
Crystallization, Structure Determination and Reticular Twinning in Iron(III) Salicylate: Fe[(HSal)(Sal)(H2O)2]


Abstract
:1. Introduction
2. Results and Discussion
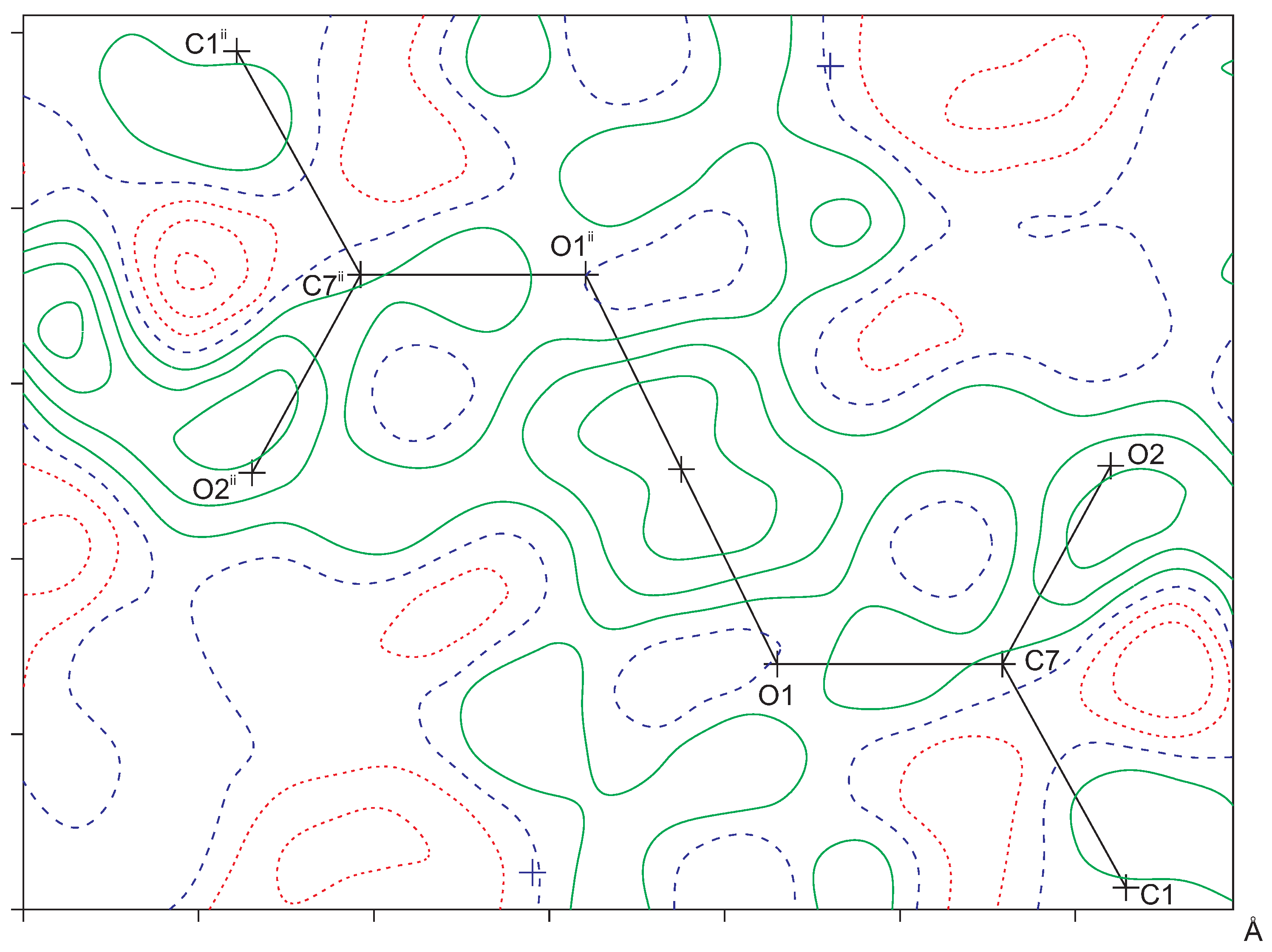
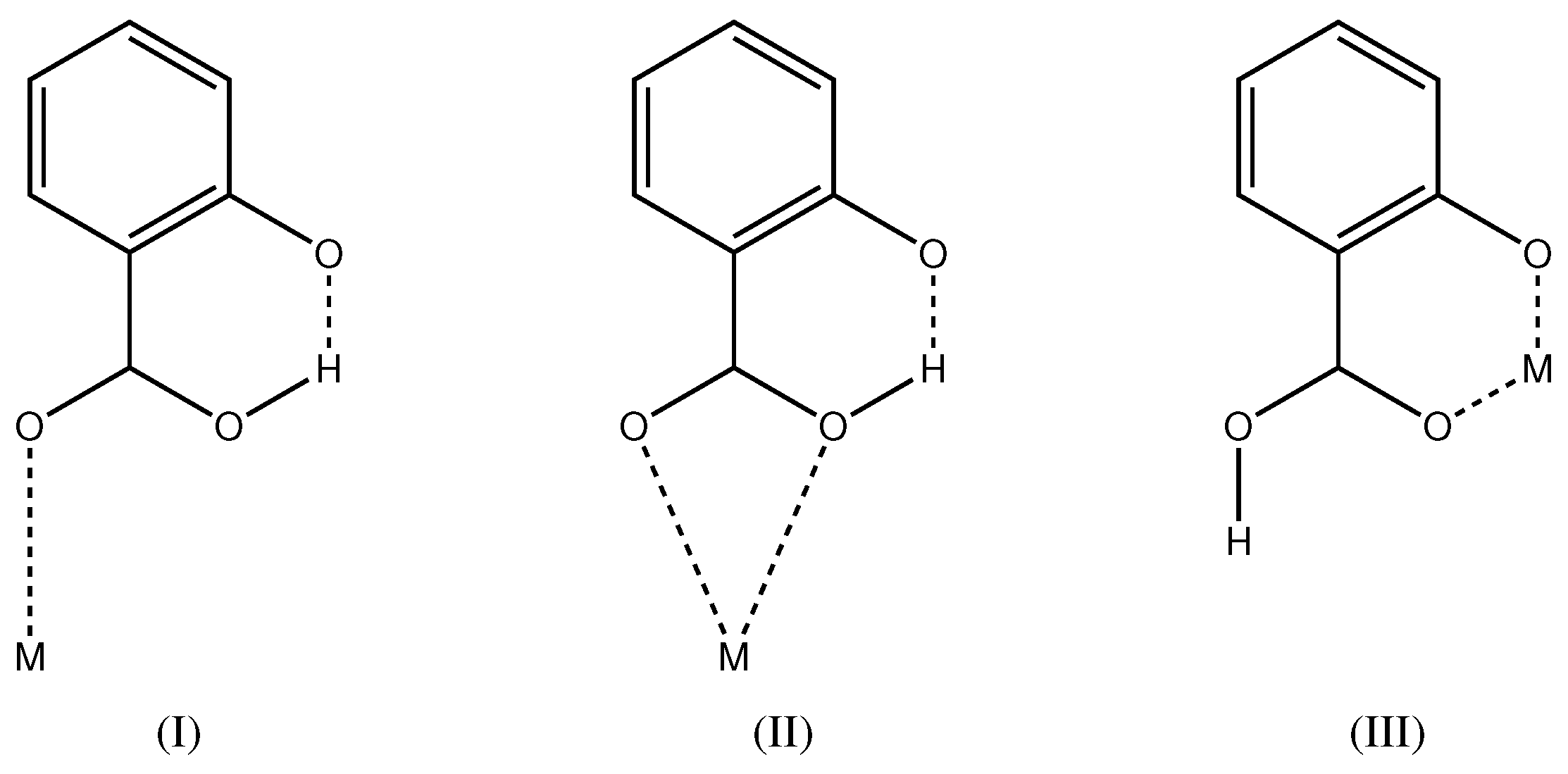
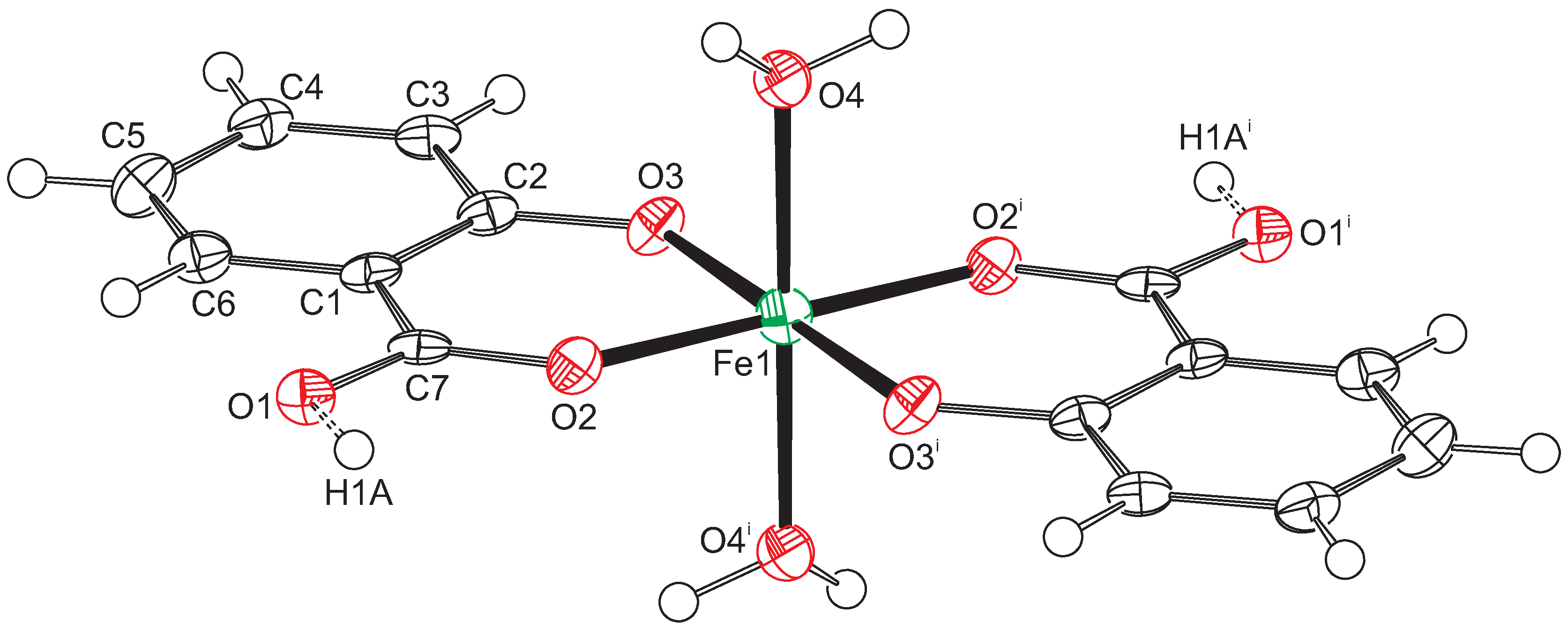
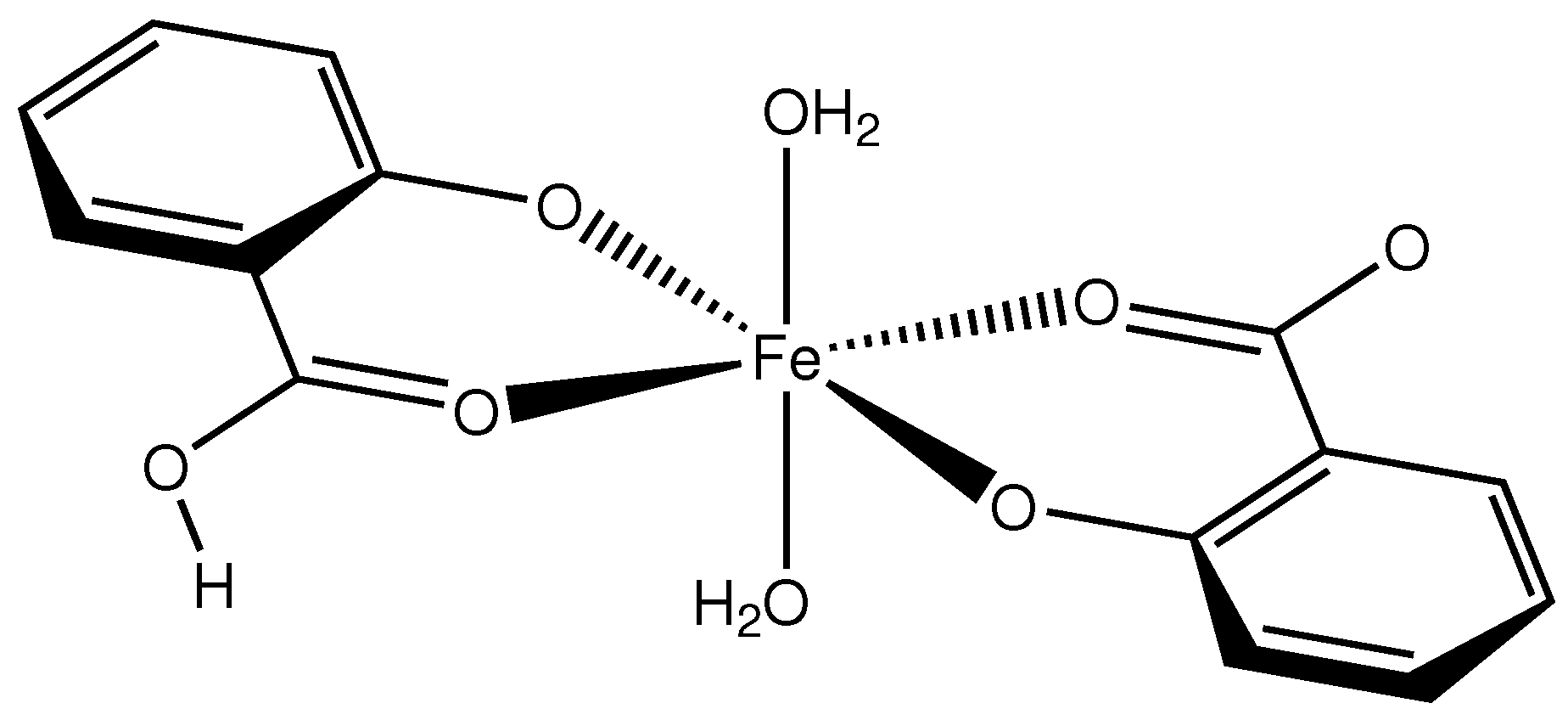
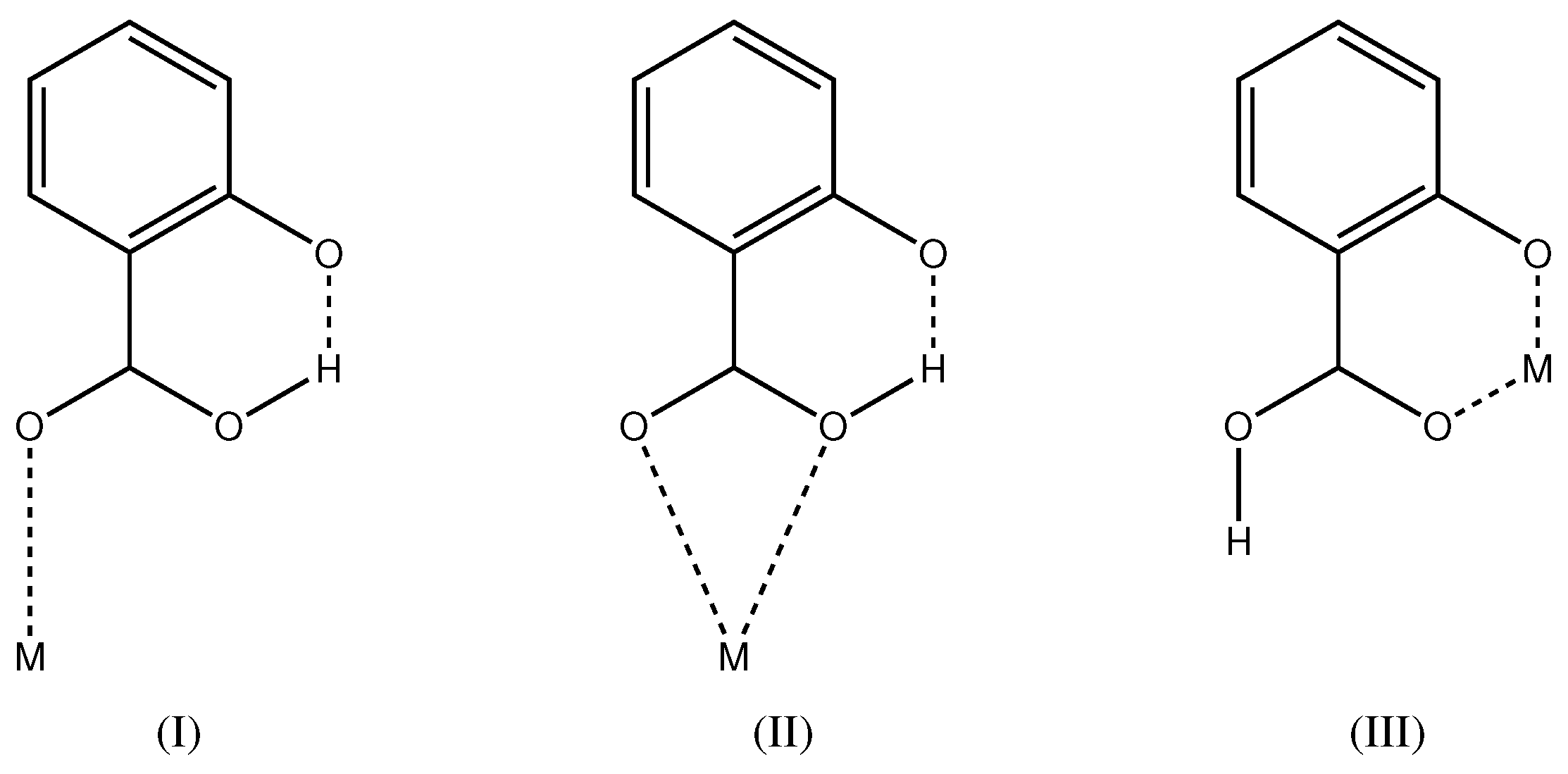
2.1. Intramolecular Interactions
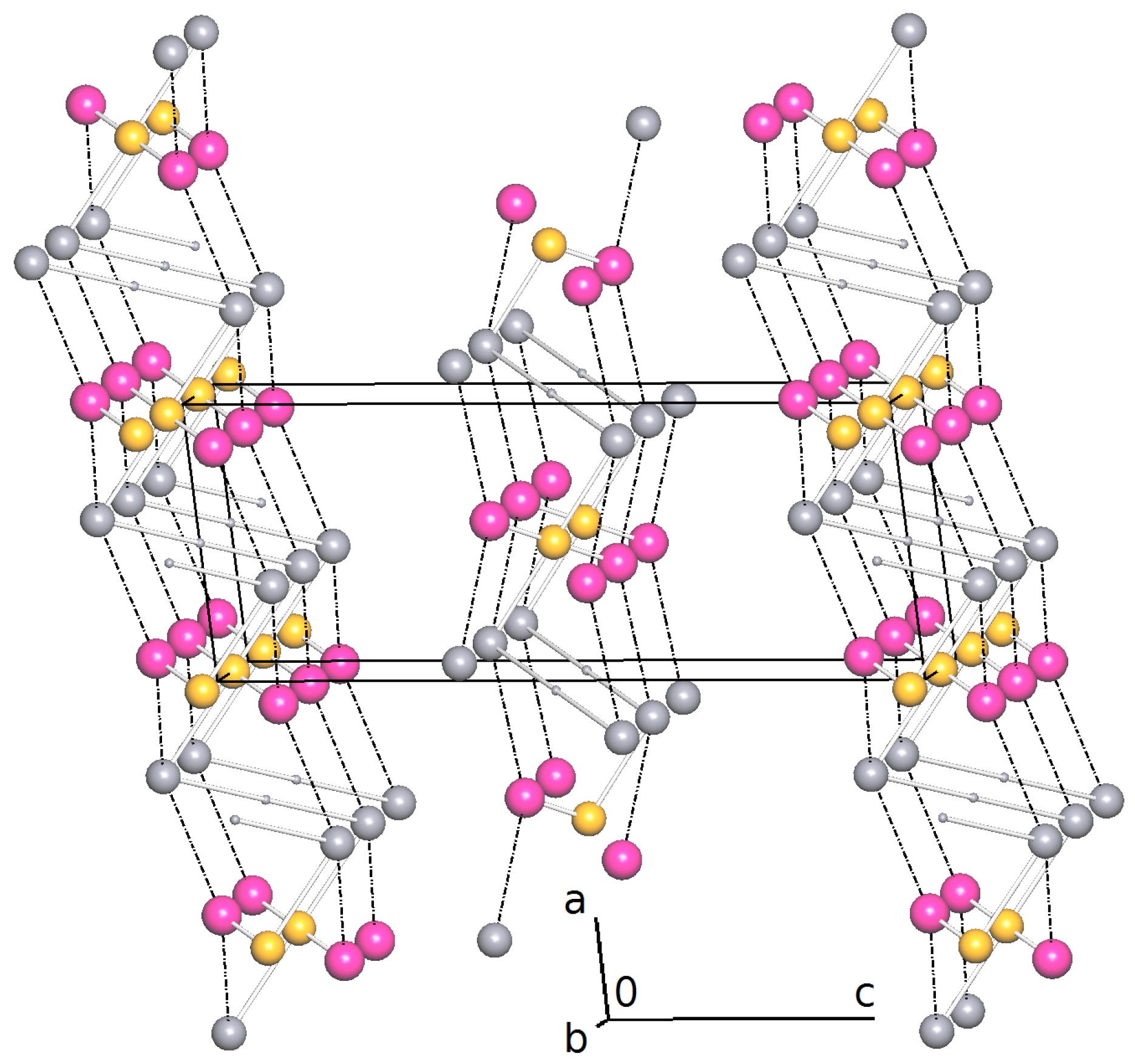
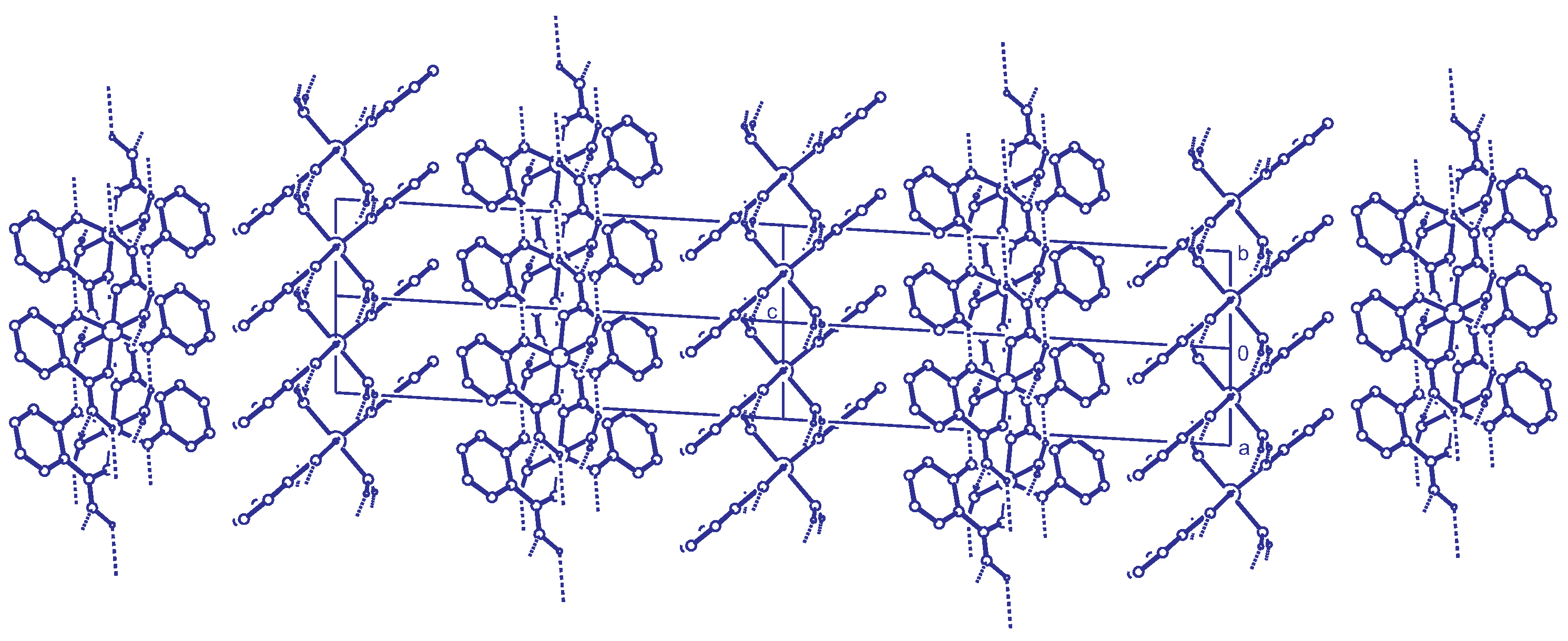
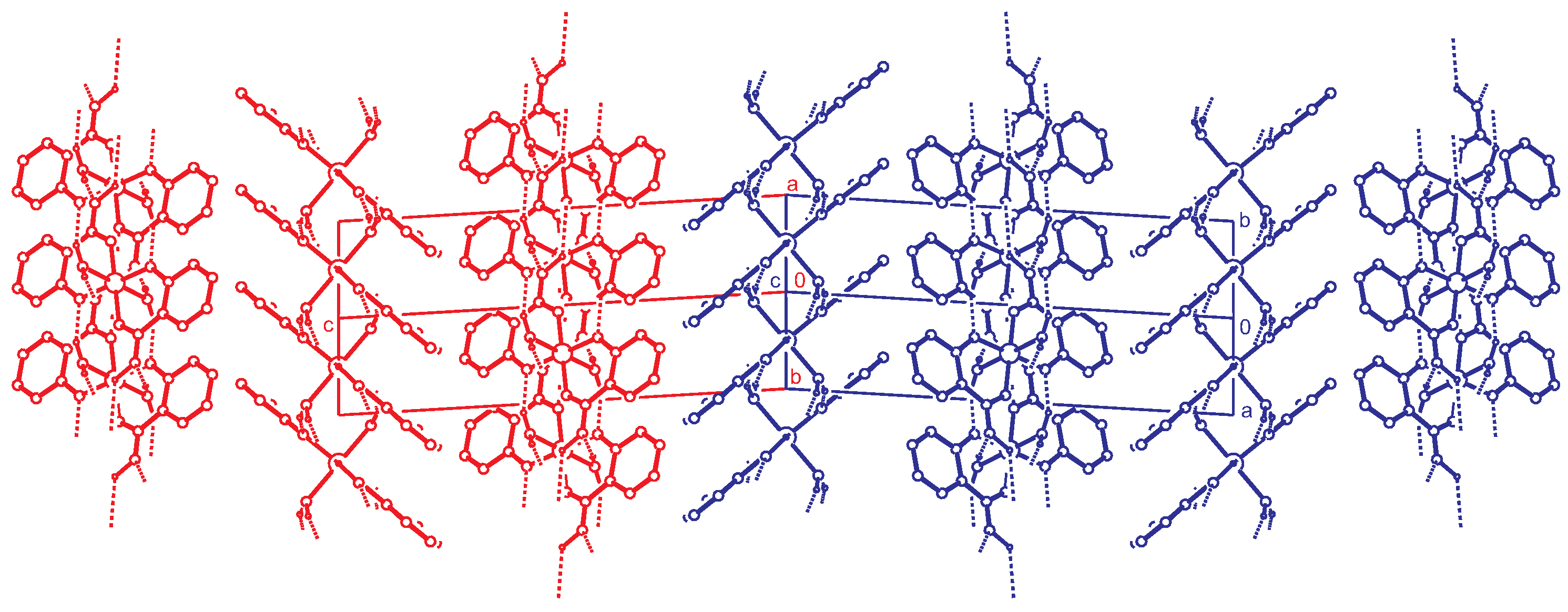
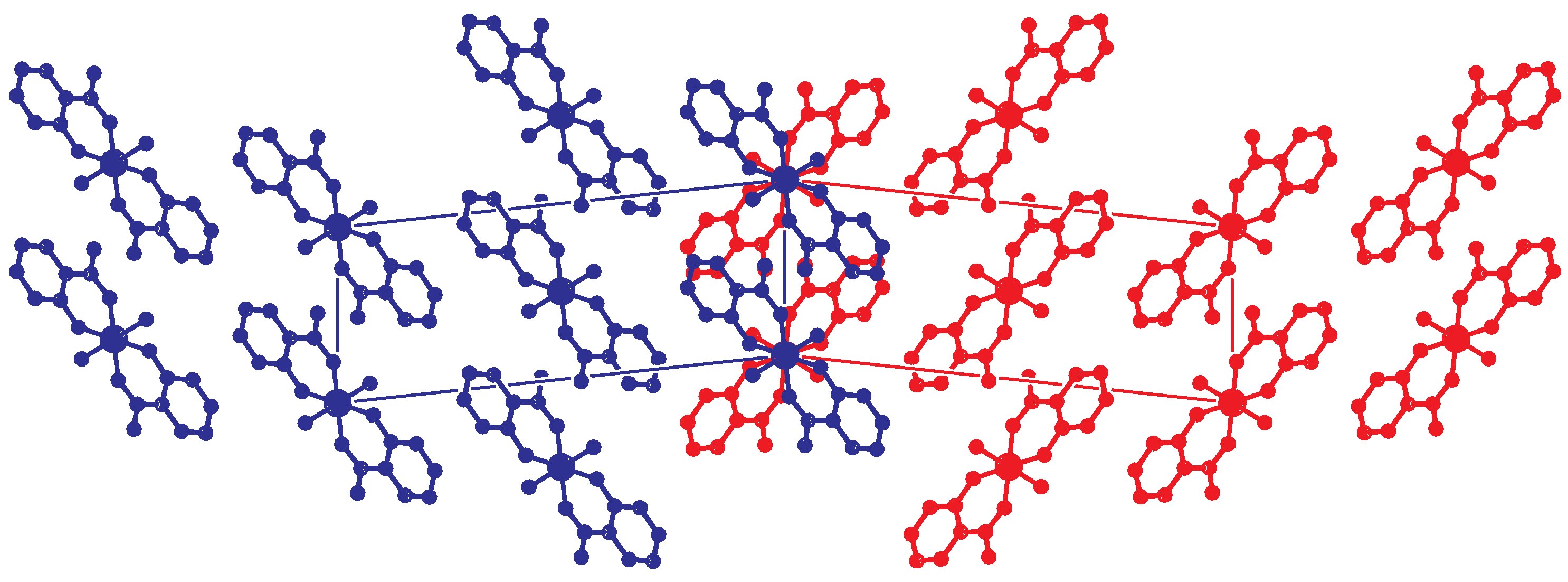
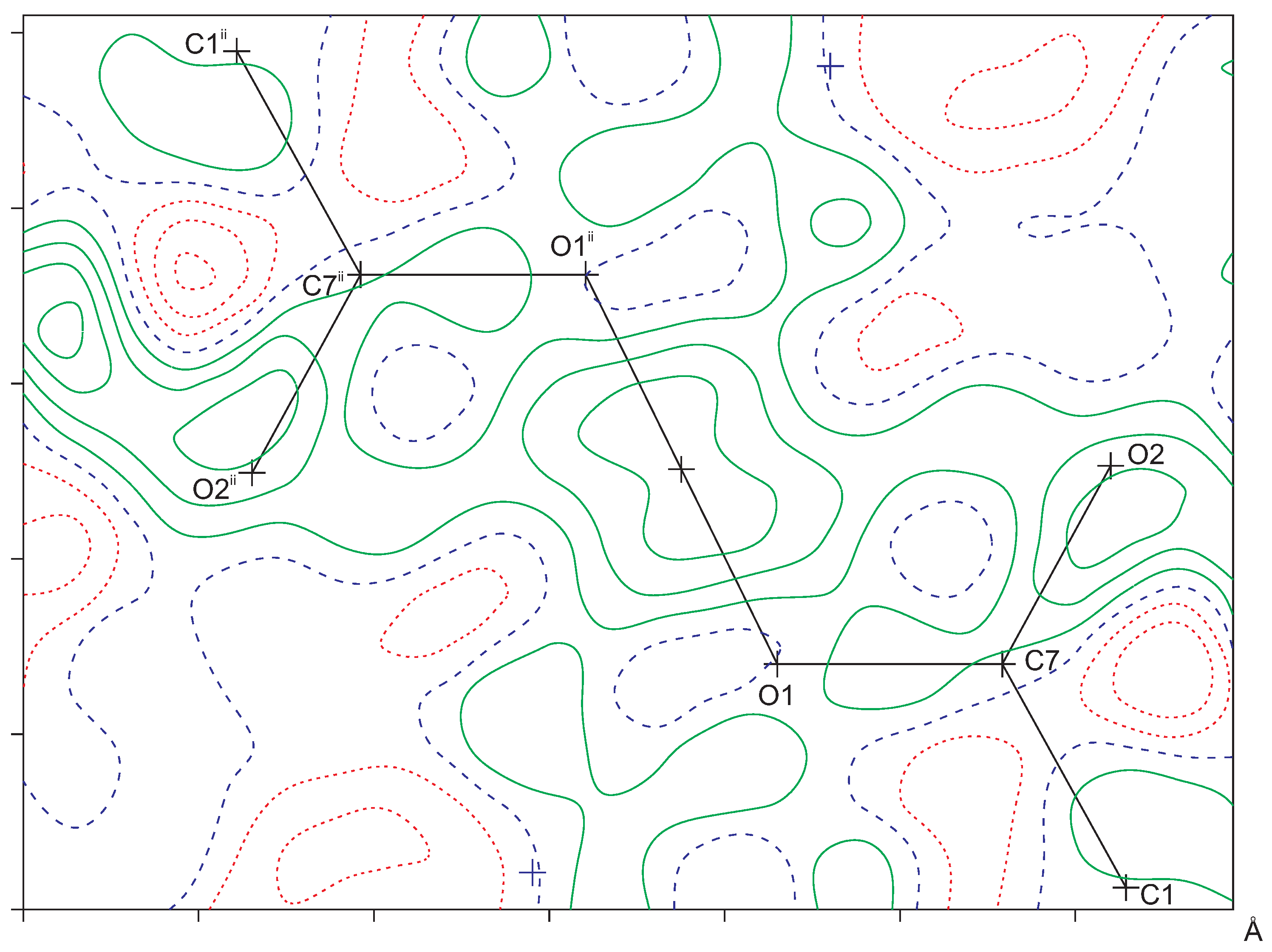
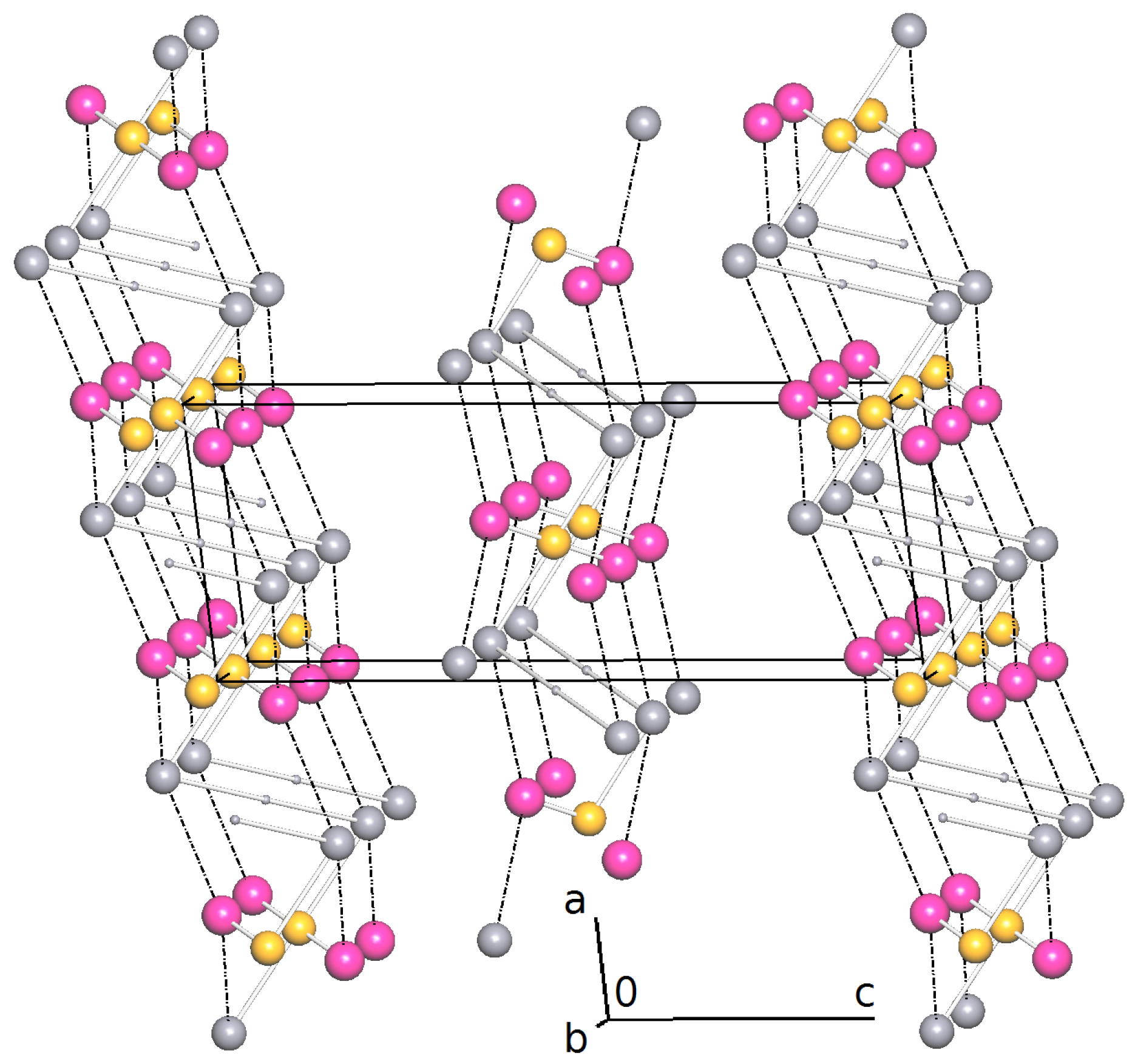
2.2. Intermolecular Interactions
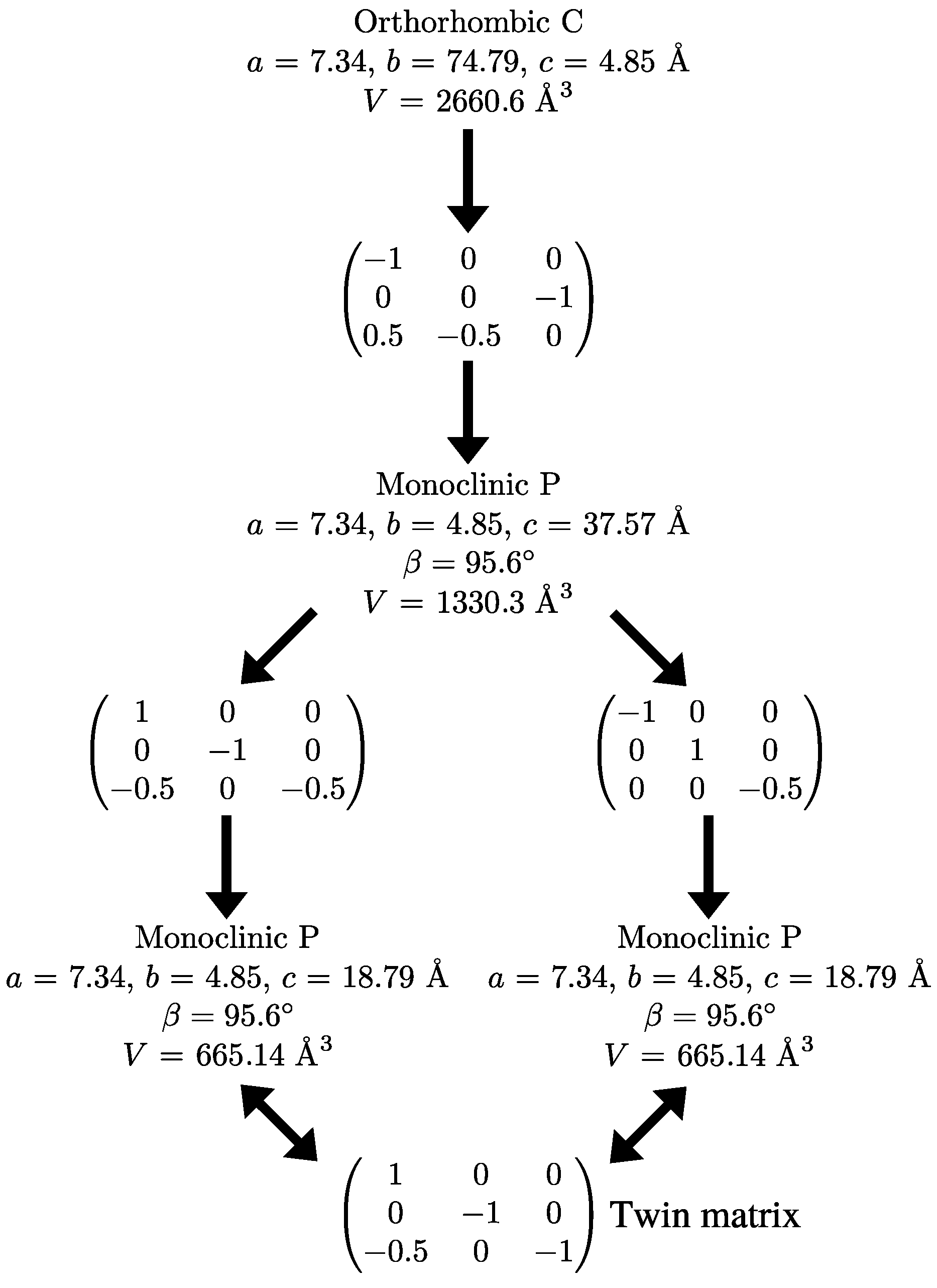
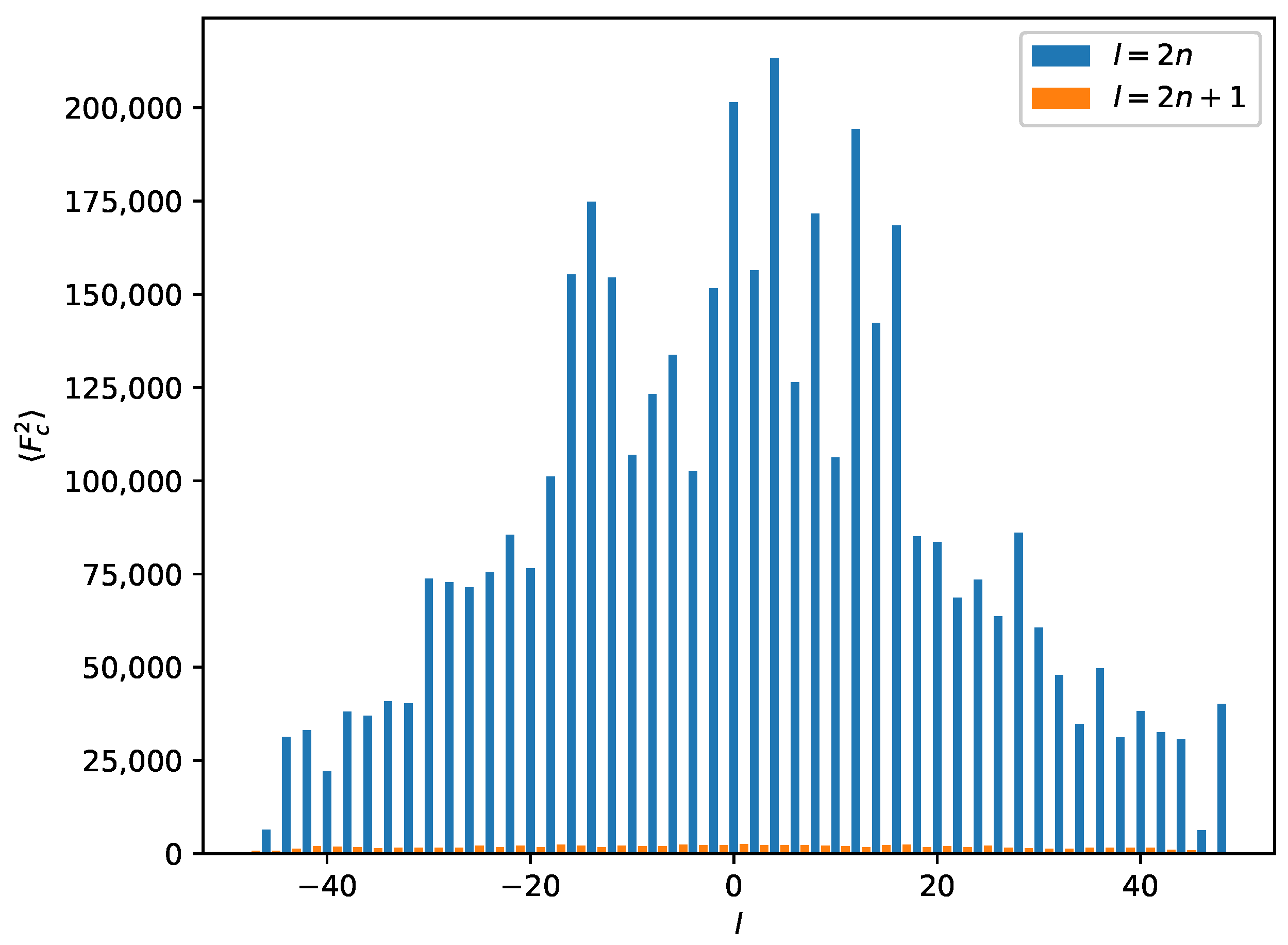
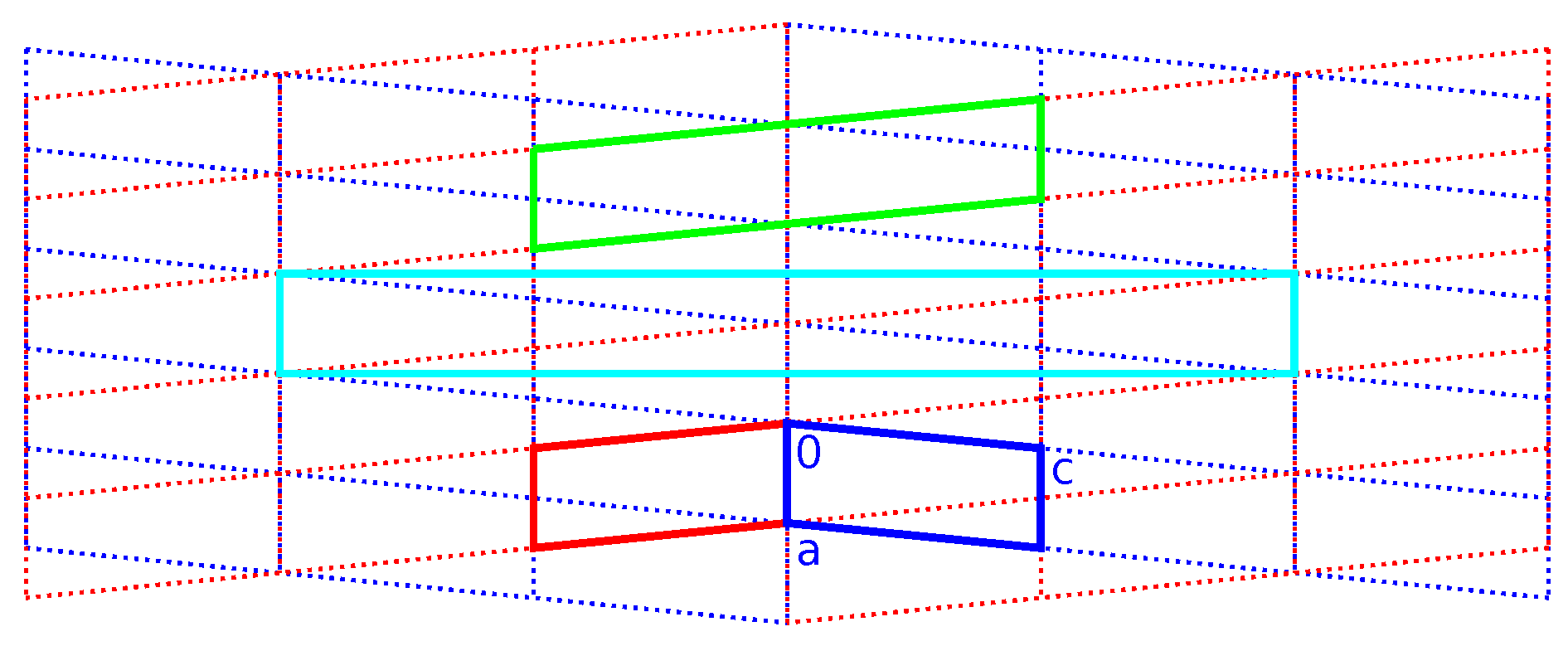
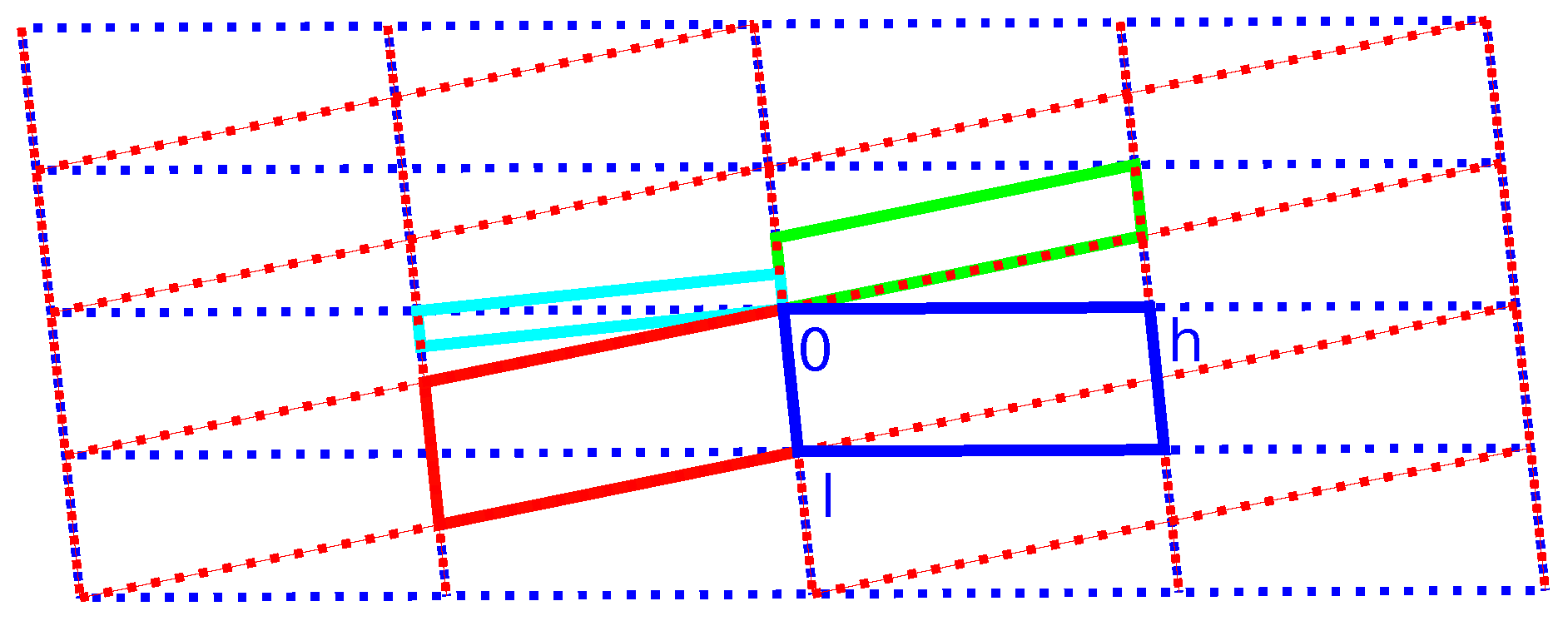
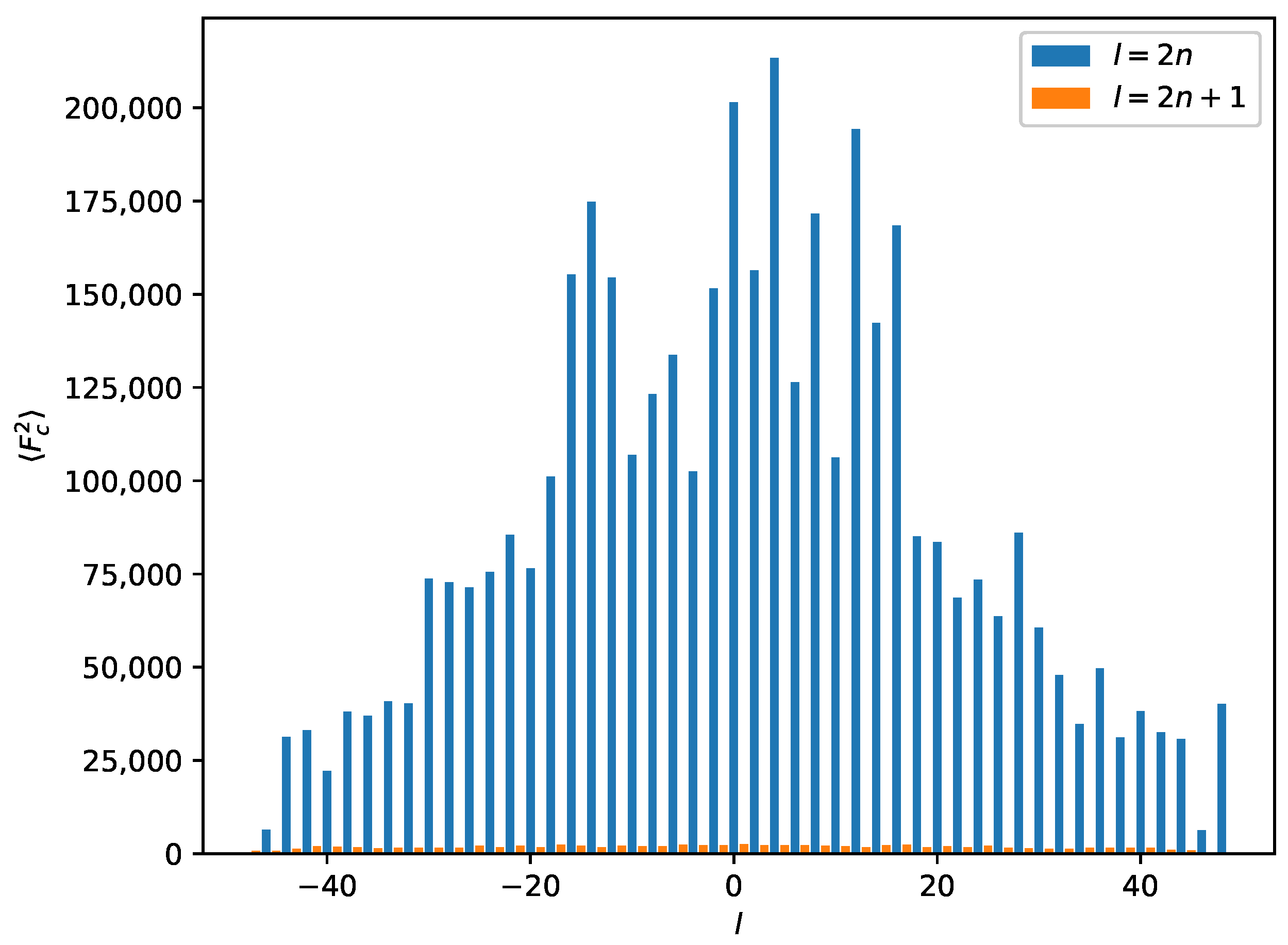
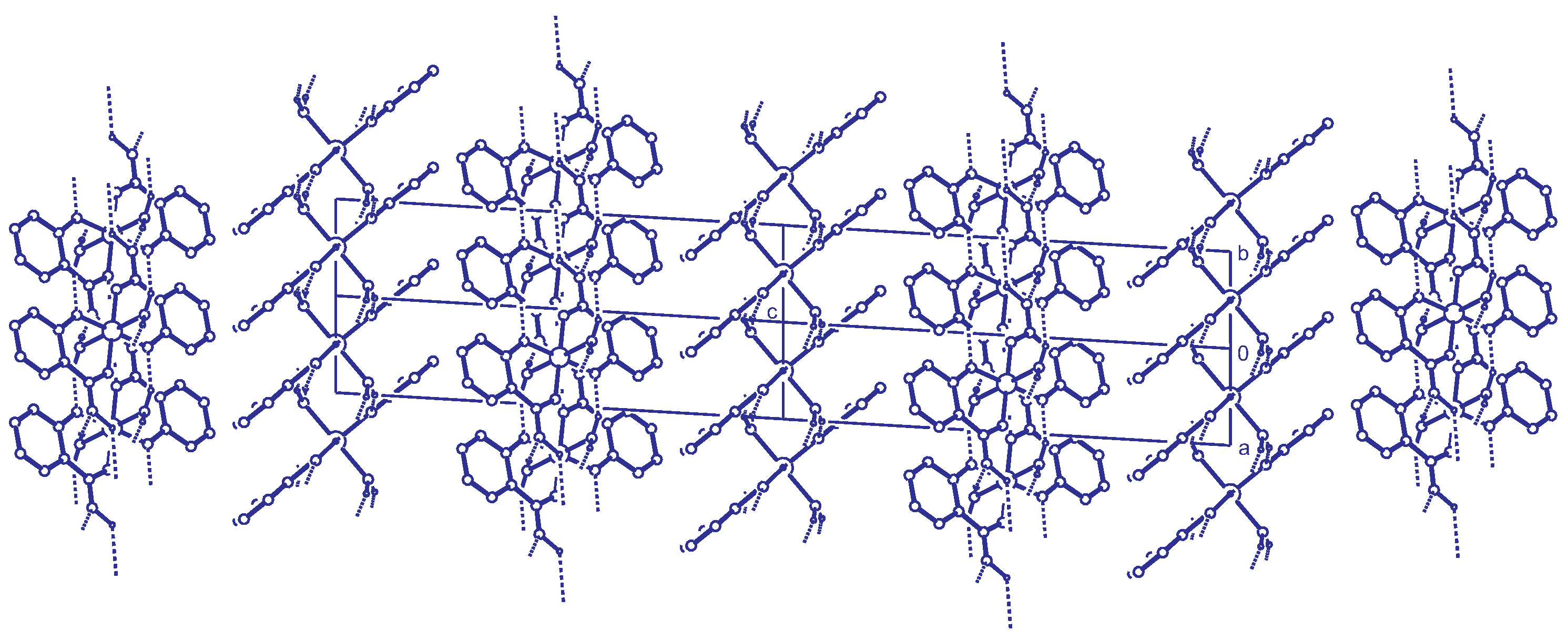
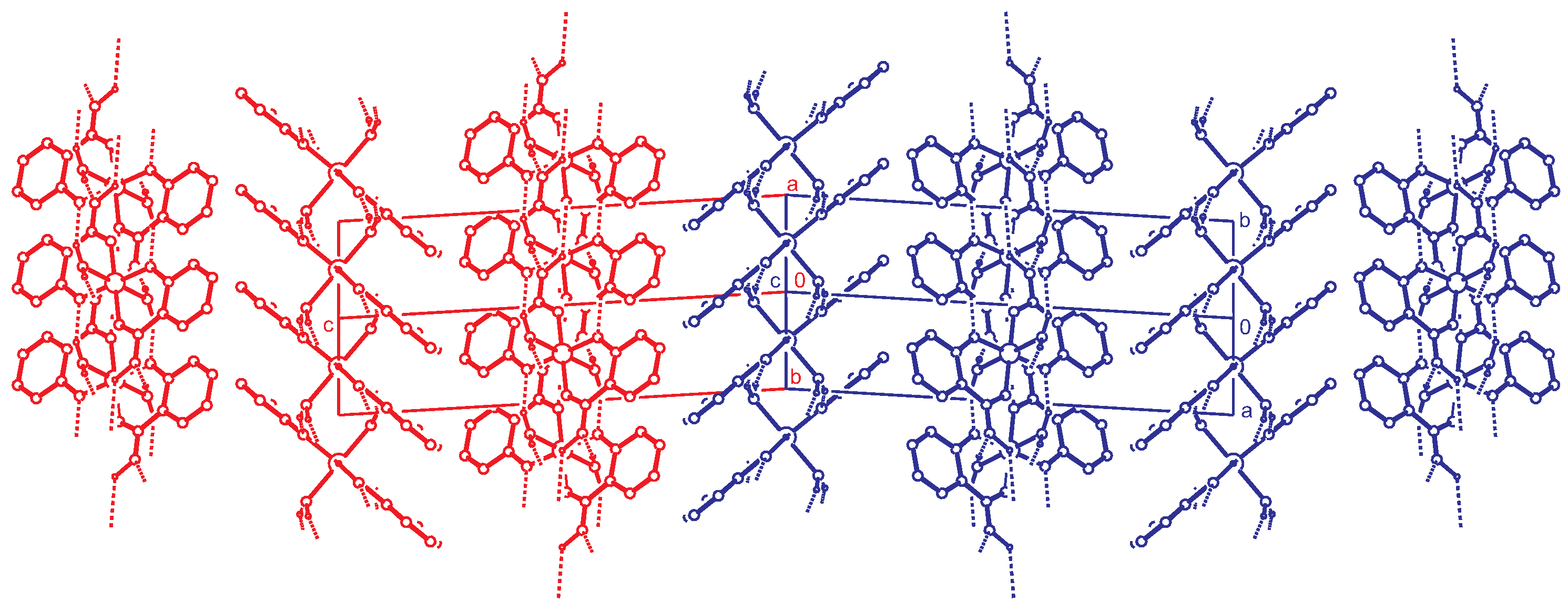
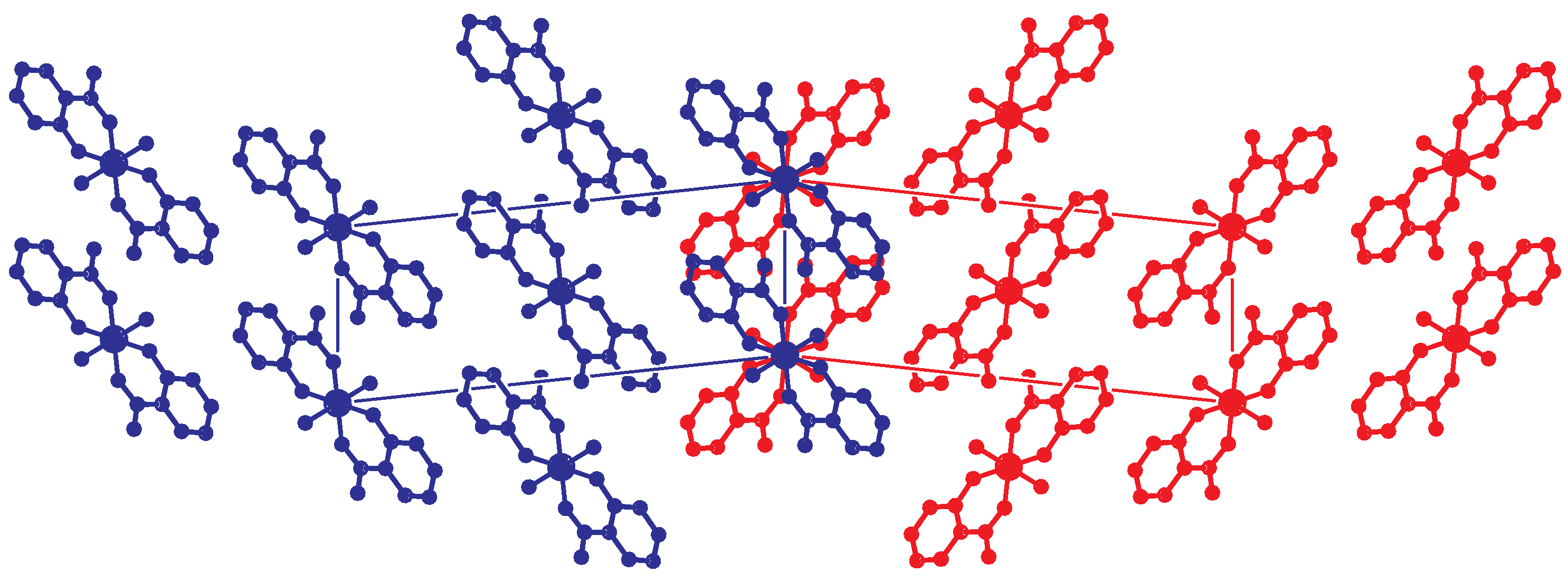
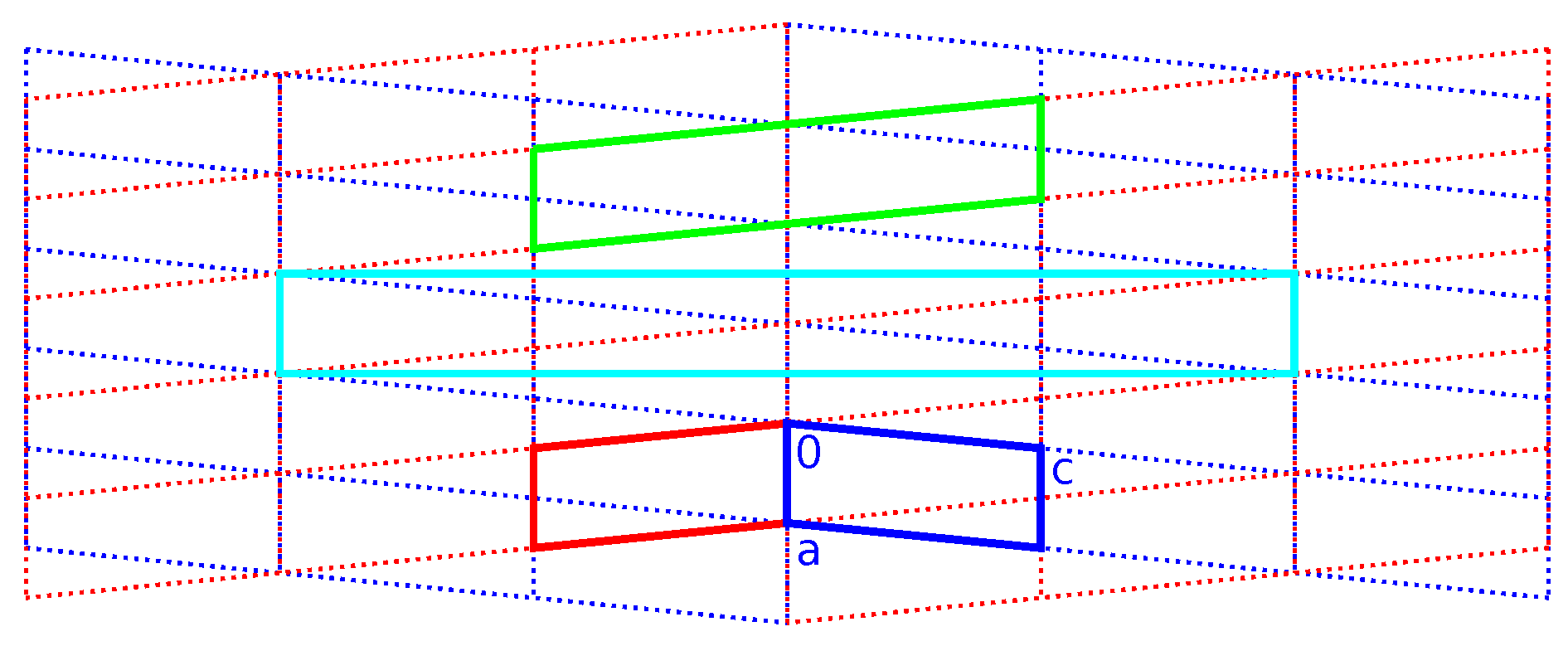
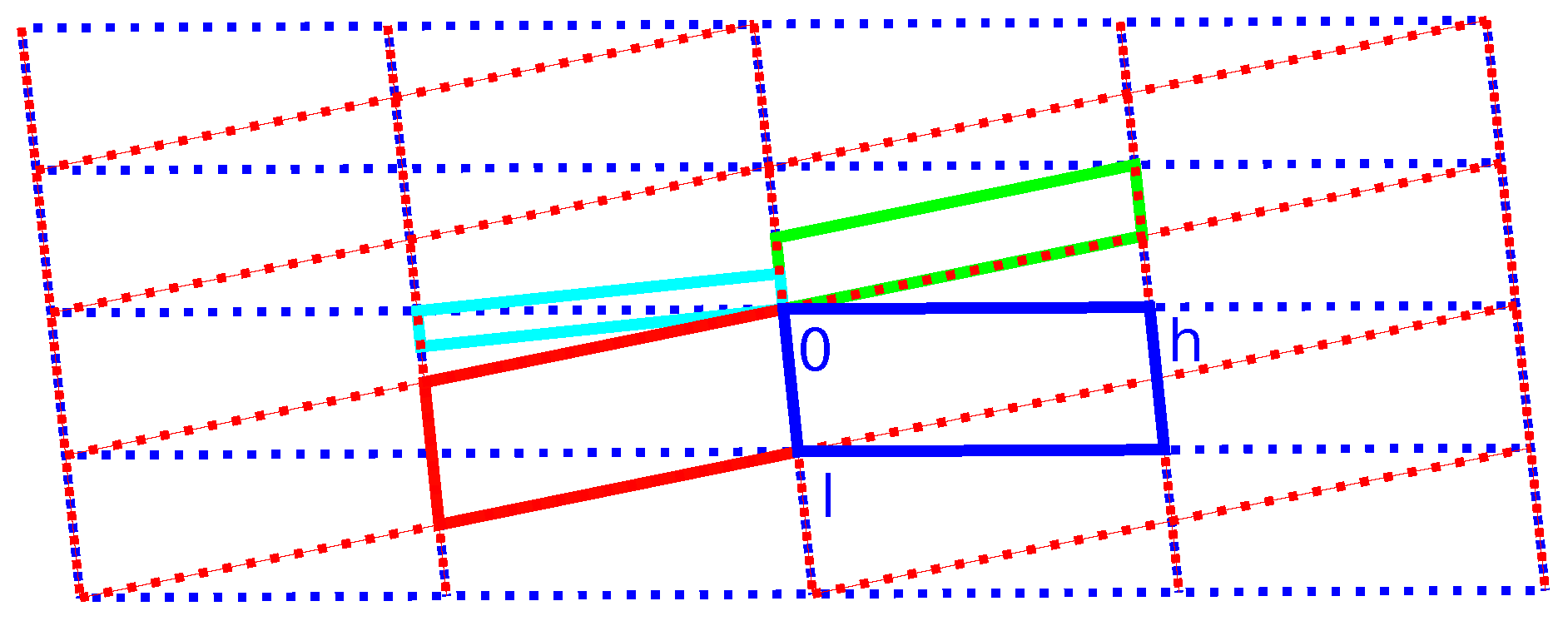
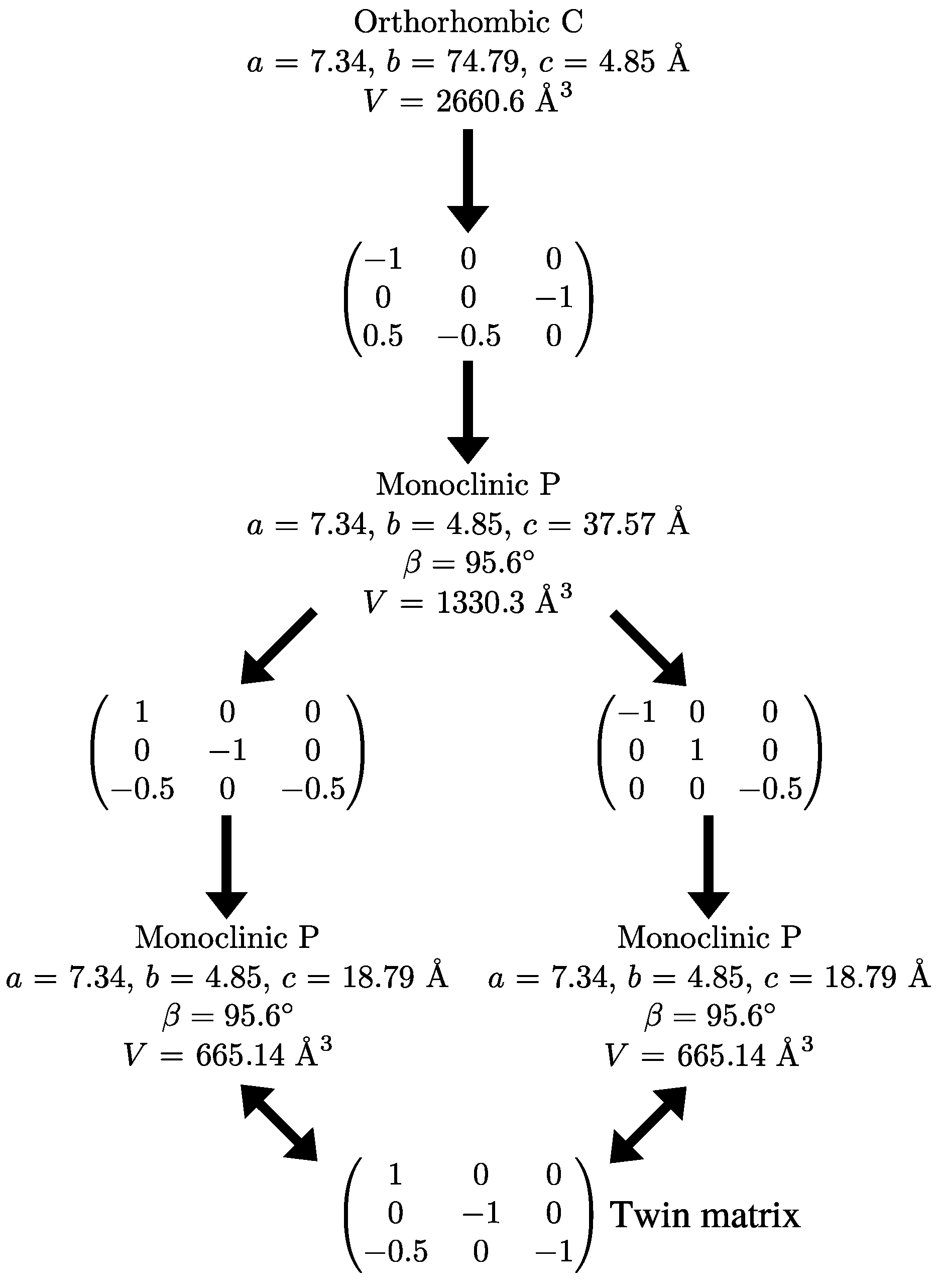
2.3. Reticular Twinning
3. Materials and Methods
3.1. Synthesis and Crystallization
3.2. X-ray Crystal Structure Determination
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| OD | Order–disorder |
| r.m.s. | root mean square |
| Salicylate di-anion () | |
| Salicylate mono-anion () | |
| Salicylic acid () |
