Abstract
The crystal structure of 3,5-bistrifluoromethylhydrocinnamic acid [systematic name: 3-[3,5-bis(trifluoromethyl)phenyl]propanoic acid], C11H8F6O2, has been determined and described. The structure was subject to the Hirshfeld surface-analysis and CE-B3LYP interaction-energies calculations. The title compound crystallises in the monoclinic P21/c space group with one molecule in the asymmetric unit. The propanoic acid side chain of the studied molecule has a bent conformation. The key supramolecular motif in the crystal structure is a centrosymmetric O–H∙∙∙O hydrogen-bonded dimer (R22(8) in the graph set notation). According to CE-B3LYP, the molecules involved in this motif exhibit the strongest pairwise interaction total energy (Etot = −67.9 kJ/mol). On the other hand, there are seven other interacting molecular pairs with significant Etot values in the range of −17 to −28 kJ/mol. In these, the energy is dominated by the dispersive contribution. A survey of the Cambridge Structural Database revealed that in other 3-phenylpropanoic acid structures, the middle dihedral angle of the propanoic acid side chain is always in the trans conformation. This contrasts the current structure where this dihedral angle is in the gauche conformation. According to the Density Functional Theory calculations in the gas phase (at the B3LYP/aug-cc-pvDZ level), the presence of the two CF3 groups (strong electron-withdrawing character) increases the population of the gauche conformers by a substituent electronic effect, and this may be a minor factor contributing to the appearance of this conformation observed in the solid state.
1. Introduction
In the investigation centred around NK1 receptor ligands, we dealt, among others, with 3,5-bistrifluoromethylhydrocinnamic acid (Scheme 1a), which is a member of a wider family of hydrocinnamic acids (3-phenylpropanoic acids, 3-PPAs, Scheme 1b). Despite its commercial availability, the crystal structure of this compound has never been reported. This appears to be a gap since 3-PPAs have an important role in synthetic, medicinal, and structural chemistry.
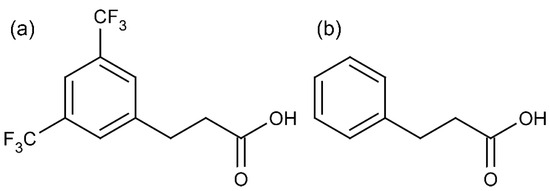
Scheme 1.
Structure of (a) 3,5-bistrifluoromethylhydrocinnamic acid and (b) 3-phenylpropanoic acid (3-PPA).
As to the synthetic aspect, 3-PPAs are valuable reagents for constructing complex organic compounds of medicinal and agricultural significance [1], including those with heterocyclic substructures. The title compound itself was used to synthesise various active compounds [2,3,4,5,6].
Regarding bioactivity, many 3-PPAs and derivatives thereof were reported to have antimicrobial activities [7,8]. Such action is also common for closely related (unsaturated) cinnamic acids [9]. 3-(3,4-dihydroxyphenyl)propanoic acid was demonstrated to modulate brain synaptic plasticity and peripheral inflammation [10]. Several 3-PPAs substituted at the aromatic ring inhibit aromatic amino acid aminotransferase of Paracoccus denitrificans [11]. Some 3-PPAs were recently identified as L-lactate transport inhibitors [12]. More specifically to the topic of this report, we showed that the title compound is a weak ligand of the human NK1 receptor [13]. Other researchers reported some moderate antibacterial activity for its close analogue (3,5-bis(trifluoromethyl)cinnamic acid) [14]. 3-phenylpropanoic acid was used as a reference model to investigate the physicochemical properties of carboxylic acid bioisosteres that are relevant to medicinal chemistry [15]. Ronacaleret (containing a 3-PPA fragment) is a clinically considered investigational calcilytic for osteoporosis treatment [16]. Furthermore, if one takes into account more complicated (branched at the acid chain) derivatives of 3-PPAs, a list of their reported bioactivities is very long. Some very recent examples include p-(benzyloxy)phenylpropanoic acids as ligands of free fatty acid receptor 1 (GPR40) [17] or 2-(aryloxy)-3-phenylpropanoic acids as agonists of peroxisome proliferator-activated receptors α and γ [18].
As to the structural aspects, many crystal structures of 3-PPAs are deposited in the Cambridge Structural Database. If we restrict ourselves only to compounds without any modification in the propanoic acid chain, the available structures are those of the parent 3-phenylpropanoic acid [15,19] and of the analogues substituted in the aromatic ring with one [19,20,21,22,23,24,25,26,27,28], two [26,29,30,31,32] or three [33,34,35] substituents. Some 3-PPA derivatives were considered in co-crystal screening for a medicinally relevant compound [36]. The conformations of certain 3-PPAs were studied by spectroscopic and theoretical methods [37,38,39].
In light of these facts that show great interest in 3-PPAs for modern chemistry, we deemed it suitable to attempt a structure determination of 3,5-bistrifluoromethylhydrocinnamic acid by using a single-crystal X-ray diffraction method. The results thereof are reported in this contribution.
2. Materials and Methods
2.1. Synthesis and Crystallization
3,5-bis(trifluoromethyl)hydrocinnamic acid was obtained from a commercial source (Sigma-Aldrich, Burlington, MA, USA) and recrystallised from methanol for single-crystal structure measurements. The synthesis of this compound has been described several times [4,5].
2.2. Single-Crystal X-ray Diffraction
Diffraction data of a single crystal of the title compound were collected at a low temperature (100 K) using mirror-monochromated Cu Kα radiation (λ = 1.54184 Å) from a Rigaku SuperNova E (dual source) four-circle diffractometer equipped with an Eos CCD detector. CrysAlis PRO software (version 1.171.40.84a)_was used for all operations, including data collection, data reduction, and multi-scan absorption correction. The structure was solved by direct methods and refined by a full-matrix least-squares technique on F2 data using SHELXTL programs [40] integrated with the OLEX2 crystallographic software [41]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms bonded to carbon atoms were inserted in calculated positions and refined isotropically using standard parameters. The H atom of the hydroxyl group was located on a difference map, and its position was freely refined. The MERCURY program [42] was applied for the graphical presentation of the molecular and crystal structures.
