Abstract
Carbon dioxide produced by human activities is one of the main contributions responsible for the greenhouse effect, which is modifying the Earth’s climate. Therefore, post-combustion CO2 capture and its conversion into high value-added chemicals are integral parts of today’s green industry. On the other hand, carbon dioxide is a ubiquitous, cheap, abundant, non-toxic, non-flammable and renewable C1 source. Among CO2 usages, this review aims to summarize and discuss the advances in the reaction of CO2, in the synthesis of cyclic carbonates, carbamates, and ureas appeared in the literature since 2017.
1. Introduction
The combustion of fossil fuels produces large amounts of waste gases. Among them, carbon dioxide is one of the main contributions responsible for the greenhouse effect, which is modifying the Earth’s climate [1]. Thus, post-combustion CO2 capture and its conversion into high value-added chemicals are integral parts of today’s green energy industry. In fact, the usage of carbon dioxide as a ubiquitous, cheap, abundant, non-toxic, non-flammable and renewable C1 source has great importance from the viewpoint of both environmental protection and resource utilization.
Carbon dioxide has two polar carbonyl bonds, but its linear shape makes it non-polar, thus it is a thermodynamically and kinetically relatively stable molecule. This feature represents the major obstacle for CO2 utilization, which still remains rather limited. The CO2 conversion mainly depends on efficient activation by appropriate catalysts, to enhance electrophilicity of the central carbon atom.
Over the past decade, the usage of CO2 as a building block to prepare valuable organic molecules has attracted increasing attention [2,3,4,5,6,7,8,9]. Even a journal completely devoted to CO2 utilization started to be published by Elsevier in 2013. Moreover, Poliakoff and Leitner proposed twelve principles of CO2 chemistry that are a set of criteria for assessing the viability of different reactions in which CO2 is the feedstock for making organic chemicals [10].
In addition to the carbon dioxide fixation [11,12], the classical carboxylation reaction [13,14,15,16,17], the reduction to carbon monoxide or formic acid [18,19,20,21], and polymerization [22,23,24], the preparation of useful heterocyclic carbonyl compounds such as cyclic carbonates, carbamates, and ureas attracted the interest of many research groups. Recently, an account has been published on the reaction of CO2 with alcohols and amines into carbonates, ureas, and carbamates in the presence of ceric oxide as the catalyst [25]. Another recent report described some porous catalysts for CO2 capture and conversions and aimed to be a guide to prepare new porous catalysts for the synthesis of cyclic carbonates [26]. Finally, a very recent review covered the synthetic strategies to cyclic urethanes from amines and CO2 and N-methylation, N-formylation from amines, CO2 and H2 [27].
Heterocyclic systems are widespread structures in natural products and artificial compounds (over 90% of new drugs contain at least one heterocyclic moiety). Heterocycles are also important building blocks for multistep synthesis. Therefore, the development of new methods for their synthesis is always welcome in the chemical community, in particular if these methods benefit from cheap, simple, and readily available starting materials. Notwithstanding the strong effort of Vessally [28,29,30,31,32,33] and others [34,35,36,37] to review large part of the synthesis of heterocycles by CO2 fixation, many papers appeared in the literature in the last years, making an update necessary. Therefore, this review aims to summarize and discuss the recent advances in the reaction of CO2, for the synthesis of cyclic carbonates, carbamates, and ureas.
The synthesis of these heterocycles can occur by two main mechanisms on molecules carrying a nucleophilic and an electrophilic site:
- The nucleophilic site (eventually enforced by a base) can add to the carbon atom, to form the corresponding carboxylate, which in turn adds at the electrophilic site to close the cycle (Scheme 1, Equation (1)).
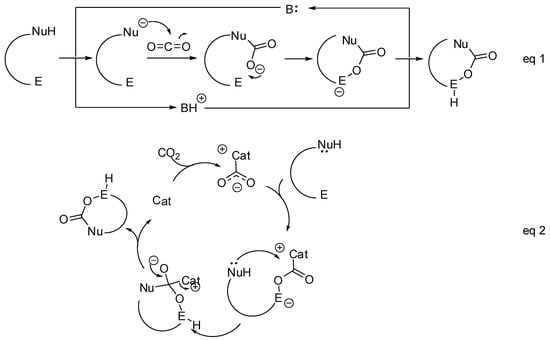 Scheme 1. General mechanistic survey.
Scheme 1. General mechanistic survey. - A catalyst is able to fix the carbon dioxide leading to a zwitterionic species. The carboxylate moiety of this activated species attacks an electrophilic site of the substrate molecule. Then the nucleophilic site closes the cycle, releasing the catalyst (Scheme 1, Equation (2)). N-heterocyclic carbenes (NHCs), N-heterocyclic olefins (NHOs), phosphorus ylides, polyoxometalates (POMs), ionic liquids (ILs), frustrated Lewis pairs (FLPs), metal-organic frameworks (MOFs) or superbases have been used for this scope.
2. Cyclic Carbonates
Cyclic carbonates find applications as electrolytes in lithium ion batteries in pharmaceuticals, products for agriculture, and as starting materials for polycarbonates [38,39,40]. Moreover, propylene carbonate is considered as a green polar aprotic solvent, because it has high boiling and flash points, low odor level and toxicity and biodegradability. The production of cyclic carbonates from CO2 is fully atom-economical and uses a safe gas, instead of toxic gases such as phosgene.
The reaction of carbon dioxide with epoxides [41,42,43,44,45] as well as with propargyl alcohols [2] has been widely reviewed in the past years. However, in the last two years a large production of research articles on this topic has appeared in the literature and they are summarized in Table 1. The generally accepted mechanism for the reaction of epoxides and carbon dioxide described in Table 1 is depicted in Scheme 2.

Table 1.
Catalysts for the reaction of epoxides and carbon dioxide (R and R1 see Scheme 2).
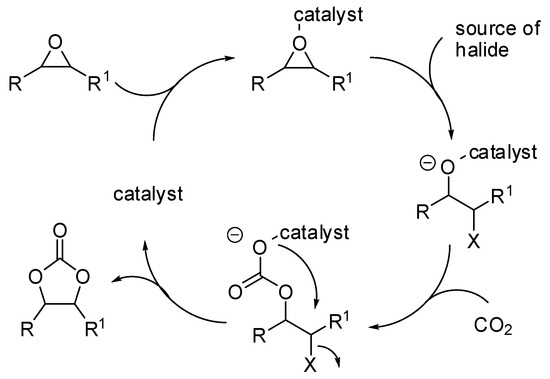
Scheme 2.
The mechanism of the addition of CO2 to epoxides.
In this mechanism, the rate determining step is the epoxide ring opening, and the nucleophilicity of the generated alkoxide species becomes significant for the attack at CO2. This pathway involves two consecutive Sn2 steps, thus the configuration is generally maintained with enantiopure epoxides. However, styrene oxide, as well as other epoxides giving stable carbocations, often gave partial racemization, which is accounted for a partial Sn1 mechanism. It should be noted that the scorpionate catalyst (entry 43) was rationally designed by authors on the basis of the mechanism and combining experimental and computational efforts [84].
A comparative study on the activity of a series of fifteen aluminum-based complexes has been recently reported, providing a useful comparison of activity metrics and explaining what are the most important features of the catalyst for cyclic organic carbonate formation [118].
Another challenging task was the research of organocatalysts as efficient as metal catalysts. In these cases, the intervention of hydrogen bond donors in the catalyst is often necessary to activate the ring opening step (see entries 24, 25, 27, 29, 34, 35, 37, 38, 39, 40, 44, 45, 46, 62, 66, 69, 70). Very recently, the activity of a diverse selection of hydrogen bond donors has been correlated to their pKa [119]. It was found that hydroxyl protons with Brønsted acidity in the range 9 < pKa < 11 gave the best catalytic performance, therefore phenol and ascorbic acid derivatives are ideal for cycloaddition reaction of epoxides and CO2. Density functional theory (DFT) calculations supported this hypothesis, low energy barriers have been calculated for the reaction catalyzed by phenols and the occurrence of aggregation between molecules of ascorbic acid further lowers the energy barriers increasing the catalytic activity. The halide source can be external (e.g., an ammonium halide salt) or internal (e.g., the counterion of the metal ion or of salt moieties in the catalyst). When no halide source is present as co-catalyst or catalyst counterion, a labile nucleophilic moiety has to be anyway present, for instance a tertiary amine (entries 17, 22, 26, 29, 35, 41, 44, 45, 56, 68, 69, 70, 75) or the carboxylate ions in histidine (entry 28) or aspartate (entry 33) catalysts.