References
- Roy, S.; Kästner, J. Synergistic Substrate and Oxygen Activation in Salicylate Dioxygenase Revealed by QM/MM Simulations. Angew. Chem. Int. Ed. 2015, 55, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.A.; Lutz, M.; Spek, A.L.; Hagen, W.R.; Weckhuysen, B.M.; van Koten, G.; Gebbink, R.J.M.K. Modeling the 2-His-1-Carboxylate Facial Triad: Ironcatecholato Complexes as Structural and Functional Models of the Extradiol Cleaving Dioxygenases. J. Am. Chem. Soc. 2007, 129, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Zhou, T. The Design of Orally Active Iron Chelators. Ann. N. Y. Acad. Sci. 2005, 1054, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.R.; Meyerhoff, M.E.; Mitchell-Koch, J.T. Salicylate Detection by Complexation with Iron(III) and Optical Absorbance Spectroscopy. An Undergraduate Quantitative Analysis Experiment. J. Chem. Educ. 2008, 85, 1658–1659. [Google Scholar] [CrossRef]
- Pozdnyakov, I.P.; Plyusnin, V.F.; Grivin, V.P.; Oliveros, E. Photochemistry of Fe(III) complexes with salicylic acid derivatives in aqueous solutions. J. Photochem. Photobiol. A Chem. 2015, 307–308, 9–15. [Google Scholar] [CrossRef]
- McBryde, W.A.E.; Rohr, J.L.; Penciner, J.S.; Page, J.A. Stability constants of three iron(III) salicylates. Can. J. Chem. 1970, 48, 2574–2586. [Google Scholar] [CrossRef]
- Park, M.V. Complex formation between iron(III) and some substituted salicylic acids. J. Chem. Soc. A 1966, 0, 816–820. [Google Scholar] [CrossRef]