2.3. Hirshfeld Surface and Pairwise Interaction Energy Analyses
The CrystalExplorer program, version 17.5, was used to calculate CE-B3LYP model energies and to perform Hirshfeld surface and energy-framework analyses [43]. The pairwise interaction energies were calculated for molecules within a 3.8 Å radius around the central molecule. The CE-B3LYP calculations were made with B3LYP/6-31G(d,p) electron densities. The default scaling factors, as reported by Mackenzie et al., were applied [44]. The wavefunctions were generated by Tonto [45]. Energy-framework representations were generated with the display threshold of 5 kJ/mol and the tube size set to 80.
2.4. Density Functional Theory Calculations
DFT calculations were performed by using the Gaussian 09 suite of programs [46]. Sets of plausible starting structures for conformers of the hydrocinnamic acid and the 3,5-bistrifluoromethylhydrocinnamic acid were manually prepared by rotating the flexible bonds in these molecules. The structures were optimised at the B3LYP/aug-cc-pvDZ level. The optimised geometries were used for harmonic frequency calculations at the same level to determine the character of the stationary points found. Duplicated minima and the structures with imaginary frequencies were discarded from further analysis. In the case of 3,5-bistrifluoromethylhydrocinnamic acid, it was possible to find many closely lying minima resulting from low-barrier rotation of the trifluoromethyl groups (in these structures, the conformation of the propionic side chain was identical). To simplify the analysis, in such cases, only one minimum (lowest energy) for a given side-chain conformation is discussed and shown. Partial atomic charges used in our discussion were those produced by the Natural Population Analysis [47], as implemented in Gaussian 09. Additional calculation variants, (1) with GD3 empirical correction for dispersion forces [48] and (2) in water with the PCM solvent model [49], were also performed, and their results and a brief discussion thereupon is given in Supplementary Materials Tables S1 and S2.
3. Results and Discussion
3.1. Crystal Structure and Hirshfeld Surface Analysis
The title compound crystallises in the monoclinic P21/c space group with one molecule in the asymmetric unit. Selected crystallographic data and refinement details are given in Table 1. The molecular structure is shown in Figure 1. All bond lengths and angles fall within normal ranges.

Table 1.
Crystallographic data and structure-refinement parameters for the title compound.
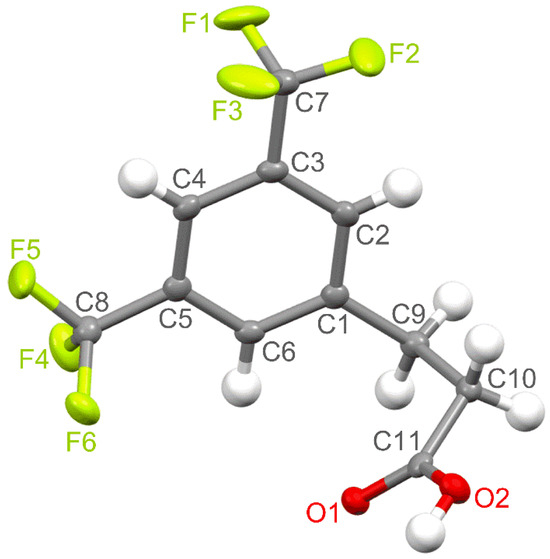
Figure 1.
The molecular structure of the title compound with the atom numbering scheme plotted with a 50% probability of displacement ellipsoids.
The benzene ring, carbon atoms of both CF3 groups and one carbon atom (C9) of the propanoic acid chain lie in an almost ideal plane. The carbon atom of one of the trifluoromethyl substituents (C7) and the C9 atom are slightly displaced from this plane by 0.044(2) Å and 0.061(2) Å, respectively. In turn, the plane formed by the propanoic acid side chain is not perfect because the C9, C10, and O2 atoms are deflected from it by 0.117(2), 0.151(2), and 0.098(2) Å, respectively. This side-chain plane is close to perpendicular to the aromatic ring plane, and the angle between them is 84.16° (αrsc). The propanoic acid part is skewed with the dihedral angles equal to 75.65° (θ1, C2−C1−C9−C10), 67.31° (θ2, C1−C9−C10−C11), and 17.39° (θ3, C9−C10−C11−O1).
The Hirshfeld surface (HS) analysis, described in detail earlier [50,51], was used to explore the non-covalent interactions between neighbouring molecules in the crystal packing of the studied compound. HS mapped with dnorm helps to visualise areas around the molecule responsible for the strongest intermolecular interactions that influence the stabilization of the crystal lattice. This surface typically contains red, blue, and white colours corresponding to interatomic contacts, which are shorter, longer, and equal to the sum of the van der Waals radii, respectively [50,51].