A quite different mechanism was proposed by Liu, Wei, Dai and co-workers, whose paper is mentioned in entries 44–45 [85]. On the basis of their results, of some experiments with modified catalysts and DFT calculations they proposed the mechanism depicted in Scheme 3.
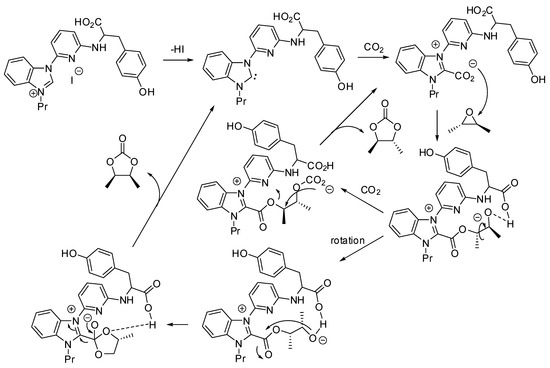
Scheme 3.
Mechanism proposed for cooperative multifunctional organocatalysts.
Conditions set up by Werner and co-workers (entry 11) [55] allowed the selective reaction of the monosubstituted oxirane in the presence of a di-substituted one. In fact, 3-(oxiran-2-yl)-7-oxabicyclo[4.1.0]heptane gave only the monocarbonate at the external epoxide moiety in 92% yield at 23 °C, and the bis-carbonate in 67% yield at 45 °C.
Moreover, the re-use of the catalyst is very important. Thus some simple catalysts were recovered by distillation or precipitation from the reaction mixture (entries 3, 6, 8, 15, 19, 26, 29, 32, 33, 35, 46), and many examples of heterogeneous catalysts have been reported (Table 1, third part). It should be particularly outlined the apparatus setup by Shi, Hu and co-workers (entry 73), who interweaved their catalyst in the stirring paddle, thus providing a very simple method to recover the catalyst [111]. In addition, the interweaved paddle could be reused more than twenty times, just increasing reaction times after the 20th time, in a gram scale reaction.
The coupling of internal epoxides and CO2 is generally more difficult for the steric hindrance of the two substituents. Thus reaction described in entries 10, 12, 13, 14, 25, 27, 31, 45 are particularly worthy of note for their good results.
In this regard, it should be mentioned the work of Kleij and co-workers, who were able to obtain trisubstituted cyclic organic carbonates with well-defined stereochemical configurations, starting from cyclic (5–8 membered) hydroxy epoxides and CO2 (Scheme 4) [120]. Syn/cis carbonates were always obtained only from addition of 25 mol% of NBu4Br. The anti/cis bicyclic carbonates instead required individual reaction conditions (different aluminum catalyst, halide source, solvent) and only five-, six-, and seven-membered cyclic epoxides reacted. Larger seven- and eight-membered cyclic epoxides could be converted to anti/trans carbonate in the presence of aluminum catalyst and dimethylaminopyridine (DMAP).
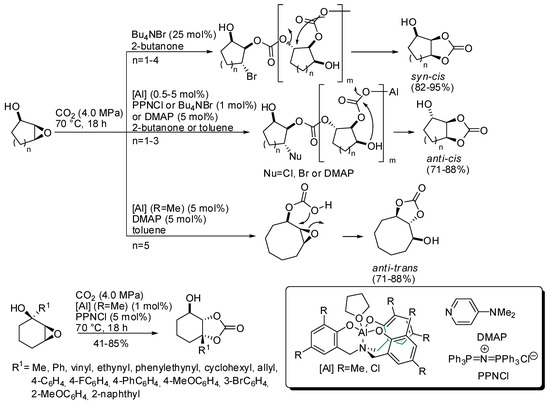
Scheme 4.
Preparation of highly substituted organic carbonates.
These different behaviors were attributed to different mechanisms of formation: syn/cis from carbonate polymer depolymerization, anti/cis from OH-assisted depolymerization (larger ring did not allow a conformation in which OH is near enough) and anti/trans from substrate-assisted activation of CO2. Then some different hydroxycyclohexene oxides were successfully transformed into anti/cis bicyclic carbonates. The reaction suffers from steric hindrance (ortho-substituted phenyl groups needed longer reaction times as 66 h, and afforded only 41% yield). Some products were then submitted for further modifications.
The style used by authors to present the results reported in Table 1 was quite different. Some were more oriented to the synthesis, other to the catalyst performance. Therefore it is not simple to compare results and efficiency at a glance. For example, turn over number (TON), a key to evaluating a catalyst, if a green, sustainable atom-economy is pursued, is often not reported. So we suggest that readers point their attention to yields, catalyst amount and CO2 pressure to evaluate the best catalyst among the eighty entries of Table 1. This difficulty in the choice of the best catalyst was also shown by D’Elia in his fine review, which showed also the harsh increase of documents in the last few years [45].
Kim and co-workers set up a divergent synthesis to cyclic carbonate and oxazolidinones in the coupling reactions between CO2 and epoxy alcohols or amines, respectively (Scheme 5) [121]. It is also worth noting that the starting materials are enantiopure and the reaction is completely enantiospecific. The divergent pathway is induced by the cocatalyst: tetrabutylammonium iodide triggers a mechanism superimposable with Scheme 2, while dimethylaminopyridine acts as a base on the amino group, which in turn attacks carbon dioxide. On the basis of this mechanism, it is clear that N-aryl groups highly influence the reactivity (electron-donating groups worked better than electron-withdrawing groups). Finally, the inversion of the stereochemistry, when oxazolidinones are prepared, allowed the synthesis of both enantiomers of 3-(arylamino)-propane-1,2-diols from a single enantiopure epoxyamine.
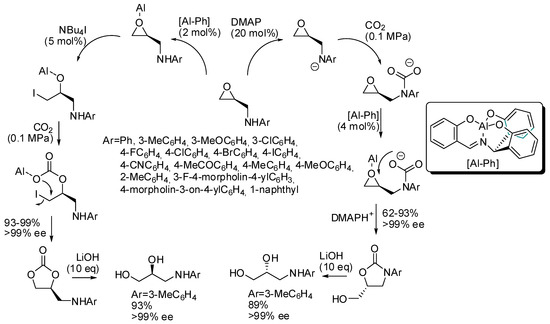
Scheme 5.
Stereodivergent synthesis starting from epoxyamines and CO2.
Reactions reported in Table 1 are general, although more or less efficient. There are also some papers that reported only one example of conversion of epoxides into carbonates. Two cases reported in Table 1 are the work of Verpoort on Zn-Co/ZIF (entry 50) [88], because it was then extended to other substrates (entry 51) [89]; and the work of Kleij and co-workers on bifunctional resorcinarenes [74], because they reported the only example for their catalyst on polymeric support (entry 60). Other catalysts have been proposed for CO2 addition to epoxides in which only one substrate was studied. For instance, Rownaghi and co-workers were able to permanently immobilize bromide on microstructured zirconium doped polyamide-imide hollow fibers crosslinked with 3-aminopropyltrimethoxysilane, followed by alkylation with 1,2-dibromoethane. This fiber acts as bifunctional catalysts for the cycloaddition of CO2 to styrene oxide (100% conversion of oxide and 98% selectivity of carbonate). This catalyst did not significantly leach ZrO2, amine, or bromine under the employed reaction conditions (120 °C, 8 h, 2.0 MPa) [122]. This work should be further developed to better compare these results with the catalytic and efficient fibers described in entry 73 [109].
The nitrogen doped ordered mesoporous polymers prepared by the Huang and Dai group showed excellent catalytic activity (conversion >95%) in the reaction of CO2 with propylene oxide (100 °C, 1.5 h, 1.2 MPa), when charged with zinc or cobalt ions [123].
The porous cationic polymers (5% wt.), obtained from condensation of tris(1,10-phenanthroline-5,6-dione)Ru(II) dichloride with ortho-aromatic amines in AlCl3 at 400 °C, allowed the conversion of propylene oxide to carbonate (almost quantitative conversion of oxide and exclusive selectivity toward carbonate, at 100 °C, 24 h, 0.1 MPa) [124].