- Chattopadhyaya, M. Effect of Substituents on the Relative Stabilities of Fe(III) Complexes of Substituted Salicylic Acids. J. Indian Chem. Soc. 1982, 59, 1416–1418. [Google Scholar]
- Furia, E.; Sindona, G. Interaction of Iron(III) with 2-Hydroxybenzoic Acid in Aqueous Solutions. J. Chem. Eng. Data 2012, 57, 195–199. [Google Scholar] [CrossRef]
- Porwal, S.K.; Furia, E.; Harris, M.E.; Viswanathan, R.; Devireddy, L. Synthetic, potentiometric and spectroscopic studies of chelation between Fe(III) and 2,5-DHBA supports salicylate-mode of siderophore binding interactions. J. Inorg. Biochem. 2015, 145, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Gibbs, G.V.; Ribbe, P.H. Quadratic Elongation: A Quantitative Measure of Distortion in Coordination Polyhedra. Science 1971, 172, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Pilati, T.; Forni, A. SYMMOL: A program to find the maximum symmetry group in an atom cluster, given a prefixed tolerance. J. Appl. Crystallogr. 1998, 31, 503–504. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Crespo-Alonso, M.; Toso, L.; Lachowicz, J.I.; Crisponi, G.; Alberti, G.; Biesuz, R.; Domínguez-Martín, A.; Niclós-Gutíerrez, J.; González-Pérez, J.M.; et al. Iron(III) and aluminium(III) complexes with substituted salicyl-aldehydes and salicylic acids. J. Inorg. Biochem. 