The red spots in Figure 2 show places where there are hydrogen-bonding interactions between neighbouring molecules. The most intensively red spots are present around the carboxyl group, which interacts with the same neighbouring moiety to form centrosymmetric O–H∙∙∙O hydrogen-bonded dimers, with an O1∙∙∙O2 intermolecular distance of 2.655(1) Å and an O1∙∙∙H2–O2 bond angle of 173.4° (Figure 3). The adjacent dimeric units are separated by the C∙∙∙O contacts of 3.196(2) Å (Figure 3). Other red spots are connected with the formation of weaker C–H∙∙∙O and C–H∙∙∙F intermolecular interactions. The detailed parameters related to hydrogen-bonding contacts in the studied crystal structure are presented in Table 2. The molecular packing is also stabilised by weak π∙∙∙π stacking interactions between parallel aromatic rings of adjacent molecules (Figure 3). The shortest contacts between symmetry-related benzene rings are 3.603(2) (C1∙∙∙C4), 3.831(2) (C2∙∙∙C4), and 3.843(2) Å (C1∙∙∙C5). However, the shortest distance between centroids of aromatic rings is much longer and equal to 5.004 Å (Cg∙∙∙Cg), indicating the formation of very weak π∙∙∙π interactions. The presence of CF3 groups further significantly affects the arrangement of the aromatic rings relative to each other. The shortest C∙∙∙F contact equals 3.651(2) Å (C6∙∙∙F5). In addition, the molecules in the crystal lattice are stabilised through C–H∙∙∙O and C–H∙∙∙F intermolecular interactions. The shortest distances between neighbouring F atoms are 3.001(2) (F2∙∙∙F2) and 3.038(2) Å (F3∙∙∙F6). Such interactions leading to the formation of specific channels between the CF3 groups of neighbouring acid molecules, extending along the a axis, can also be considered stabilising for the crystal structure [52,53].
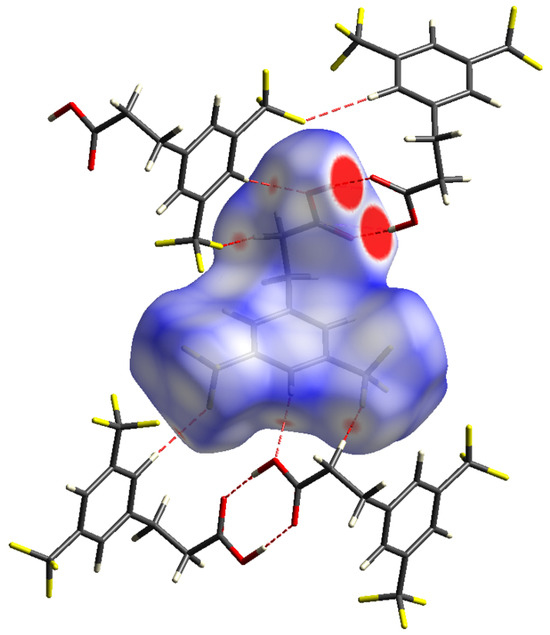
Figure 2.
Hirshfeld surface for 3,5-bistrifluoromethylhydrocinnamic acid with the dnorm property mapped (colour coded in the range from −0.1533 (red) to 1.1510 (blue) au), highlighting neighbouring molecules associated with the hydrogen-bonding contacts.
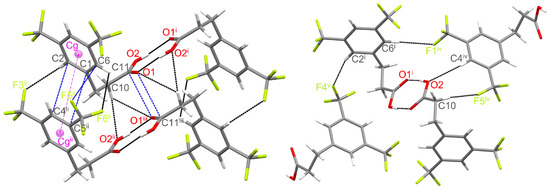
Figure 3.
Fragments of the crystal structure showing the intermolecular interactions between neighbouring molecules (Symmetry codes: (i) −x + 1, −y + 1, −z + 1; (ii) x − 1, y, z; (iii) −x, −y + 1,−z + 1; (iv) −x + 1, y − , −z + ; (v) x + 1, −y + , z + ). The geometric details of the interactions are given in Table 2.
Table 2.
Geometrical parameters (Å, °) for the hydrogen-bond interactions in the studied compound (symmetry codes are as in the caption to Figure 3).
The percentage contributions of interatomic contacts to the HS area are shown in Figure 4. The most numerous interactions in the structure of 3,5-bistrifluoromethylhydrocinnamic acid belong to the F∙∙∙H and F∙∙∙F contacts, which both account for 57.2% of the total HS area. The other significant interatomic contacts are C∙∙∙H, O∙∙∙H and H∙∙∙H, contributing in sum 34.8%. The remaining interatomic interactions are negligible.
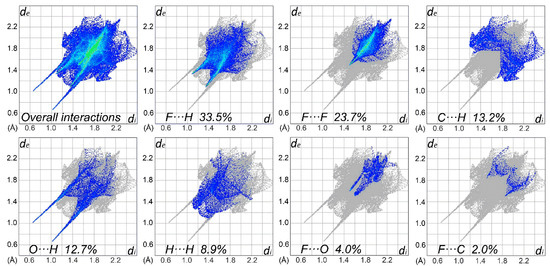
Figure 4.
Two-dimensional fingerprint plots for overall interactions and important individual interatomic contacts, including the reciprocal contacts.
3.2. Intermolecular Interaction Energies and Energy Frameworks
In order to get insight into crystal packing on a ‘whole-of-molecules’ level, we performed a calculation of CrystalExplorer (CE) model energies [44,54] for the pairs present in the crystal structure. In the CE model, the interaction energy (Etot) for selected pairs of molecules is constructed as a sum of four contributions scaled by scale factors (k):
Etot = keleEele + kpolEpol + kdisEdis + krepErep
The terms in the equation are those accounting for electrostatic (Eele), polarization (Epol), dispersion (Edis), and repulsion (Erep) contributions to energy. The scale factors (k) were calibrated so that the total energies agree with the quantum mechanical results at a selected level of theory [44,54].
Within a 3.8 Å radius from a central molecule with symmetry code x, y, z (marked as 0 in Figure 5), there are 13 neighbour molecules. Among these, it is possible to discern eight non-redundant pairs. The interaction energetics in these pairs are shown in Table 3. As expected, according to the CE-B3LYP calculation, the strongest pairwise interaction was found for the pair (molecules 0 and a in Figure 5), which is bonded by classical H-bonds between the carboxylic acid groups. Here, the total interaction energy amounts to −67.9 kJ/mol and is dominated by the electrostatic term. A much weaker but still significant interaction energy of −28.1 kJ/mol was found for two pairs of molecules from neighbouring unit cells, related by translation along the a-axis (molecules 0 and b1 or b2, Figure 5). In this case, the interaction energy comes almost exclusively from the dispersion term, with the electrostatic contribution of negligible magnitude. This is consistent with the type of contacts found in these pairs (weak π∙∙∙π, C6−H6∙∙∙F6ii, C10−H10A∙∙∙O2ii and C2−H2A∙∙∙F3ii contacts).