Dufaud and co-workers presented a study on the binding strength of cavitand receptors of ammonium salts. Thus, tetraphosphonate showed a higher binding strength toward the ammonium cation. However, the presence of acidic phenol groups, which cooperatively activated the hexene oxide in a triphosphonate cavitand host, gave quantitative yields of cyclic carbonate at atmospheric pressure of CO2 and 100 °C. Moreover, Bu4NI turned out to be the most efficient halide donor [125].
Propylene carbonate was also obtained from propylene oxide at atmospheric CO2 pressure and at ambient temperature, by using various metal carbamates, such as Ti(O2CNEt2)4, Al(O2CNR2)3 (R=Et, i-Pr), Cu(O2CNEt2)2 and Sn(O2CNEt2)4, in combination with NBu4X (X=Br or Cl) as a cocatalyst [126]. With different catalyst and cocatalyst amounts, conversion up to 71% and selectivity up to >99% were reached. Solid catalysts were prepared from titanium and zirconium carbamates with amorphous silica, but these heterogeneous catalysts were less efficient than the homogeneous ones.
Graphitic carbon nitride (prepared with 1:1 urea–thiourea mixture) treated with aqueous H2SO4 produced a catalyst bifunctional acidic (–SO3H) and basic (–NH2) sites. The concentration of H2SO4 influenced the number of sites and the highest catalytic activity was reached with 60 wt. % acid. At 100 °C and 1.0 MPa CO2 pressure for 1 h, the synthesis of cyclic carbonates from epichlorohydrin (92.8% conversion, 99.2% selectivity) and propylene oxide (61.5% conversion, 99.3% selectivity), was obtained [127]. The catalyst was easily recovered and recycled six times with negligible loss of activity.
Seven alkanol amine catalysts derived from reactants containing two or three epoxy moieties and secondary amines were synthesized by Chung and co-workers [128]. Among them, bis(methylpiperazinyl)triol (1.4 mol%, at 1 MPa, 120 °C, 3 h) revealed the best catalyst in the formation of propylene carbonate (98% yield). Under milder conditions (5.6 mol% catalyst 0.5 MPa, 100 °C, 8 h) yield decreased to 90%. Comparing all the set of catalysts, the catalyst performance was affected by the number of hydroxyl and amine groups, and by the synergistic effects of these groups as demonstrated by DFT calculations of the stable conformational state. The proposed reaction mechanism is depicted in Scheme 6.
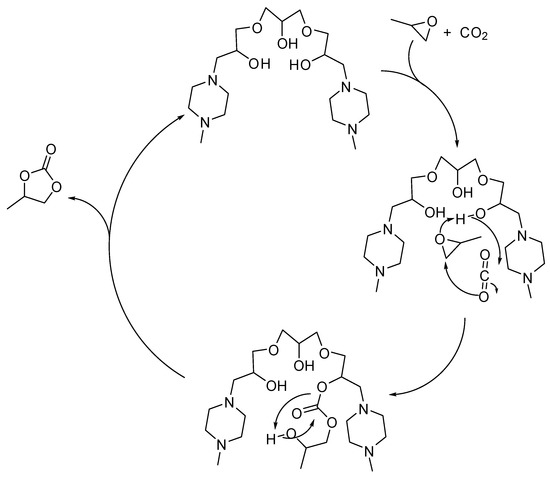
Scheme 6.
Plausible mechanism of the cycloaddition of CO2 to propylene oxide catalyzed by bis(methylpiperazinyl)triol.
The synthesis of 1,2-butylene carbonate from 1,2-butylene oxide was obtained in a selectivity of 76% and 64% yield at 135 °C and 7.5 MPa pressure of CO2 in 20 h in the absence of organic solvent with a ceria-lanthana-zirconia/graphene nanocomposite catalyst [129].
The merit of the synthesis of carbonates from internal epoxides has already been outlined above. Recently, polyethylene glycol (PEG-400) and KI (4 mol%) were found able to catalyze the reaction of methyl soyate epoxide to the corresponding carbonated fatty acid methyl esters in 99% yield at 120 °C with 3.0 MPa pressure of CO2 in 20 h. Longer reaction times (30 h) are requested to obtain the methyl linoleate tricarbonate [130]. However, for these catalysts further studies would be required to better understand their importance in this reaction.
The kinetic resolution of epoxides was employed to prepare enantioenriched epoxides [131]. Recently, Ema and co-workers reported the kinetic resolution of epoxides with CO2 by a chiral macrocyclic organocatalyst (catalyst-1, Scheme 7) [132]. The s factor {(ln[1-c(1+ee)])/(ln[1-c(1-ee)]), where c is the conversion} is satisfactory for disubstituted epoxides (9–13), but it is low for terminal epoxides (2.5–4.1). These values make this reaction very interesting for internal epoxides, taking also into account the trouble in performing the synthesis of 4,5-disubstituted 1,3-dioxolan-2-ones. The X-ray analysis showed multiple hydrogen-bonding sites for the enantioselective activation of epoxides into the chiral cavity of the catalyst. DFT calculations confirmed a classical mechanism as depicted in Scheme 2.
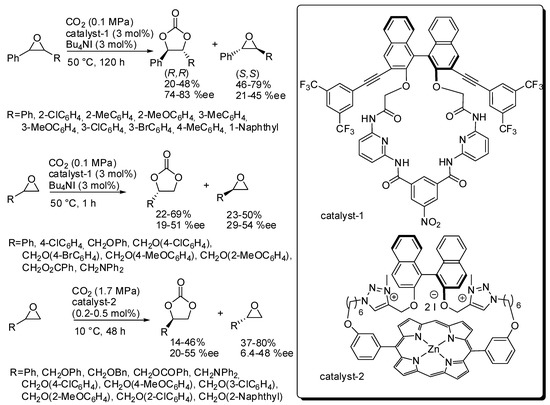
Scheme 7.
Kinetic resolution of epoxides with organocatalyst.
Later they reported another macrocyclic organocatalyst (catalyst-2, Scheme 7) but its efficiency was comparable for terminal epoxides (s = 1.7–5.0). However, with respect to catalyst-1, catalyst-2 showed some advantages, that are the low catalyst loading (only 0.2–0.5 mol%) and the opposite resolution of the epoxide and some drawbacks, that are higher CO2 pressure and longer reaction times [133].
On the other hand, Meggers and co-workers set up reaction conditions for the kinetic resolution of chiral terminal epoxides with s factors up to 16.6 [134]. They employed an iridium(III) complex (1 mol%) of iridium catalyst and 1.5 mol% of NEt4Br as the co-catalyst (Scheme 8). It should be noted that no polymerization side reaction is observed. Authors provided a mechanism in agreement with Scheme 2. While the decrease of enantiomeric excess with the progress in conversion was expected, the decrease of the s factor with increasing conversion was surprising, but authors did not give a satisfactory explanation for this phenomenon. Moreover, they did not also explain the different enantiomers obtained with simple alkyl epoxides.
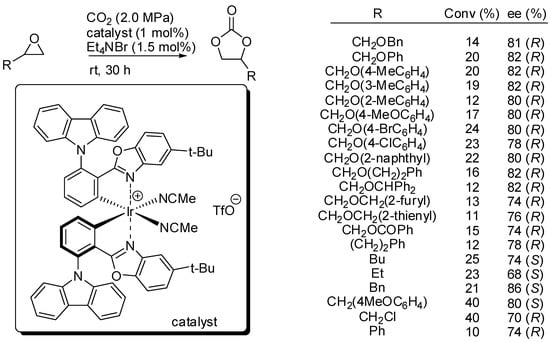
Scheme 8.
Kinetic resolution of epoxides with iridium catalyst.
The synthesis of six membered cyclic carbonates from the addition of CO2 to oxetanes is more difficult, because the four-membered ring is less reactive than epoxide and for the higher thermodynamic stability of the polymers with respect to the cyclic carbonates. Therefore, examples of this reaction are less common in the literature. In the last two years isolate examples are reported:
Tassaing, Jérôme et al. studied the influence of the main reaction parameters (organocatalytic mixture, pressure, and temperature) on the yield and the selectivity in the product distribution of the reaction of CO2 with oxetanes with an organocatalytic combination of ammonium salts and aromatic alcohols [135]. Cyclic carbonate was favored at lower temperatures, pressures, and conversions, as well as in the absence of alcohol. It should also be noted that substituted oxetanes were unreactive. They also investigated the mechanism of the synthesis of the six membered cyclic carbonate using DFT calculations performed at the M06-2X/6-311G(d,p) level. The mechanism is not far from that accepted for epoxides (Scheme 9).
