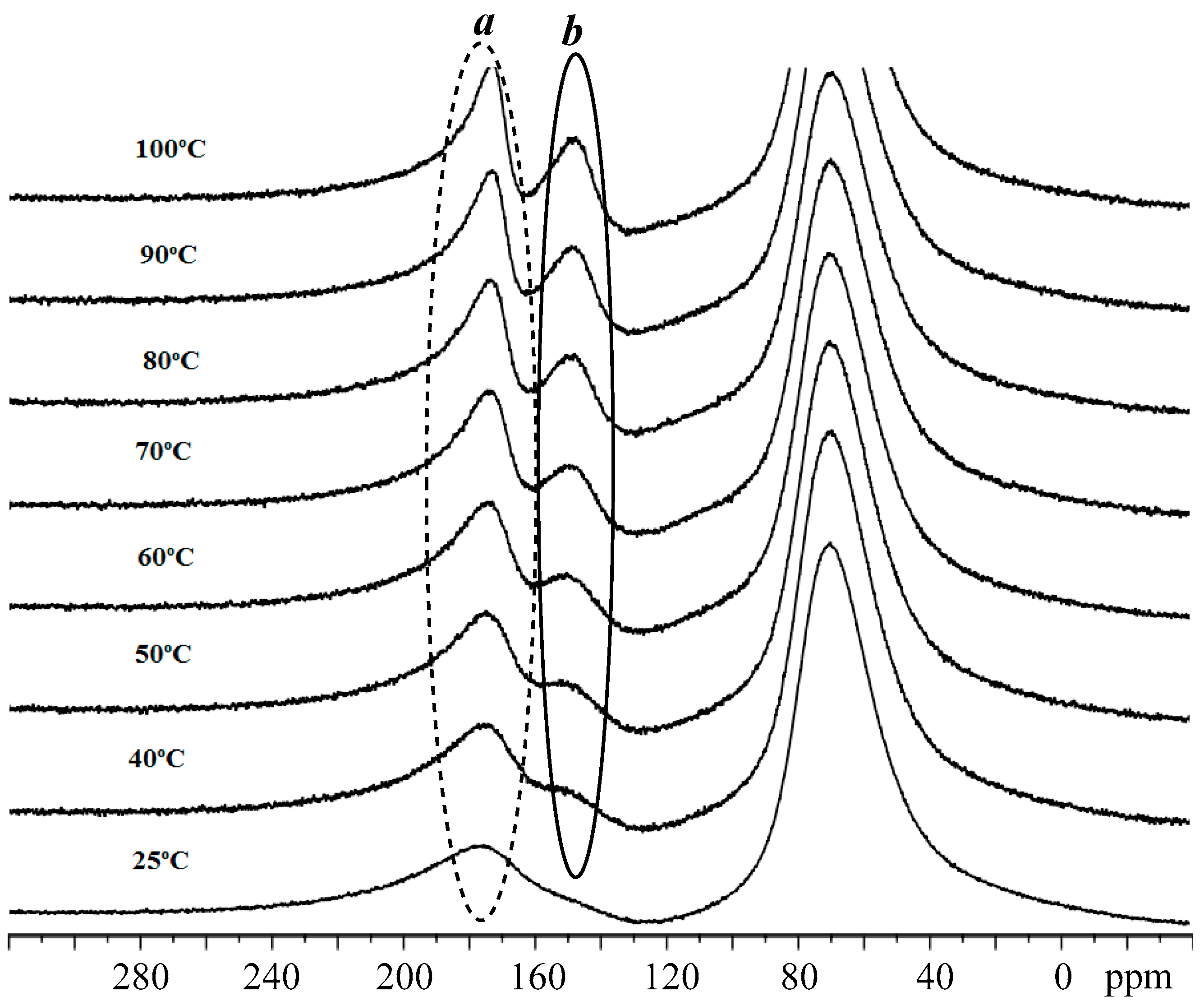
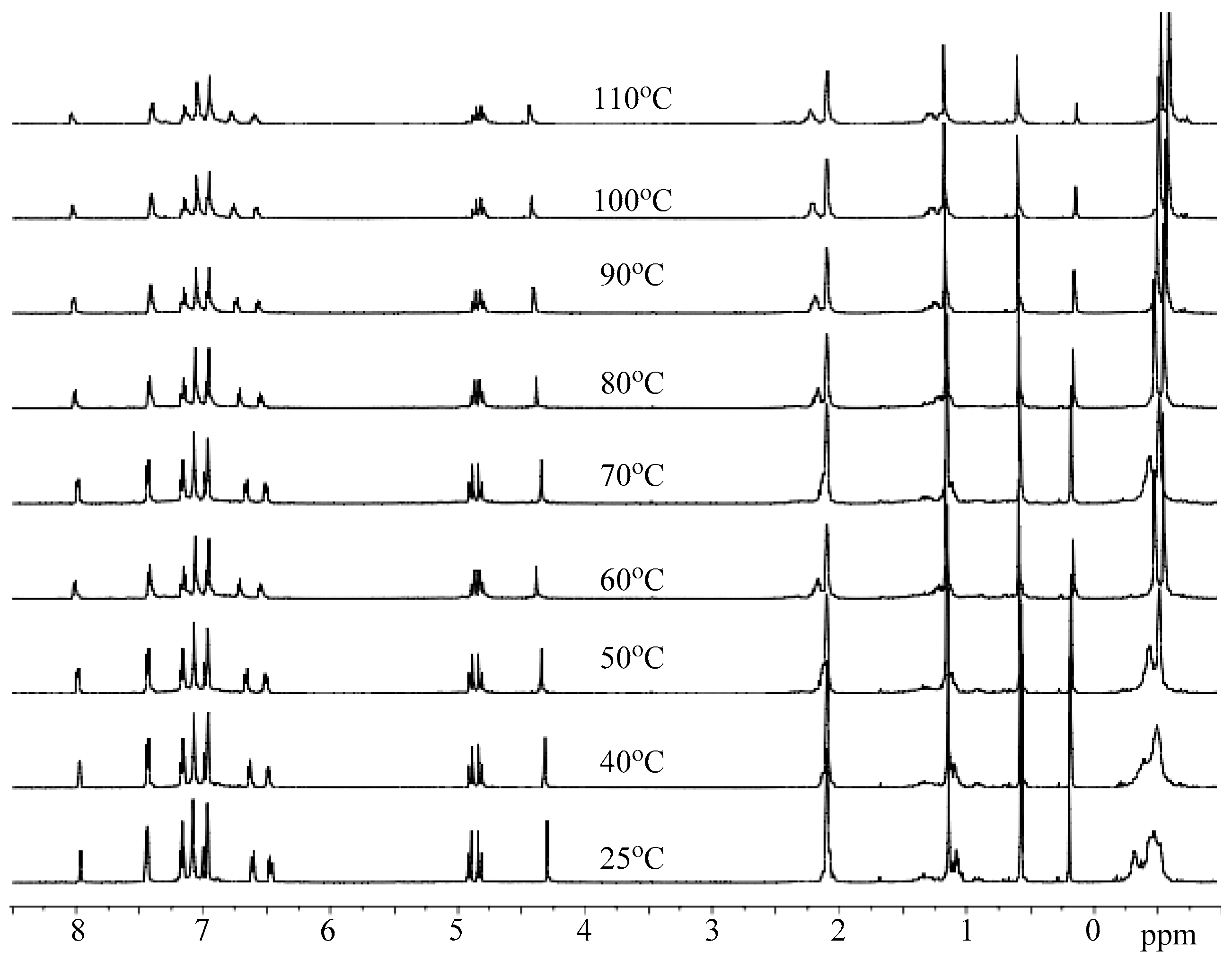
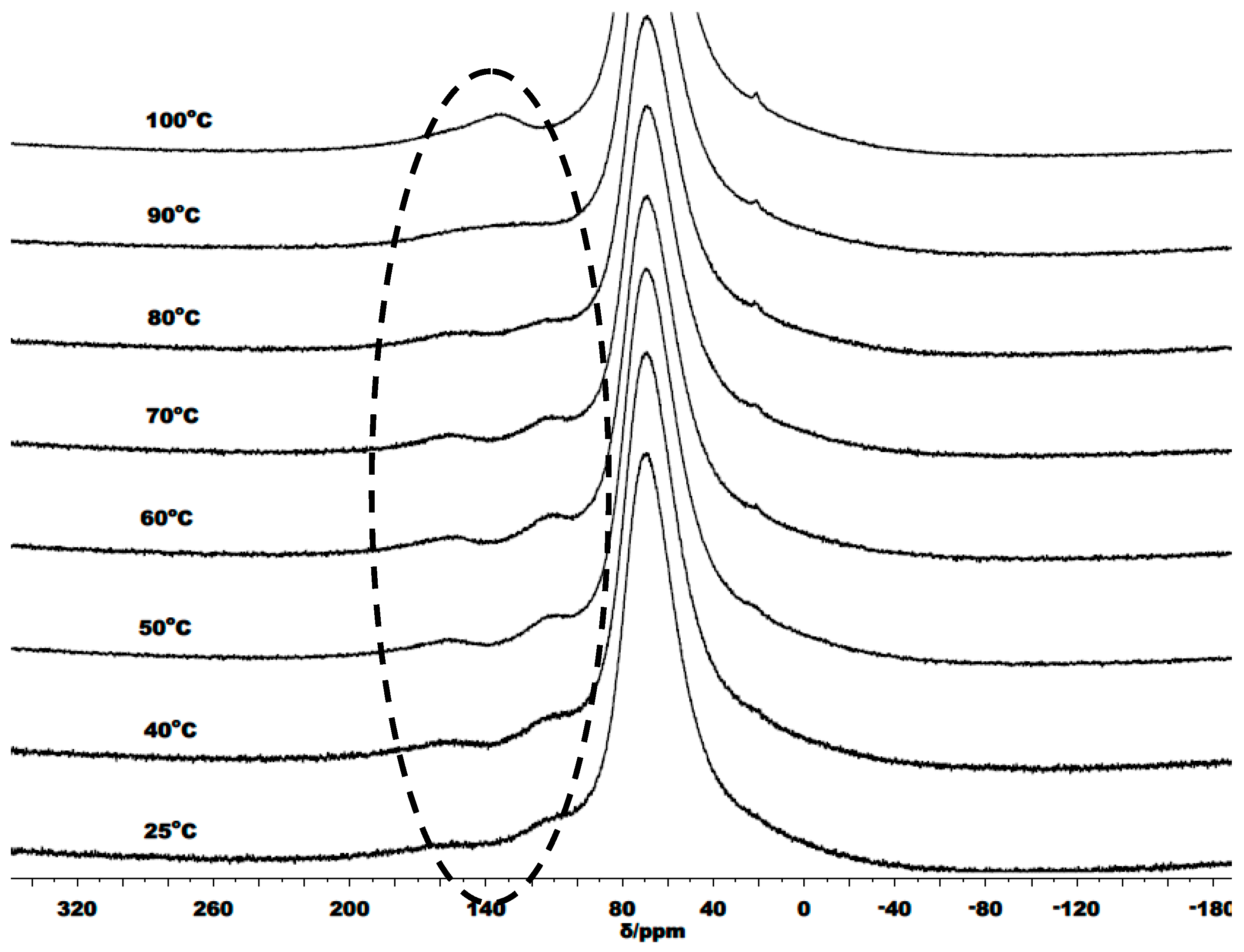
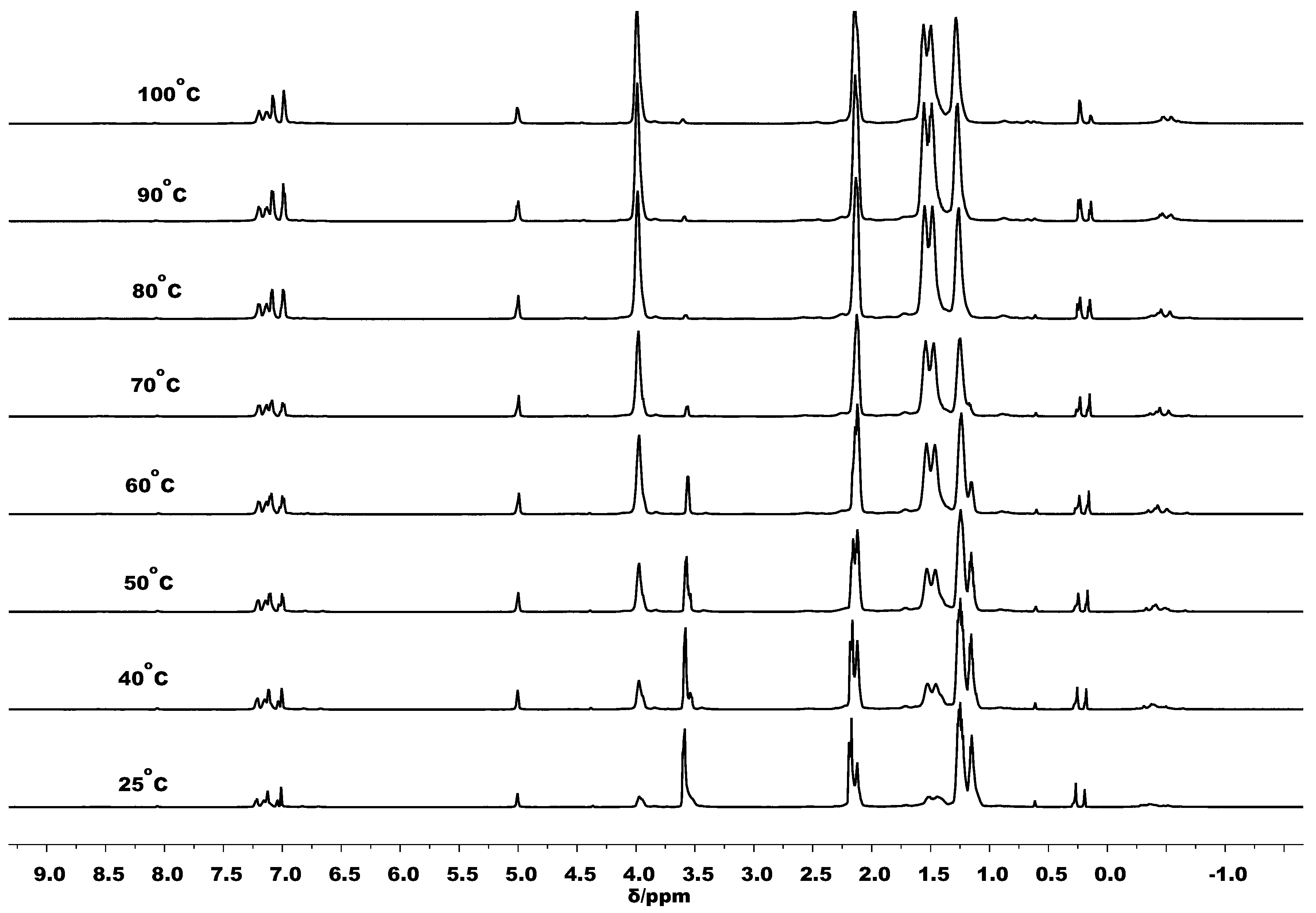
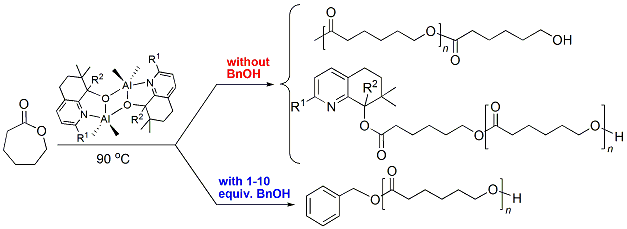
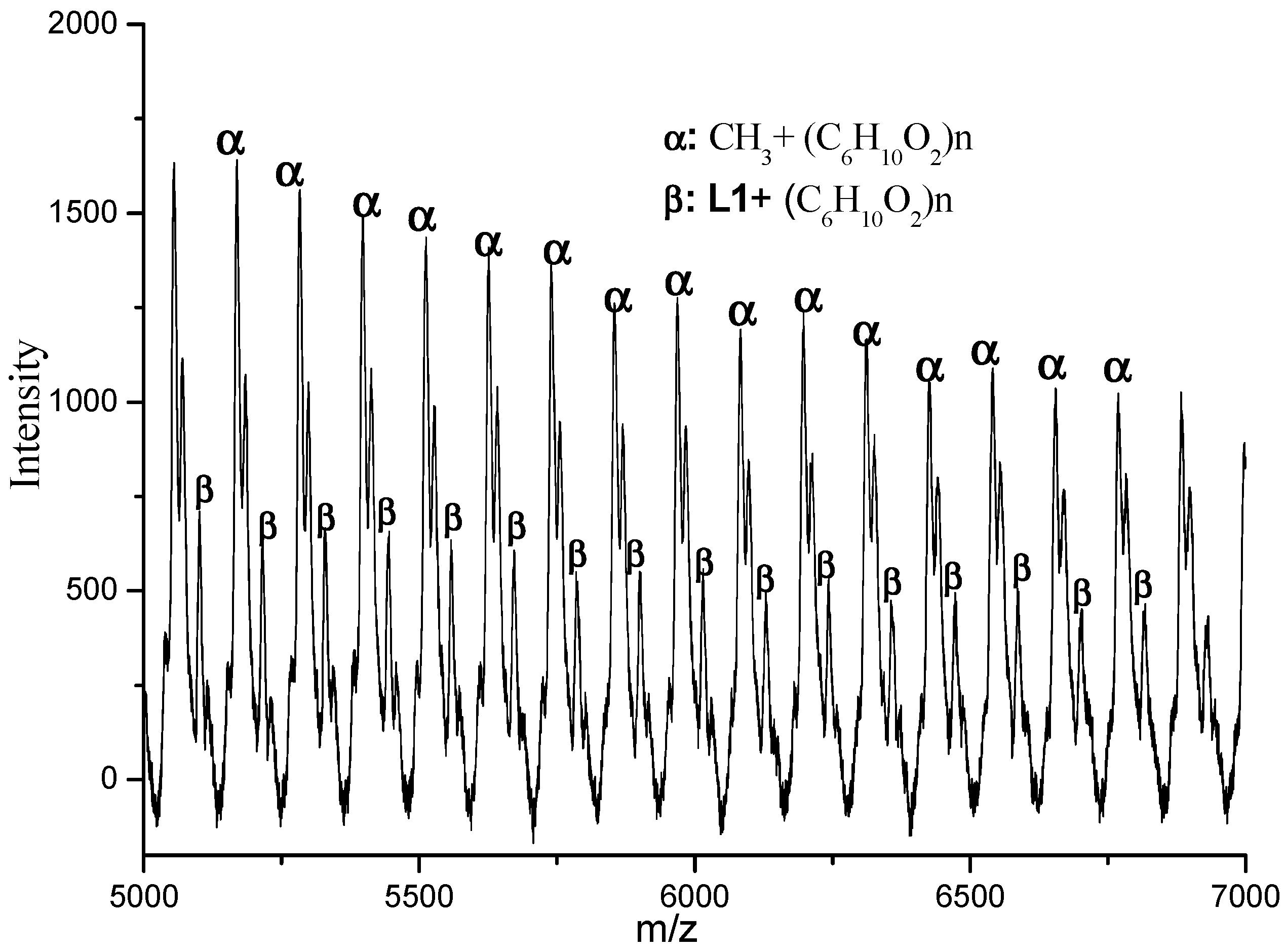
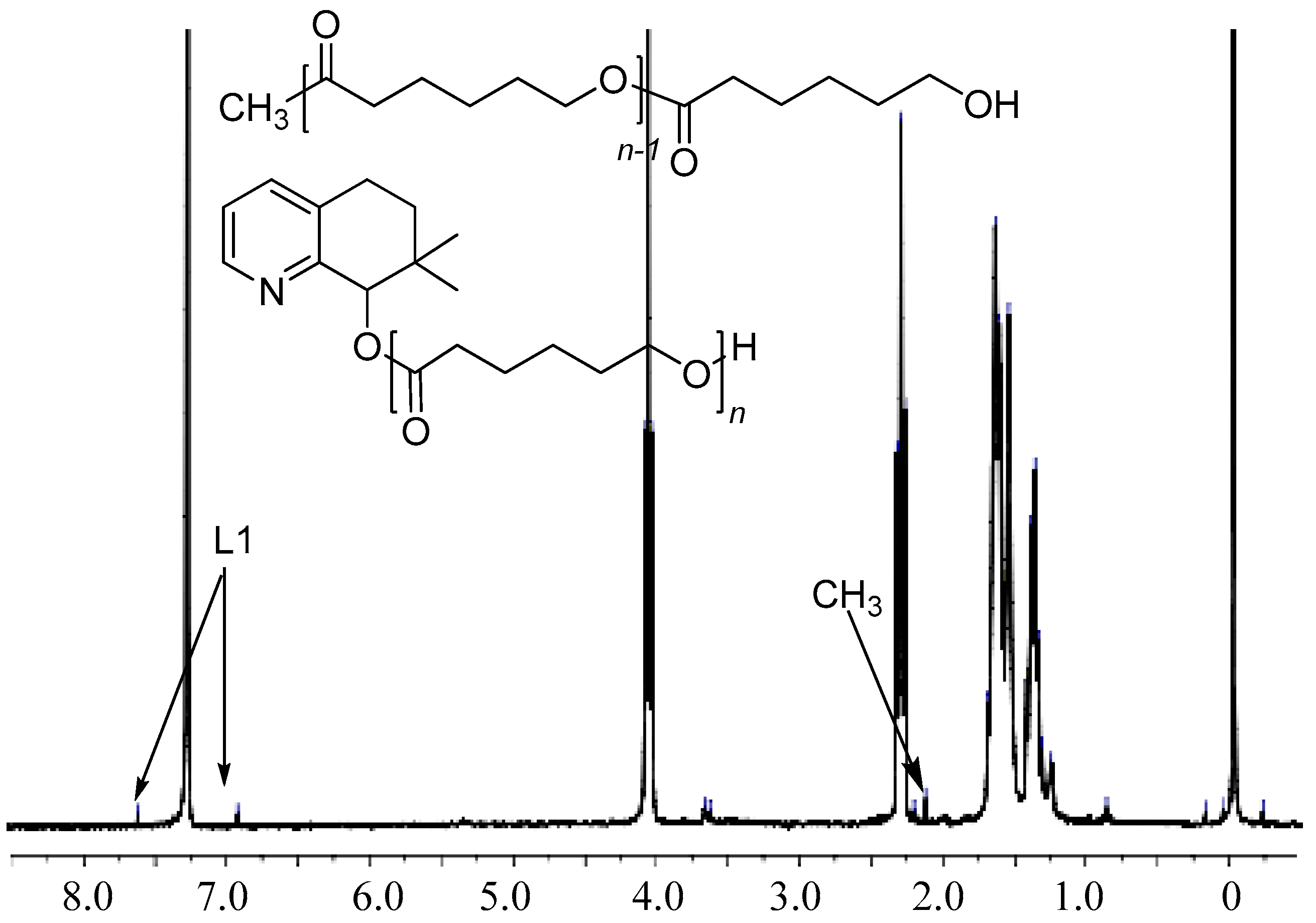
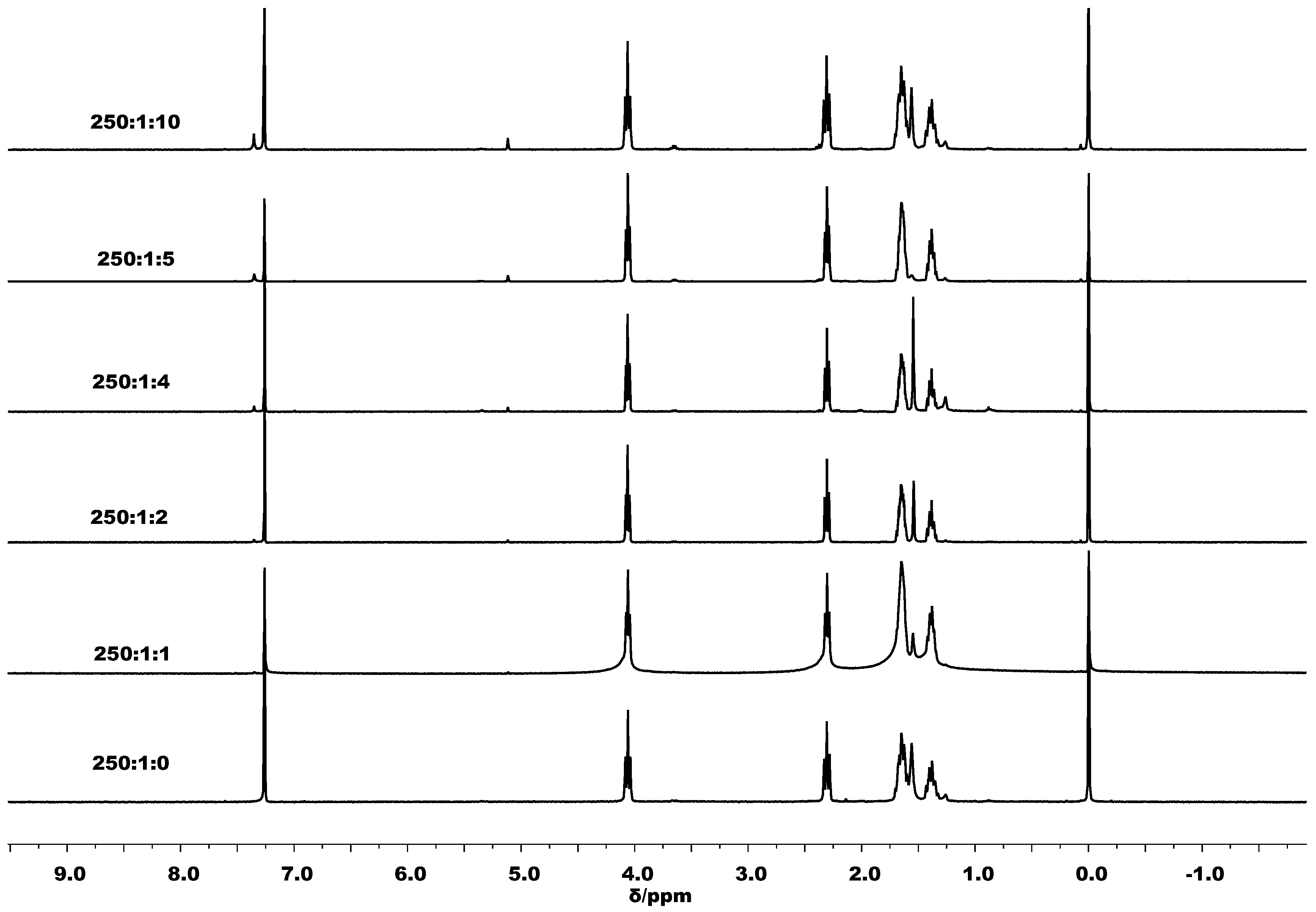
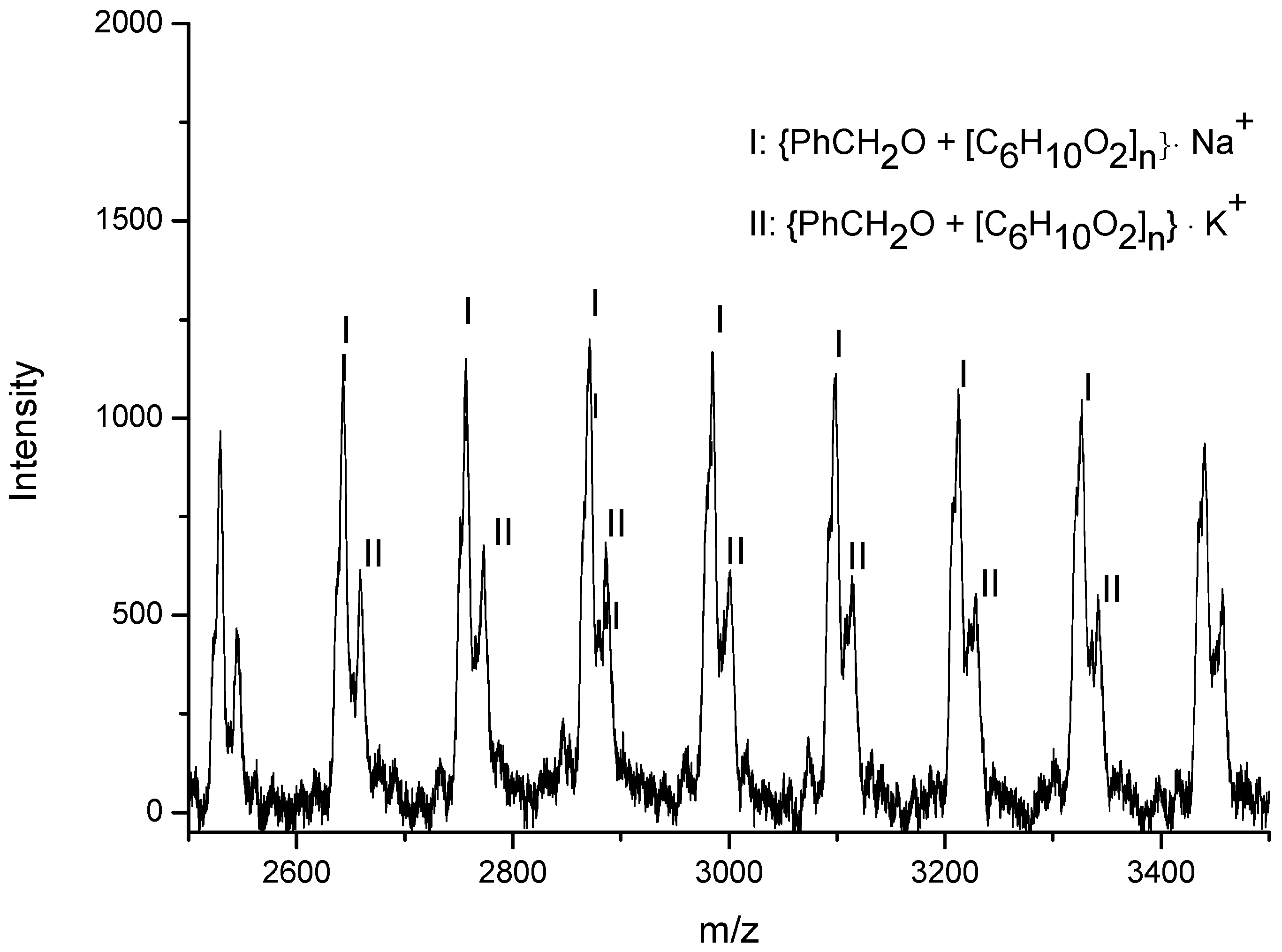
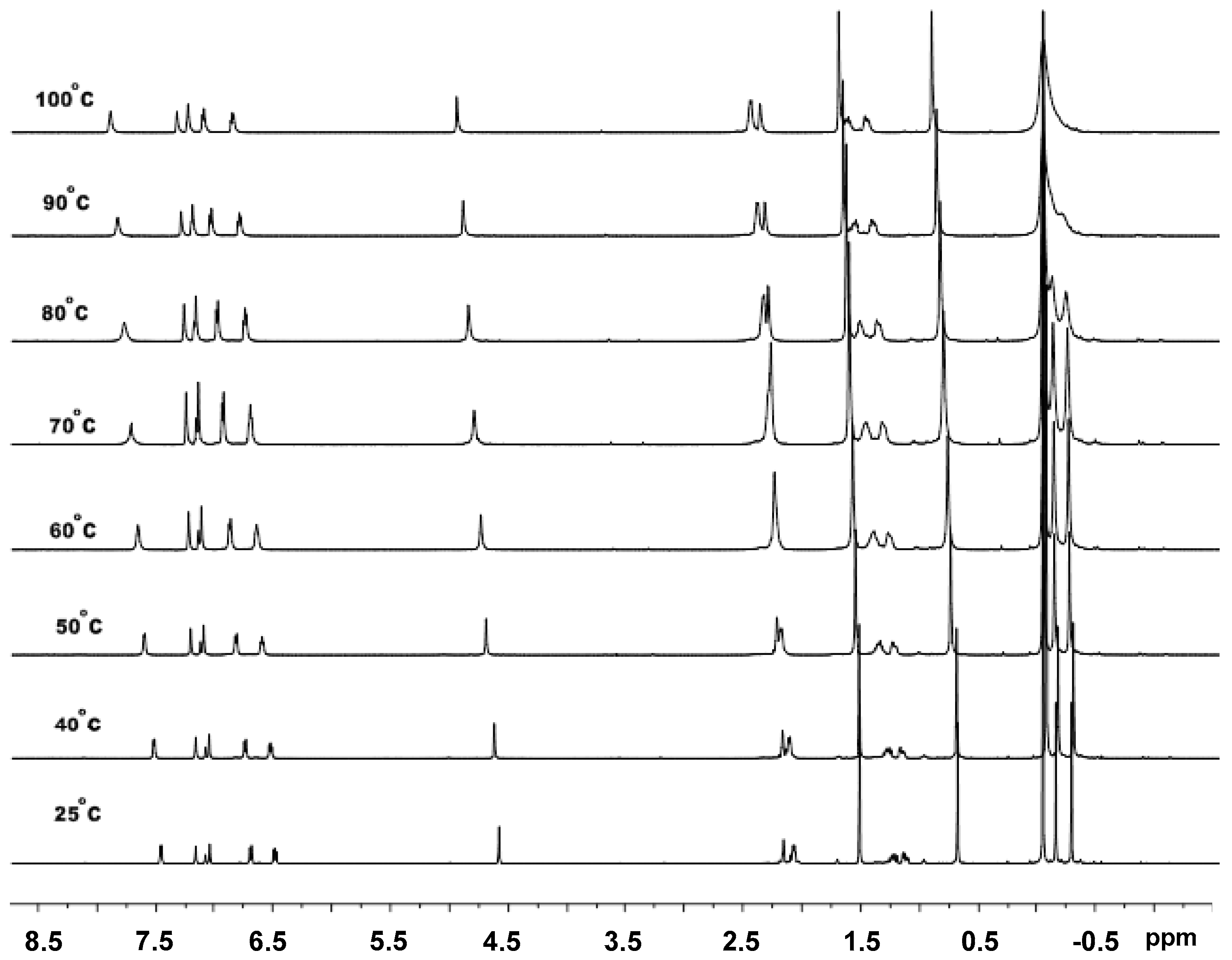
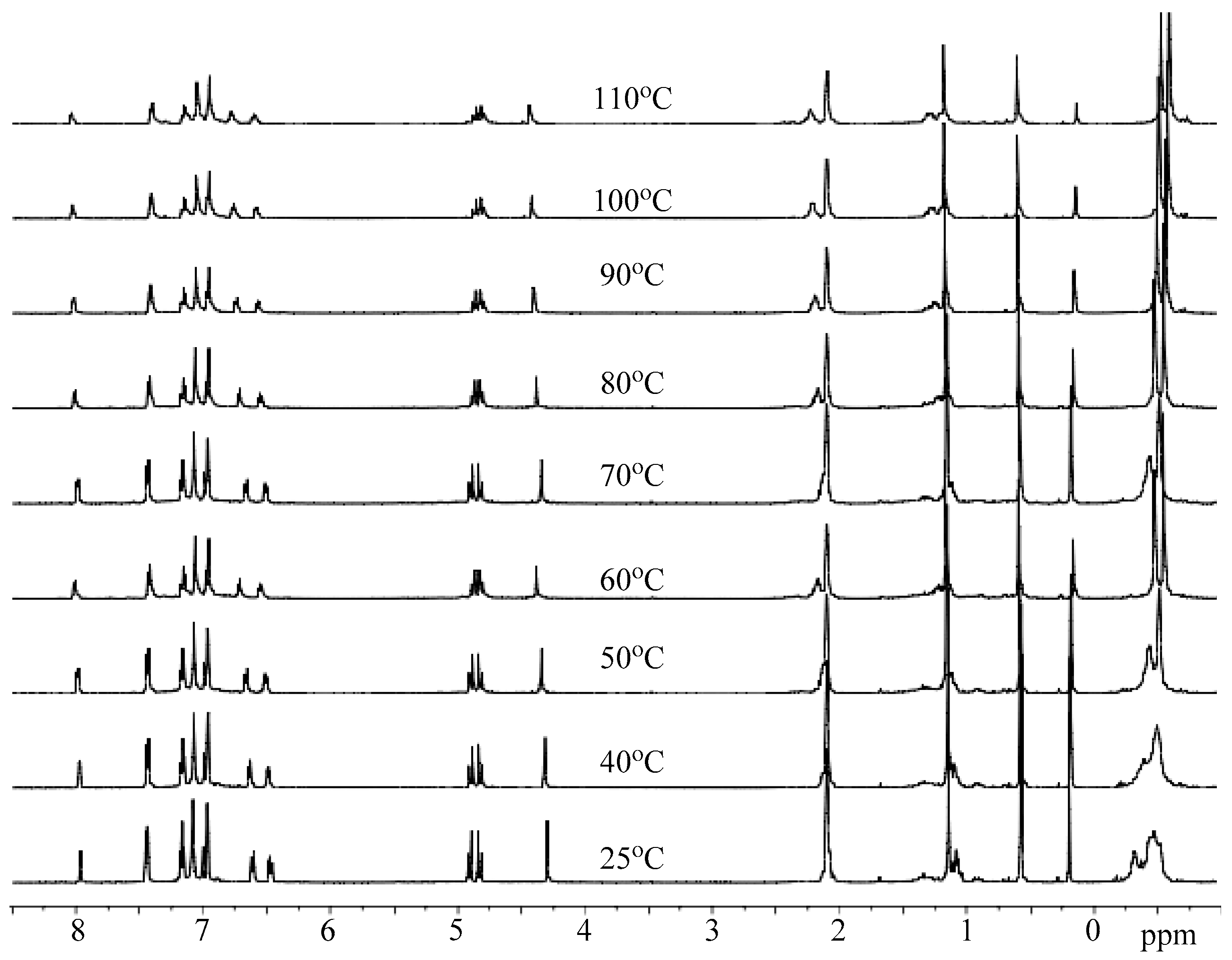
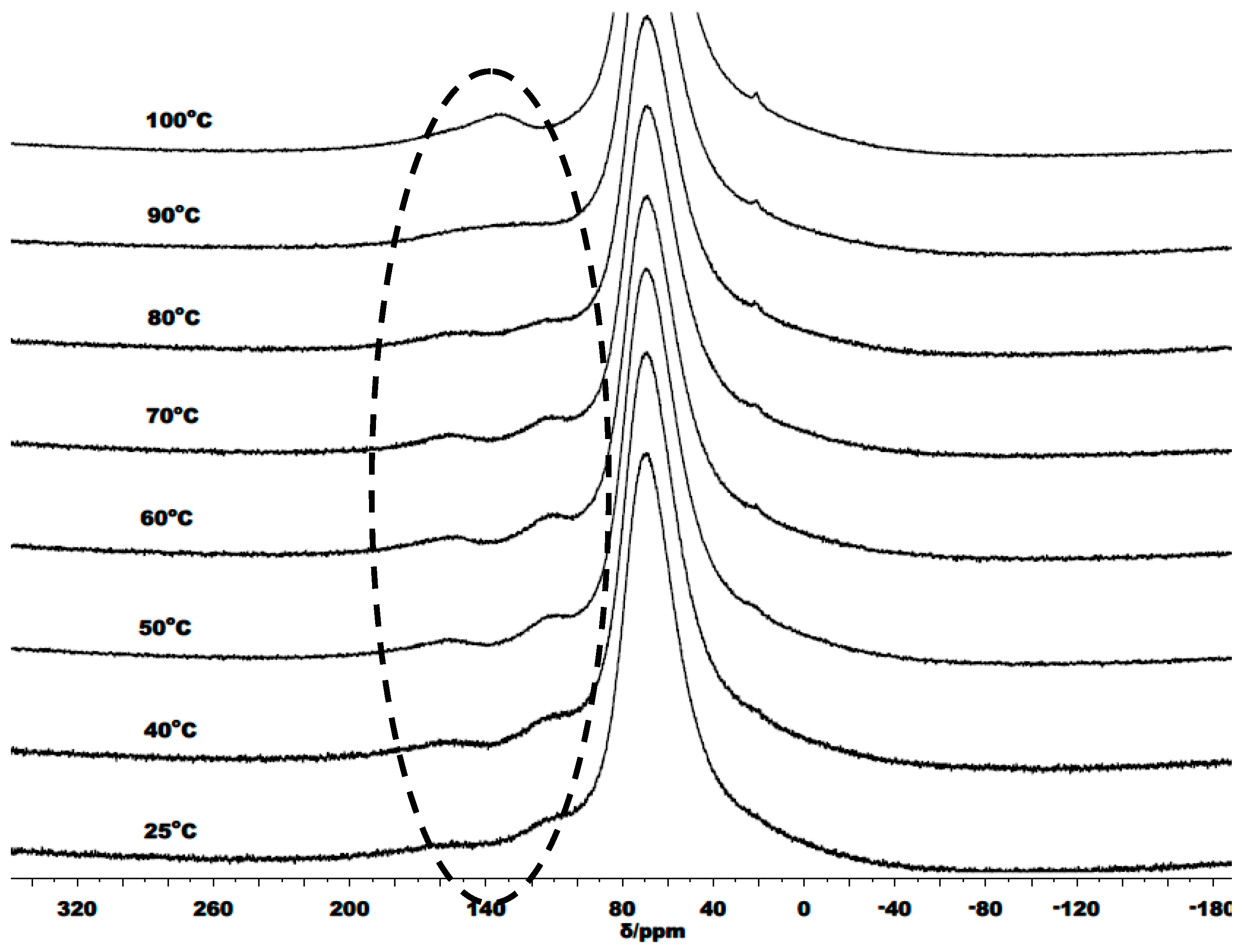
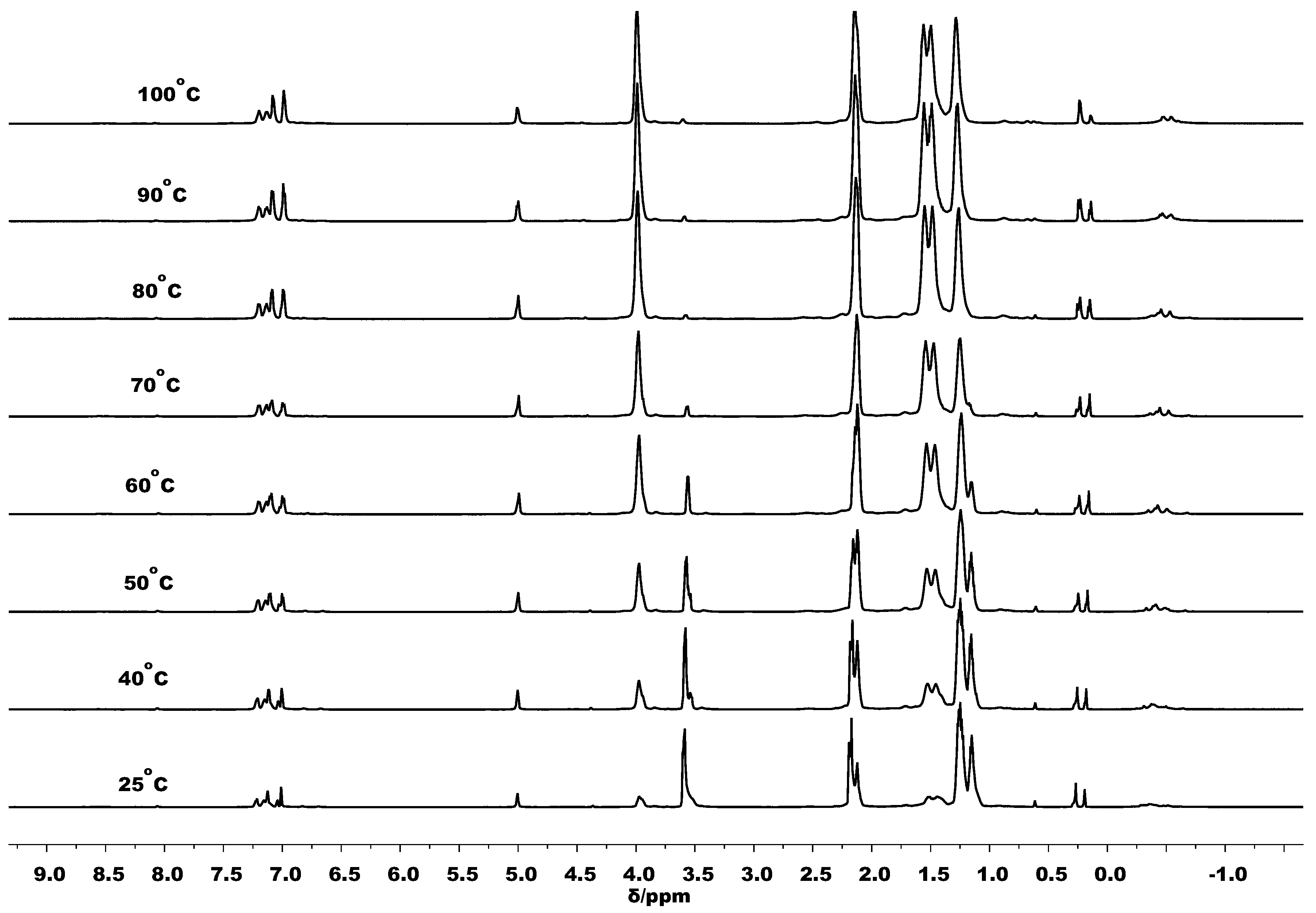
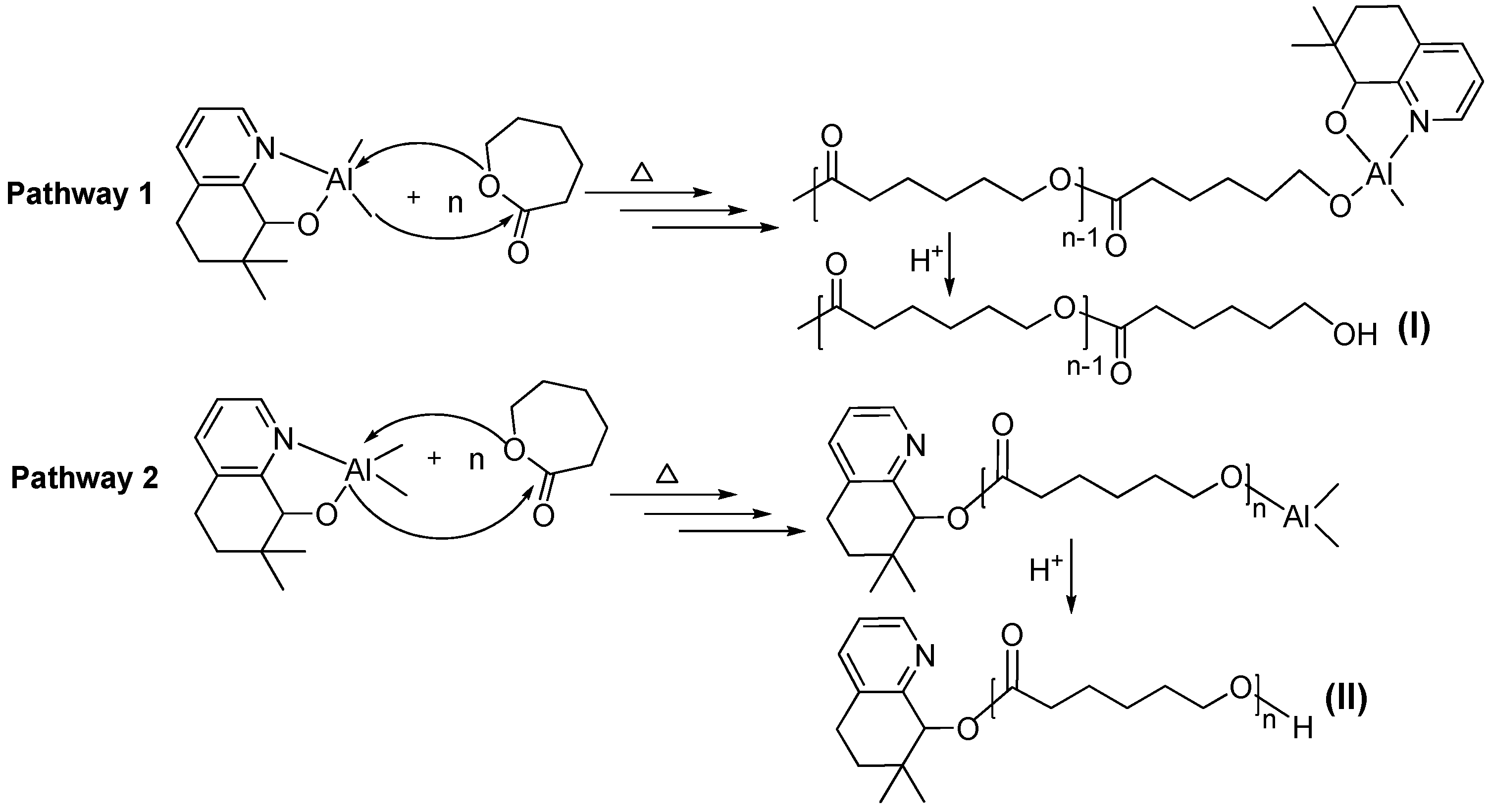
3.3. Mechanistic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Reddy, M.M.; Vivekanandhan, S.; Misra, M.; Bhatia, S.K.; Mohanty, A.K. Biobased plastics and bionanocomposites: Current status and future opportunities. Prog. Polym. Sci. 2013, 38, 1653–1689. [Google Scholar] [CrossRef]
- Singhvi, M.; Gokhale, D. Biomass to biodegradable polymer (PLA). RSC Adv. 2013, 3, 13558–13568. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Park, H.-M.; Ha, C.-S. Bio-nanocomposites for food packaging applications. Prog. Polym. Sci. 2013, 38, 1629–1652. [Google Scholar] [CrossRef]
- Armentano, I.; Bitinis, N.; Fortunati, E.; Mattioli, S.; Rescignano, N.; Verdejo, R.; Lopez-Manchado, M.A.; Kenny, J.M. Multifunctional nanostructured PLA materials for packaging and tissue engineering. Prog. Polym. Sci. 2013, 38, 1720–1747. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Baciu, D.; Gounari, E.; Steriotis, T.; Charalambopoulou, G.; Bikiaris, D. Biocompatible Nanobioglass Reinforced Poly(ɛ-Caprolactone) Composites Synthesized via In Situ Ring Opening Polymerization. Polymers 2018, 10, 381. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Zhou, S. Aluminum alkyl complexes: Synthesis, structure, and application in ROP of cyclic esters. Dalton Trans. 2016, 45, 4471–4485. [Google Scholar] [CrossRef] [PubMed]
- Arbaoui, A.; Redshaw, C. Metal catalysts for ɛ-caprolactone polymerization. Polym. Chem. 2010, 1, 801–826. [Google Scholar] [CrossRef]
- Spassky, N.; Wisniewski, M.; Pluta, C.; Le Borgne, A. Highly stereoelective polymerization of rac-(d,l)-lactide with a chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective ring-opening polymerization of meso-Lactide: Synthesis of syndiotactic poly(lactic acid). J. Am. Chem. Soc. 1999, 121, 4072–4073. [Google Scholar] [CrossRef]