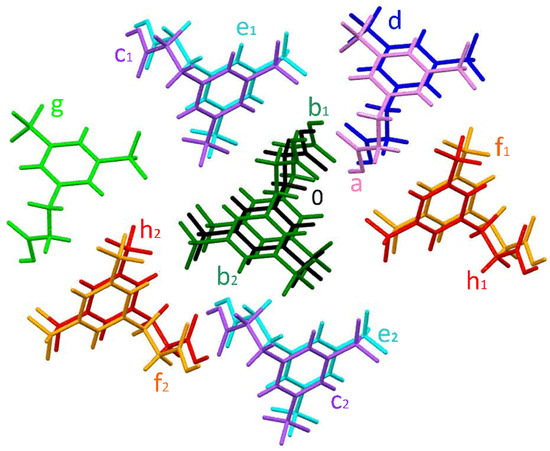
Figure 5.
Colour coding of neighbouring molecules in the studied structure in relation to the central molecule (black).
Table 3.
CE-B3LYP interaction energies (kJ/mol) of the molecular pairs in the first coordination sphere of the title compound in the crystal structure (scaling factors for the energy contributions are kele = 1.057, kpol = 0.740, kdisp = 0.871 and krep = 0.618).
There are three further types of pairs (molecules 0 and c, 0 and d, and 0 and e) for which the total interaction energy is predicted to be slightly lower than −20 kJ/mol. The electrostatic contribution in these is cancelled out by the repulsion, and so, the dispersive term again dominates. In pairs (molecules 0 with c1 and c2 in Figure 5) related by a two-fold screw axis parallel to the b-axis and translation along this axis, the shortest contacts include the CPh−H∙∙∙O (i.e., C4iv−H4iv∙∙∙O2) and C−H∙∙∙F (i.e., C10−H10B∙∙∙F5iv) interactions, and the total interaction energy is −19.3 kJ/mol. A very similar Etot value of −19 kJ/mol is found for the pair of molecules (0 and d, Figure 5) that are related by inversion and translation along the a-axis. The shortest contacts between these molecules involve carboxyl groups (C11∙∙∙O1iii and C10−H10A∙∙∙O1iii). In turn, the shortest C−H∙∙∙F contact and the pairwise energy interaction equal to −17.4 kJ/mol are present for the two molecule pairs (0 with e1 and e2, Figure 5) related by the 21 screw axis and translation along the b-axis. The remaining three types of pairs (involving molecules f, g, and h, Figure 5) are characterised by a much lower Etot value below −10 kJ/mol.
An energy-framework analysis (Figure 6) shows that the crystal packing energetics are anisotropic and dominated by electrostatic contribution. It is clearly seen that the thickest cylinders of the total energies are those corresponding to the thick cylinders of the Coulomb energies. On the other hand, the contribution of dispersion forces is still important, as it provides non-negligible stabilization (as judged by the relative thickness of the cylinders) in pairs with no electrostatic stabilization.
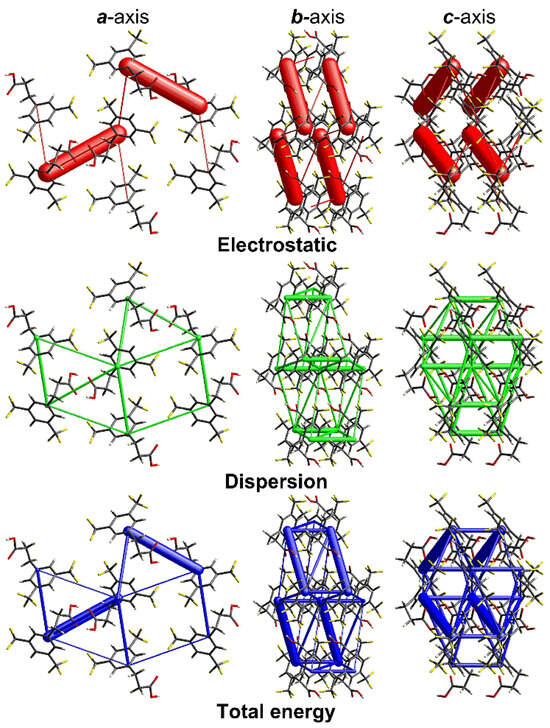
Figure 6.
Energy-framework plots for the title compound. The electrostatic and dispersion energy components and the total energy interaction are shown separately in projections along the a, b, and c-axis. The cylinders are lines joining the molecular centroids, and their thickness is proportional to the magnitude of a given interaction. The tube size and the cut-off energy are set to 80 and 5 kJ/mol, respectively. The plots were produced with CrystalExplorer [43].
3.3. Comparison with Crystal Structures of Other 3-Phenylpropanoic Acids (3-PPAs)
For comparative purposes, we analysed the available structures of other 3-phenylpropanoic acids (Table 4). 3-phenylpropanoic acid salts (3-phenylpropionates and others), metal complexes, co-crystals, inclusion complexes, and solvates were excluded from this analysis.
Table 4.
Selected geometrical parameters of 3-PPAs crystal structures. The entries are sorted by ascending with the ring-plane/chain-plane αrsc angle.
