
Synthesis and Crystallographic Characterisation of Pyridyl- and Indoleninyl-Substituted Pyrimido[1,2-b]Indazoles



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Instrumentation
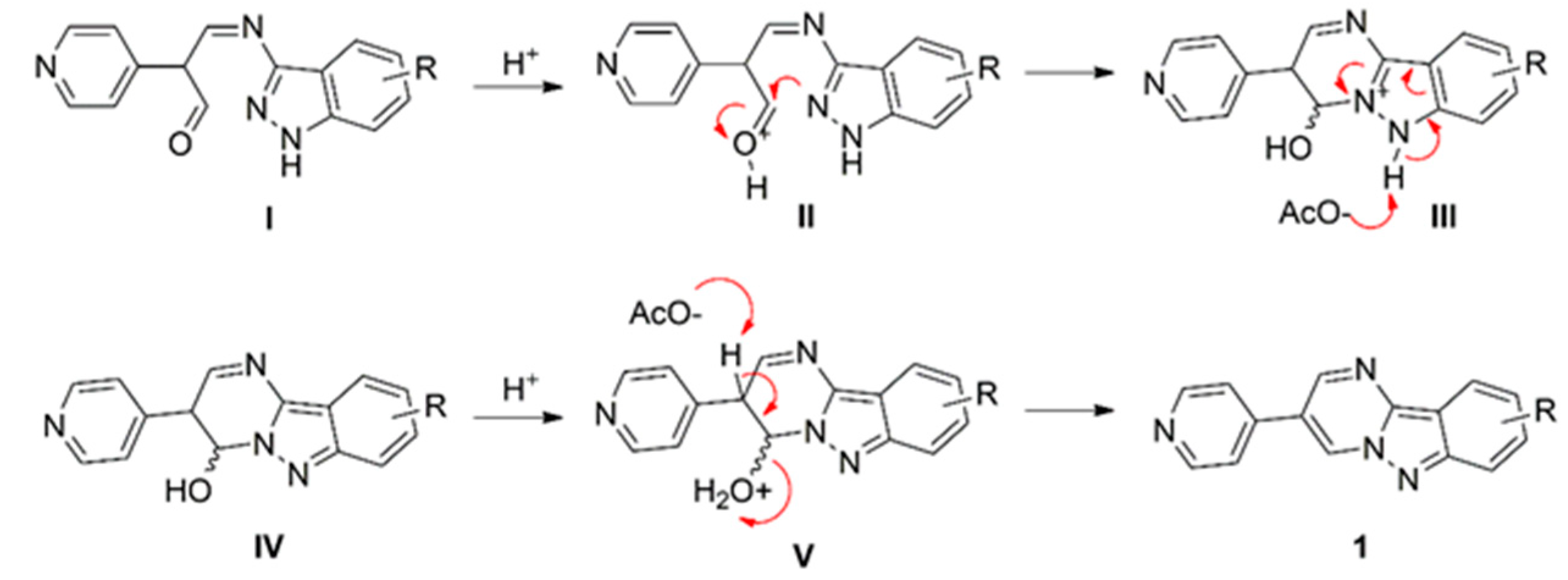
2.2. Synthetic Procedure for the Preparation of 1a–d
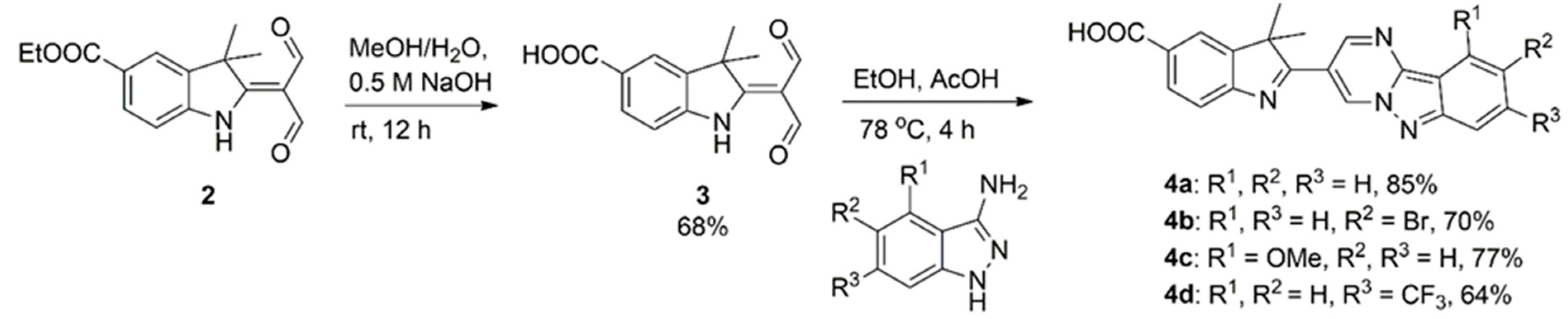
2.3. Synthetic Procedure for the Preparation of 3
2.4. General Procedure for the Preparation of 4a–d
2.5. X-ray Crystallography
2.6. Computational Details
3. Result and Discussion
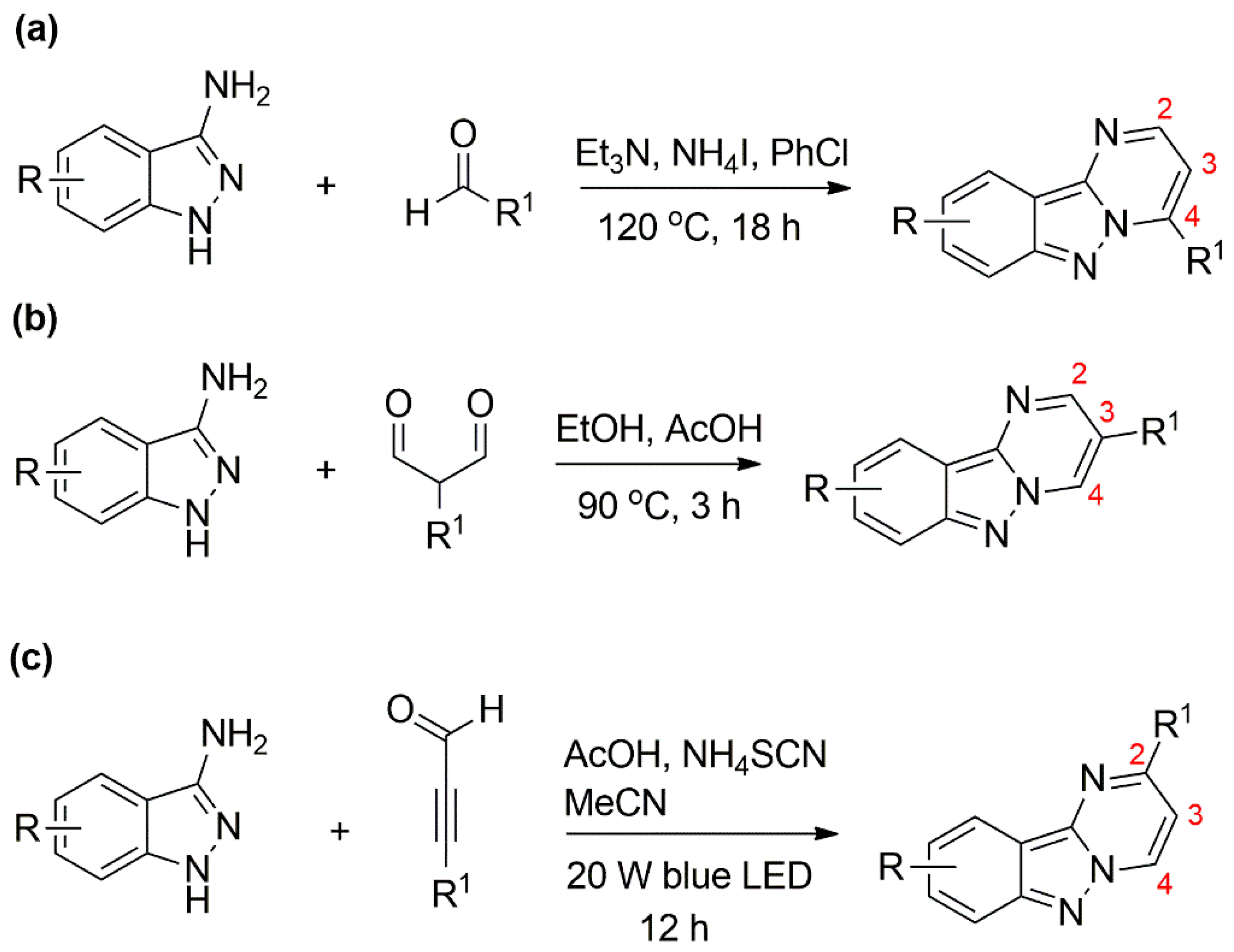
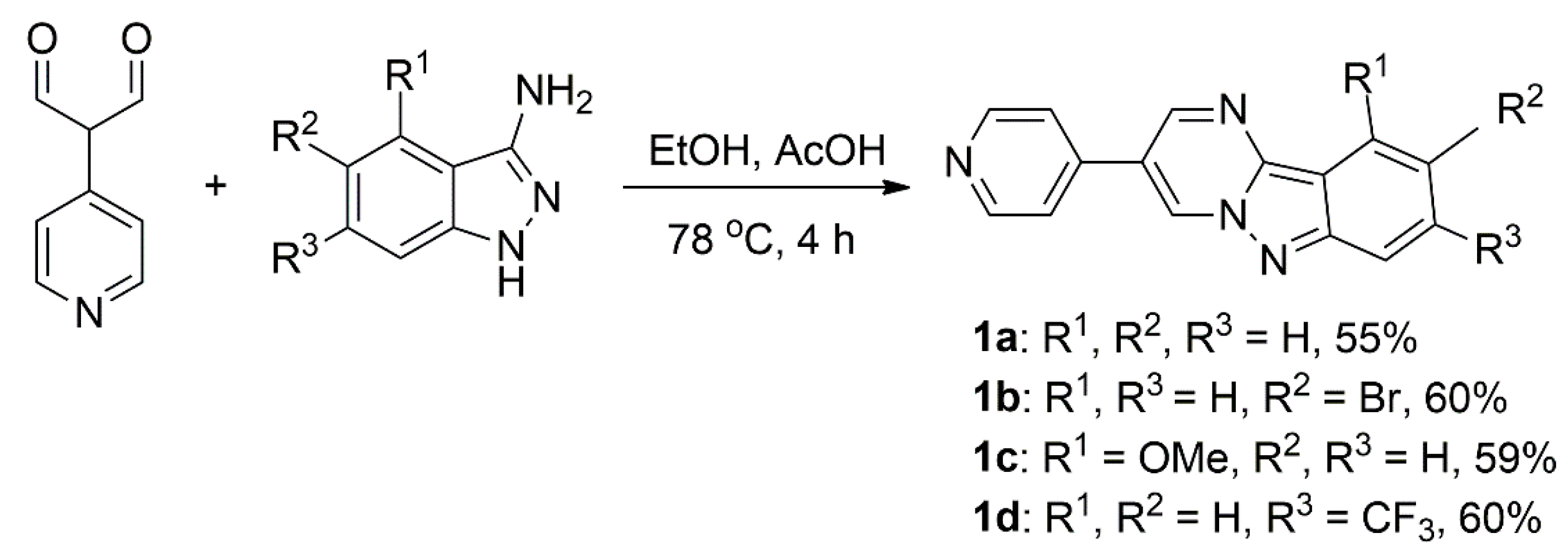
3.1. Synthesis and Characterisation
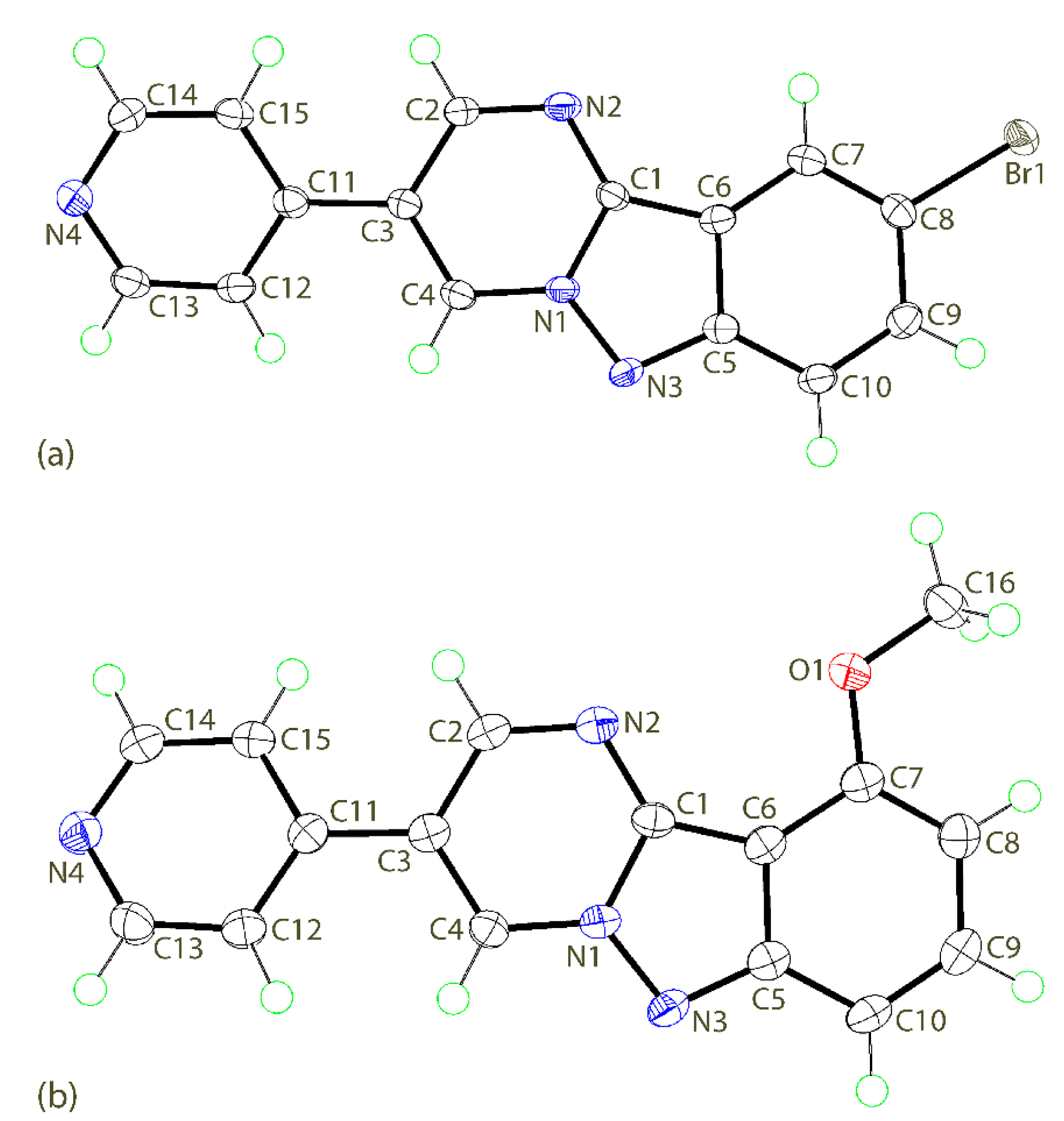
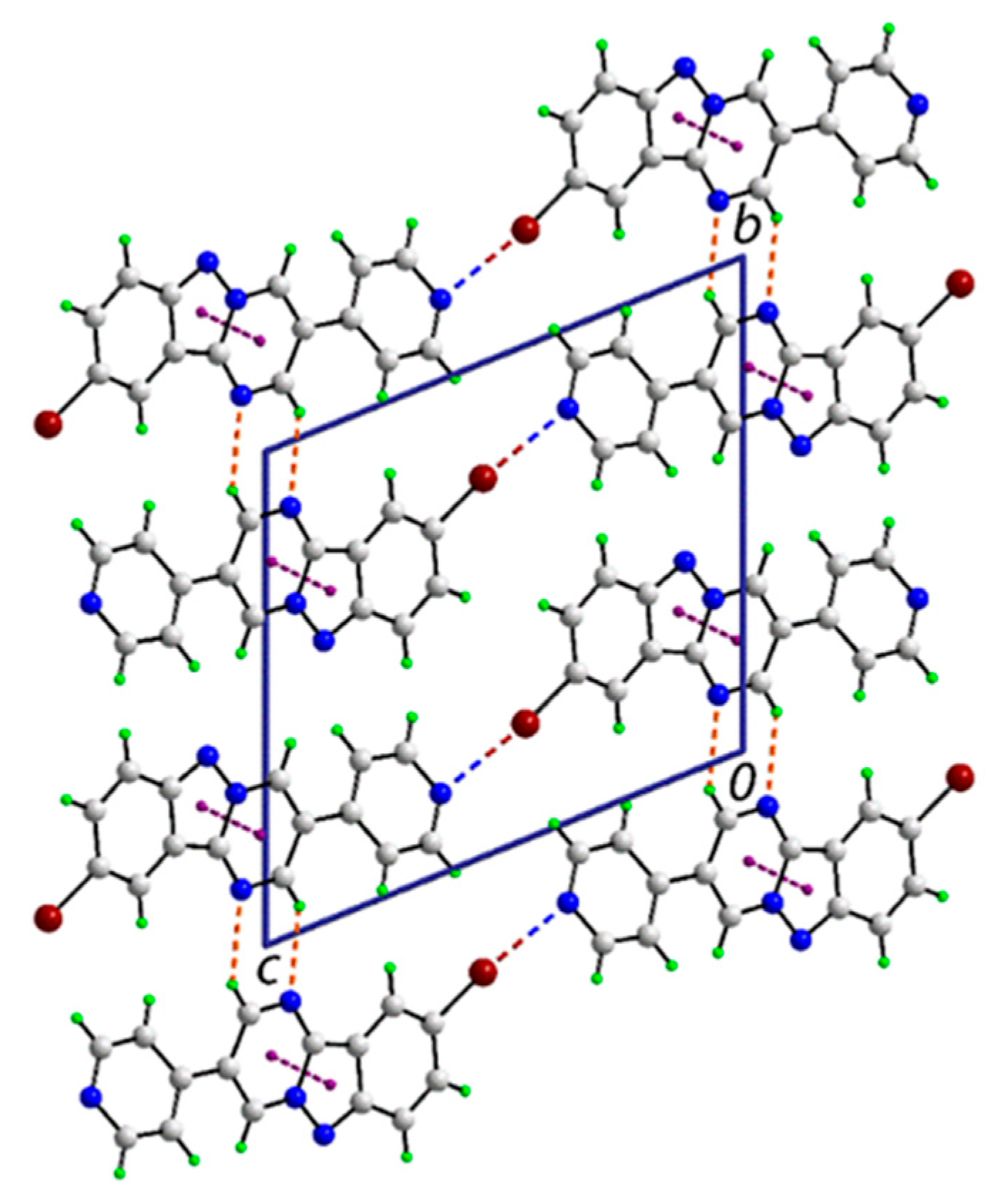
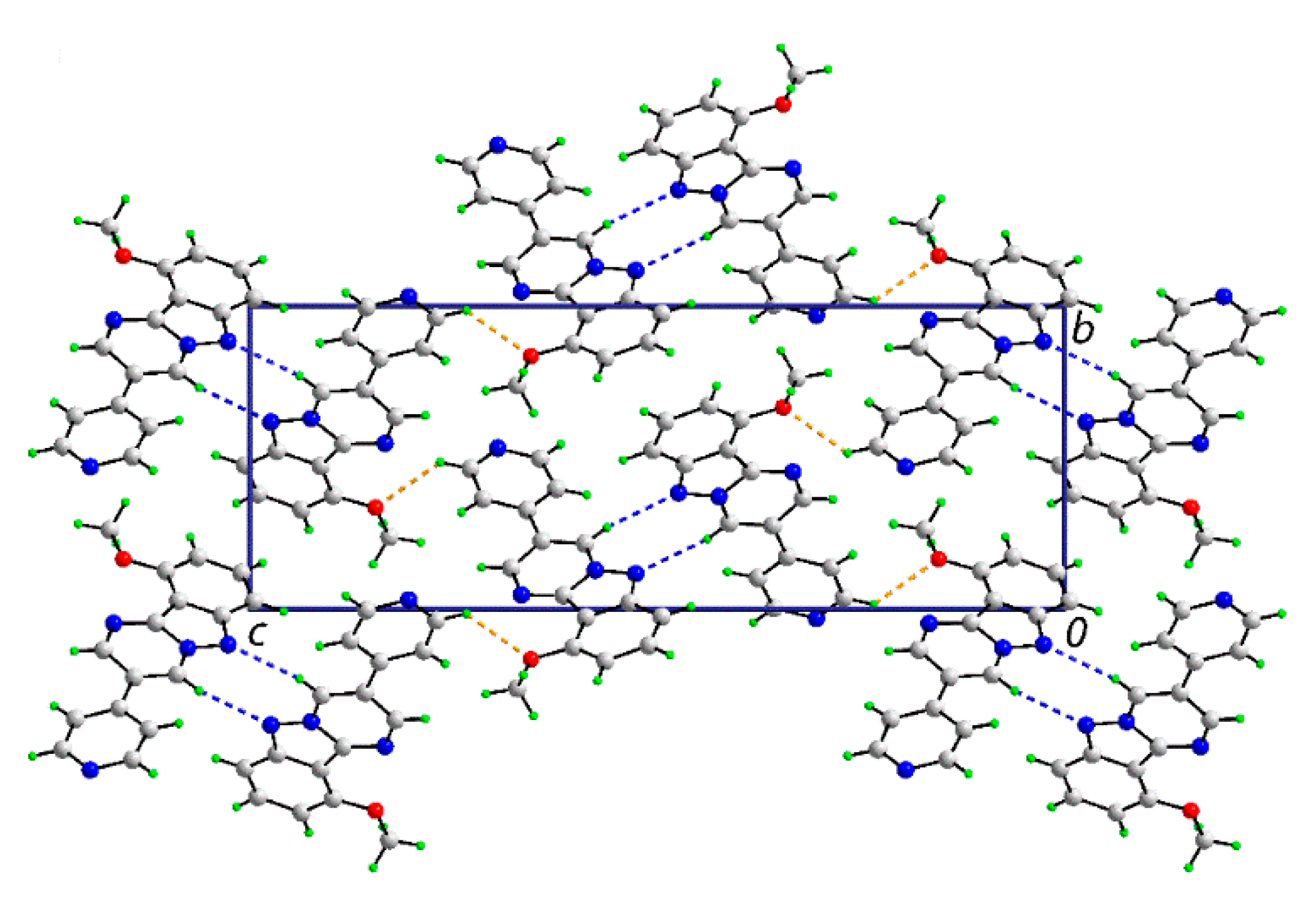
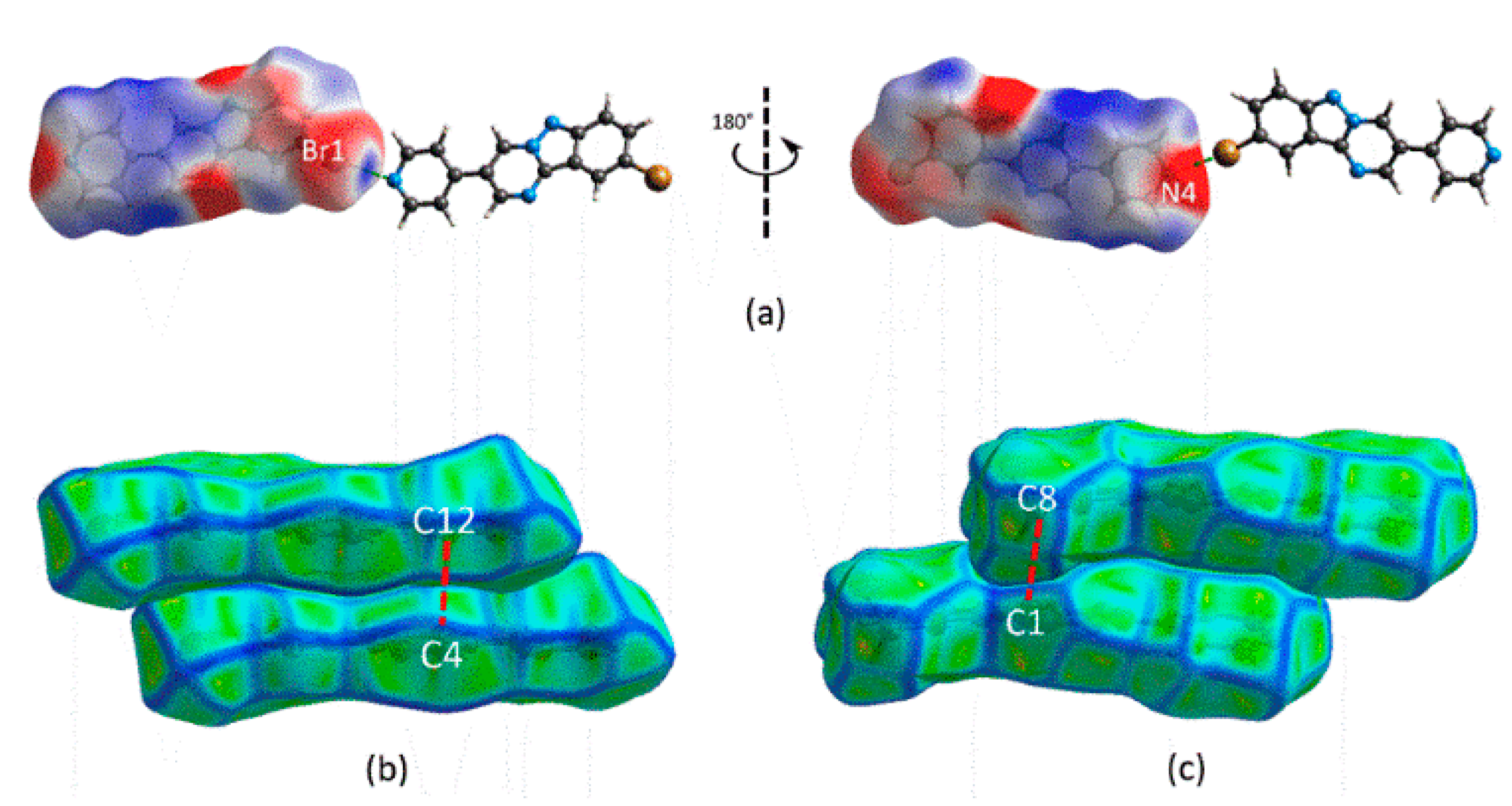
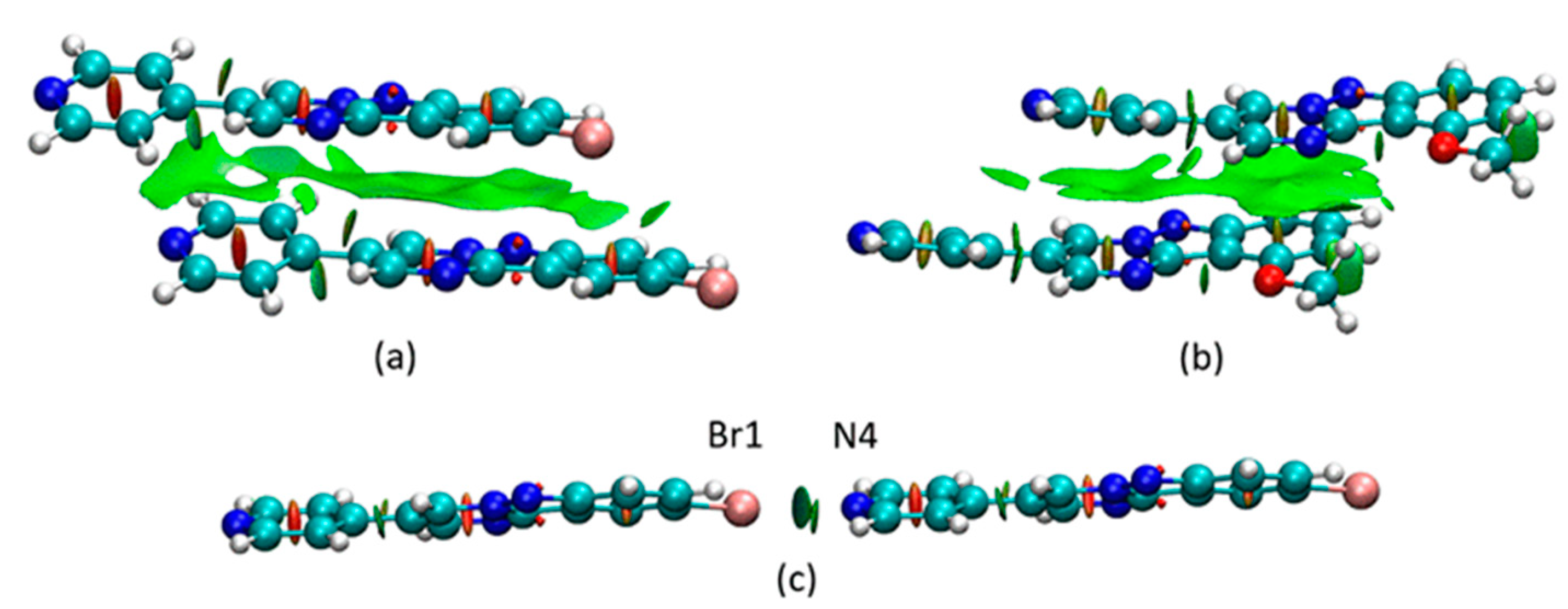
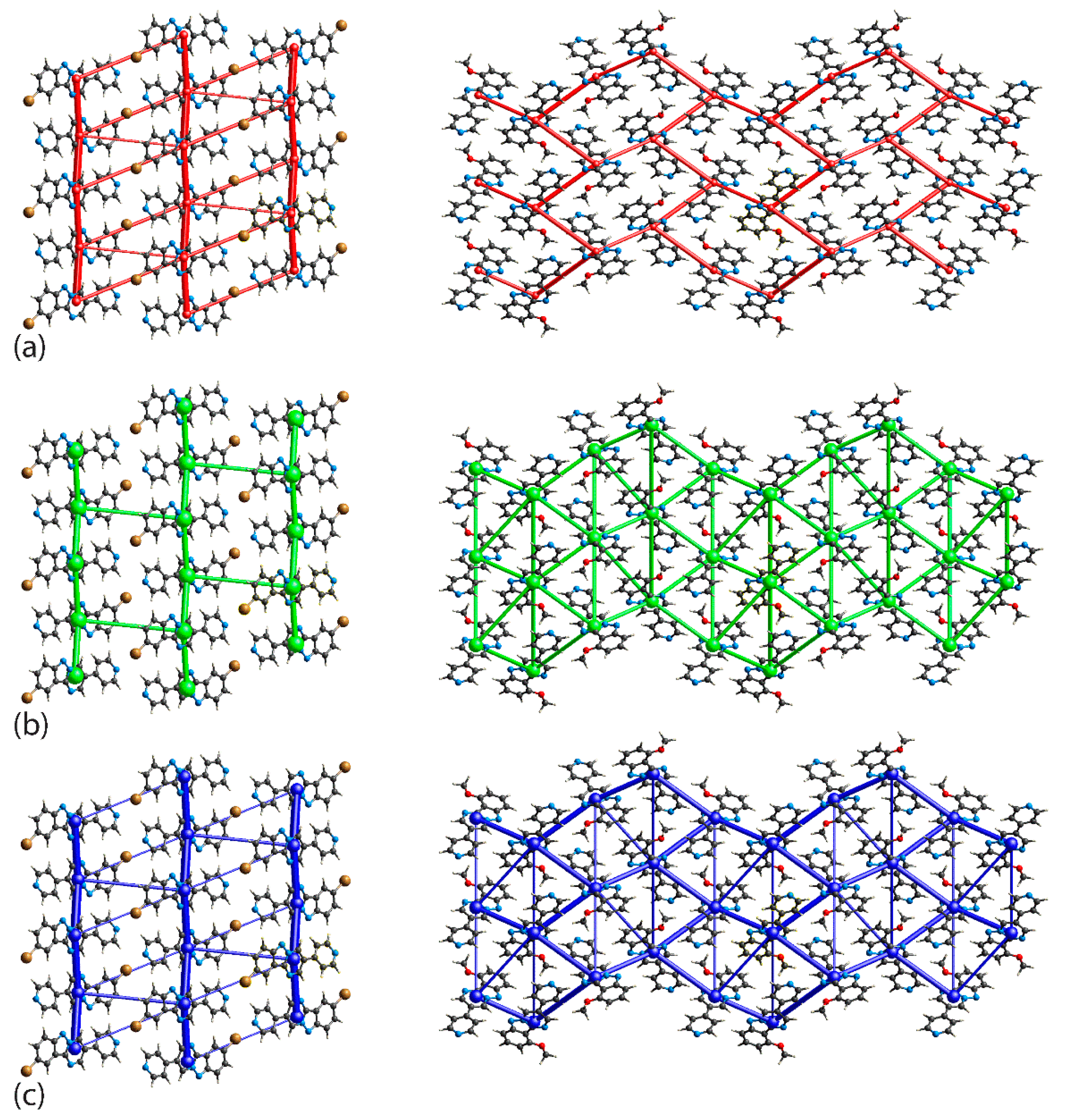
3.2. Crystal and Molecular Structures
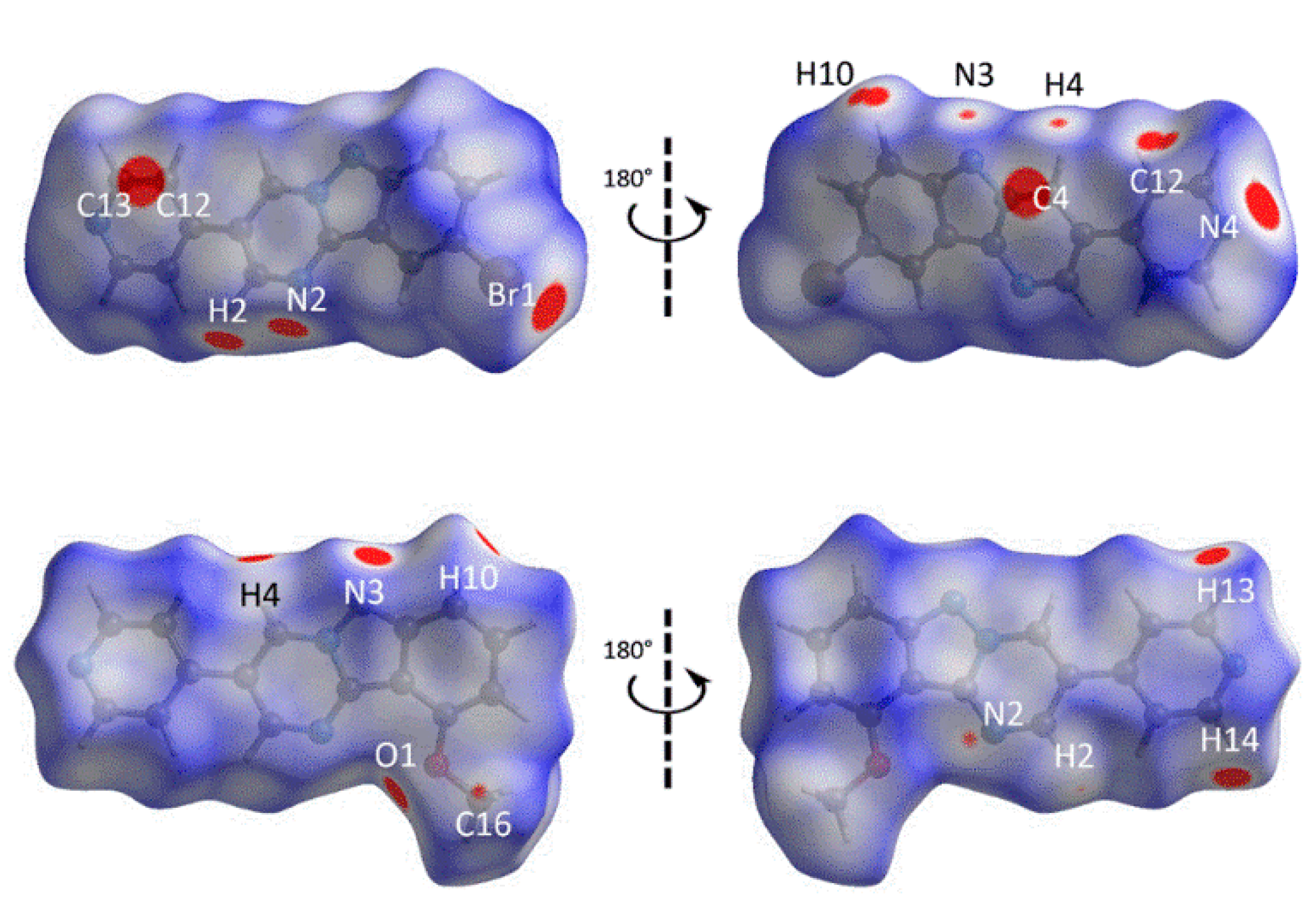
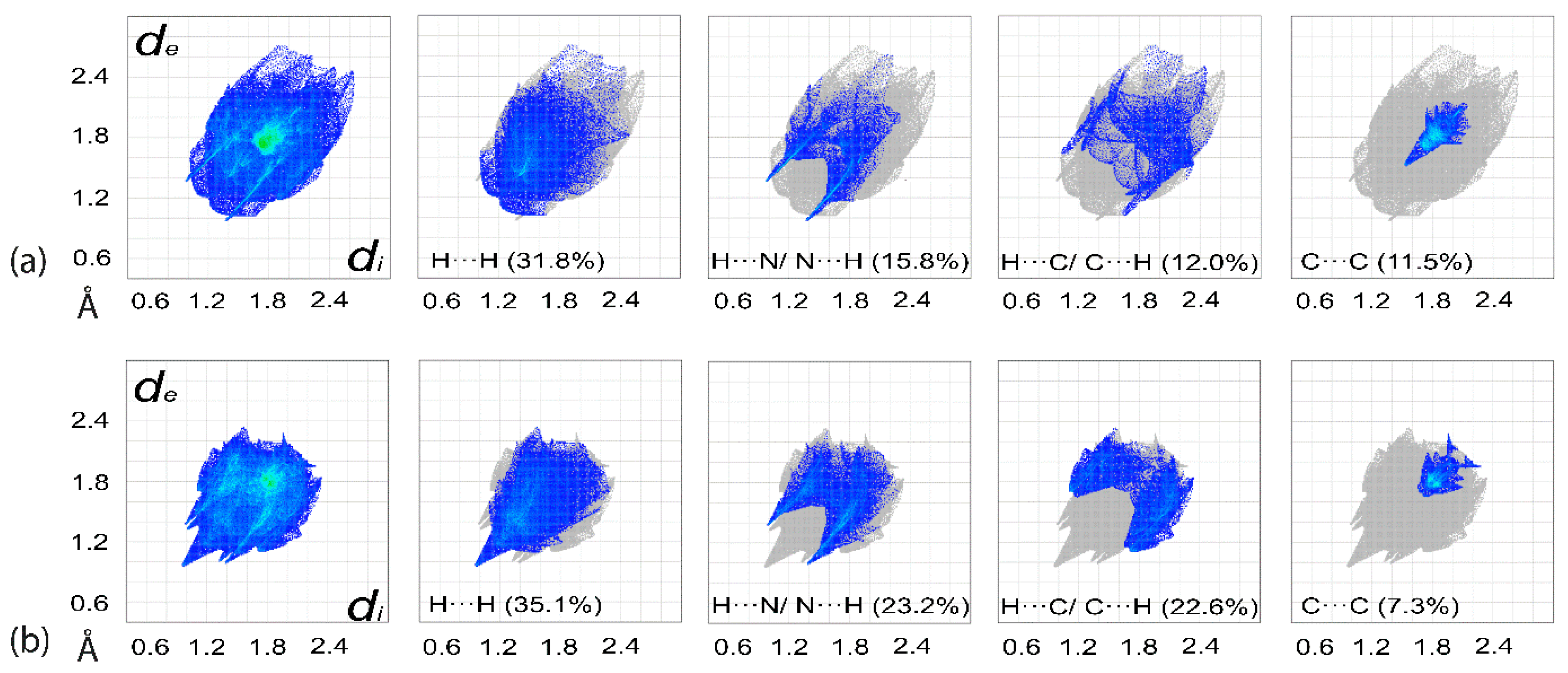
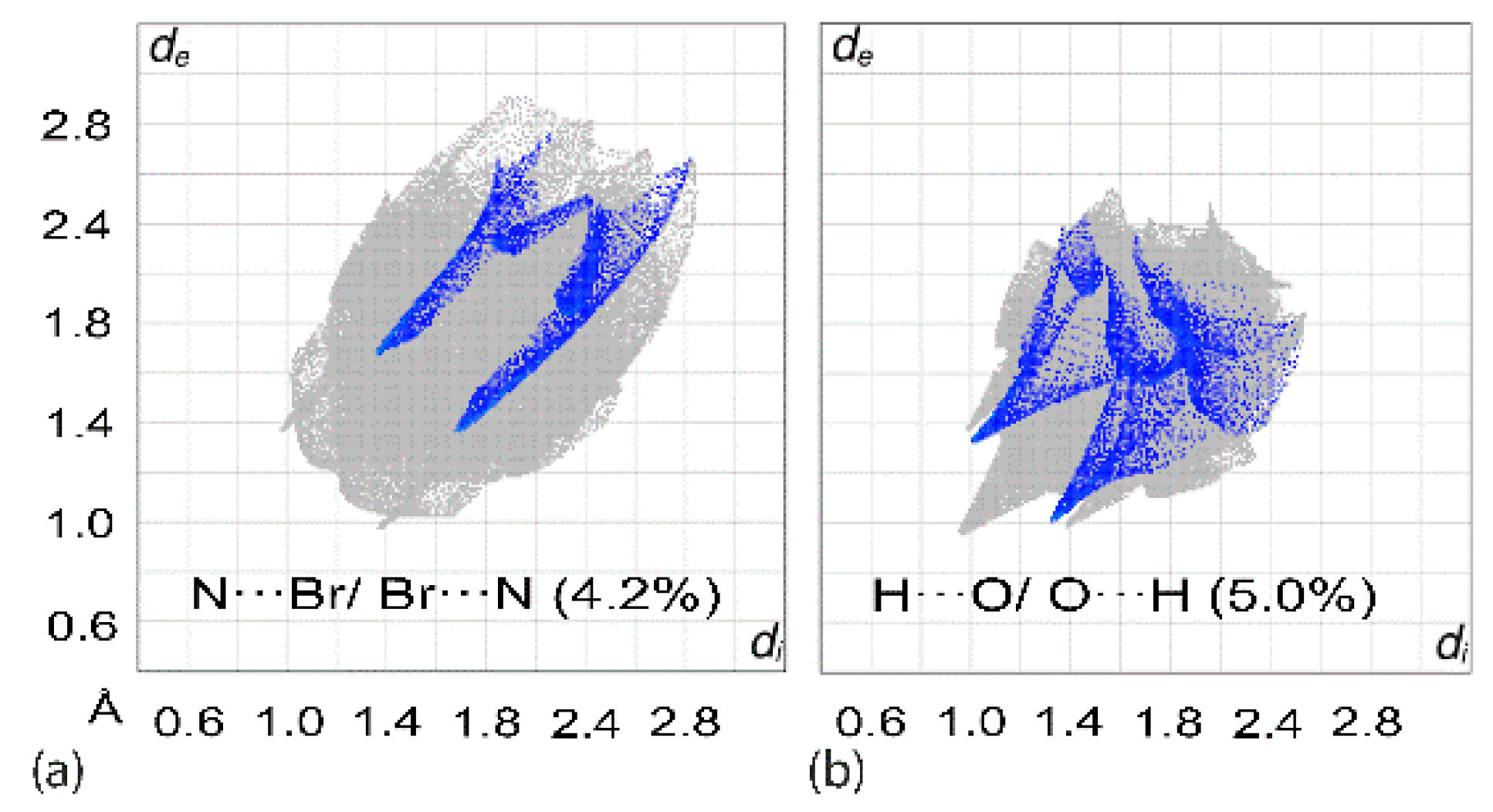
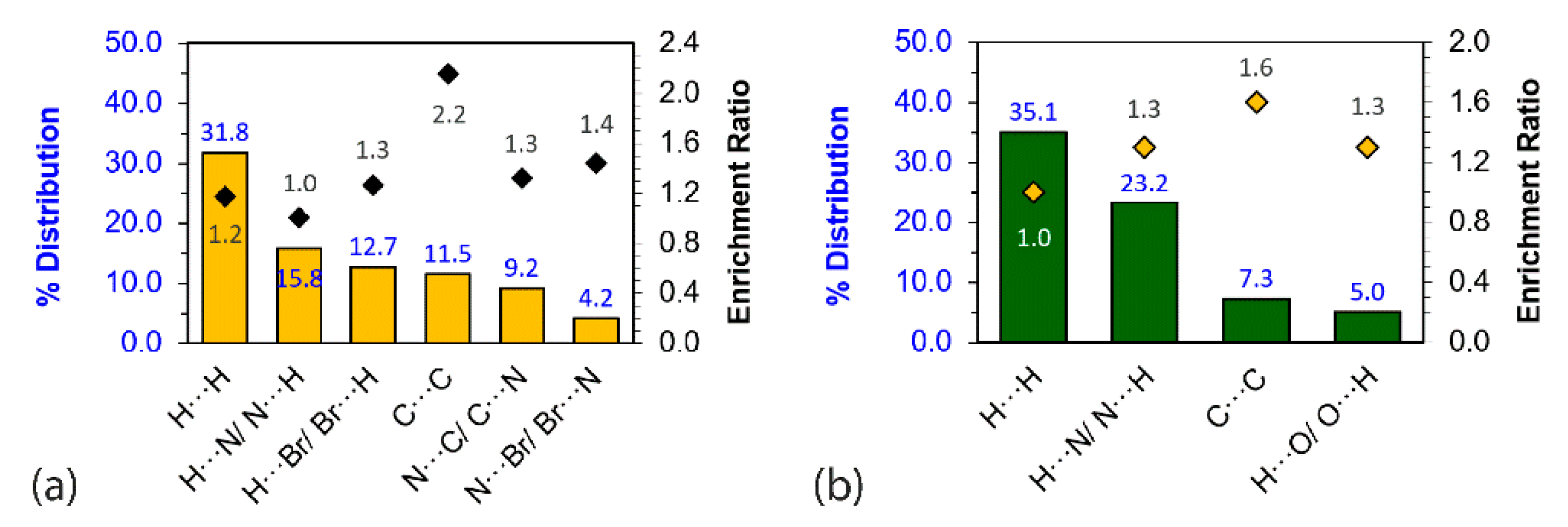
3.3. Computational Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balwe, S.G.; Jeong, Y.T. An approach towards the synthesis of novel fused nitrogen tricyclic heterocyclic scaffolds via GBB reaction. Org. Biomol. Chem. 2018, 16, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Han, X.; Tong, P.; Zhang, Z.; Shen, H.; Guo, Y.; Bai, S. Aerobic α,β-C(sp3)–H bond difunctionalization and C–N bond cleavage of triethylamine: Difunctional ammonium iodide enabling the regioselective synthesis of 4-arylpyrimido[1,2-b]indazoles. Org. Lett. 2019, 21, 6074–6078. [Google Scholar] [CrossRef] [PubMed]
