Abstract
A series of silylated 2-aminopyrimidines Me(4−n)Si(NHpyr)n (Me = methyl, NHpyr = pyrimid-2-ylamino, n = 1, 2, 3, 4), i.e., compounds 1, 2, 3, and 4, respectively, was prepared from a series of the respective chlorosilanes Me(4−n)SiCln and 2-aminopyrimidine. Triethylamine was used as a sacrificial base. Compounds 1, 2, 3, and 4 are solid at room temperature. They were analyzed using 1H, 13C, 29Si NMR, and Raman spectroscopy, and their molecular structures were confirmed by single-crystal X-ray diffraction analyses. All structures exhibit intramolecular van der Waals contacts between the silicon atom and one nitrogen atom of the pyrimidine moiety. Thus, their Si coordination spheres can be interpreted as [4+n] coordinated capped tetrahedra. Intermolecular hydrogen bonds (N–H···N bridges between the Si-bound amino groups and the non-Si-capping pyrimidine N atoms) are a constant contributor to the solid-state structures of these compounds. Furthermore, compounds 2 and 4 exhibit N–H···N bridges which involve 50% of their Si-capping N atoms as hydrogen bridge acceptors. Consequently, 50% of the non-Si-capping pyrimidine N atoms are stabilized by C–H···N contacts. As a result of a particularly dense network of intermolecular hydrogen bridges, the melting point of Si(NHpyr)4 (compound 4) is higher than 300 °C.
1. Introduction
Silylated amines, also known as aminosilanes, are compounds with at least one Si–N bond. Because of the trifunctionality of the nitrogen atom, this generic class of compounds involves silylated amines with one N-bound silyl group (I in Figure 1, aminosilanes with terminal amino groups), two (II, referred to as silazanes), or even three Si–N bonds per N atom (III, trisilylamines). They have been the focus of many research projects for several decades, driven, e.g., by some of their inherent properties. Their elemental composition (Si, C, N in particular) is speaking for their investigation as precursors for SiNC or SiONC ceramic materials, e.g., [1,2]. Further aspects of their materials chemistry involve the use of silazanes in coatings [3]. Their mutual feature, the Si–N bond, also offers entry into further fields of silicon chemistry which make use of this synthon. For example, CO2 can insert into the Si–N bond with the formation of carbamoyloxysilanes (e.g., IV) [4,5,6]. The latter are interesting starting materials for the synthesis of isocyanates and silicones in a CO2-consuming process [5,7,8]. The insertion of isocyanates in Si–N bonds may afford silylated urea derivatives (e.g., V) [9]. The insertion of heteroallenes into Si–N bonds may also serve as an entry into silicon coordination chemistry (i.e., the syntheses of Si compounds with hexa- or pentacoordinate Si atom, VI [10] and VII [11], respectively). As part of those studies and of related investigations in our group, we synthesized a portfolio of aminosilanes and other compounds with Si–N bond(s), including silylated amines [6,9,12], imines [13], and cyclic aminosilanes [14], and explored their reactions with CO2 or isocyanates. In addition, we have already investigated the reactivity of silyl morpholine towards a variety of heteroallenes [15].
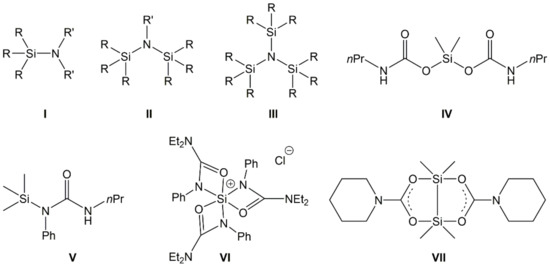
Figure 1.
Generic representation of aminosilanes (I), silazanes (II), and trisilylamines (III) (with R being various substituents, e.g., alkyl, aryl, H, silyl, and R′ being various substituents, except from silyl) as well as examples of compounds obtained from the insertion of CO2 (examples IV and VII) or phenylisocyanate (examples V and VI) into Si–N bonds of aminosilanes.
Guanidinosilanes (or silicon guanidinates) represent another class of Si–N compounds with interesting properties. Guanidinates (the mono-anionic forms of guanidine derivatives) may form simple silylguanidines (VIII [16], IX [17], X [18], XI [19], XII [20], Figure 2), some are capable of acting as chelating ligands in hypercoordinate Si complexes (XIII [21], XIV [22]), and some are capable of stabilizing low-coordinate Si compounds (e.g., XV [23], XVI [24]).
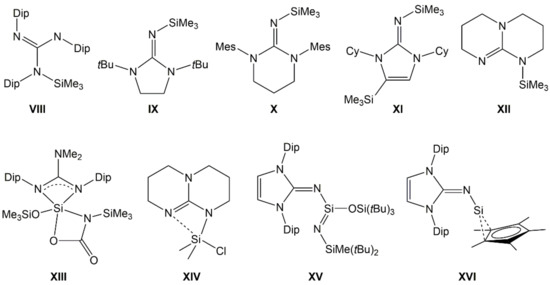
Figure 2.
Examples of silylated guanidinates with tetracoordinate (VIII–XII), hypercoordinate (XIII, XIV) and lower coordinate (XV), and low valent (XVI) Si atom. (Dip = 2,6-diisopropylphenyl, Mes = mesityl, Cy = cyclohexyl).
Furthermore, some guanidinates are also capable of bridging Si atoms with other metal atoms in heteronuclear complexes (e.g., XVII [25], XVIII [20], XIX [26], Figure 3). For some representatives of Si-guanidinates, it has also been shown that their Si–N bond may be prone to CO2 insertion [22,27]. The silicon compounds of aromatic guanidines (2-aminopyrimidine derivatives), however, are scarcely explored. Compounds XX [28], XXI [29], and XXII [30] are some isolated examples of compounds which feature the silylated 2-aminopyrimidine motif. This served as motivation for us to explore the first systematic series of 2-aminopyrimidine-derived silanes Me(4−n)Si(NHpyr)n (Me = methyl, NHpyr = pyrimid-2-ylamino, n = 1, 2, 3, 4), i.e., compounds 1, 2, 3, and 4, respectively.
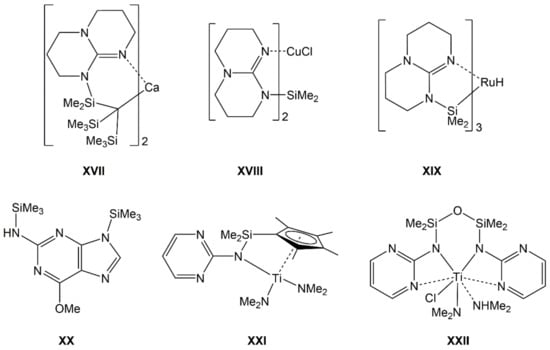
Figure 3.
Top: Examples of silylated guanidinates in heteronuclear Si-metal complexes with Si–linker–metal connection (XVII), without other Si–metal connections (XVIII), and with Si–metal bond (XIX). Bottom: Crystallographically characterized compounds with silylated 2-aminopyrimidine motif (mono-anionic bound to Si in XX, di-anionic bound to Si, and Ti in XXI and XXII).