- Tang, Z.H.; Chen, X.S.; Pang, X.; Yang, Y.K.; Zhang, X.F.; Jing, X.B. Stereoselective polymerization of rac-lactide using a monoethylaluminum schiff base complex. Biomacromolecules 2004, 5, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Radano, C.P.; Baker, G.L.; Smith, M.R. Stereoselective polymerization of a racemic monomer with a racemic catalyst: Direct preparation of the polylactic acid stereocomplex from racemic lactide. J. Am. Chem. Soc. 2000, 122, 1552–1553. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Sun, W.-H.; Wang, L.; Redshaw, C. Dimethylaluminium aldiminophenolates: Synthesis, characterization and ring-opening polymerization behavior towards lactides. Dalton Trans. 2012, 41, 11587–11596. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Ge, J.L.; Ma, H.Y. Aluminum methyl, alkoxide and α-alkoxy ester complexes supported by 6,6′-dimethylbiphenyl-bridged salen ligands: Synthesis, characterization and catalysis for rac-lactide polymerization. Dalton Trans. 2016, 45, 6682–6695. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, N.; Fujiki, M.; Nomura, K. Ring-opening polymerization of various cyclic esters by Al complex catalysts containing a series of phenoxy-imine ligands: Effect of the imino substituents for the catalytic activity. J. Mol. Catal. A Chem. 2008, 292, 67–75. [Google Scholar] [CrossRef]
- Iwasa, N.; Katao, S.; Liu, J.Y.; Fujiki, M.; Furukawa, Y.; Nomura, K. Notable effect of fluoro substituents in the imino group in ring-opening polymerization of ε-caprolactone by Al complexes containing phenoxyimine ligands. Organometallics 2009, 28, 2179–2187. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile copolymerization of glycolide and rac-lactide by dimethyl(salicylaldiminato)aluminum compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- García-Valle, F.M.; Tabernero, V.; Tomas, C.; Mosquera, M.E.G.; Cano, J.; Milione, S. Biodegradable PHB from rac-β butyrolactone: Highly controlled ROP mediated by a pentacoordinated aluminum complex. Organometallics 2018, 37, 837–840. [Google Scholar] [CrossRef]