2013, 128, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zarembowitch, J.; Kahn, O.; Jaud, J.; Galy, J. Versatility of iron(III) upon coordination with the binucleating ligand N, N′-bis-(2-hydroxy, 3-carboxybenzilidene)1,2-diaminoethane. Inorg. Chim. Acta 1982, 65, L35–L36. [Google Scholar] [CrossRef]
- Brown, I.D. The Chemical Bond in Inorganic Chemistry; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, G. Covalent versus Electrostatic Nature of the Strong Hydrogen Bond: Discrimination among Single, Double, and Asymmetric Single-Well Hydrogen Bonds by Variable-Temperature X-ray Crystallographic Methods in β-Diketone Enol RAHB Systems. J. Am. Chem. Soc. 2004, 126, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Walker, B.; Michaelides, A. Quantum nature of the hydrogen bond. Proc. Natl. Acad. Sci. USA 2011, 108, 6369–6373. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farajtabar, A.; Gharib, F. Solvent effect on protonation constants of salicylic acid in mixed aqueous organic solutions of DMSO. Monatsheft Chem.-Chem. Mon. 2010, 141, 381–386. [Google Scholar] [CrossRef]
- García, M.; Ramis, G.; Mongay, C. Spectrophotometric determination of protonation constants of monoprotic systems in strong acid and strong basic media. Spectrochim. Acta Part A Mol. Spectrosc. 1982, 38, 1005–1009. [Google Scholar] [CrossRef]
- Ma, Z.; Lu, W.; Liang, B.; Pombeiro, A.J.L. Synthesis, characterization, photoluminescent and thermal properties of zinc(II) 4′-phenyl-terpyridine compounds. New J. Chem. 2013, 37, 1529–1537. [Google Scholar] [CrossRef]