2. Materials and Methods
2.1. General Considerations
All reactions were carried out under dry argon atmosphere using standard Schlenk techniques [31,32]. Details of the chemicals used and purification methods applied can be found in the supporting information (cf. Table S1). NMR spectra (of CDCl3 solutions with tetramethylsilane, TMS, as an internal standard) were recorded either on a BRUKER DPX 400 spectrometer (Bruker Biospin, Rheinstetten/Karlsruhe, Germany) at 400.13 MHz, 100.61 MHz, and 79.49 MHz or on a BRUKER AVANCE III 500 MHz spectrometer (Bruker Biospin, Rheinstetten/Karlsruhe, Germany) at 500.13 MHz, 125.76 MHz, and 99.36 MHz for 1H, 13C, and 29Si, respectively. The spectra are shown in the supporting information (Figures S1–S15). Raman spectra (cf. Figure S16 in the supporting information) were measured with a Bruker FT-Raman spectrometer RFS 100/S (Bruker Optics, Ettlingen, Germany). The device works with an air-cooled Nd:YAG-Laser with a wavelength of 1064 nm and a nitrogen-cooled detector. The samples (dried in vacuo; according to 1H and 13C NMR, they were devoid of solvent of crystallization) were filled in glass tubes and sealed with PTFE-paste. The intensity is listed as follows: vs (very strong) 80–100% of the highest signal, followed by s (strong), m (medium), w (weak), and vw (very weak), descending further in intensity in 20% increments with respect to the strongest signal. The boiling points of liquid samples were measured as described in [33]. Melting points were measured using a Polytherm A hot stage microscope (Wagner & Munz, München, Germany) with an attached 52II thermometer from Fluke. For single-crystal X-ray diffraction studies, data collection was performed on a STOE IPDS2 image plate diffractometer (STOE & Cie, Darmstadt, Germany) equipped with a low-temperature device with Mo-Kα radiation (λ = 0.71073 Å) using ω scans. Software for data collection: X-AREA, cell refinement: X-AREA, and data reduction: X-RED [34]. Preliminary structure models were derived by direct methods [35]. Non-hydrogen atoms were refined anisotropically in full-matrix least-squares cycles based on F2 for all reflections using SHELXL 2019/3 [36]. C-bound hydrogen atoms were included isotropically in the models in idealized positions constrained to the pivot atoms (riding model). N-bound hydrogen atoms were located as residual electron density peaks and were refined isotropically without restraints. Graphics of the molecular structures were generated with ORTEP-3 (version 2014.1) [37,38] and POV-RAY (version 3.7) [39].
2.2. Syntheses and Characterization
Compound 1 (pyrimid-2-ylaminotrimethylsilane). 2-Aminopyrimidine (4.44 g, 47 mmol) and triethylamine (4.89 g, 48 mmol) were dissolved in 50 mL of THF, whereupon chlorotrimethylsilane (4.99 g, 46 mmol) was added with stirring, and the mixture was heated to reflux for 4 h. After stirring at room temperature overnight, the resultant suspension was filtered, and from the filtrate all volatiles were removed under reduced pressure (condensation into a cold trap) to afford 4.03 g (24 mmol, 52%) of crude product 1, which was contaminated with small amounts of 2-aminopyrimidine. To this crude product, 2 mL of CDCl3 was added to obtain a saturated solution for an initial NMR analysis (general reaction control). In the presence of this solvent, suitable crystals for single-crystal X-ray diffraction were obtained after standing for several days at room temperature. The crystals were washed with n-pentane and dried under reduced pressure. To remove traces of THF and 2-aminopyrimidine, the product was recrystallized from CHCl3. A product of sufficient purity (according to NMR spectroscopy) was obtained along an analogous route using excess triethylamine and chlorotrimethylsilane: 4.38 g (46 mmol) 2-aminopyrimidine, 7.35 g (72 mmol) triethylamine, and 7.75 g (71 mmol) chlorotrimethylsilane yielded 5.25 g (31 mmol, 68%,) of 1. Melting point: 58 °C; boiling point: 88 °C/0.9 Torr; Raman: ν (cm−1) 3086 (w), 3032 (w), 2959 (s), 2901 (vs), 1584 (w), 1087 (m), 992 (w), 925 (w), 871 (vw), 855 (vw), 788 (vw), 641 (vw), 626 (w), 392 (vw), 296 (vw), 250 (vw), 230 (vw), 219 (vw), 203 (vw), 182 (vw), and 103 (m); 1H NMR (CDCl3, 400 MHz): δ (ppm) 0.29 (s, 9 H, SiMe3), 4.88 (s (br), 1 H, NH), 6.55 (t, 4.8 Hz, 1 H, H5), and 8.26 (d, 4.8 Hz, 2 H, H4,6); 13C NMR (CDCl3, 100 MHz): δ (ppm) −0.4 (SiMe3), 111.1 (C5), 157.9 (C4,6), and 164.5 (C2); and 29Si NMR (CDCl3, 79 MHz): δ (ppm) 4.7.
Compound 2 (bis(pyrimid-2-ylamino)dimethylsilane). 2-Aminopyrimidine (7.38 g, 78 mmol) and triethylamine (7.96 g, 79 mmol) were dissolved in 80 mL of THF, whereupon dichlorodimethylsilane (5.04 g, 39 mmol) was added with stirring, and the mixture was heated to reflux for 6 h. After stirring at room temperature overnight, the suspension was filtered, and all volatiles were removed from the filtrate under reduced pressure to afford 6.94 g (28 mmol, 72%) of crude product 2. To this crude product, 2 mL of CDCl3 was added to obtain a saturated solution for an initial NMR analysis (general reaction control). In the presence of this solvent, suitable crystals for single-crystal X-ray diffraction were obtained after standing for several days at room temperature. The crystals were washed with n-pentane. To remove traces of THF, the product was recrystallized from CHCl3. Melting point: 134 °C; Raman: ν (cm−1) 3121 (vw), 3088 (w), 3052 (w), 3027 (w), 3001 (vw), 2965 (vw), 2905 (vw), 1580 (w), 1133 (vw), 1079 (w), 986 (vw), 871 (vs), 780 (vw), 707 (vw), 651 (vw), 587 (vw), 458 (vw), 410 (vw), 194 (w), 120 (vs), and 78 (vw); 1H NMR (CDCl3, 400 MHz): δ (ppm) 0.56 (s, 6 H, SiMe2), 5.74 (s (br), 2 H, NH), 6.55 (t, 4.8 Hz, 2 H, H5), and 8.25 (d, 4.8 Hz, 4 H, H4,6); 13C NMR (CDCl3, 100 MHz) δ (ppm) −1.0 (SiMe2), 111.4 (C5), 157.9 (C4,6), and 164.1 (C2); and 29Si NMR (CDCl3, 79 MHz): δ (ppm) −5.6.
Compound 3 (tris(pyrimid-2-ylamino)methylsilane). 2-Aminopyrimidine (5.77 g, 61 mmol) and triethylamine (6.17 g, 61 mmol) were dissolved in 80 mL of THF, whereupon trichloromethylsilane (2.99 g, 20 mmol) was added with stirring, and the mixture was heated to reflux for 1 h. After stirring at room temperature overnight, the suspension was filtered, and half of the solvent was removed from the filtrate under reduced pressure. Crystals, suitable for single-crystal X-ray diffraction, were obtained after standing one night at room temperature. The crystals were isolated by decantation, washed with n-pentane, and briefly dried in vacuo. Yield: 2.60 g (3.6 mmol, 36%) of the THF solvate (3)2 · (THF). Melting point: 134 °C (decomp.); Raman: ν (cm−1) 3156 (vw), 3121 (vw), 3088 (w), 3052 (w), 3027 (w), 3000 (vw), 2913 (vw), 1580 (w), 1133 (vw), 1079 (w), 986 (vw), 871 (vs), 815 (vw), 780 (vw), 763 (vw), 651 (vw), 587 (vw), 458 (w), 410 (w), 194 (w), 120 (vs), and 78 (vw); and for NMR characterization, the compound was recrystallized from CHCl3. 1H NMR (CDCl3, 400 MHz): δ (ppm) 0.81 (s, 3 H, SiMe), 6.26 (s (br), 3 H, NH), 6.62 (t, 4.8 Hz, 3 H, H5), and 8.30 (d, 4.8 Hz, 6 H, H4,6); 13C NMR (CDCl3, 100 MHz): δ (ppm) −1.3(SiMe2), 111.9 (C5), 158.1 (C4,6), and 163.6 (C2); 29Si NMR (CDCl3, 79 MHz): δ (ppm) −24.2.