- Shi, T.; Luo, W.L.; Liu, S.F.; Li, Z.B. Controlled random copolymerization of rac-lactide and caprolactone by well-designed phenoxyimine Al complexes. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 611–617. [Google Scholar] [CrossRef]
- Shi, T.; Zheng, Q.; Zuo, W.; Liu, S.; Li, Z. Bimetallic aluminum complexes supported by bis(salicylaldimine) ligand: Synthesis, characterization and ring-opening polymerization of lactide. Chin. J. Polym. Sci. 2018, 36, 149–156. [Google Scholar] [CrossRef]
- Maisse-François, A.; Azora, L.; Schmitta, A.-L.; Coquela, A.; Brelotc, L.; Welter, R.; Bellemin-Laponnaz, S.; Dagorne, S. Structural diversity and versatility for organoaluminum complexes supported by mono- and di-anionic aminophenolate bidentate ligands. J. Organomet. Chem. 2012, 696, 4248–4256. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, H.Y. Aluminum complexes of bidentate phenoxy-amine ligands: Synthesis, characterization and catalysis in ring-opening polymerization of cyclic esters. J. Organomet. Chem. 2013, 731, 23–28. [Google Scholar] [CrossRef]
- Chen, L.; Li, W.; Yuan, D.; Zhang, Y.; Shen, Q.; Yao, Y. Syntheses of Mononuclear and Dinuclear Aluminum Complexes Stabilized by Phenolato Ligands and Their Applications in the Polymerization of ε Caprolactone: A Comparative Study. Inorg. Chem. 2015, 54, 4699–4708. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ouyang, H.; Chen, L.J.; Yuan, D.; Zhang, Y.; Yao, Y. A Comparative Study on Dinuclear and Mononuclear Aluminum Methyl Complexes Bearing Piperidyl–Phenolato Ligands in ROP of Epoxides. Inorg. Chem. 2016, 55, 6520–6524. [Google Scholar] [CrossRef] [PubMed]
- Roymuhury, S.K.; Chakraborty, D.; Ramkumar, V. Aluminium complexes bearing N,O-aminophenol ligands as efficient catalysts for the ring opening polymerization of lactide. Eur. Polym. J. 2015, 70, 203–214. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Wang, L.; Redshaw, C.; Sun, W.-H. Dialkylaluminium 2-imidazolylphenolates: Synthesis, characterization and ring-opening polymerization behavior towards lactides. J. Organomet. Chem. 2014, 750, 65–73. [Google Scholar] [CrossRef]