The vast majority of the analysed derivatives, with only a few exceptions, crystallised in monoclinic space groups having a two-fold screw axis and differing only in the sliding plane. The crystal structures of the analysed 3-PPAs exhibit the R22(8) H-bond motif common for carboxylic acids [58,59], and the title compound is no exception here. In all but two structures, this motif is associated with the formation of cyclic dimers by the carboxylic groups of the propanoic acid side chains. The exceptions (WIKRUE and VASDOM) are structures of derivatives with two different carboxylic acid side chains in one molecule. In WIKRUE, the dimers are formed between the two different side chains located para to each other (propanoic acid to 2-oxyethanoic acid). In VASDOM, the cyclic dimer is formed by prop-2-enoic acid side chains, while the propanoic acid fragment is involved through its COOH group in noncentrosymmetric hydrogen-bond interactions with the C=CH−C=O moiety of the neighbouring prop-2-enoic acid part.
A second point for comparison is the overall molecule’s shape (mutual orientation of the ring and the side chain). In the structure presented herein, the side-chain plane is close to perpendicular to the aromatic ring’s plane. In the previously published 3-phenylpropanoic acids (Table 4), the mutual arrangement of the ring and side chain (αrsc angle) is found to be either in one plane or almost perpendicular (as in the title structure) or in the middle between these two extremities. No clear pattern of a substituent effect on this behaviour is easily recognizable. Interestingly, in the crystal structures of the parent unsubstituted 3-phenylpropanoic acid (YASFIJ, YASFIJ01, and YASFIJ02), there are two crystallographically independent molecules (marked in Table 4 as A and B), and each has a different geometry.
Another point for analysis is the conformation of the propanoic acid side chain. In the structure presented herein, the side chain is bent and skewed, giving the (c,sc,sp) conformation of the θ1, θ2, and θ3 dihedral angles, respectively. For the other 3-PPAs presented in Table 4, the conformations of the propanoic acid side chain are either (p,ap,p) or (c,ap,p), and the synclinal conformation of the θ2 dihedral angle, as in the title compound’s structure, is not found.
3.4. Quantum Chemical Conformational Analysis
The quantum chemical (DFT) conformational analysis was performed for the title compound and its parent analogue, hydrocinnamic acid. The calculations were made in the gas phase at the B3LYP/aug-cc-pvDZ level (the results of the additional calculation variants are shown and briefly discussed in the Supplementary Materials, Tables S1 and S2). The aim of this modelling was to see if the appearance of the synclinal conformation of the dihedral angle θ2 may be related to the substituent effect of the CF3 groups on the conformational properties of the molecule. The conformers found for both molecules are shown in Figure 7, and their geometric and energetic parameters are displayed in Table 5.
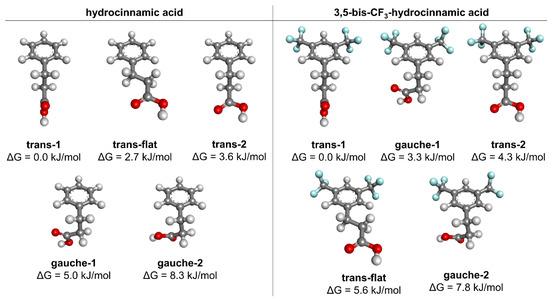
Figure 7.
Conformers of the hydrocinnamic and 3,5-bis-trifluoromethylhydrocinnamic acids, found from calculations in the gas phase at the B3LYP/aug-cc-pvDZ level of theory.
Table 5.
Geometric and energetic parameters for the conformers of the unsubstituted hydrocinnamic acid and 3,5-bis-trifluoromethylhydrocinnamic acid (B3LYP/aug-cc-pvDZ level, gas phase).
In both cases, the trans-1 (c,ap,sp) conformer is the global minimum. The second most stable conformer in the unsubstituted hydrocinnamic acid is trans-flat (sp,ap,sp) (ΔG = 2.7 kJ/mol). The gauche-1 conformer (c,sc,sp) is fourth in the ranking, with ΔG = 5.0 kJ/mol. The introduction of 3,5-bis-CF3 substitution reverses this order of conformer stabilities. In this substituted analogue, the gauche-1 is the second lowest in energy (ΔG = 3.3 kJ/mol), while the trans-flat conformer is fourth in the ranking (ΔG = 5.6 kJ/mol).
A possible explanation for this stability gain of the gauche-1 conformer may be derived from the fact that the CF3 substituent strongly withdraws electrons from the Cortho atom and increases the positive partial charge on the Hortho atom (partial charges from the natural population analysis shown in Figure 8). This results in increased attractive interaction between the carbonyl oxygen and the Hortho (shortening of the distance between these atoms, Figure 8) and adds some stabilization for the gauche-1 conformer. Such an explanation is consistent with another earlier experimental report (NMR measurements in solution), in which electron-withdrawing substituents were shown to increase the gauche population of 3-(para-substituted)phenylpropanoic acids [60].
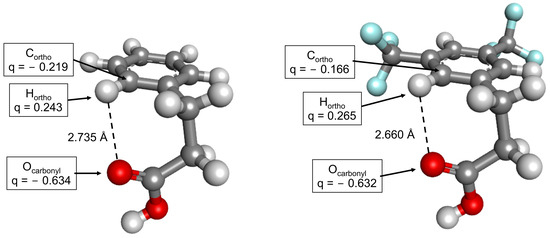
Figure 8.
Gauche-1 conformers of the hydrocinnamic (left) and 3,5-bistrifluoromethylhydrocinnamic acids (right). The partial atomic charges of the selected atoms (indicated by arrows) are shown in the boxes. The charges are derived from natural population analysis.
It seems then that 3,5-bis-CF3 substitution increases the population of the gauche-1 conformer and decreases the population of the trans-flat conformer in comparison with the unsubstituted hydrocinnamic acid. This could be one of the factors that favour the appearance of the gauche-1 conformer in the solid-state structure of the 3,5-bis-CF3 analogue. On the other hand, since the discussed conformational energetic differences are relatively small, the substituent effect is likely not a key factor but rather one of the contributors here, with most of the influence coming from the supramolecular interactions.