Compound 4 (tetrakis(pyrimid-2-ylamino)silane). 2-Aminopyrimidine (6.72 g, 71 mmol) and triethylamine (7.28 g, 72 mmol) were dissolved in 80 mL of THF, whereupon tetrachlorosilane (3.06 g, 18 mmol) was added with stirring, and the mixture was heated to reflux for 1 h (and 40 mL of additional THF was added). After stirring at room temperature overnight, the suspension was filtered, and all volatiles were removed from the filtrate under reduced pressure (50 °C water bath, ≤1 mbar). First, a slightly yellowish solution was obtained. According to NMR spectroscopy, this liquid still contained THF, which was not removed under the conditions applied. Upon storing this solution at room temperature for two days, slightly yellow crystals and a colorless liquid (THF) formed. The solid was isolated by decantation and dried in vacuum to afford 2.22 g (5 mmol, 28%) of a white solid. Whereas NMR and Raman spectroscopy were carried out with this product obtained from THF, crystals for the X-ray diffraction analysis of 4 were obtained along a transsilylation route: Heating of 1 (2.05 g, 12 mmol) with tetrachlorosilane (0.52 g, 3 mmol) in 100 mL anisole for 4 h and slowly cooling to room temperature afforded a few crystals of the anisole solvate of 4. Melting point: >300 °C; Raman: ν (cm−1) 3158 (vw), 3121 (vw), 3088 (w), 3052 (w), 3027 (w), 3000 (vw), 1580 (w), 1133 (vw), 1079 (w), 986 (vw), 871 (vs), 651 (vw), 521 (vw), 458 (w), 410 (w), 194 (w), and 120 (vs); 1H NMR (CDCl3, 400 MHz): δ (ppm) 5.48 (s (br), 4 H, NH), 6.59 (t, 4.8 Hz, 4 H, H5), and 8.29 (d, 4.8 Hz, 8 H, H4,6); 13C NMR (CDCl3, 100 MHz): δ (ppm) 111.5 (C5), 158.3 (C4,6), and 163.2 (C2); and 29Si NMR (CDCl3, 79 MHz): δ (ppm) −52.5.
Attempt of the reaction of 1 with CO2. A sample of 1 (ca. 20 mg) was dissolved in about 1 mL of THF-d8 in an NMR tube. With a long cannula, CO2 was introduced into this solution (slow bubbling) for 20 min and NMR spectra were recorded afterwards (cf. Figures S13–S15). The three NMR spectra show the signals of the starting material 1. According to the 29Si NMR spectrum, besides SiMe4, this is still the sole Si compound in this sample. In the 1H and 13C spectra, some additional signals are present which can be assigned to free 2-aminopyrimidine and CO2 (the latter is assigned to the 13C NMR signal at 126 ppm). No signal for any new carbonyl moiety (such as a carbamate) could be found.
3. Results and Discussion
3.1. Syntheses
The herein reported silylated derivatives of 2-aminopyrimidine (H2Npyr), i.e., compounds 1, 2, 3, and 4, could be synthesized, starting from the respective chlorosilanes and triethylamine (as HCl scavenger) in THF as the solvent with good yields (Scheme 1). The recrystallization of compounds 1 and 2 (from chloroform) and compound 3 (from THF) afforded crystals suitable for single-crystal X-ray diffraction analyses. As compound 4 exhibits very poor solubility in these solvents, an alternative synthesis path (a transsilylation of 1 and SiCl4 in anisole as the solvent) was pursued to obtain crystals suitable for X-ray diffraction (Scheme 1, bottom).
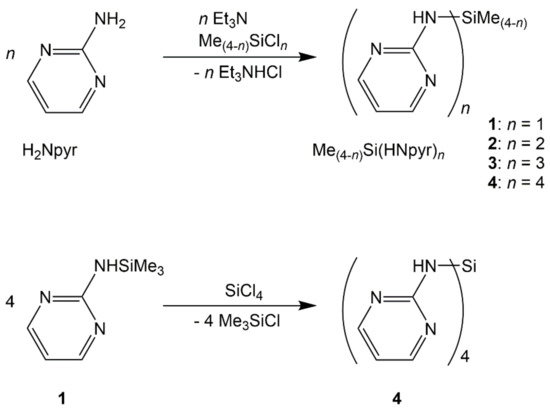
Scheme 1.
Triethylamine supported syntheses of compounds 1, 2, 3, and 4 as well as transsilylation of 1 and SiCl4 with formation of 4.
3.2. Raman Spectroscopy
In the Raman spectra, besides the typical ν(C-Harom) slightly above 3000 cm−1 and ν(C-Haliph.) slightly below 3000 cm−1, specific bands for the pyrimidine units are present (see Table 1, assignment of bands taken from [40]). Whereas corresponding vibrations are found at the same wavenumbers for compounds 2, 3, and 4, the respective data of 1 are slightly different. We attribute this to the pronounced flexibility of the molecules of compound 1. Whereas the former three are bound to more than just one neighbor molecules via N–H···N hydrogen bridges and their Si atom represents a bridge/node in this network, in compound 1, only two molecules are combined into dimers via N–H···N hydrogen bridges, and their Si atoms are part of terminal groups (vide infra).

Table 1.
Specific Raman bands for the pyrimidine unit in all compounds in cm−1.
3.3. Molecular Structures
The molecular structures of compounds 1–4 in their solid forms exhibit common features of the Si coordination spheres, which are based on formally covalent Si–N bonds (involving the amino group of the 2-aminopyrimidine building block) and additional remote Si···N interactions with pyrimidine N atoms. The typical bond length of Si–N bonds in silylated guanidines is 1.76(2) Å for 40 examples of compounds with tetracoordinate Si at the tricoordinate guanidine N and 1.66(2) Å for 73 examples of compounds with tetracoordinate Si at the dicoordinate guanidine N (a search in CSD 2023 version 5.44). Because of the latter group, the average Si–N bond length in silylated guanidines is shorter than Si–N bonds in general (1.739 ± 0.030 Å [41]), but the former is more representative for the herein presented compounds. With reference to the van der Waals radii of silicon and nitrogen (2.10 and 1.55 Å, respectively), any Si-N distance below 3.65 Å is an indication of atomic interaction [42]. Particular individual features of the molecular structures of compounds 1, 2, 3, and 4 (Table 2 and Table 3) will be discussed in the following.
Table 2.
Selected bond lengths [Å] and angles [deg] in the Si coordination spheres of compounds 1, 2, 3, and 4. Prominent values within groups of corresponding angles are shaded in grey.
Table 3.
Selected interatomic distances [Å] of the remote N···Si coordination in compounds 1, 2, 3, and 4.
Compound 1 (monoclinic, P21/c) crystallizes with two independent molecules in the asymmetric unit (Figure 4, Table 2, Table 3 and Table A1). The silicon atoms are situated in slightly distorted tetrahedral coordination spheres with bond angles in the ranges 102.94(8)–111.10(10)° and 104.00(8)–111.27(8)° around Si1 and Si2, respectively. The Si–N bond lengths of the two independent molecules are essentially equal (1.749(1) Å, Table 2). They are in the expected range for Si–N bonds in general and for those in guanidines with silylated tricoordinate N atom. For each Si atom, the Si···N distance to one of the pyrimidine nitrogen atoms is markedly below the sum of van der Waals radii (3.064 Å for Si1···N2 and 3.004 Å for Si2···N6, Table 3). These N atoms are capping a tetrahedral face of the Si coordination sphere, thus creating a remote [4+1]-coordination. This affects the angles between the four covalently Si-bound substituents: The sum of C-Si-C and C-Si-N angles of the capped tetrahedral face (332.7 and 333.6° at Si1 and Si2, respectively) exceeds the 3-fold tetrahedral angle (328.5°), and the noticeably smallest angle in the Si coordination sphere (highlighted in Table 2) is spanned by the Si–N bond and the Si–C bond trans to the N-capped tetrahedral face. The other pyrimidine N atoms, which are not involved in remote Si···N coordination, are involved in intermolecular N–H···N hydrogen bridges (vide infra).
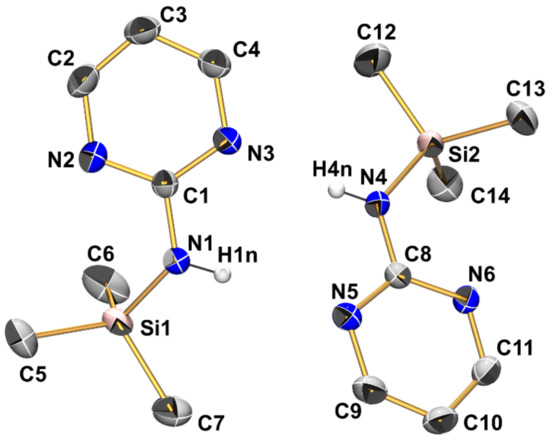
Figure 4.