- Yakaiah, T.; Kurumurthy, C.; Lingaiah, B.P.V.; Narsaiah, B.; Pamanji, R.; Velatooru, L.R.; Rao, J.V.; Gururaj, S.; Parthasarathy, T.; Sridhar, B. GdCl3 promoted synthesis of novel pyrimidine fused indazole derivatives and their anticancer activity. Med. Chem. Res. 2012, 21, 4261–4273. [Google Scholar] [CrossRef]
- Bharate, S.B.; Mahajan, T.R.; Gole, Y.R.; Nambiar, M.; Matan, T.T.; Kulkarni-Almeida, A.; Balachandran, S.; Junjappa, H.; Balakrishnan, A.; Vishwakarma, R.A. Synthesis and evaluation of pyrazolo[3,4-b]pyridines and its structural analogues as TNF-α and IL-6 inhibitors. Bioorg. Med. Chem. 2008, 16, 7167–7176. [Google Scholar] [CrossRef] [PubMed]
- Jismy, B.; El Qami, A.; Pišlar, A.; Frlan, R.; Kos, J.; Gobec, S.; Knez, D.; Abarbri, M. Pyrimido[1,2-b]indazole derivatives: Selective inhibitors of human monoamine oxidase B with neuroprotective activity. Eur. J. Med. Chem. 2021, 209, 112911. [Google Scholar] [CrossRef] [PubMed]
- Chino, A.; Seo, R.; Amano, Y.; Namatame, I.; Hamaguchi, W.; Honbou, K.; Mihara, T.; Yamazaki, M.; Tomishima, M.; Masuda, N. Fragment-based discovery of pyrimido[1,2-b] indazole PDE10A inhibitors. Chem. Pharm. Bull. 2018, 66, 286–294. [Google Scholar] [CrossRef]
- Han, X.; Pin, S.S.; Burris, K.; Fung, L.K.; Huang, S.; Taber, M.T.; Zhang, J.; Dubowchik, G.M. Synthesis and structure–activity relationship of imidazo[1,2-a]benzimidazoles as corticotropin-releasing factor 1 receptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 4029–4032. [Google Scholar] [CrossRef] [PubMed]