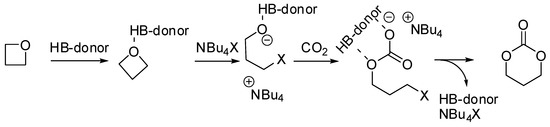
Scheme 9.
Mechanism of the reaction of oxetane with CO2.
Very recently, Dove and Coulembier found trimethylene carbonate as byproduct in the co-polymerization of oxetane and CO2 catalyzed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene and I2 [136]. However, trimethylene carbonate was the sole product, albeit in low yields (30% after 5 days) when 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis-[tris(dimethylamino)-phosphoranylidenamino]-2λ5,4λ5-catenadi(phosphazene) was used as a cocatalyst together with iodine. Authors affirmed that the strong complexation between the two catalysts was very likely the reason of this selectivity.
The cycloaddition of propargylic alcohols with CO2 is another manner to prepare five-membered cyclic carbonates. As for the addition to epoxides, this is also a 100% atom economical reaction. Moreover, the resulting α-alkylidene cyclic carbonates are important compounds with many applications in organic synthesis. The reaction has been widely studied in the past years and reviewed in the literatures cited in the introduction, thus here we restrict the description to the last two years again (Table 2). The reaction is generally catalyzed by a base which deprotonates the alcohol to start the catalytic cycle, as described in Scheme 10. Sometimes the alcoholate can open the cyclic carbonate leading to an oxoalkyl acyclic carbonate.
Table 2.
Catalysts for the reaction of propargyl alcohols and carbon dioxide (R, R1 and R2 see Scheme 10).
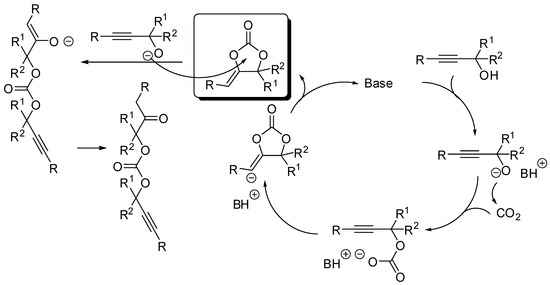
Scheme 10.
Mechanism of the cycloaddition of propargylic alcohols with CO2.
A detailed mechanistic investigation was recently performed by Tassaing’s research group for the conversion of 2-methyl-3-butyn-2-ol [142]. Authors found an influence of the temperature, pressure, solvent, and catalyst nature and loading on the rates and yields of the reaction. Moreover, in contrast to our previous results [143], they did not detect other secondary products. Finally they confirmed the mechanism depicted in Scheme 10 by DFT calculation at the M06-2X/6-311G(d, p) level. Then, the same research group tested a series of n-tetrabutylammonium organic salts as catalysts for the same reaction [144]. In particular, tetrabutylammonium acetate and azide allowed yields up to 98% in less than 10 h at 80 °C and 3.0 MPa under solvent-free conditions. Once more, they supported their data by kinetic studies and mechanism by DFT calculations.
The mechanism described in Scheme 10 involved, after cyclization a vinyl anion intermediate, which sequentially underwent protonation. However, this anion could be trapped by every electrophile. In fact, under palladium catalysis it was trapped by aryl halides [145]. The reaction suffered from steric factors. In fact, 2-chloro-1-iodobenzene and 1,2,4-trichloro-5-iodobenzene gave 45% and 6% yields, respectively. Iodomethane gave the corresponding product, albeit in only 31% yield. Among the propargyl alcohol tested, only 2-methyl-4-(pyridin-2-yl)but-3-yn-2-ol gave no reaction. The reaction exclusively gave the stereoisomer in which the Ar group arising from the aryl halide is located trans to the oxygen attached to the double bond. Authors after some control experiments proposed the mechanism depicted in Scheme 11.
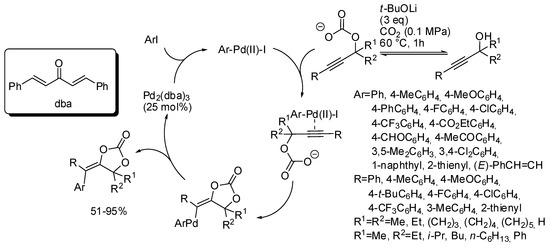
Scheme 11.
Arylcarboxylation of propargylic alcohols with CO2 and aryl halides.
The reaction of diols and CO2 under basic conditions is another method to obtain cyclic carbonates of different ring size. Ongoing their study on this reaction [146], Buchard and co-workers reported the synthesis of 5-, 6-, 7- and 8-membered cyclic carbonates using 2,2,6,6-tetramethylpiperidine as the base catalyst (Scheme 12, Equation (1)) [147]. As expected, larger cycles were recovered with increasing amounts of polymers.
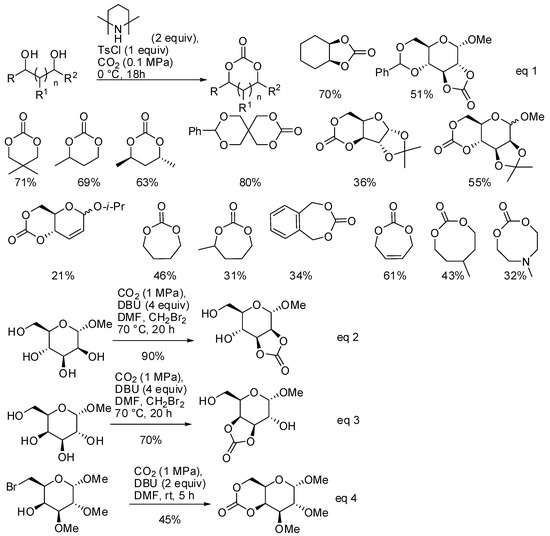
Scheme 12.
Synthesis of 5-, 6-, 7- and 8-membered cyclic carbonates.
From DFT calculations, authors suggested a mechanism in which CO2 is attacked by the alcoholate, then tosylation of the carbonate occurred and, finally, the second alcoholate moiety closed the cycle by addition/elimination process. It should be noted that sugar diols also reacted, which gave poor yields with the classical reaction with phosgene.
Another reaction of sugar diols and CO2 was introduced by Feng, Gnanou and co-workers. They already found that unprotected α-methyl D-glucopyranoside reacted with CO2, leading to water soluble oligoglycocarbonates [148]. Further studies from the same group showed that the reaction of CO2 with cis vicinal hydroxy groups of sugars afforded instead 5-membered bicyclic glycocarbonates, in particular in the cis-2,3 or cis-3,4 positions of methyl α-D-mannopyranoside and methyl α-D-galactopyranoside, respectively (Scheme 12, Equations (2)–(4)) [149]. Also, a six-membered bicyclic glycocarbonate was prepared from methyl 6-bromo-6-deoxy2,3-di-O-methyl α-D-galactopyranoside. The cyclic glycocarbonates were further allowed to react in good yield with amines or polymerized.
These two interesting procedures summarize two environmental benign features: the use of CO2 as starting material and the avoidance of phosgene or its derivatives
3. Cyclic Carbamates
Carbamates are widely employed in agricultural chemistry, medicinal chemistry, and polyurethane preparation. In particular, cyclic carbamates (2-oxazolidinones, 2-oxazinanones) are the core structures of many valuable drugs. For a long time phosgene was the classical C1 synthon for their synthesis; but its high toxicity has led researchers to explore greener C1 sources. Thus the use of CO2 has been gaining increasing importance.
Many strategies for CO2-based preparation of cyclic carbamates parallel those already seen above: for instance the cycloaddition to aziridines, oxetanes, or amino epoxides or the addition to alkenes, alkynes, and propargylic amines or alcohols. Most of these syntheses are already reviewed in the literature cited in the introduction, but in the last two years other papers appeared in the literature and they are collected in this section.
The synthesis of substituted 2-oxazolidinones by coupling of CO2 with aziridines is another reaction which allows the chemical fixation of CO2 with 100% atom efficiency. A first example has been reported in the previous section (Scheme 5) [106]. Other syntheses of substituted 2-oxazolidinones by coupling of CO2 with aziridines published in the last two years are collected in Table 3.
Table 3.
Catalysts for the reaction of aziridines and carbon dioxide (R, R1, and R2 see Scheme 13).
While regioselectivity is not a problem with epoxides, because the oxygen atoms are indistinguishable, the synthesis of oxazolidinones often gives mixture of regioisomers (Scheme 13). The most striking difference with the mechanism of epoxides is the higher nucleophilicity of the nitrogen, which give raise a zwitterionic intermediate.