Molecular structures of the two crystallographically independent molecules of compound 1 in the crystal structure, shown with displacement ellipsoids at the 50% probability level and labels. C-bound hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles are listed in Table 2 and Table 3.
The crystal structure of compound 2 (triclinic, P) comprises one molecule in the asymmetric unit (Figure 5, Table 2, Table 3 and Table A1). This structure has recently been reported by Merzweiler and Lechner [43]. As our own structure determination afforded a model with the enhanced precision of bond lengths and angles, we refer to our data in the discussion. As found for compound 1, the Si–N bonds in 2 (1.738(1) Å and 1.744(1) Å) are in the expected range for Si–N bonds. The silicon atom is in a tetrahedral coordination sphere with angles between 104.54(5) and 112.33(5)°. As a result of the presence of two pyrimidylamino groups, the molecule of 2 features two Si···N contacts below the sum of the van der Waals radii (Si1···N2 3.061 Å, Si1···N5 3.045 Å). As a common feature of 1 and 2, the smallest angles in the silicon coordination sphere (highlighted in Table 2) are also the Me-Si-N angles which involve the methyl group trans to the tetrahedral face capped by a pyrimidine N atom of this entity.
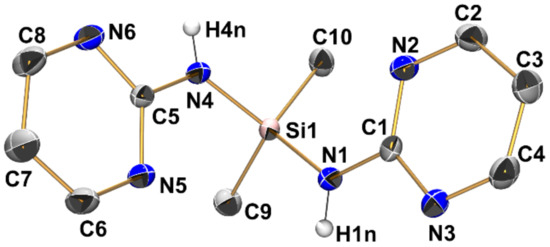
Compound 3 (triclinic, P) crystallizes with two independent aminosilane molecules and one THF molecule in the asymmetric unit (Figure 6, Table 2, Table 3 and Table A1). For both molecules, the Si–N bond lengths are in the range from 1.733(1) Å to 1.744(1) Å, which is within the expected range for general Si–N bonds. The silicon atoms in both molecules have a markedly distorted tetrahedral coordination sphere with angles ranging between 104.11(6) and 115.77(6)°, with the N-Si-N angles at the lower end (around 105°) and the N-Si-C angles at the upper end, which is essentially in accordance with VSEPR. Furthermore, this rather pronounced difference between these groups of angles is a result of the molecular conformation in which each molecule of 3 features three Si···N contacts below the sum of the van der Waals radii between its Si atom and one pyrimidine nitrogen atom of each pyrimidine moiety. All of these Si···N contacts are established by capping the tetrahedral faces trans to the Si–N bonds, which results in mutual widening of the C-Si-N and compressing of the N-Si-N angles. Interestingly, for each molecule of 3, one of these Si···N contacts is markedly longer than the other two (Table 3). While the pyrimidine moieties involved in this remote coordination are all involved in the same type of N–H···N hydrogen bonding in the crystal packing (vide infra), the remote N···Si coordination of one out of three N···Si interactions per molecule is markedly off from capping the center of a tetrahedral face. This is best reflected by the different N-Si-N angles spanned by the capping N atom and the Si–N bond trans to the capped face (molecule 1: N9-Si1-N1 157.04(5)°, N3-Si1-N4 154.85(5)°, N6-Si1-N7 147.92(5)°; molecule 2: N15-Si2-N16 154.15(5)°, N12-Si2-N13 154.61(5)°, N18-Si2-N10 144.36(5)°).

Figure 6.
Molecular structures of the two crystallographically independent molecules of compound 3 in the crystal structure, shown with displacement ellipsoids at the 50% probability level and labels. C-bound hydrogen atoms and the solvent of crystallization (THF) are omitted for clarity. Selected interatomic distances (Å) and angles are listed in Table 2 and Table 3.
Compound 4 (monoclinic, P21/c) crystallizes with three aminosilane molecules and three anisole molecules in the asymmetric unit (Figure 7, Table 2, Table 3 and Table A1). All Si–N bond lengths are in the narrow range of 1.7153(14) Å to 1.7272(13) Å (in accordance with the expected range of Si–N bond lengths in general). In the order 1 > 2 > 3 > 4, systematic shortening of the Si–N bonds is observed, which causes slight but increasing deviations of the Si–N bond lengths from the expected values for compounds with tetracoordinate Si at the tricoordinate guanidine N (for compound 4 in particular). We attribute this to the Si–N bond shortening as a result of a greater number of electronegative substituents at Si (especially in compound 4), whereas many silylated guanidines with Si–C bonds (such as compounds VIII, XII, and XX) contribute to the average Si–N bond length. The silicon atoms are situated in distorted tetrahedral coordination spheres with angles ranging from 107.28(6)° to 113.35(6)°. This distortion is less pronounced than in compound 3 as the Si coordination sphere is more symmetric (SiN4), and this is true for the wider coordination sphere as well. Each of the three crystallographically independent molecules of 4 features four intramolecular Si···N contacts with interatomic distances around 3 Å, thus each of the tetrahedral faces is capped similarly. Nonetheless, a systematic feature of distortion of the tetrahedral coordination sphere is encountered, which is particularly pronounced for molecules 1 and 3 of the asymmetric unit: Two out of six N-Si-N angles are markedly wider than the other four (highlighted in Table 2). These wider angles are those of the tetrahedron which do not feature an N–H bond pointing along the tetrahedral edge. Even though there are no intramolecular N–H···N hydrogen bonds, intramolecular N–H → N orientation systematically supports N-Si-N bond angles < 109.5° in the otherwise tetrahedral SiN4 coordination sphere. These attractive interactions may also contribute to the shorter Si–N bonds in compound 4 in particular.
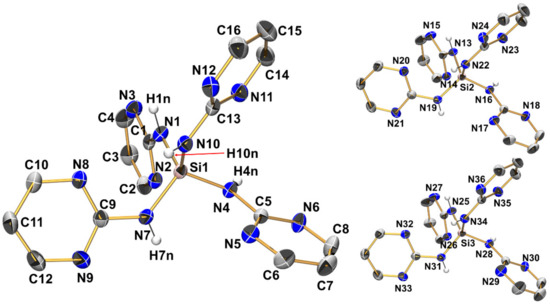
Figure 7.
Molecular structures of the three crystallographically independent molecules of compound 4 in the crystal structure, shown with displacement ellipsoids at the 50% probability level. For one molecule, as a representative example, all labels are shown, whereas for the other two molecules only Si and N atoms are labeled. C-bound hydrogen atoms and the solvent of crystallization (anisole) are omitted for clarity. Selected interatomic distances (Å) and angles are listed in Table 2 and Table 3.
3.4. Intermolecular N–H···N Hydrogen Bonding
In addition to the Si···N contacts (cf. Section 3.3), the pyrimid-2-ylamino groups of compounds 1–4 establish intermolecular N–H···N bridges in these solid-state structures. Comparison of the four silanes reveals that intermolecular contacts via N–H···N bridges are always established by sets of two of such interactions (each silane molecule acting as a hydrogen bridge donor and as a hydrogen bridge acceptor in the one and the other case). The structures under investigation exhibit three different motifs of that dimeric N–H···N bridging (we call them types A, B, C, Figure 8). Type A: the NH group and pyrimidine N atom of one pyrimidylamino group of molecule 1 interact with the pyrimidine N atom and NH group, respectively, of one pyrimidylamino group of molecule 2. This interaction pattern results in the formation of an NHNCNHNC eight-membered ring (R motif [44]). Type B: the NH group and pyrimidine N atom of two individual pyrimidylamino groups of molecule 1 interact with the pyrimidine N atom and NH group, respectively, of two individual pyrimidylamino groups of molecule 2. This interaction pattern results in the formation of an NHNCNSiNHNCNSi twelve-membered ring (R motif). Type C: This represents an arrangement intermediate between types A and B. The NH group and pyrimidine N atom of two individual pyrimidylamino groups of molecule 1 interact with the pyrimidine N atom and NH group, respectively, of one pyrimidylamino group of molecule 2. This interaction pattern results in the formation of an R motif with an NHNCNSiNHNC ten-membered ring.