- Ndakala, A.J.; Gessner, R.K.; Gitari, P.W.; October, N.; White, K.L.; Hudson, A.; Fakorede, F.; Shackleford, D.M.; Kaiser, M.; Yeates, C.; et al. Antimalarial Pyrido[1,2-a]benzimidazoles. J. Med. Chem. 2011, 54, 4581–4589. [Google Scholar] [CrossRef]
- Yakaiah, T.; Lingaiah, B.P.V.; Narsaiah, B.; Kumar, K.P.; Murthy, U.S.N. GdCl3 catalysed Grieco condensation: A facile approach for the synthesis of novel pyrimidine and annulated pyrimidine fused indazole derivatives in single pot under mild conditions and their anti-microbial activity. Eur. J. Med. Chem. 2008, 43, 341–347. [Google Scholar] [CrossRef]
- Li, L.; Xu, H.; Dai, L.; Xi, J.; Gao, L.; Rong, L. An efficient metal-free cascade process for the synthesis of 4-arylpyrimido[1,2-b]indazole-3-carbonitrile derivatives. Tetrahedron 2017, 73, 5358–5365. [Google Scholar] [CrossRef]
- Palaniraja, J.; Roopan, S.M.; Rayalu, G.M. One-pot synthesis of highly functionalized pyrimido[1,2-b]indazoles via 6-endo-dig cyclization. RSC Adv. 2016, 6, 24610–24616. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, J.; Lin, J.; Zhang, Z.; Wu, S.; He, Q.; Cao, H. Controllable site-selective construction of 2- and 4-substituted pyrimido[1,2-b]indazole from 3-aminoindazoles and ynals. J. Org. Chem. 2021, 86, 9107–9116. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.M.; Balwe, S.G.; Lim, K.T.; Jeong, Y.T. A novel three-component method for the synthesis of spiro[chromeno [4′,3′:4,5] pyrimido[1,2-b] indazole-7,3′-indoline]-2′,6(9H)-dione. Tetrahedron 2017, 73, 2806–2813. [Google Scholar] [CrossRef]