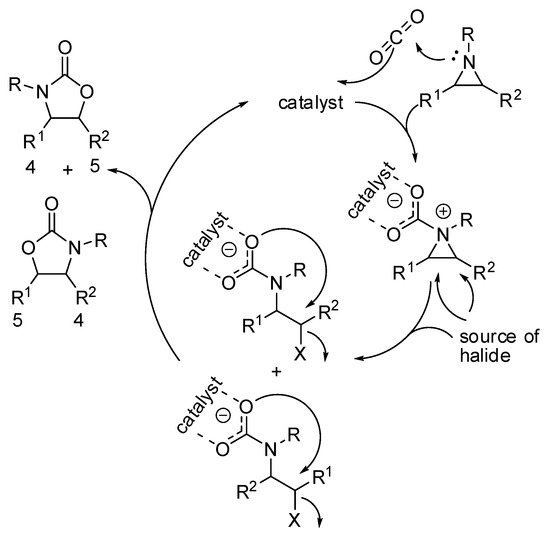
Scheme 13.
A simplified mechanism of the addition of CO2 to aziridines.
The positive charge on the nitrogen atom generally favors the attack of the halide source on the carbon which better carries the positive charge, leading to the five-substituted product from N,C-disubstituted aziridines. Recently, Pinhas and co-workers reported DFT calculations at the B3LYP/6-31+G(d,p) level of theory, kinetic studies and experiments in order to elucidate the mechanism [151]. Their calculations demonstrated that the reaction could not proceed at room temperature without a catalyst. Moreover, their data were unable to distinguish between two possible pathways involving either initial capture of CO2 or ring opening by X− from the catalyst. In fact, both mechanisms are influenced by polar solvents. The reaction is zero order with respect to aziridine, thus isolate aziridine is not involved in the rate determining step, which was the addition of CO2.
The synthesis of oxazolidin-2-ones from unsaturated amines by using CO2 has attracted much attention in the last years [152]. A study of the reaction mechanism with the ωB97XD functional theory was conducted by Yuan and co-workers under Ag(I) catalysis [153]. Authors discovered that the substrate is incorporated into the AgNO3 salt, followed by isomerization (Scheme 14). Then this intermediate attacks CO2 leading to a carbamate intermediate. The five-membered versus the six-membered ring closure has transition states the energy of which is influenced by the diazabicycloundecene (DBU) amount (an increasing of DBU amount increases the energy gap between the two transition states, favoring the oxazolidinone). Moreover, solvents with larger proton affinities also favor the formation of oxazolidinones.
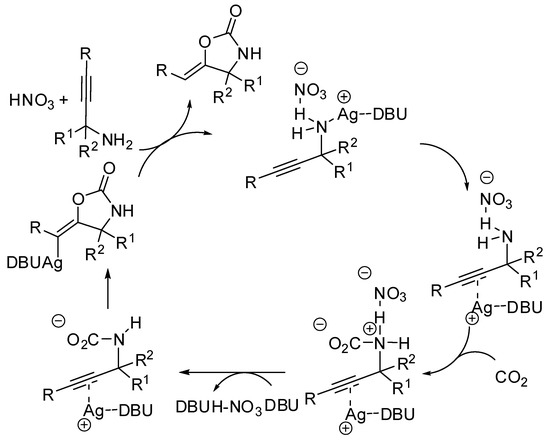
Scheme 14.
Mechanism of diazabicycloundecene (DBU)-AgNO3-catalyzed CO2 incorporation into propargylic amine.
The papers regarding propargylamines and published in the last two years are collected in Table 4.
Table 4.
Catalysts for the reaction of propargylamines and carbon dioxide (R, R1, R2 see Scheme 14, NR3 indicates the nitrogen substituent).
With the catalyst reported in entry 1, some homopropargylic amines were tested (Scheme 15) [134]. However, at 40 °C and with 5 mol% of catalyst loading only 36–37% yields were recovered, albeit the starting amine was completely consumed. Authors also observed that, in the absence of CO2, internal alkynes were unreactive, while terminal alkynes gave some side reactions. Thus, in order to improve yield, they tested a homopropargylamine with an internal (phenyl-substituted) alkyne moiety. However, CO2 was not incorporated and dehydropyrrole was favored over the carboxylative cyclization. The same reaction was observed with 2-alkynylaniline, which led to indole.
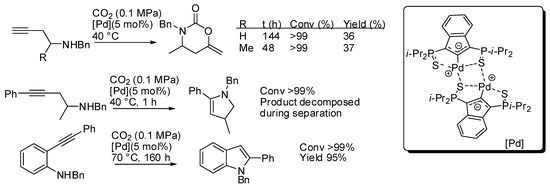
Scheme 15.
Other reactions carried out with the catalyst described in Table 4 entry 1.
The reactions described in entries 3 [156] and 8 [161] are very interesting, because they were carried out under air. All reactions described in entry 8 were carried out under these conditions, while only three different oxazolidinones were prepared in 66–95% yields with cyanuric acid. Actually, the 2-oxazolidinone syntheses in low-concentration CO2 in air are rare (See also: [163,164]).
The observation by Fujita and co-workers [140] that some amounts of tautomer 2-oxazolinone was found under their reaction conditions, prompted them to set up the best conditions for this tautomerization (5 mol% of Bu4NOH, at 80 °C for 6 h).
As shown in Table 4, N-alkyl propargylamines are generally employed in these reactions, because N-aryl derivatives undergo alkyne hydroarylation more easily than CO2 incorporation owing to the lower nucleophilicity of the nitrogen atom. Only in Table 4, entry 1, N-p-methoxyphenylpropargylamine was reported to react in 74% yield, but simple N-phenylpropargylamine did not give any oxazolidinone [134]. In the recent literature, however, an interesting work was performed by Zhou and his research group, who were able to carry out a four component one-pot sequence reaction to give enantioenriched oxazolidinones (Scheme 16) [165]. Authors separately tested the preparation of the enantioenriched propargyl amines and their cyclization, and, when the best conditions were found, they successfully performed the one-pot reaction. The absolute configuration of the chiral 2-oxazolidinone was determined to be R by X-ray analysis. The reaction was scaled up to 4.0 mmol scale with only 5 mol % of copper catalyst, and product was achieved in 84% yield and 94% ee.
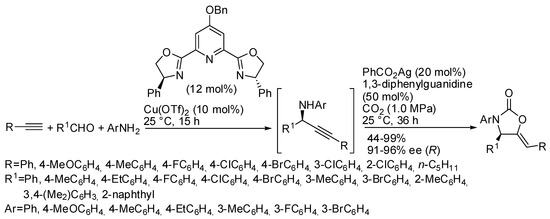
Scheme 16.
Tandem asymmetric aldehyde-alkyne-amine coupling-carboxylative cyclization one-pot sequence.
Finally, the CO2-fixation to propargyl amines-pyridine co-polymers promoted by a two-fold amount of DBU should be mentioned [166,167]. The polyoxazolidinone was obtained in 100% conversion (86% yields) in 48 h, even under air. In the same paper also three examples of propargyl amines are reported and it should be noted that N-benzyl-2-methyl-4-(pyridin-2-yl)but-3-yn-2-amine afforded a E/Z mixture of oxazolidinone that is the only example in the two years covered by this review.
Another three-component reaction involving propargylic amines, CO2 and N-bromosuccinimide (NBS) allowed the stereoselective synthesis of (E)-bromovinyloxazolidinones [168]. The reaction was catalyzed by silver acetate in the presence of a base. Among the bases, a guanidine derivative was found the most efficient (Scheme 17). The reaction mechanism is similar to that reported in Scheme 14, but the vinylsilver intermediate is trapped by bromine atom and not by hydrogen from the amine. This is another example of vinyl anion trapping by an electrophile different from hydrogen as already reported in Section 2 (Scheme 11) [145]. It should also be noted that in this reaction only the (E)-vinylsilver isomer is formed.
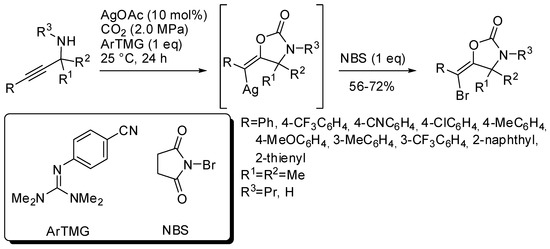
Scheme 17.