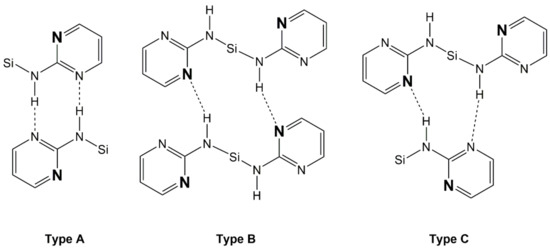
Figure 8.
Schematic representation of the intermolecular N–H···N interactions encountered with the crystal packings of compounds 1, 2, 3, and 4: Type A with formation of NHNCNHNC eight-membered rings, type B with formation of NHNCNSiNHNCNSi twelve-membered rings, and type C with formation of NHNCNSiNHNC ten-membered rings. The pyrimidine N atoms, which are involved in remote N···Si coordination, are pronounced by font size and bold style.
Within the group of compounds under investigation, hydrogen bond systems of type A were encountered with different compounds (1, 2, and 3, cf. Figure 9, Figure 10 and Figure 11, respectively), whereas the other two types were observed in one case each (type B in compound 2, Figure 10, and type C in compound 4, Figure 12).

Figure 9.
Graphical representation of intramolecular N···Si and intermolecular N–H···N interactions (blue and red lines, respectively) of compound 1 in the crystal structure. C-bound hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles are listed in Table 3 and Table 4.
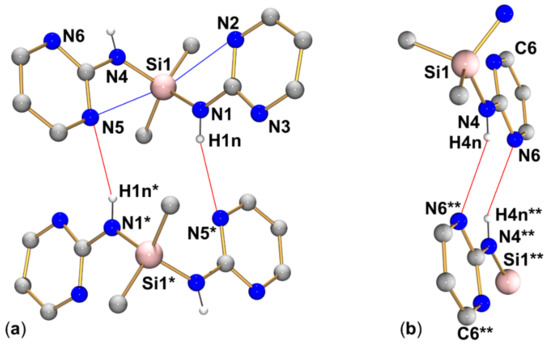
Figure 10.
Graphical representation of intramolecular N···Si and intermolecular N–H···N interactions (blue and red lines, respectively) of compound 2 in the crystal structure (R motif in (a) and R motif in (b)). C-bound hydrogen atoms are omitted for clarity. (The molecule labeled with asterisks * is generated from the asymmetric unit by symmetry operation −x, 1−y, 2−z; the fragment of the molecule labeled with two asterisks ** is generated from the asymmetric unit by symmetry operation 1−x, 1−y, 1−z). Selected interatomic distances (Å) and angles are listed in Table 3 and Table 4.
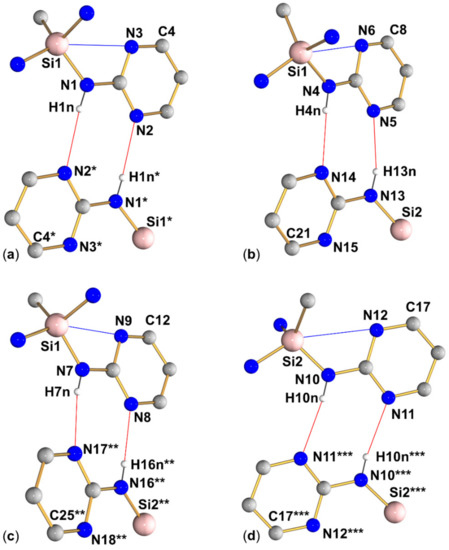
Figure 11.
Graphical representation of intramolecular N···Si and intermolecular N–H···N interactions (blue and red lines, respectively) of compound 3 in the crystal structure. (Subfigures (a–d) show the four R motifs encountered in this structure.) C-bound hydrogen atoms are omitted for clarity. (The molecular fragment labeled with asterisks * is generated from the asymmetric unit by symmetry operation 2−x, 1−y, −z; the fragment labeled with two asterisks ** is generated from the asymmetric unit by translation x + 1, y, z; the fragment labeled with three asterisks *** is generated from the asymmetric unit by symmetry operation 1−x, 1−y, 1−z). Selected interatomic distances (Å) and angles are listed in Table 3 and Table 4.
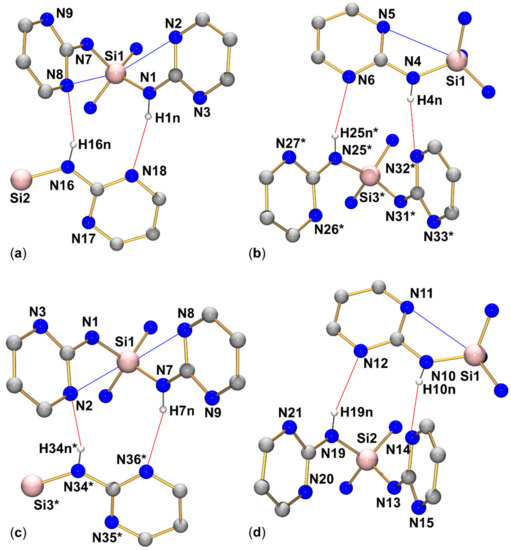
Figure 12.
Graphical representation of intramolecular N···Si and intermolecular N–H···N interactions (blue and red lines, respectively) of compound 4 in the crystal structure. (Subfigures (a–d) show the four R motifs encountered in this structure.) C-bound hydrogen atoms are omitted for clarity. (The molecular fragments labeled with asterisks * are generated from the asymmetric unit by translation x−1, y, z). Selected interatomic distances (Å) and angles are listed in Table 3 and Table 4.
The packing in favor of the formation of interactions of type A can be reasoned by the possibility of the formation of a pair of N–H···N bridges in which the H-bond donor meets the acceptor at the ideal angle with respect to the orientation of the H-bond acceptor’s lone pair. In principle, this should lead to near linear arrangements of the atom sequence N–H···N…C(γ) (C(γ) being the pyrimidine C atom in γ position with respect to the hydrogen bond acceptor pyrimidine N atom). The variability in the N-N-C angles of these arrays (see Table 4) indicates that this hydrogen bonding motif offers some flexibility to adapt to further packing requirements.
Table 4.
Selected interatomic distances [Å] and angles [deg] associated with N–H···N hydrogen bridges in compounds 1, 2, 3, and 4. The symmetry codes (asterisks * at the atom labels) used with compounds 2, 3, and 4 are the same as those listed in the caption of the figure which depicts that particular compound (Figure 10, Figure 11 and Figure 12, respectively).
Intermolecular packing types B and C provide some interesting insights into these and related silanes with this kind of remote Si coordination in which the tetrahedral Si coordination sphere is capped by pyridine (or related) N atom(s). Even though pyrimid-2-ylamino groups always offer an N-located lone pair for Si-coordination and a second N-located lone pair for other interactions such as N–H···N hydrogen bridges, the latter proved capable of competing with the remote N···Si-coordination by interacting with the same N atom and thus leaving the other pyrimidine N atom available for further packing interactions. This finding complements the insight that 2-pyridylaminosilanes of the type R2Si(NH-pyridyl)2 may sacrifice their possibility of N···Si-coordination in favor of using the pyridine N atom for establishing intermolecular packing interactions. In detail, Me2Si(NH-pyridyl)2 features remote coordination, whereas Ph2Si(NH-pyridyl)2 utilizes pyridine N atoms for other interactions [45].