- Sumrit, P.; Chuawong, P.; Nanok, T.; Duangthongyou, T.; Hormnirun, P. Aluminum complexes containing salicylbenzoxazole ligands and their application in the ring-opening polymerization of rac-lactide and ε-caprolactone. Dalton Trans. 2016, 45, 9250–9266. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, W.S.; Liu, J.Y.; Li, Y.S. Living ring-opening homo- and copolymerisation of ε-caprolactone and L-lactide by cyclic β-ketiminato aluminium complexes. Dalton Trans. 2014, 43, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Bouyahyi, M.; Roisnel, T.; Carpentier, J.-F. Aluminum Complexes of Bidentate Fluorinated Alkoxy-Imino Ligands: Syntheses, Structures, and Use in Ring-Opening Polymerization of Cyclic Esters. Organometallics 2012, 31, 1458–1466. [Google Scholar] [CrossRef]
- Liang, L.-C.; Chen, F.-Y.; Huang, M.-H.; Cheng, L.-C.; Li, C.-W.; Lee, H.M. Aluminium complexes of bidentate N,O- and N,N-ligands derived from oxidative functionalization of amido phosphines: Synthesis, structure and reactivity. Dalton Trans. 2010, 39, 9941–9951. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-L.; Lin, Y.-F.; Jiang, M.-T.; Lu, W.-Y.; Vandavasi, J.K.; Wang, L.-F.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y. Improvement in aluminum complexes bearing schiff bases in ring opening polymerization of ε caprolactone: A five-membered-ring system. Organometallics 2017, 36, 1936–1945. [Google Scholar] [CrossRef]
- Francis, J.A.; Bott, S.G.; Barron, A.R. Aluminium compounds containing bidentate ligands: Chelate ring size and rigid conformation effects. J. Chem. Soc. Dalton Trans. 1998, 3305–3310. [Google Scholar] [CrossRef]
- Francis, J.A.; Bott, S.G.; Barron, A.R. Sterically crowded aryloxides of aluminum: Intramolecular coordination of bidentate ligands. J. Organomet. Chem. 2000, 597, 29–37. [Google Scholar] [CrossRef]
- Galvez-Ruiz, J.C.; Noth, H.; Flores-Parra, A. Organometallic Aluminum Compounds Derived from 2-(1,3,5-Dithiazinan-5-yl)ethanol Ligands. Inorg. Chem. 2003, 42, 7569–7578. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Han, S.Y.; Kim, J.H.; Kang, Y.Y.; Lee, J.; Kim, Y. Monomeric or dimeric aluminum complexes as catalysts for cycloaddition between CO2 and epoxides. Eur. J. Inorg. Chem. 2015, 2323–2329. [Google Scholar] [CrossRef]