- Seth, P.; Ghosh, S.; Figuerola, A.; Ghosh, A. Trinuclear heterometallic CuII-MnII complexes of a salen type Schiff base ligand: Anion dependent variation of phenoxido bridging angles and magnetic coupling. Dalton Trans. 2014, 43, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Palanisami, N.; Rajakannu, P.; Murugavel, R. Non-covalently aggregated zinc and cadmium complexes derived from substituted aromatic carboxylic acids: Synthesis, spectroscopy, and structural studies. Inorg. Chim. Acta 2013, 405, 522–531. [Google Scholar] [CrossRef]
- Terada, T.; Hirabayashi, K.; Liu, D.; Nakamura, T.; Wakimoto, T.; Matsumoto, T.; Tatsumi, K. [3:1] Site-Differentiated [4Fe-4S] Clusters Having One Carboxylate and Three Thiolates. Inorg. Chem. 2013, 52, 11997–12004. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Allen, F.H. The Cambridge Structural Database in retrospect and prospect. Angew. Chem. Int. Ed. 2014, 53, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, P.; Tomas, A.; Nguyen-Huy, D.; Viossat, B. Crystal structure of aquachloro (salicylato)(1,10-phenanthroline)-copper(II), C19H15CICuN2O4. Z. Kristallogr. New Cryst. Struct. 2000, 215, 521–522. [Google Scholar] [CrossRef]