3.5. Reaction with CO2
CO2 can insert into Si–N bonds with the formation of carbamoyloxysilanes in certain cases (e.g., IV) [1,5,6]. Even though CO2 insertion into the Si–N bonds of arylamines is not as facile as the insertion into the Si–N bonds of alkylamines [8], additional N-donor bases may support the reaction of Si–N bonds and CO2 [8] by increasing the CO2 concentration in the solution [46] or because N-donor bases are assumed to catalyze the reaction [47,48]. Therefore, we investigated the reaction of 1 with CO2. Introducing gaseous CO2 into a solution of 1 in THF-d8 for 20 min did not show any formation of carbamoyloxysilanes according to the NMR spectra which only show 1, free 2-aminopyrimidine, and CO2 (at δ 126 ppm in the 13C NMR spectrum). This result indicates that CO2 insertion into the Si–N bonds of this arylamine is hampered, and the additional N-donor sites (of the Si-bound and of the free 2-aminopyrimidine) do not drive this reaction to a facile level under ambient conditions (room temperature, CO2 bubbling into a THF solution of the aminosilane). The exploration of the reaction conditions required for utilizing CO2 insertion into the Si–N bonds of pyrimidylaminosilanes will thus be subject to future studies.
4. Conclusions
Four silylated 2-aminopyrimidines were synthesized, crystallized, and analyzed using X-ray diffraction. Silanes with related pyridine motifs (such as pyridine-2-olate and pyridine-2-thiolate) involve their pyridine N atoms in remote N···Si coordination [49,50,51,52]. In the case of some representatives with pyridine-2-thiolate, rather strong N-Si-bonding with the formation of hexacoordinate Si complexes was found [51,53,54]. With the pyridine-2-amino group at the Si atom, both the presence and absence of remote N···Si coordination were encountered [45]. The series of silanes 1–4 with the related pyrimidine-2-amino motif exhibits remote N···Si coordination for all representatives, but the additional interactions possible because of the second lone pair donor N atom in the aromatic ring open a portfolio of intermolecular packing interactions via N–H···N hydrogen bridges. Interestingly, in some cases, the remotely N···Si coordinating pyrimidine N atom serves as the hydrogen bridge acceptor. With more than one pyrimidylamino group bound per Si atom, these compounds may form highly linked networks through hydrogen bonds. In this regard, the compound Si(NHPyr)4 (compound 4) may become interesting for high-temperature applications as the melting point is higher than 300 °C. Moreover, functional groups with hydrogen bridge donor or acceptor qualities may be adsorbed in silica aerogels (e.g., the sorption of mefenamic acid [55]), a feature which is of importance for drug delivery investigations. Vice versa, functional groups with hydrogen bridge donor or acceptor qualities may alter the sorbent properties of porous materials. In this regard, pyrimidylaminosilanes may become interesting starting materials for the tuning of properties in non-hydrolytic Si–N-based sol–gel processes [56,57,58]. For a further exploration of coordination chemistry (such as the use of these silanes as ligands in main group or transition metal chemistry), stronger intermolecular packing may pose an obstacle for providing well-soluble ligands and molecular complexes resulting therefrom. Thus, the preparation of coordination compounds (such as analogs of the Pb and Sn complexes obtained from pyrid-2-ylaminosilanes [45] or Pd and Cu complexes obtained from silicon pyridine-2-olates [49,52]) may pose challenges. The additional functional groups (the second pyrimidine N atom in combination with the still present Si-NH group) may give rise to heteronuclear complexes which may form 3D networks, a group of compounds which is yet to be explored. The 2-aminopyrimidine motif itself has already entered that field of research, e.g., in Cu(II)-containing MOFs [59] and in coordination networks with Ag(I) centers [60].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13070990/s1, NMR spectra (1H, 13C{1H}, and 29Si{1H} of CDCl3 solutions of compounds 1 (Figures S1–S3), 2 (Figures S4–S6), 3 (Figures S7–S9), and 4 (Figures S10–S12); 1H, 13C{1H}, and 29Si{1H} of a THF-d8 solution of compound 1 in the presence of 2-aminopyrimidine and CO2 (Figures S13–S15)), Raman spectra of compounds 1–4 (Figure S16) as well as details (suppliers, purification if applicable) of the chemicals used in this study (Table S1) and parameters of NMR data acquisition (Table S2).
Author Contributions
Conceptualization, M.H.; funding acquisition, E.K.; investigation, M.H. and J.W.; writing—original draft preparation, M.H. and J.W.; writing—review and editing, M.H., E.K., and J.W.; visualization, M.H. and J.W. All authors have read and agreed to the published version of the manuscript.
Funding
Part of this work was performed within the research group “Chemical utilization of carbon dioxide with aminosilanes (CO2-Sil)” that is financially supported by the European Union (European social fund, ESF), the Ministry of Science and Art of Saxony (SMWK), and the Sächsische Aufbaubank (SAB), Project number 100310482.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
CCDC 2256578 (2), 2256579 (1), 2256580 ((3)2 · (THF)), and 2256581 (4 · (anisole)) contain the supplementary crystal data for this article. These data can be obtained free-of-charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures/ (accessed on 15 April 2023).
Acknowledgments
The authors are grateful to Beate Kutzner and Regina Moßig (TU Bergakademie Freiberg, Institut für Anorganische Chemie) for NMR (B.K.) and Raman spectroscopy (R.M.).
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Table A1.
Crystallographic data from data collection and refinement for compounds 1, 2, 3 (THF solvate), and 4 (anisole solvate).
Table A1.
Crystallographic data from data collection and refinement for compounds 1, 2, 3 (THF solvate), and 4 (anisole solvate).
| Parameter | 1 | 2 1 | (3)2 · (THF) 2 | 4 · (Anisole) 3 |
|---|---|---|---|---|
| Formula | C7H13N3Si | C10H14N6Si | C30H38N18OSi2 | C23H24N12OSi |
| Moiety formula | C7H13N3Si | C10H14N6Si | 2 (C13H15N9Si), C4H8O | C16H16N12Si, C7H8O |
| Mr | 167.29 | 246.36 | 722.96 | 512.63 |
| T (K) | 180(2) | 180(2) | 180(2) | 180(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | triclinic | triclinic | monoclinic |
| Space group | P21/c | P | P | P21/c |
| a (Å) | 9.4337(3) | 8.6757(4) | 10.0640(3) | 15.9611(2) |
| b (Å) | 19.5412(6) | 8.8740(4) | 11.1921(3) | 21.1674(3) |
| c (Å) | 10.7349(3) | 9.0989(4) | 18.6757(5) | 22.6832(3) |
| α (°) | 90 | 64.211(3) | 72.898(2) | 90 |
| β (°) | 100.794(3) | 75.463(3) | 89.000(2) | 97.740(1) |
| γ (°) | 90 | 79.194(3) | 63.346(2) | 90 |
| V (Å3) | 1943.92(10) | 608.08(5) | 1780.70(9) | 7593.81(18) |
| Z | 8 | 2 | 2 | 12 |
| ρcalc (g·cm−1) | 1.14 | 1.35 | 1.35 | 1.35 |
| μMoKα (mm−1) | 0.2 | 0.2 | 0.2 | 0.1 |
| F (000) | 720 | 260 | 760 | 3216 |
| θmax (°), Rint | 28.0, 0.0377 | 28.0, 0.0232 | 28.0, 0.0319 | 27.0, 0.0428 |
| Completeness | 100% | 99.8% | 99.9% | 99.9% |
| Reflns collected | 17,865 | 9797 | 33,018 | 130,089 |
| Reflns unique | 4699 | 2937 | 8598 | 16,561 |
| Restraints | 0 | 0 | 24 | 0 |
| Parameters | 214 | 165 | 527 | 1150 |
| GoF | 1.097 | 1.080 | 1.063 | 1.108 |
| R1, wR2 [I > 2σ(I)] | 0.0367, 0.0876 | 0.0308, 0.0770 | 0.0385, 0.0959 | 0.0407, 0.0939 |
| R1, wR2 (all data) | 0.0495, 0.0957 | 0.0336, 0.0792 | 0.0462, 0.1019 | 0.0506, 0.1010 |
| Largest peak/hole (e·Å−3) | 0.24, −0.23 | 0.25, −0.25 | 0.28, −0.31 | 0.27, −0.35 |
1 One of the two methyl groups was refined disordered (rotational disorder about the Si–C bond) with site occupancies of 0.40(2) and 0.60(2). 2 This structure features two disordered moieties. The solvent of crystallization (a THF molecule) was refined disordered over two sites with site occupancies of 0.771(4) and 0.229(4). Furthermore, the pyrimidino group of one of the two crystallographically independent molecules of 3 was refined disordered over two sites with site occupancies of 0.914(2) and 0.086(2). 3 The asymmetric unit of this structure comprises three molecules of 4 and three molecules of the solvent of crystallization (anisole). The latter consist of (i) a couple of two anisole molecules connected by C–H···C(π) stacking (with site occupancy of 0.5), which is located near a center of inversion and thus disordered by symmetry (thus furnishing full site occupancy); (ii) an adjacent anisole molecule, which is slightly disordered over two sites (site occupancy factors refined to 0.407(13) and 0.593(13); for stable refinement of this less pronounced disorder, the benzene rings of this molecule were treated as idealized hexagons); and (iii) an additional (remote) anisole molecule which did not show disorder.