Synthesis of (E)-bromovinyloxazolidinones.
Also allylamines have been employed for carbon dioxide fixation into cyclic carbamates.
The reactions had three components and also involved alkyl halides and the presence of light, because generally the alkyl halide is homolitically broken by an irradiated metal catalyst. For instance tris(bipyridine)ruthenium(II) chloride allowed the synthesis of many oxazolidinones (Scheme 18, Equation (1)), as well as 3-benzyl-6-phenyl-6-(2,2-difluoro-3-ethoxy-3-oxopropyl)-1,3-oxazinan2-one and 3-benzyl-6-phenyl-5-(2,2difluoro-3-ethoxy-3-oxopropyl)-1,3-oxazinan-2-one (54 and 38% yields, respectively) [169]. The reaction of primary allylamines, allylamines with electron withdrawing nitrogen protecting groups or aliphatic R2 substituents failed as well as the synthesis of cyclic carbonates from allyl alcohols and of oxazinone from 2-(prop-1-en-2-yl)aniline. The reaction was scaled up to 1.31 g of recovered product. Some manipulations of the products were also successfully carried out. Then the same research group carried out a similar reaction under palladium catalysis and found the same features, except for primary allylamines, which smoothly reacted. (Scheme 18, Equation (2)) [170]. Scheme 18 also reported the mechanism surmised by authors for the reaction. Both reactions were scaled up to gram-scale and oxazolidinone was recovered in 84% yield under ruthenium catalysis and 77% yield under palladium catalysis. Products of both reactions were also manipulated to give open-chain compounds.
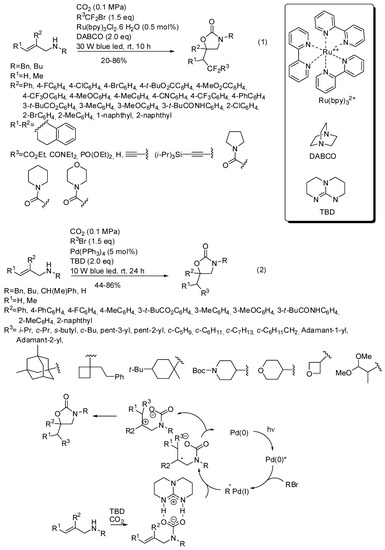
Scheme 18.
Visible-light photoredox induced reaction of allylamines with CO2.
He and co-workers demonstrated that, in the presence of 1,5,7-triazabicyclo[4,4,0]dec-5-ene TDB as the base, the metal catalyst is unnecessary with perfluoroalkyl halides [171]. They carried out some experiments to elucidate the mechanism and from the evidence they proposed the mechanism depicted in Scheme 19.
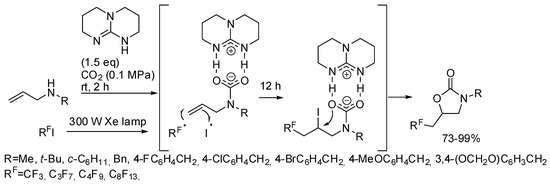
Scheme 19.
Visible-light photoredox induced reaction of allylamines with CO2 and perfluoro compounds.
The N-heterocyclic carbene copper and silver complexes supported on graphene or nanotubes, introduced by Chen and co-workers (see also Table 1, entry 72 and Table 2, entry 5), allowed the synthesis of oxazolidinones in a three-component reaction (Scheme 20) [115]. Copper complexes gave generally higher yields.
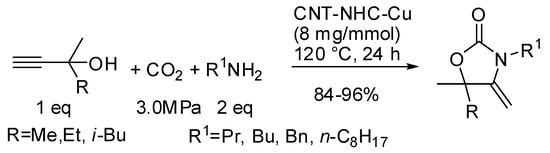
Scheme 20.
Three-component coupling of CO2, primary amine, and various propargyl alcohols.
An alternative and attractive strategy to produce oxazolidinones from less-toxic and easily available starting materials is a three-component reaction of CO2, epoxides, and amines. However, this reaction suffers from large amounts of epoxides because at least a molecule is transformed into a diol by-product (Scheme 21). Two alternative pathways have been suggested:
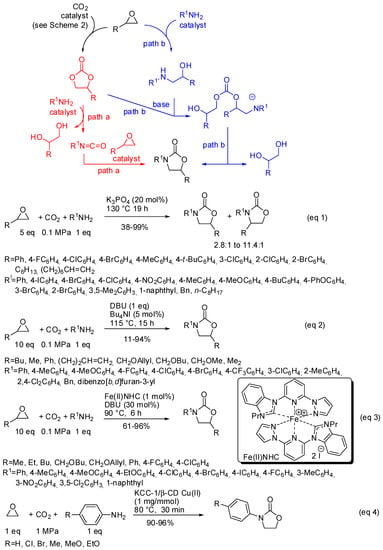
Scheme 21.
Three-component reactions among CO2, amines and epoxides.
- the first one involved the formation of cyclic carbonate by the classical mechanism (see Scheme 2), then amine opened the carbonate by releasing of diol and formation of a isocyanate, which in turn added another molecule of epoxide (path a);
- the second one involved the interaction between the cyclic carbonate and the amino-alcohol from nucleophilic opening of epoxide by amine(path b).
For instance, potassium phosphate was found to be a highly active catalyst in this reaction mainly for styrene oxide derivatives (Scheme 21, Equation (1)) [172]. A mixture of four- and five-substituted oxazolidinones is generally obtained. However, aliphatic epoxides with a long chain alkyl group (R = C6H13, (CH2)6CH=CH2) afforded a single regioisomer albeit in 45% and 38% yields, respectively. Also p-nitroaniline, benzyl- and octyl-amines gave a single regioisomer. In all cases, the five-substituted oxazolidinone is recovered. Both isocyanates and aminoalcohols gave the products when submitted to the reaction conditions.
Diazabicycloundecene, instead, allowed the reaction of aliphatic epoxides. However, increasing the bulkiness of the epoxides yields lowered and cyclohexene oxide led to unidentified mixture of products (Scheme 21, Equation (2)) [173]. The analysis of the reaction course by GC-MS did not show the presence of isocyanate intermediates. Also, a Fe(II) N-heterocyclic carbene (NHC) complex was found able to perform this reaction (Scheme 21, Equation (3)) [174]. Authors did not attempt reaction with internal epoxides, and also found that styrene oxides gave lower yields than aliphatic epoxides. Some experiments, carried out to elucidate the mechanism, suggested path b for the reaction.
KCC-1 nanoparticles have been already encountered in this review in Table 4, entry 9. On this fibrous material was supported Cu (II)-β-cyclodextrin and the obtained catalyst afforded oxazolidinones from reaction of anilines, CO2 and ethylene oxide (Scheme 21, Equation (4)) [175]. Albeit restricted only to ethylene oxide and p-substituted anilines, this reaction is interesting because it did not use epoxide excess. In fact, in the proposed mechanism, authors affirmed that the diol by-product could be captured by the catalyst. Then, a molecule of aniline could nucleophilically remove it to give the ethanolamine intermediate for a further catalytic cycle.
Conversely from CO, which can favorably add the ethanolamine [176], the direct reaction between amino alcohols and CO2 is thermodynamically disfavored. To shift the equilibrium to 2-oxazolidinones dehydrating agents or auxiliaries are often needed, with generation of waste. Among dehydrating agents in the last two years, He and co-workers proposed propargyl alcohols and published some papers with different catalysts to carry out this reaction [177,178,179].
The catalyst or the catalyst mixture should activate the OH groups both of propargyl alcohol and of amino alcohol by hydrogen bonds and the CO2, as well as coordinate the triple bond and act as a Lewis acid on the carbonyl group. In all the papers, yields of oxazolidinones and ketones by-product are comparable. Authors performed control experiments, DFT calculation, kinetic and NMR studies and from these data suggested the reaction pathway depicted in Scheme 22 [177], which was confirmed in the other papers. The first proposed catalyst was the couple Ag2O and tetramethylguanidine (Scheme 22) [177]. The reaction was also applied to diols; 1-phenylethan-1,2-diol and 3-phenoxypropan-1,2-diol gave the corresponding cyclic carbonates in 95% and 58%, respectively.