- Jadhav, A.M.; Balwe, S.G.; Jeong, Y.T. An efficient protocol for synthesis of novel polyheterocyclic chromeno pyrimido[1,2-b]indazolone derivatives using [Et3NH][HSO4] as a reusable catalyst under solvent-free conditions. Tetrahedron Lett. 2019, 60, 151251. [Google Scholar] [CrossRef]
- Zhou, Y.; Lou, Y.; Wang, Y.; Song, Q. Oxidant-controlled divergent transformations of 3-aminoindazoles for the synthesis of pyrimido[1,2-b]-indazoles and aromatic nitrile-derived dithioacetals. Org. Chem. Front. 2019, 6, 3355–3359. [Google Scholar] [CrossRef]
- Balwe, S.G.; Shinde, V.V.; Rokade, A.A.; Park, S.S.; Jeong, Y.T. Green synthesis and characterization of silver nanoparticles (Ag NPs) from extract of plant Radix Puerariae: An efficient and recyclable catalyst for the construction of pyrimido[1,2-b]indazole derivatives under solvent-free conditions. Catal. Commun. 2017, 99, 121–126. [Google Scholar] [CrossRef]
- Kong, W.; Zhou, Y.; Song, Q. Lewis-acid promoted chemoselective condensation of 2-aminobenzimidazoles or 3-aminoindazoles with 3-ethoxycyclobutanones to construct fused nitrogen heterocycles. Adv. Synth. Catal. 2018, 360, 1943–1948. [Google Scholar] [CrossRef]
- Palaniraja, J.; Roopan, S.M.; Rayalu, G.M.; Al-Dhabi, N.A.; Arasu, M.V. A metal-free regioselective multicomponent approach for the synthesis of free radical scavenging pyrimido-fused indazoles and their fluorescence studies. Molecules 2016, 21, 1571. [Google Scholar] [CrossRef]
- Annareddygari, S.; Kasireddy, V.R.; Reddy, J. Transition-metal-free N-arylation: A general approach to aza-fused poly-heteroaromatics. J. Heterocycl. Chem. 2019, 56, 3267–3276. [Google Scholar] [CrossRef]