- Li, M.; Chen, M.; Chen, C.L. Ring-opening polymerization of rac-lactide using anilinotroponebased aluminum complexes-sidearm effect on the catalysis. Polymer 2015, 64, 234–239. [Google Scholar] [CrossRef]
- Sun, W.-H.; Shen, M.; Zhang, W.J.; Huang, W.; Liu, S.F.; Redshaw, C. Methylaluminium 8-quinolinolates: Synthesis, characterization and use in ring-opening polymerization (ROP) of ε-caprolactone. Dalton Trans. 2011, 40, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.; Rajendran, N.; Liang, T.; Sun, W.-H. Thermo-enhanced ring-opening polymerization of ε-caprolactone: The synthesis, characterization, and catalytic behavior of aluminum hydroquinolin-8-olates. Dalton Trans. 2017, 46, 7833–7843. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Save, M.; Schappacher, M.; Soum, A. Controlled Ring-Opening Polymerization of Lactones and Lactides Initiated by Lanthanum Isopropoxide, General Aspects and Kinetics. Macromol. Chem. Phys. 2002, 203, 889–899. [Google Scholar] [CrossRef]
- Tseng, H.-C.; Chiang, M.Y.; Lu, W.-Y.; Chen, Y.-J.; Lian, C.-J.; Chen, Y.-H.; Tsai, H.-Y.; Lai, Y.-C.; Chen, H.-Y. A closer look at ε-caprolactone polymerization catalyzed by alkyl aluminum complexes: The effect of induction period on overall catalytic activity. Dalton Trans. 2015, 44, 11763–11773. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Chen, E.Y.-X. Neutral, three-coordinate, chelating diamide aluminum complexes: Catalysts/initiators for synthesis of telechelic oligomers and high polymers. Organometallics 2002, 21, 1438–1442. [Google Scholar] [CrossRef]
- Yu, R.-C.; Hung, C.-H.; Huang, J.-H.; Lee, H.-Y.; Chen, J.-T. Four- and five-Coordinate aluminum ketiminate complexes: Synthesis, characterization, and ring-opening polymerization. Inorg. Chem. 2002, 41, 6450–6455. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, S.; Zhu, X.; Zhou, S.; Mu, X.; Huang, Z.; Hong, D. Aluminum Complexes Bearing N-Protected 2-Amino- or 2-Imino-Functionalized Pyrrolyl Ligands: Synthesis, Structure, and Catalysis for Preparation of Pyrrolyl-End-Functionalized Polyesters. Organometallics 2016, 35, 2621–2629. [Google Scholar] [CrossRef]