Interestingly, in the previously published gas-phase spectroscopic and theoretical conformational investigations for 3-PPAs [37,39], only gauche-1 (a minor conformer) and trans-1 (a dominating conformer) conformations of the side chain were mentioned, and the trans-flat conformation was not reported at all. This is in contrast to the presence of the trans-flat conformer in solid-state structures of 3-PPAs and our gas-phase QM results, according to which this conformer is a significant low-energy minimum.
4. Conclusions
In the present work, we have structurally characterised 3,5-bistrifluoromethylhydrocinnamic acid, which is a member of the important family of 3-phenylpropanoic acids. The key structural motif in the analysed crystal is centrosymmetric O–H∙∙∙O hydrogen-bonded dimer (R22(8) motif). CE-B3LYP calculations show that the molecules involved in this motif exhibit the strongest pairwise interaction (Etot = −67.9 kJ/mol), but seven other interacting molecular pairs have significant Etot values in the range of −17 kJ/mol to −28 kJ/mol. In these latter pairs, the energy is dominated by the dispersive contribution. On surveying the CSD database, it has been found that other 3-PPA crystals do not exhibit the gauche conformation of the middle dihedral angle of the propanoic acid side chain, which in turn, is observed in the current structure. DFT calculations in the gas phase show that strong electron-withdrawing CF3 substituents increase the population of the gauche conformation for the discussed dihedral angle, probably via the electronic substituent effect. It seems that this may be some minor factor contributing to the appearance of this conformation in the solid state. Experimental and theoretical work to systematically explore the substituent effect on the conformations of 3-PPAs is underway.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/cryst14040342/s1, Table S1. Geometric and energetic parameters for the conformers of the unsubstituted hydrocinnamic acid and 3,5-bis-trifluoromethylhydrocinnamic acid (B3LYP/aug-cc-pvDZ level, gas-phase, with Grimme D3 correction for dispersion); Table S2. Geometric and energetic parameters for the conformers of the unsubstituted hydrocinnamic acid and 3,5-bis-trifluoromethylhydrocinnamic acid (B3LYP/aug-cc-pvDZ level, with PCM solvent [water] model).
Author Contributions
Conceptualization, P.F.J.L. and K.Ł.; methodology, P.F.J.L. and K.Ł.; investigation, P.F.J.L. and K.Ł.; resources, P.F.J.L. and K.Ł. writing—original draft preparation, P.F.J.L. and K.Ł.; writing—review and editing, P.F.J.L. and K.Ł.; visualization, P.F.J.L. and K.Ł.; funding acquisition, P.F.J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre in Poland, grant No. 2016/23/D/NZ7/03636.
Data Availability Statement
CCDC 2337717 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures (accessed on 5 March 2024).
Acknowledgments
The calculations were performed at Świerk Computing Centre, National Centre for Nuclear Research, Świerk, Poland. The authors are most grateful for the computational grant at this facility.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Korneev, S. Hydrocinnamic Acids: Application and Strategy of Synthesis. Synthesis 2013, 45, 1000–1015. [Google Scholar] [CrossRef]
- Teall, M.; Harrison, T.; Moseley, J.D.; Owens, A.P.; Sadowski, S.; Cascieri, M.A. Linear amides as substance P antagonists. Bioorg. Med. Chem. Lett. 1996, 6, 1585–1588. [Google Scholar] [CrossRef]
- Ryckmans, T.; Balançon, L.; Berton, O.; Genicot, C.; Lamberty, Y.; Lallemand, B.; Pasau, P.; Pirlot, N.; Quéré, L.; Talaga, P. First dual NK1 antagonists–serotonin reuptake inhibitors: Synthesis and SAR of a new class of potential antidepressants. Bioorg. Med. Chem. Lett. 2002, 12, 261–264. [Google Scholar] [CrossRef]
- Li, F.-N.; Kim, N.-J.; Paek, S.-M.; Kwon, D.-Y.; Min, K.H.; Jeong, Y.-S.; Kim, S.-Y.; Park, Y.-H.; Kim, H.-D.; Park, H.-G.; et al. Design, synthesis, and biological evaluation of novel diarylalkyl amides as TRPV1 antagonists. Bioorg. Med. Chem. 2009, 17, 3557–3567. [Google Scholar] [CrossRef]
- Lemhadri, M.; Doucet, H.; Santelli, M. Direct synthesis of 3-arylpropionic acids by tetraphosphine/palladium catalysed Heck reactions of aryl halides with acrolein ethylene acetal. Tetrahedron 2004, 60, 11533–11540. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, B.-W.; Lee, H.U.; Choi, D.-K.; Yoon, S.-H. Synthesis of Clovamide Analogues That Inhibit NO Production in Activated BV-2 Microglial Cells. Biol. Pharm. Bull. 2017, 40, 1475–1482. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shaala, L.A.; Youssef, D.T.A.; Alzughaibi, T.A.; Elhady, S.S. Antimicrobial Chlorinated 3-Phenylpropanoic Acid Derivatives from the Red Sea Marine Actinomycete Streptomyces coelicolor LY001. Mar. Drugs 2020, 18, 450. [Google Scholar] [CrossRef] [PubMed]