References
- Kroke, E.; Li, Y.; Konetschny, C.; Lecomte, E.; Fasel, C.; Riedel, R. Silazane derived ceramics and related materials. Mater. Sci. Eng. Rep. 2000, 26, 97–199. [Google Scholar] [CrossRef]
- Völger, K.; Hauser, R.; Kroke, E.; Riedel, R.; Ikuhara, Y.; Iwamoto, Y. Synthesis and characterization of novel non-oxide sol-gel derived mesoporous amorphous Si-C-N membranes. J. Ceram. Soc. Jpn. 2006, 114, 567–570. [Google Scholar] [CrossRef]
- Krüger, C.; Rochow, E. Polyorganosilazanes. J. Polym. Sci. Part A Gen. Pap. 1964, 2, 3179–3189. [Google Scholar] [CrossRef]
- Kraushaar, K.; Wiltzsch, C.; Wagler, J.; Böhme, U.; Schwarzer, A.; Roewer, G.; Kroke, E. From CO2 to Polysiloxanes: Di(carbamoyloxy)silanes Me2Si[(OCO)NRR’]2 as Precursors for PDMS. Organometallics 2012, 31, 4779–4785. [Google Scholar] [CrossRef]
- Kraushaar, K.; Schmidt, D.; Schwarzer, A.; Kroke, E. Chapter Four—Reactions of CO2 and CO2 Analogs (CXY with X, Y=O, S, NR) with Reagents Containing Si–H and Si–N Units. In CO2 Chemistry; Aresta, M., van Eldik, R., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 117–162. [Google Scholar] [CrossRef]
- Herbig, M.; Gevorgyan, L.; Pflug, M.; Wagler, J.; Schwarzer, S.; Kroke, E. CO2 Capture with Silylated Ethanolamines and Piperazines. ChemistryOpen 2019, 9, 894–902. [Google Scholar] [CrossRef]
- Wiltzsch, C.; Kraushaar, K.; Schwarzer, A.; Kroke, E. CO2 as an Oxygen Source for Polysiloxanes—Preparation, Crystal Structure and Thermal Decomposition of Two Novel Silylcarbamates. Z. Naturforsch. B 2011, 66, 917–922. [Google Scholar] [CrossRef]
- Gründler, F.; Scholz, H.; Herbig, M.; Schwarzer, S.; Wagler, J.; Kroke, E. Formation of Aromatic O-Silylcarbamates from Aminosilanes and Their Subsequent Thermal Decomposition with Formation of Isocyanates. Eur. J. Inorg. Chem. 2021, 2021, 2211–2224. [Google Scholar] [CrossRef]
- Kraushaar, K.; Herbig, M.; Schmidt, D.; Wagler, J.; Böhme, U.; Kroke, E. Insertion of phenyl isocyanate into mono- and diaminosilanes. Z. Naturforsch. B 2017, 72, 909–921. [Google Scholar] [CrossRef]
- Schöne, D.; Gerlach, D.; Wiltzsch, C.; Brendler, E.; Heine, T.; Kroke, E.; Wagler, J. A Distorted Trigonal Antiprismatic Cationic Silicon Complex with Ureato Ligands: Syntheses, Crystal Structures and Solid State 29Si NMR Properties. Eur. J. Inorg. Chem. 2010, 2010, 461–467. [Google Scholar] [CrossRef]
- Ryll, C.; Kraushaar, K.; Wagler, J.; Brendler, E.; Kroke, E. Disilanes with Pentacoordinate Si Atoms by Carbon Dioxide Insertion into Aminodisilanes: Syntheses, Molecular Structures, and Dynamic Behavior. ACS Omega 2022, 7, 9527–9536. [Google Scholar] [CrossRef]
- Herbig, M.; Böhme, U.; Kroke, E. Lactamomethylsilanes—Synthesis, Structures, and Reactivity towards CO2 and Phenylisocyanate. Z. Anorg. Allg. Chem. 2019, 645, 377–387. [Google Scholar] [CrossRef]
- Herbig, M.; Kroke, E. Synthesis and spectroscopic properties of iminosilanes. Chem. Data Collect. 2023, 44, 100992. [Google Scholar] [CrossRef]
- Herbig, M.; Böhme, U.; Schwarzer, A.; Kroke, E. Formation of 1-aza-2-silacyclopentanes and unexpected products from the insertion of phenylisocyanate into 2,2-dimethyl-1-(trimethylsilyl)-1-aza-2-sila-cyclo-pentane. Main Group Met. Chem. 2018, 41, 11–19. [Google Scholar] [CrossRef]
- Herbig, M.; Böhme, U.; Kroke, E. Insertion of CO2 and related heteroallenes into the Si-N-bond of methyl(N-morpholino)silanes. Inorg. Chim. Acta 2018, 473, 20–28. [Google Scholar] [CrossRef]
- Jin, G.; Jones, C.; Junk, P.; Lippert, K.; Roseab, R.; Stasch, A. Synthesis and characterisation of bulky guanidines and phosphaguanidines: Precursors for low oxidation state metallacycles. New J. Chem. 2009, 33, 64–75. [Google Scholar] [CrossRef]
- Lysenko, S.; Daniliuc, C.; Jones, P.; Tamm, M. Tungsten alkylidyne complexes with ancillary imidazolin-2-iminato and imidazolidin-2-iminato ligands and their use in catalytic alkyne metathesis. J. Organomet. Chem. 2013, 744, 7–14. [Google Scholar] [CrossRef]
- Deka, H.; Fridman, N.; Eisen, M. A Sacrificial Iminato Ligand in the Catalytic Cyanosilylation of Ketones Promoted by Organoactinide Complexes. Inorg. Chem. 2022, 61, 3598–3606. [Google Scholar] [CrossRef]
- Seidel, F.; Tomizawa, I.; Nozaki, K. Expedient Synthetic Identification of a P-Stereogenic Ligand Motif for the Palladium-Catalyzed Preparation of Isotactic Polar Polypropylenes. Angew. Chem. Int. Ed. 2020, 59, 22591–22601. [Google Scholar] [CrossRef]
- Oakley, S.; Coles, M.; Hitchcock, P. Poly(guanidyl)silanes as a new class of chelating, N-based ligand. Dalton Trans. 2004, 33, 1113–1114. [Google Scholar] [CrossRef]
- Mück, F.; Baus, J.; Ulmer, A.; Burschka, C.; Tacke, R. Reactivity of the Donor-Stabilized Guanidinatosilylene [ArNC(NMe2)NAr]Si[N(SiMe3)2] (Ar = 2,6-Diisopropylphenyl). Eur. J. Inorg. Chem. 2016, 2016, 1660–1670. [Google Scholar] [CrossRef]
- von Wolff, N.; Lefèvre, G.; Berthet, J.; Thuéry, P.; Cantat, T. Implications of CO2 Activation by Frustrated Lewis Pairs in the Catalytic Hydroboration of CO2: A View Using N/Si+ Frustrated Lewis Pairs. ACS Catal. 2016, 6, 4526–4535. [Google Scholar] [CrossRef]
- Reiter, D.; Frisch, P.; Wendel, D.; Hörmann, F.; Inoue, S. Oxidation reactions of a versatile, two-coordinate, acyclic iminosiloxysilylene. Dalton Trans. 2020, 49, 7060–7068. [Google Scholar] [CrossRef]
- Inoue, S.; Leszczyn’ska, K. An Acyclic Imino-Substituted Silylene: Synthesis, Isolation, and its Facile Conversion into a Zwitterionic Silaimine. Angew. Chem. Int. Ed. 2012, 51, 8589–8593. [Google Scholar] [CrossRef]
- Coles, M.; Sözerli, S.; Smith, J.; Hitchcock, P.; Day, I. An Ether-Free, Internally Coordinated Dialkylcalcium(II) Complex. Organometallics 2009, 28, 1579–1581. [Google Scholar] [CrossRef]
- Smart, K.; Grellier, M.; Coppel, Y.; Vendier, L.; Mason, S.; Capelli, S.; Albinati, A.; Montiel-Palma, V.; Muñoz-Hernández, M.; Sabo-Etienne, S. Nature of Si−H Interactions in a Series of Ruthenium Silazane Complexes Using Multinuclear Solid-State NMR and Neutron Diffraction. Inorg. Chem. 2014, 53, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.; Strunden, T.; Greb, L. Silicon Carbamates by CO2 Fixation: Brønsted Acid Labile Precursor of a Lewis Superacid. Inorg. Chem. 2022, 61, 15693–15698. [Google Scholar] [CrossRef]