- Volovenko, Y.M.; Chuiguk, V.A. Pyrimido[1,2-b]indazoles from 3-aminoindazoles and β-diketones. Chem. Heterocycl. Compd. 1974, 10, 859–860. [Google Scholar] [CrossRef]
- Geng, X.; Xu, Z.; Cai, Y.; Wang, L. Visible-light-driven multicomponent cyclization by trapping a 1,3-vinylimine ion intermediate: A direct approach to pyrimido[1,2-b]indazole derivatives. Org. Lett. 2021, 23, 8343–8347. [Google Scholar] [CrossRef]
- Zhou, J.; Li, W.; Zheng, H.; Pei, Y.; Liu, X.; Cao, H. Visible light-induced cascade cyclization of 3-aminoindazoles, ynals, and chalcogens: Access to chalcogen-containing pyrimido[1,2-b]-indazoles. Org. Lett. 2021, 23, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Ramle, A.Q.; Fei, C.C.; Tiekink, E.R.T.; Basirun, W.J. Indoleninyl-substituted pyrimido[1,2-b]indazoles via a facile condensation reaction. RSC Adv. 2021, 11, 24647–24651. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction; CrysAlis PRO; Rigaku Oxford Diffraction: Oxfordshire, UK, 2017.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography; Springer: Berlin/Heidelberg, Germany, 2003; pp. 142–151. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ramle, A.Q.; Khaledi, H.; Hashim, A.H.; Mingsukang, M.A.; Mohd Arof, A.K.; Ali, H.M.; Basirun, W.J. Indolenine—dibenzotetraaza [14] annulene Ni (II) complexes as sensitizers for dye—sensitized solar cells. Dyes Pigm. 2019, 164, 112–118. [Google Scholar] [CrossRef]