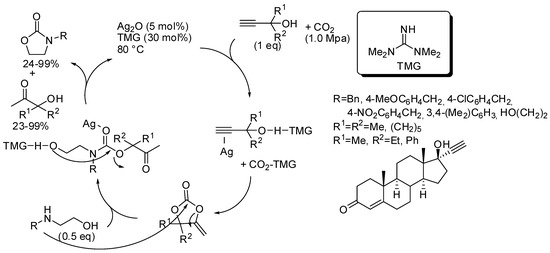
Scheme 22.
Cascade reaction of propargyl alcohols, carbon dioxide, and 2-aminoethanols.
Then, the same research group proposed another catalytic mixture to perform the same reaction, that are: CuI (5 mol%) 1,10-phenantroline (5 mol%) t-BuOK (10 mol%), CO2 (0.5 MPa), 80 °C, 12 h [178]. Yields ranged 18–95% for hydroxyketones and 24–97% for the same oxazolidinones reported in Scheme 22, as well as the diols. Only 1,2-propanediol was added extra. The third catalyst was 1,5,7-triazabicylo[4.4.0]dec-5-ene trifluoroethanol (15 mol%), CO2 (0.1 MPa), 80 °C, 12 h [179]. For the same set of products, yields ranged 23–99% for hydroxyketones and 24–99% for the oxazolidinones and cyclic carbonates. The easy recovering of this last catalyst allowed its recycle and the target product was still obtained in 75% yield, after five reuses.
Repo and co-workers used tosyl chloride as the auxiliary [180]. The most important feature of this reaction is the formation and the stabilization of the carbamate species before the competing N-tosylation reaction, which is then irreversible (Scheme 23). Lower reaction temperatures and higher CO2 pressure favored the oxazolidinone formation. The presence of a good leaving group such as in 3-chloropropan-2-ol-1-amine favors the 6-membered ring notwithstanding the higher stability of the 5-membered ring. In fact, propan-1,2-diol-1-amine gave mainly the five-membered ring. Starting from chiral compounds the Sn2 nature of the reaction allowed the preparation of enantioenriched compounds in good purity. The mechanism was then studied by DFT calculation confirming author’s hypothesis.
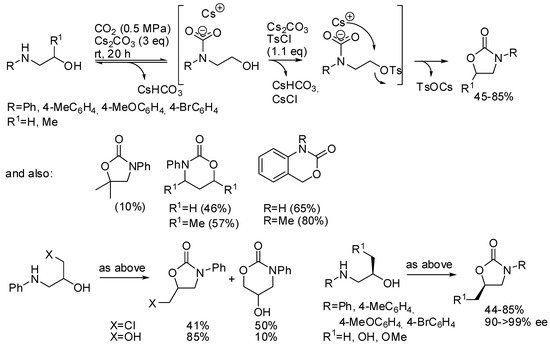
Scheme 23.
Synthesis of cyclic carbamates from amino alcohols.
Finally, oxazolidinones could be obtained in high yields from a three-component reaction among CO2, 1,2-dichloroethane and aromatic amines catalyzed by N-heterocyclic carbene obtained in situ from an ionic liquid (Scheme 24) [181]. However, the reaction with 2,3-dichlorobutane afforded the corresponding 4,5-dimethyl-3-aryloxazolidin-2-ones in very low yields (28–40%). On the other hand, 1,2-dichloropropane gave good yields (about 70%) at higher temperatures, but without regioselectivity and a mixture of 5-methyl-3-aryloxazolidin-2-one and 4-methyl-3-aryloxazolidin-2-one was always recovered. The reaction occurred also with CO2 diluted in water, air and nitrogen. The catalyst was used five times without significant deactivation. Interestingly the reaction was extended to other dichloroalkanes. Many six-membered 3-aryl-1,3-oxazinan-2-ones (19 examples) were obtained in 70–93% yields at 90 °C for 6 h. The seven-membered 3-aryl-1,3-oxazepan-2-one was also formed but in lower yield (22–35%, 3 examples). Larger cycles such as those from 1,5-dichloropentane, 1,6-dichlorohexane and 1,10-dichlorodecane were not obtained, but ω-chloroalkylcarbamates were recovered in 63, 55, 37%, respectively. Some experiments were carried out to elucidate the mechanism depicted in Scheme 24. Authors also demonstrated by means of control experiments that carbonate ions were not the C1 source of the reaction.
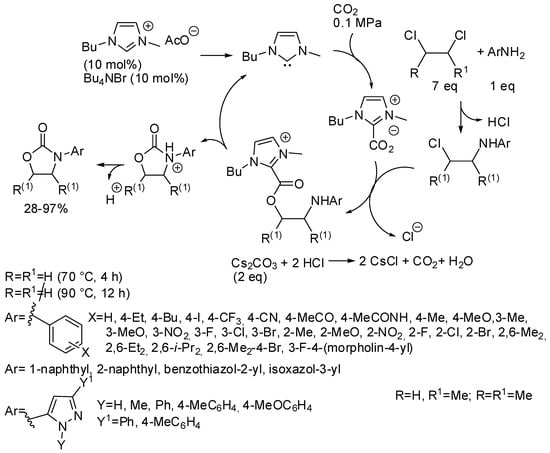
Scheme 24.
Synthesis of oxazolidinones from 1,2-dichloroethanes.
These last two reactions have been employed also for larger cyclic carbamates than oxazolidinones. Very recently, an enantioselective cyclization was developed to prepare six-membered cyclic carbamates from homoallylic amines (Scheme 25) [182]. The catalyst was studied in order to have Brønsted basicity enough to avoid amine to form an achiral carbamate salt intermediate. Some amounts of water favored the catalyst-carbamic acid complex, but an excess allowed the formation of an unreactive crystalline ligand. The optimum amount was 90 ppm. Allylic amines gave low enantiomeric excess under these conditions. Unsubstituted homoallylamines (R1 = H) gave low yield (43%) and ee (13%). The enantiomeric catalyst gave the enantiomeric product in comparable yield and selectivity. The absolute configuration was determined by X-ray analysis.
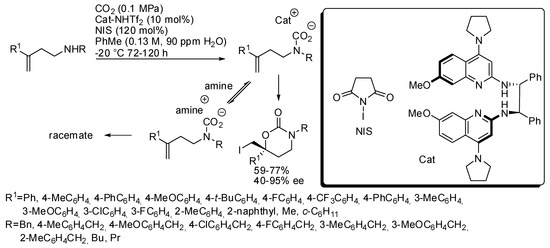
Scheme 25.
Synthesis of enantioenriched 1,3-oxazinan-2-ones.
Other six-membered rings, recently obtained by CO2 fixation, are some azo-linked 4H-benzo[d][1,3]oxazine-2,4-diones from azo-linked aminobenzoic acids [183]. The catalyst was called by authors CuO@RHA/MCM-41 nanocomposite and was prepared from CuO nanoparticles and a MCM-41 matrix obtained from rice husk ash (RHA). The best yields were obtained with electron-withdrawing substituted substrates. This heterogeneous catalyst was recycled up to six times without significant loss of activity. In the proposed mechanism (Scheme 26), Cu ions act as a Lewis acid to activate CO2. Then a carbamate ion is formed, which in turn undergoes intramolecular reaction and dehydration to give the product.
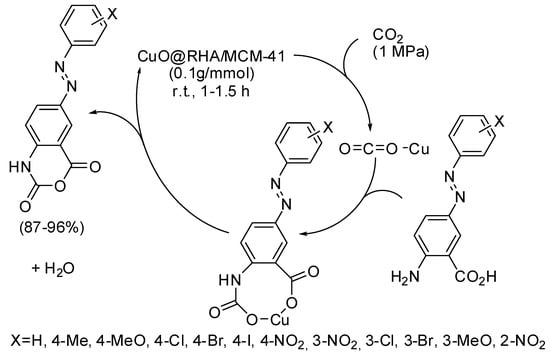
Scheme 26.
Synthesis of 4H-benzo[d][1,3]oxazine-2,4-diones.
Another cyclic carbamate, which shows biological and pharmaceutical activities, is 1,3,4-oxadiazole-2(3H)-one. Its synthesis could be performed by 1,3-dipolar cycloaddition of nitrile imine with CO2. However, the low reactivity of carbon dioxide toward 1,3-dipoles and the fast dimerization of nitrile imines make this reaction rare. In the last two years three papers appeared in the literature anyway. In 2017, Zhang found that CsF/18-crown-6 are able to enhance both the reactivity of CO2 as a 1,3-dipolarophile and the in situ formation of nitrile imines from hydrazone chloride (Scheme 27, Equation (1)) [184]. Among the prepared products, a potential drug for Parkinson’s disease therapy and the commercial herbicide oxadiazon were obtained in 89% and 88% isolated yield, respectively. The same procedure can be successfully applied to the synthesis of 3,4-thiadiazol-2(3H)-one from COS.