- Kociok-Köhn, G.; Molloy, K.; Price, G.; Smith, D. Structural characterisation of trimethylsilyl-protected DNA bases. Supramol. Chem. 2008, 20, 697–707. [Google Scholar] [CrossRef]
- Bertolasi, V.; Boaretto, R.; Chierotti, M.; Gobettoc, R.; Sostero, S. Synthesis, characterization and reactivity of new complexes of titanium and zirconium containing a potential tridentate amidinato-cyclopentadienyl ligand. Dalton Trans. 2007, 36, 5179–5189. [Google Scholar] [CrossRef]
- Oberthür, M.; Arndt, P.; Kempe, R. Synthesis and Structure of Mononuclear Titanium Complexes Containing ansa-Aminopyridinato Ligands. Chem. Ber. 1996, 129, 1087–1091. [Google Scholar] [CrossRef]
- Herzog, S.; Dehnert, J. Eine rationelle anaerobe Arbeitsmethode. Z. Chem. 1964, 4, 1–11. [Google Scholar] [CrossRef]
- Böhme, U. Inertgastechnik; De Gruyter: Oldenbourg, Germany, 2020. [Google Scholar] [CrossRef]
- Herbig, M.; Kroke, E. Low Cost Apparatus for Rapid Boiling Point Determination of Small Air Sensitive Samples under Inert Atmosphere. Thermochim. Acta 2017, 654, 81–84. [Google Scholar] [CrossRef]
- Cie, S. X-RED and X-AREA; Stoe & Cie: Darmstadt, Germany, 2009. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2007, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Cason, C.; Fröhlich, T.; Lipka, C. POV-RAY (Version 3.7); Persistence of Vision Raytracer Pty. Ltd.: Williamstown, Australia, 1994–2004. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies; Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Kaftory, M.; Kapon, M.; Botoshansky, M. The Structural Chemistry of Organosilicon Compounds. In The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Wiley & Sons: Chichester, UK, 1998; pp. 181–265. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Merzweiler, K.; Lechner, B.-D. (Martin-Luther-Universität Halle-Wittenberg, Halle, Germany). Private communication, 2022. Private Communication to the Cambridge Structure Database. deposition number CCDC 2182492, CSD refcode CEKWIC. [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Wagner, C.; Merzweiler, K. Novel Dinuclear Tin(II) and Lead(II) Compounds with 2-Pyridyl Functionalized Silylamido Ligands. Z. Anorg. Allg. Chem. 2017, 643, 938–944. [Google Scholar] [CrossRef]
- Pérez, E.; Santos, R.; Gambardella, M.; de Macedo, L.; Rodrigues-Filho, U.; Launay, J.; Franco, D. Activation of Carbon Dioxide by Bicyclic Amidines. J. Org. Chem. 2004, 69, 8005–8011. [Google Scholar] [CrossRef]
- Breederveld, H. The reaction of dialkylaminosilanes with carbon dioxide and with carbon disulphide. Recl. Trav. Chim. Pays-Bas 1962, 81, 276–278. [Google Scholar] [CrossRef]
- Cragg, R.; Lappert, M. Amino-derivatives of metals and metalloids. Part IV. Aminosilylation and aminophosphination of some unsaturated substrates. J. Chem. Soc. A 1966, 82–85. [Google Scholar] [CrossRef]
- Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics 2018, 6, 119. [Google Scholar] [CrossRef]
- Kuß, S.; Brendler, E.; Wagler, J. Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn)—[4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn. Crystals 2022, 12, 1802. [Google Scholar] [CrossRef]
- Seidel, A.; Weigel, M.; Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. Molecular Structures of the Silicon Pyridine-2-(thi)olates Me3Si(pyX), Me2Si(pyX)2 and Ph2Si(pyX)2 (py = 2-Pyridyl, X = O, S), and Their Intra- and Intermolecular Ligand Exchange in Solution. Crystals 2022, 12, 1054. [Google Scholar] [CrossRef]
- Seidel, A.; Gericke, R.; Brendler, E.; Wagler, J. Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2. Inorganics 2023, 11, 2. [Google Scholar] [CrossRef]
- Baus, J.; Burschka, C.; Bertermann, R.; Guerra, C.; Bickelhaupt, F.; Tacke, R. Neutral Six-Coordinate and Cationic Five-Coordinate Silicon(IV) Complexes with Two Bidentate Monoanionic N,S-Pyridine-2-thiolato(-) Ligands. Inorg. Chem. 2013, 52, 10664–10676. [Google Scholar] [CrossRef] [PubMed]
- Wächtler, E.; Gericke, R.; Kutter, S.; Brendler, E.; Wagler, J. Molecular structures of pyridinethiolato complexes of Sn(II), Sn(IV), Ge(IV), and Si(IV). Main Group Met. Chem. 2013, 36, 181–191. [Google Scholar] [CrossRef]
- Khodov, I.; Sobornova, V.; Mulloyarova, V.; Belov, K.; Dyshin, A.; de Carvalho, L.B.; Tolstoy, P.; Kiselev, M. Exploring the Conformational Equilibrium of Mefenamic Acid Released from Silica Aerogels via NMR Analysis. Int. J. Mol. Sci. 2023, 24, 6882. [Google Scholar] [CrossRef]
- Rovai, R.; Lehmann, C.W.; Bradley, J.S. Non-Oxide Sol–Gel Chemistry: Preparation from Tris(dialkylamino)silazanes of a Carbon-Free, Porous, Silicon Diimide Gel. Angew. Chem. Int. Ed. 1999, 38, 2036–2038. [Google Scholar] [CrossRef]
- Lippe, K.; Wagler, J.; Kroke, E.; Herkenhoff, S.; Ischenko, V.; Woltersdorf, J. Cyclic Silylcarbodiimides as Precursors for Porous Si/C/N Materials: Formation, Structures, and Stabilities. Chem. Mater. 2009, 21, 3941–3949. [Google Scholar] [CrossRef]
- Cheng, H.; Li, Y.; Kroke, E.; Herkenhoff, S. In situ synthesis of Si2N2O/Si3N4 composite ceramics using polysilyloxycarbodiimide precursors. J. Eur. Ceram. Soc. 2013, 33, 2181–2189. [Google Scholar] [CrossRef]
- Hossain, A.; Dey, A.; Seth, S.K.; Ray, P.P.; Ballester, P.; Pritchard, R.G.; Ortega-Castro, J.; Frontera, A.; Mukhopadhyay, S. Enhanced Photosensitive Schottky Diode Behavior of Pyrazine over 2-Aminopyrimidine Ligand in Copper(II)-Phthalate MOFs: Experimental and Theoretical Rationalization. ACS Omega 2018, 3, 9160–9171. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhang, N.; Huang, R.-B.; Zheng, L.-S. Series of Ag(I) Coordination Complexes Derived from Aminopyrimidyl Ligands and Dicarboxylates: Syntheses, Crystal Structures, and Properties. Cryst. Growth Des. 2010, 10, 3699–3709. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).