- Sakurai, T.; Sundaralingam, M.; Jeffrey, G.A. A nuclear quadrupole resonance and x-ray study of crystal structure of 2,5-dichloroaniline. Acta Crystallogr. 1963, 16, 354–363. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen...halogen interactions—are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms. J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Interaction and polarization energy relationships in σ-hole and π-hole bond. Crystals 2020, 10, 76. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Cao, D. The Role of Shape Complementarity in the Protein-Protein Interactions. Sci. Rep. 2013, 3, 3271. [Google Scholar] [CrossRef]
- Lee, S.M.; Lo, K.M.; Tan, S.L.; Tiekink, E.R.T. (Tris{2-[(5-chloro-2-oxidobenzylidene-κO)amino-κN]ethyl}amine-κN)ytterbium(III): Crystal structure and Hirshfeld surface analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 1390–1395. [Google Scholar] [CrossRef] [Green Version]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1b | 1c |
|---|---|---|
| Formula | C15H9BrN4 | C16H12N4O |
| Molecular weight | 325.17 | 276.30 |
| Crystal size/mm3 | 0.03 × 0.04 × 0.34 | 0.03 × 0.07 × 0.31 |
| Colour | yellow | yellow |
| Crystal system | triclinic | orthorhombic |
| Space group | P | P212121 |
| a/Å | 4.44212(11) | 5.35980(8) |
| b/Å | 12.0067(2) | 9.27255(15) |
| c/Å | 12.4922(3) | 24.9428(4) |
| a/° | 112.154(2) | 90 |
| β/° | 90.006(2) | 90 |
| γ/° | 93.8346(19) | 90 |
| V/Å3 | 615.47(3) | 1239.63(3) |
| Z | 2 | 4 |
| Dc/g cm−3 | 1.755 | 1.480 |
| μ/mm−1 | 4.499 | 0.788 |
| Measured data | 15400 | 9411 |
| θ range/° | 3.8–67.7 | 3.5–67.7 |
| Unique data | 2531 | |
| Observed data (I ≥ 2.0σ(I)) | 2420 | 2476 |
| No. parameters | 181 | 192 |
| R, obs. data; all data | 0.027; 0.028 | 0.028; 0.029 |
| a; b in weighting scheme | 0.041; 0.400 | 0.044; 0.250 |