- Li, H.; Debuigne, A.; Jrome, R.; Lecomte, P. Synthesis of Macrocyclic poly(ɛ-caprolactone) by intramolecular cross-Linking of unsaturated end groups of chains precyclic by the initiation. Angew. Chem. Int. Ed. 2006, 45, 2264–2267. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sierra, M.L.; Oliver, J.P. Synthesis and Spectroscopic Studies of Alkylaluminum Alkoxides Derived from Optically Active Alcohols. Crystal and Molecular Structure of Monomeric Dimethylaluminum (2S,3R)-(+)-4-(Dimethylamino)-l,2-diphenyl-3-methyl-2-butanoxide. Organometallics 1994, 13, 4285–4293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C1 | C4 | |
|---|---|---|
| Empirical formula | C26H40Al2N2O2 | C13H19AlNOCl |
| Formula weight | 466.56 | 267.72 |
| Crystal color | colorless | colorless |
| Temperature (K) | 293(2) | 293(2) |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Crystal system | monoclinic | monoclinic |
| space group | P2/n | P21/n |
| a (Å) | 9.762(2) | 10.934(5) |
| b (Å) | 9.773(2) | 9.902(4) |
| c (Å) | 13.676(3) | 13.019(6) |
| α (°) | 90.00 | 90.00 |
| β (°) | 90.39(3) | 100.391(5) |
| γ (°) | 90.00 | 90.00 |
| Volume (Å3) | 1304.7(5) | 1386.5(11) |
| Z | 2 | 4 |
| Dcalc (Mg m−3) | 1.188 | 1.283 |
| µ (mm−1) | 0.136 | 0.323 |
| F (000) | 504.0 | 568.0 |
| Crystal size (mm) | 0.49 × 0.29 × 0.21 | 0.64 × 0.38 × 0.16 |
| θ range (°) | 5.14–54.96 | 4.49–54.94 |
| Limiting indices | −12 ≤ h ≤ 11 −12 ≤ k ≤ 12 −17 ≤ l ≤ 17 | −14 ≤ h ≤ 14 −12 ≤ k ≤ 12 −16 ≤ l ≤ 16 |
| No.of rflns collected | 13159 | 14473 |
| No. of unique rflns Rint | 2963 (0.0301) | 3144 (0.0526) |
| Completeness to θ (%) | 99.0% | 99.3% |
| Goodness-of-fit on F2 | 1.198 | 1.137 |
| Final R indices [I > 2σ (I)] | R1 = 0.0372, wR2 = 0.1078 | R1 = 0.0652, wR2 = 0.1592 |
| R indices (all data) | R1 = 0.0429, wR2 = 0.1246 | R1 = 0.0678, wR2 = 0.1621 |
| Largest diff. peak and hole (eÅ−3) | 0.44/−0.48 | 0.36/−0.34 |
| C1 | C4 | |
|---|---|---|
| Bond lengths (Å) | ||
| Al(1)–N(1) | 2.122(13) | 2.194(3) |
| Al(1)–O(1) | 1.876(11) | 1.856(18) |
| Al(1)–O(1i) | 1.953(11) | 1.966(2) |
| Al(1)–C(12) | 1.997(15) | 1.977(3) |
| Al(1)–C(13) | 1.983(16) | 1.985(3) |
| O(1)–C(8) | 1.427(15) | 1.398(5) |
| Bond angles (°) | ||
| N(1)–Al(1)–O(1) | 79.14(5) | 78.73(9) |