- Banti, C.; Giannoulis, A.; Kourkoumelis, N.; Owczarzak, A.; Kubicki, M.; Hadjikakou, S. Silver(I) compounds of the anti-inflammatory agents salicylic acid and p-hydroxyl-benzoic acid which modulate cell function. J. Inorg. Biochem. 2015, 142, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.F.; Griffith, W.P.; White, A.J.; Williams, D.J. A new bonding mode for salicylate: The X-ray crystal structure of (pyH)[MoO2(Hsal)(sal)]. Polyhedron 1992, 11, 2711–2712. [Google Scholar] [CrossRef]
- Palanisami, N.; Prabusankar, G.; Murugavel, R. A novel dimeric copper salicylate with an undissociated COOH group: Synthesis and crystal structure of [Cu2(HSal)(Sal)(2,2′-bpy)2](ClO4). Inorg. Chem. Commun. 2006, 9, 1002–1006. [Google Scholar] [CrossRef]
- Baroni, T.E.; Bembenek, S.; Heppert, J.A.; Hodel, R.R.; Laird, B.B.; Morton, M.D.; Barnes, D.L.; Takusagawa, F. Hydrogen bonding in tungsten (VI) salicylate free acids. Coord. Chem. Rev. 1998, 174, 255–282. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Herbst-Irmer, R. Crystal Structure Refinement: A Crystallographer’s Guide to SHELXL; Chapter 7: Twinning; Oxford University Press: Oxford, UK, 2006; pp. 106–149. [Google Scholar]
- Dornberger-Schiff, K. Reinterpretation of pseudo-orthorhombic diffraction patterns. Acta Crystallogr. 1966, 21, 311–322. [Google Scholar] [CrossRef]
- Le Page, Y. Mallard’s law recast as a Diophantine system: fast and complete enumeration of possible twin laws by [reticular][pseudo] merohedry. J. Appl. Crystallogr. 2002, 35, 175–181. [Google Scholar] [CrossRef]
- Arend, H.; Connelly, J. Tetramethoxysilane as gel forming agent in crystal growth. J. Cryst. Growth 1982, 56, 642–644. [Google Scholar] [CrossRef]