- Narayana, K.J.P.; Prabhakar, P.; Vijayalakshmi, M.; Venkateswarlu, Y.; Krishna, P.S.J. Biological activity of phenylpropionic acid isolated from a terrestrial Streptomycetes. Pol. J. Microbiol. 2007, 56, 191–197. [Google Scholar] [PubMed]
- Guzman, J. Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef]
- Okamoto, A.; Ishii, S.; Hirotsu, K.; Kagamiyama, H. The Active Site of Paracoccus denitrificans Aromatic Amino Acid Aminotransferase Has Contrary Properties: Flexibility and Rigidity. Biochemistry 1999, 38, 1176–1184. [Google Scholar] [CrossRef]
- Bosshart, P.D.; Kalbermatter, D.; Bonetti, S.; Fotiadis, D. The making of a potent L-lactate transport inhibitor. Commun. Chem. 2021, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.F.J.; Matalińska, J. NK1 receptor binding of a few low molecular weight 3,5-bistrifluoromethylbenzene derivatives. Acta Pol. Pharm. Drug Res. 2023, 80, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Galano, J.J.; Alías, M.; Pérez, R.; Velázquez-Campoy, A.; Hoffman, P.S.; Sancho, J. Improved Flavodoxin Inhibitors with Potential Therapeutic Effects against Helicobacter pylori Infection. J. Med. Chem. 2013, 56, 6248–6258. [Google Scholar] [CrossRef] [PubMed]
- Lassalas, P.; Gay, B.; Lasfargeas, C.; James, M.J.; Tran, V.; Vijayendran, K.G.; Brunden, K.R.; Kozlowski, M.C.; Thomas, C.J.; Smith, A.B.; et al. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem. 2016, 59, 3183–3203. [Google Scholar] [CrossRef] [PubMed]
- Widler, L.; Altmann, E.; Beerli, R.; Breitenstein, W.; Bouhelal, R.; Buhl, T.; Gamse, R.; Gerspacher, M.; Halleux, C.; John, M.R.; et al. 1-Alkyl-4-phenyl-6-alkoxy-1 H -quinazolin-2-ones: A Novel Series of Potent Calcium-Sensing Receptor Antagonists. J. Med. Chem. 2010, 53, 2250–2263. [Google Scholar] [CrossRef] [PubMed]
- Kuranov, S.O.; Luzina, O.A.; Onopchenko, O.; Pishel, I.; Zozulya, S.; Gureev, M.; Salakhutdinov, N.F.; Krasavin, M. Exploring bulky natural and natural-like periphery in the design of p-(benzyloxy)phenylpropionic acid agonists of free fatty acid receptor 1 (GPR40). Bioorg. Chem. 2020, 99, 103830. [Google Scholar] [CrossRef] [PubMed]
- Laghezza, A.; Pochetti, G.; Lavecchia, A.; Fracchiolla, G.; Faliti, S.; Piemontese, L.; Di Giovanni, C.; Iacobazzi, V.; Infantino, V.; Montanari, R.; et al. New 2-(Aryloxy)-3-phenylpropanoic Acids as Peroxisome Proliferator-Activated Receptor α/γ Dual Agonists Able To Upregulate Mitochondrial Carnitine Shuttle System Gene Expression. J. Med. Chem. 2013, 56, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Chattopadhyay, B.; Mukherjee, M.; Mukherjee, A.K. Structure Analysis of Molecular Compounds with Z ′ = 2 Using Laboratory X-ray Powder Diffraction Data: 3-Phenylpropionic Acid and Its Derivatives. Cryst. Growth Des. 2012, 12, 466–474. [Google Scholar] [CrossRef]
- Begum, N.S.; Jain, M.; Chandrasekhar, S.; Venkatesan, K. Structure of 3-(2-hydroxyphenyl)propionic acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1992, 48, 1076–1078. [Google Scholar] [CrossRef]
- Glusker, J.P.; Zacharias, D.E.; Carrell, H.L. Structure refinement and molecular packing of p-chloro-trans-cinnamic acid and β-(p-chlorophenyl)propionic acid. J. Chem. Soc. Perkin Trans. 2 1975, 4, 68–74. [Google Scholar] [CrossRef]
- Okabe, N.; Suga, T. 3-(p-Hydroxyphenyl)propionic Acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1995, 51, 1324–1325. [Google Scholar] [CrossRef]
- Gerkin, R.E. 3-(2-Methylphenyl)propanoic acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1999, 55, 1857–1859. [Google Scholar] [CrossRef]
- Christiansen, E.; Urban, C.; Merten, N.; Liebscher, K.; Karlsen, K.K.; Hamacher, A.; Spinrath, A.; Bond, A.D.; Drewke, C.; Ullrich, S.; et al. Discovery of Potent and Selective Agonists for the Free Fatty Acid Receptor 1 (FFA 1/GPR40), a Potential Target for the Treatment of Type II Diabetes. J. Med. Chem. 2008, 51, 7061–7064. [Google Scholar] [CrossRef]
- Guan, J.-N.; Kong, X.-J.; Xu, B.; Liang, J.-H.; Song, N. 3-[4-(Trifluoromethyl)phenyl]propanoic acid. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o844. [Google Scholar] [CrossRef]
- Das, U.; Chattopadhyay, B.; Mukherjee, M.; Mukherjee, A.K. Crystal structure and electronic properties of three phenylpropionic acid derivatives: A combined X-ray powder diffraction and quantum mechanical study. Chem. Phys. Lett. 2011, 501, 351–357. [Google Scholar] [CrossRef]
- Tan, S.-S.; Wen, H.-L.; Liu, C.-B.; Peng, X.-P.; Li, X.-S. Crystal structure of 3-(4-(carboxymethoxy)phenyl)propanoic acid, C11H12O5. Z. Für Krist. New Cryst. Struct. 2007, 222, 137–138. [Google Scholar] [CrossRef]
- Lu, L.; Wu, W.P.; Ma, A.Q.; Xie, B.; Wu, Y. Synthesis, structural, and luminescence of two coordination polymers. Russ. J. Coord. Chem. 2015, 41, 573–578. [Google Scholar] [CrossRef]