| N(1)–Al(1)–O(1i) | 154.49(5) | 158.62(8) |
| Al(1)–O(1)–C(8) | 121.91(8) | 121.8(3) |
| Al(1)–N(1)–C(9) | 113.13(9) | 110.97(18) |
| O(1)–Al(1)–O(1i) | 77.25(5) | 79.93(8) |
| C(12)–Al(1)–C(13) | 123.79(7) | 134.10(13) |
| Proton Type | δ (ppm) in Toluene-d8 | δ (ppm) in Benzene-d6 | δ (ppm) in Chloroform-d |
|---|---|---|---|
| Py-CH | 7.44 (1H), 6.63 (1H),6.43 (1H) | 7.29 (1H), 6.43 (1H),6.23 (1H) | 8.24 (1H), 7.79 (1H), 7.50 (1H) |
| Cy-CH | 4.58 (1H) | 4.49 (1H) | 4.92 (1H) |
| Cy-CH2 | 2.04 (2H), 1.20–1.11 (2H) | 1.86 (2H), 1.08 (1H),0.96 (1H) | 2.88 (2H), 1.94–1.84 (1H),1.74–1.66 (1H) |
| Cy-CH3 | 1.51 (3H), 0.67 (3H) | 1.45 (3H), 0.58 (3H) | 1.50 (3H), 0.82 (3H) |
| Al-CH3 | −0.12 (3H), −0.26 (3H) | −0.12 (3H), −0.25 (3H) | −0.62, −0.66, −0.67 |
| Run | ɛ-CL:Al:BnOH | T (°C) | t (min) | Solvent | Conv. (%) b | Mn (×104) c | Mn (calc) (×104) d | PDI c | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 250:1:1 | 25 | 10 | Toluene | 0 | ||||
| 2 | 250:1:1 | 40 | 10 | Toluene | 0 | ||||
| 3 | 250:1:1 | 60 | 10 | Toluene | 47 | 1.11 | 1.35 | 1.27 | 705 |
| 4 | 250:1:1 | 80 | 10 | Toluene | 85 | 1.61 | 2.43 | 1.47 | 1275 |
| 5 | 250:1:1 | 90 | 10 | Toluene | >99 | 1.89 | 2.83 | 1.61 | 1485 |
| 6 | 250:1:1 | 100 | 10 | Toluene | 95 | 1.84 | 2.72 | 1.72 | 1425 |
| 7 | 250:1:1 | 110 | 10 | Toluene | 92 | 1.84 | 2.63 | 1.76 | 1380 |
| 8 | 250:1:1 | 90 | 3 | Toluene | 43 | 1.04 | 1.24 | 1.21 | 2150 |
| 9 | 250:1:1 | 90 | 5 | Toluene | 63 | 1.35 | 1.81 | 1.41 | 1890 |
| 10 | 250:1:1 | 90 | 7 | Toluene | 86 | 1.18 | 2.46 | 1.43 | 1843 |
| 11 | 250:1:0 | 90 | 10 | Toluene | 0 | ||||
| 12 | 250:1:0 | 90 | 20 | Toluene | 0 | ||||
| 13 | 250:1:0 | 90 | 30 | Toluene | 15 | 0.27 | 0.43 | 1.15 | 75 |
| 14 | 250:1:2 | 90 | 10 | Toluene | 96 | 1.02 | 2.75 | 1.47 | 1440 |
| 15 | 250:1:4 | 90 | 10 | Toluene | 85 | 0.61 | 2.43 | 1.37 | 1275 |
| 16 | 250:1:5 | 90 | 10 | Toluene | 82 | 0.51 | 2.35 | 1.47 | 1230 |
| 17 | 250:1:10 | 90 | 10 | Toluene | 69 | 0.28 | 1.98 | 1.15 | 1035 |
| 18 | 100:1:1 | 90 | 10 | Toluene | >99 | 0.75 | 2.83 | 1.40 | 594 |
| 19 | 300:1:1 | 90 | 10 | Toluene | 93 | 1.58 | 2.66 | 1.47 | 1674 |
| 20 | 400:1:1 | 90 | 10 | Toluene | 63 | 2.11 | 1.81 | 1.57 | 1512 |