- Duisenberg, A.J.M. Indexing in single-crystal diffractometry with an obstinate list of reflections. J. Appl. Crystallogr. 1992, 25, 92–96. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. Sect. D 2010, 66, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, A.M.M.; Xian, X.; Kroon-Batenburg, L.M.J. EVAL15: A diffraction data integration method based on ab initio predicted profiles. J. Appl. Crystallogr. 2010, 43, 70–82. [Google Scholar] [CrossRef]
- Sheldrick, G. TWINABS, Version 2012/1; University of Göttingen: Göttingen, Germany, 2012. [Google Scholar]
- Herbst-Irmer, R.; Sheldrick, G.M. Refinement of Twinned Structures with SHELXL97. Acta Crystallogr. Sect. B 1998, 54, 443–449. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced rigid-bond restraints. Acta Crystallogr. Sect. A 2012, 68, 448–451. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| –⋯ O1 | 1.24 Å | 1.24 Å | 2.476(8) Å | 180 |
| –⋯ O1 | 0.95 Å | 1.87 Å | 2.808(8) Å | 170 |
| –⋯ O3 | 0.95 Å | 1.81 Å | 2.704(9) Å | 156 |
| Molecular Formula | ||
|---|---|---|
| Formula weight | 365.09 | |
| Temperature | 110(2) K | |
| Wavelength | 1.54184 Å (CuK) | |
| Crystal system, space group | Monoclinic, | |
| Unit cell dimensions | a = 7.3392(11) | = 96.090(9) |
| b = 4.8493(6) | ||
| c = 18.794(3) | ||
| Volume | 665.11(15) Å | |
| Z, Calculated density | 2, 1.823 g/cm | |
| Absorption coefficient | 9.53 mm | |