| Rw, obs. data; all data | 0.069; 0.070 | 0.074; 0.075 |
| Range of residual electron density peaks/eÅ−3 | −0.77–0.43 | −0.19–0.212 |
| A | H | B | H···B | A···B | A–H···B | Symmetry Operation |
|---|---|---|---|---|---|---|
| 1b | ||||||
| C2 | H2 | N2 | 2.46 | 3.298(3) | 146 | 2 + x, y, 1 + z |
| C8 | Br1 | N4 | 1.895(2) | 3.0613(19) | 171.72(7) | 2 + x, y, 1 + z |
| Cg(N1,N3,C1,C5,C6) | Cg(N1,N2,C1–C4) | 3.4764(11) | 0.07(10) | 1 + x, y, z | ||
| C4 | C12 | 3.074(3) | 1 + x, y, z | |||
| 1c | ||||||
| C14 | H14 | O1 | 2.44 | 3.161(2) | 133 | 1 − x, ½ + y, 1½ − z |
| C4 | H4 | N3 | 2.51 | 3.435(2) | 163 | ½ + x, 1½ − y, 2 − z |
| Contact | Distance (Å) | ∑vdw(X···Y) | Symmetry Operation |
|---|---|---|---|
| 1b | |||
| Br1···N4 | 3.06 | 3.40 | 2 + x, y, 1 + z |
| N2···H2 | 2.36 | 2.64 | −1 − x, −y, −z |
| N3···H4 | 2.56 | 2.64 | −x, 1 − y, −z |
| C4···C12 | 3.07 | 3.40 | 1 + x, y, z |
| C12···H10 | 2.65 | 2.79 | −x, 1 − y, −z |
| C13···H10 | 2.67 | 2.79 | −x, 1 − y, −z |
| 1c | |||
| O1···H14 | 2.35 | 2.61 | 1 − x, −½ + y, 3/2 − z |
| N4···H2 | 2.61 | 2.64 | 2 − x, ½ + y, 3/2 − z |
| N3···H4 | 2.39 | 2.64 | −½ + x, 1½ − y, 2 − z |
| C13···H10 | 2.78 | 2.79 | 1½ + x, 1½ − y, 2 − z |
| C1···C8 | 3.40 | 3.40 | 1 + x, y, z |
| N2···H16c | 2.58 | 2.79 | 1 + x, y, z |
| Close Contact | Eelectrostatic | Epolarization | Edispersion | Erepulsion | Eint | Symmetry Operation |
|---|---|---|---|---|---|---|
| 1b | ||||||
| Cg2(C4)···Cg3(C12) | −39.9 | −2.9 | −91.4 | 103.3 | −60.1 | 1 + x, y, z |
| C4–H4···N3 (×2) + | −34.6 | −5.9 | −34.7 | 48.1 | −41.4 | −x, 1 − y, −z |