| 21 | 500:1:1 | 90 | 10 | Toluene | 51 | 2.23 | 1.46 | 1.49 | 1530 |
| 22 | 750:1:1 | 90 | 10 | Toluene | 23 | 2.51 | 0.67 | 1.35 | 1035 |
| 23 | 250:1:1 | 90 | 10 | Bulk | >99 | 0.81 | 2.83 | 1.64 | 1485 |
| 24 | 250:1:1 | - | 10 | Hexane | Trace | ||||
| 25 | 250:1:1 | - | 10 | THF | Trace | ||||
| 26 | 250:1:1 | - | 10 | CH2Cl2 | 9 | 0.40 | 0.27 | 2.03 | 135 |
| 27 e | 250:1:1 | 90 | 30 | Bulk | trace | ||||
| 28 e | 250:1:1 | 60 | 30 | Hexane | 83 | 1.85 | 1.90 | ||
| 29 e | 250:1:1 | 60 | 30 | THF | 42 | 0.80 | 1.20 | ||
| 30 e | 250:1:1 | 35 | 30 | CH2Cl2 | 29 | 0.59 | 1.16 |
| Run | Pro-Initiator (R1, R2, R3) | Conv. (%) b | Mn (×104) c | Mn (calc.) (×104) d | PDI c | TOF (h−1) |
|---|---|---|---|---|---|---|
| 1 | C1 (H, H, Me) | >99 | 1.89 | 2.83 | 1.61 | 1485 |
| 2 | C2 (H, H, Et) | 95 | 1.35 | 2.72 | 1.32 | 1425 |
| 3 | C3 (H, H, i-Bu) | 91 | 1.21 | 2.60 | 1.25 | 1365 |
| 4 | C4 (Cl, H, Me) | 83 | 1.54 | 2.38 | 1.55 | 1245 |
| 5 | C5 (H, Me, Me) | 97 | 0.73 | 2.78 | 1.30 | 1455 |
| 6 | C6 (Cl, Me, Me) | 82 | 1.24 | 2.35 | 1.48 | 1230 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhang, W.; Solan, G.A.; Liang, T.; Sun, W.-H. Bimetallic Aluminum 5,6-Dihydro-7,7-dimethyl quinolin-8-olates as Pro-Initiators for the ROP of ε-CL; Probing the Nuclearity of the Active Initiator. Polymers 2018, 10, 764. https://doi.org/10.3390/polym10070764
Zhang Q, Zhang W, Solan GA, Liang T, Sun W-H. Bimetallic Aluminum 5,6-Dihydro-7,7-dimethyl quinolin-8-olates as Pro-Initiators for the ROP of ε-CL; Probing the Nuclearity of the Active Initiator. Polymers. 2018; 10(7):764. https://doi.org/10.3390/polym10070764
Chicago/Turabian StyleZhang, Qiurui, Wenjuan Zhang, Gregory A. Solan, Tongling Liang, and Wen-Hua Sun. 2018. "Bimetallic Aluminum 5,6-Dihydro-7,7-dimethyl quinolin-8-olates as Pro-Initiators for the ROP of ε-CL; Probing the Nuclearity of the Active Initiator" Polymers 10, no. 7: 764. https://doi.org/10.3390/polym10070764
APA StyleZhang, Q., Zhang, W., Solan, G. A., Liang, T., & Sun, W.-H. (2018). Bimetallic Aluminum 5,6-Dihydro-7,7-dimethyl quinolin-8-olates as Pro-Initiators for the ROP of ε-CL; Probing the Nuclearity of the Active Initiator. Polymers, 10(7), 764. https://doi.org/10.3390/polym10070764