| F(000) | 374 | |
| Crystal size | 25 × 25 × 125 μm3 | |
| 0.51 Å | ||
| Limiting indices | −7 7 | |
| −4 4 | ||
| −18 19 | ||
| Refl. collected/unique/observed | 3505/1246/1033 | |
| Completeness to = 51.35 | 99.0% | |
| Number of parameters | 107 | |
| Number of restraints | 93 | |
| R1/wR2 | 0.0678/0.1730 | |
| R1/wR2 [all refl.] | 0.0862/0.1901 | |
| Goodness of Fit | 1.073 | |
| Twin fraction (BASF) | 0.461(4) | |
| Residual density [min/max] | −0.48/0.71 eÅ | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Horn, J.A.; Souvignier, B.; Lutz, M. Crystallization, Structure Determination and Reticular Twinning in Iron(III) Salicylate: Fe[(HSal)(Sal)(H2O)2]. Crystals 2017, 7, 377. https://doi.org/10.3390/cryst7120377
Van der Horn JA, Souvignier B, Lutz M. Crystallization, Structure Determination and Reticular Twinning in Iron(III) Salicylate: Fe[(HSal)(Sal)(H2O)2]. Crystals. 2017; 7(12):377. https://doi.org/10.3390/cryst7120377
Chicago/Turabian StyleVan der Horn, Jitschaq Anne, Bernd Souvignier, and Martin Lutz. 2017. "Crystallization, Structure Determination and Reticular Twinning in Iron(III) Salicylate: Fe[(HSal)(Sal)(H2O)2]" Crystals 7, no. 12: 377. https://doi.org/10.3390/cryst7120377
APA StyleVan der Horn, J. A., Souvignier, B., & Lutz, M. (2017). Crystallization, Structure Determination and Reticular Twinning in Iron(III) Salicylate: Fe[(HSal)(Sal)(H2O)2]. Crystals, 7(12), 377. https://doi.org/10.3390/cryst7120377