- Bugenhagen, B.; Al Jasem, Y.; AlAzani, M.; Thiemann, T. Crystal structure of 3-(2,5-dimethoxyphenyl)propionic acid. Acta Crystallogr. Sect. E Crystallogr. Commun. 2015, 71, o337–o338. [Google Scholar] [CrossRef]
- Liu, W.; Ren, W.; Li, J.; Shi, Y.; Chang, W.; Shi, Y. A Ligand-Directed Catalytic Regioselective Hydrocarboxylation of Aryl Olefins with Pd and Formic Acid. Org. Lett. 2017, 19, 1748–1751. [Google Scholar] [CrossRef]
- Pan, R.; Shi, C.; Zhang, D.; Tian, Y.; Guo, S.; Yao, H.; Lin, A. Nickel-Catalyzed Reductive 1,2-Dialkynylation of Alkenes Bearing an 8-Aminoquinoline Directing Group. Org. Lett. 2019, 21, 8915–8920. [Google Scholar] [CrossRef]
- Drebushchak, T.N.; Boldyreva, E.V.; Fucke, K. Hydrogen bonding of catechol groups in the crystal structure of dihydrocaffeic acid. J. Struct. Chem. 2013, 54, 368–372. [Google Scholar] [CrossRef]
- Antonenko, T.A.; Shpakovsky, D.B.; Berseneva, D.A.; Gracheva, Y.A.; Dubova, L.G.; Shevtsov, P.N.; Redkozubova, O.M.; Shevtsova, E.F.; Tafeenko, V.A.; Aslanov, L.A.; et al. Cytotoxic activity of organotin carboxylates based on synthetic phenolic antioxidants and polycyclic bile acids. J. Organomet. Chem. 2020, 909, 121089. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Cordes, D.B.; Slawin, A.M.Z.; O’Hagan, D. Total Synthesis of a Reported Fluorometabolite from Streptomyces sp. TC1 Indicates an Incorrect Assignment. The Isolated Compound Did Not Contain Fluorine. J. Nat. Prod. 2014, 77, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Jaivel, N.; Uvarani, C.; Rajesh, R.; Velmurugan, D.; Marimuthu, P. Correction to Natural Occurrence of Organofluorine and Other Constituents from Streptomyces sp. TC1. J. Nat. Prod. 2015, 78, 343. [Google Scholar] [CrossRef]
- Fucke, K.; Myz, S.A.; Shakhtshneider, T.P.; Boldyreva, E.V.; Griesser, U.J. How good are the crystallisation methods for co-crystals? A comparative study of piroxicam. New J. Chem. 2012, 36, 1969. [Google Scholar] [CrossRef]
- Dickinson, J.A.; Joireman, P.W.; Randall, R.W.; Robertson, E.G.; Simons, J.P. Conformational Structures of 3-Phenyl-1-propionic Acid, Its p -Hydroxy Derivative, and Its Hydrated Clusters. J. Phys. Chem. A 1997, 101, 513–521. [Google Scholar] [CrossRef]
- Martinez, S.J.; Alfano, J.C.; Levy, D.H. The electronic spectroscopy of tyrosine and phenylalanine analogs in a supersonic jet: Acidic analogs. J. Mol. Spectrosc. 1991, 145, 100–111. [Google Scholar] [CrossRef]
- Joireman, P.W.; Kroemer, R.T.; Pratt, D.W.; Simons, J.P. Conformationally induced rotation of a molecular electronic transition moment. J. Chem. Phys. 1996, 105, 6075–6077. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Proceedings of the International Conference on Computational Science 2003, Petersburg, Russia, 2–4 June 2003; pp. 142–151. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01 2013; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 7, 3814. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Chopra, D.; Row, T.N.G. Role of organic fluorine in crystal engineering. CrystEngComm 2011, 13, 2175. [Google Scholar] [CrossRef]
- Dey, D.; Bhandary, S.; Thomas, S.P.; Spackman, M.A.; Chopra, D. Energy frameworks and a topological analysis of the supramolecular features in in situ cryocrystallized liquids: Tuning the weak interaction landscape via fluorination. Phys. Chem. Chem. Phys. 2016, 18, 31811–31820. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, B.; Spiteller, M. CCDC 900046: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2017. [Google Scholar]
- Cody, V.; Erman, M.; De Jarnette, F.E. Crystal Structure of the Active Thyroxine Analogue, 3,5-Diiodothyropropionic Acid (3-(4-Hydroxy-phenoxy-3,5-di-iodophenyl) Propanoic Acid). J. Chem. Res. 1977, 126, 1444. [Google Scholar]
- Ramesh, P.; Sreenivasulu, C.; Kishore, D.R.; Srinivas, D.; Gorantla, K.R.; Mallik, B.S.; Satyanarayana, G. Recyclable Aliphatic Nitrile-Template Enabled Remote meta -C–H Functionalization at Room Temperature. J. Org. Chem. 2022, 87, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Deepa, P.; Vijay Solomon, R.; Angeline Vedha, S.; Kolandaivel, P.; Venuvanalingam, P. The nature of hydrogen bonding in R 2 2 (8) crystal motifs—A computational exploration. Mol. Phys. 2014, 112, 3195–3205. [Google Scholar] [CrossRef]
- Allen, F.H.; Samuel Motherwell, W.D.; Raithby, P.R.; Shields, G.P.; Taylor, R. Systematic analysis of the probabilities of formation of bimolecular hydrogen-bonded ring motifs in organic crystal structures. New J. Chem. 1999, 23, 25–34. [Google Scholar] [CrossRef]
- Spassov, S.L.; Simova, S.D. Conformational analysis of 3-arylpropanoic acids and their methyl esters by 1H nuclear magnetic resonance spectroscopy. J. Chem. Soc. Perkin Trans. 2 1978, 7, 1113. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).