| C10–H10···C12 (×2) + | ||||||
| C10–H10···C13 (×2) | ||||||
| C2–H2···N2 (×2) | −28.3 | −5.0 | −30.2 | 42.3 | −33.8 | −1 − x, −y, −z |
| C8–Br1···N4 | −15.3 | −1.7 | −8.1 | 26.4 | −8.1 | 2 + x, y, 1 + z |
| 1c | ||||||
| Cg1(C1)···Cg4(C8) + | H14 | O1 | 2.44 | 3.161(2) | 133 | 1 − x, ½ + y, 1½ − z |
| C16–H16c···N2 | ||||||
| C14–H14···O1 | −19.3 | −4.6 | −19.5 | 26.1 | −24.6 | 1 − x, ½ + y, 1½−z |
| C4–H4···N3 | −14.7 | −4.2 | −19.7 | 22.1 | −22.2 | ½ + x, 1½ − y, 2 − z |
| C2–H2···N4 | −10.5 | −2.2 | −13.3 | 12.2 | −16.8 | 2 − x, ½ + y, 1½ − z |
| C10–H10···C13 | −3.3 | −1.0 | −11.4 | 14.4 | −5.3 | −1½ + x, 1½ − y, 2 − z |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramle, A.Q.; Tan, S.L.; Tiekink, E.R.T. Synthesis and Crystallographic Characterisation of Pyridyl- and Indoleninyl-Substituted Pyrimido[1,2-b]Indazoles. Crystals 2022, 12, 1283. https://doi.org/10.3390/cryst12091283
Ramle AQ, Tan SL, Tiekink ERT. Synthesis and Crystallographic Characterisation of Pyridyl- and Indoleninyl-Substituted Pyrimido[1,2-b]Indazoles. Crystals. 2022; 12(9):1283. https://doi.org/10.3390/cryst12091283
Chicago/Turabian StyleRamle, Abdul Qaiyum, Sang Loon Tan, and Edward R. T. Tiekink. 2022. "Synthesis and Crystallographic Characterisation of Pyridyl- and Indoleninyl-Substituted Pyrimido[1,2-b]Indazoles" Crystals 12, no. 9: 1283. https://doi.org/10.3390/cryst12091283
APA StyleRamle, A. Q., Tan, S. L., & Tiekink, E. R. T. (2022). Synthesis and Crystallographic Characterisation of Pyridyl- and Indoleninyl-Substituted Pyrimido[1,2-b]Indazoles. Crystals, 12(9), 1283. https://doi.org/10.3390/cryst12091283