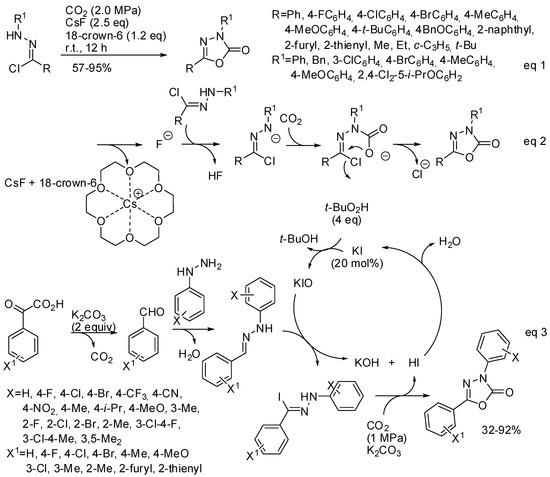
Scheme 27.
Synthesis of 1,3,4-oxadiazole-2(3H)-one.
The reaction mechanism was studied by Fernández-Herrera, Merino and co-workers at the SMD/M06-2X/def2-TZVP level. They found that the reaction proceeds by a three-step mechanism and not by 1,3-dipolar cycloaddition (Scheme 27, Equation (2)), thus explaining how the low reactivity of carbon dioxide toward 1,3-dipoles is overcome. The rate-determining step is the final five-membered ring closure. However, authors affirmed that increasing the concentration and the temperature the concerted pathway becomes more likely [185].
Very recently, a new synthesis of substituted 1,3,4-oxadiazol-2(3H)-ones from aryl hydrazines and α-oxocarboxylic acids, with KI as the catalyst, has been developed (Scheme 27, Equation (3)) [186]. Authors made some control experiments and they found that the extra pressurized CO2 is necessary for obtaining good yield (with the CO2 only released by the α-oxocarboxylic acid, the product was obtained in 38% yield); the hypoiodite ions is a key active species to give the hydrazone iodide; and that methyl acrylate instead of CO2 led to a cycloadduct in 74% yield, thus suggesting a cycloaddition reaction also with CO2. However, it is surprising that after this evidence, authors did not start from more available aldehydes instead of α-oxocarboxylic acids.
Finally, the synthesis of 2-benzoxazolone (60% yield) from 2-aminophenol in the presence of tributylamine should be mentioned [187]. Reaction details will be discussed in the next section (Scheme 31).
4. Cyclic Ureas
Ureas framework is present in natural products, agricultural pesticides, herbicides, and pharmaceuticals. The synthesis of urea itself was the founder of modern organic chemistry. However, the selective and efficient synthesis of substituted ureas and in particular of cyclic ureas from CO2 and amines is still challenging [188,189].
Quinazoline-2,4(1H,3H)-diones, important intermediates in the synthesis of pharmaceuticals, can be obtained from reaction of 2-aminobenzonitriles and CO2. This synthesis is particularly important because of the direct utilization of carbon dioxide as well as the high atom economy. In the last two years many different methods have been reported in the literature. For instance, the bi-functional graphitic carbon nitride prepared by Samanta and Srivastava [127] was efficient only in dimethylformamide (90.2% yield) or dimethylsulfoxide (93.2% yield), with 50 mg/mmol of catalyst, at 130 °C and 2.5 MPa CO2 pressure, after 12 h in the reaction of 2-aminobenzonitrile. The aprotic dipolar solvent should enhance the activation of the CO2 molecules as well as favor the abstraction of an acidic proton from the NH2 group of 2-aminobenzonitrile. Examples of reaction with larger applicability are collected in Table 5.
Table 5.
Catalysts for the reaction of 2-aminobenzonitriles and CO2.
The accepted mechanism for this reaction is depicted in Scheme 28. Many reactions showed a relationship between the pKa of the basic catalyst and the reaction rate. For example, in ionic liquid the pKa = 14.7 (the acidity of the quinazoline-2,4-dione product) is the borderline between the base-catalyzed reaction and the quinazoline-2,4-dione ion catalysis [190]. A further computational study was conducted at M06-2X level and 6-31G (d) basis set utilizing organic bases as the catalyst, which confirmed the mechanism [191]. Other studies on the mechanism were carried out with the M06 functional owing to its recognized ability in the description of organometallic chemistry with noncovalent interactions [192].
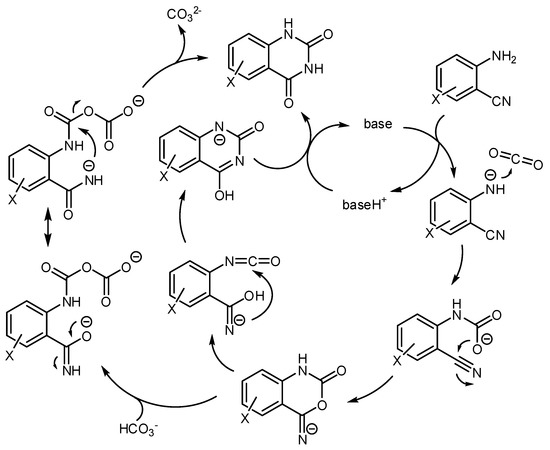
Scheme 28.
Mechanism of the reaction of aminobenzonitriles and CO2 catalyzed by bases.
Cesium carbonate was chosen as the catalyst and the LanL2DZ basis set was used for Cs atoms and the 6–311 + G(d,p) basis set was used to describe other atoms. The energy involved in different reaction pathways was calculated and the most favorable pathway is very similar to the depicted in Scheme 28. A “naked nitrogen ion” is necessary for the attack to CO2 and the rearrangement 6-imino-1,3-oxazinen-2-one to pyrimidine-2,4(1H,3H)-dione occurs intramolecularly. In this study, the isocyanate intermediate is not found along the reaction pathway, but the complex A was invoked as the key intermediate for the rearrangement.
Fujita reported just an example of pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (footnote to entry 1, Table 5) [160], but almost at the same time Zhang and co-workers published a detailed study on the synthesis of many dihydropyrimidine-2,4(1H,3H)-diones fused with heterocycles (Scheme 29) [193].
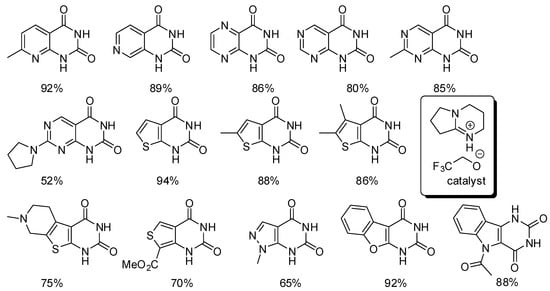
Scheme 29.
Synthesis of heterocycle-fused pyrimidine-2,4-(1H,3H)-diones.
An ionic liquid was used both as the catalyst and the solvent (6 mmol/mmol substrate). The reaction temperatures (60–90 °C) and times (3–96 h) clearly depended from the substrate. Among the various amino-carbonitrile heterocycles, only 2-amino-1H-indole-3-carbonitrile and 3-amino-1Hindole-2-carbonitrile did not react. However, N-acetyl-3-amino-1H-indole-2-carbonitrile gave the expected product, suggesting that an N-H group on the heterocycle prevented the reaction. In fact, no other free N-H heterocycle was tested.
Both CO2 and RNC are classical C1 sources and their use in a multicomponent reaction is an attractive way to prepare heterocycles introducing carbonyl and imine moiety at the same time. However, the difference in kinetic and thermodynamic stability of both C1-reactants makes their contemporary use rare, because are scarce the catalytic system which sufficiently activates CO2 and tunes the RNC insertion. In particular, the reaction of 2-haloanilines, RNC, and CO2 could give rise to two reaction products: 2-amino-4H-benzo[d][1,3]oxazin-4-one and 4-imino-1,4-dihydro2H-benzo[d][1,3]oxazin-2-one. Tuning both regio- and chemoselectivity to give the second product is particularly interesting, because it spontaneously rearranges to quinazoline-2,4-(1H,3H)-diones (see Scheme 28). Palladium catalysis was able to perform this cascade reaction affording N3-subsituted quinazoline-2,4-(1H,3H)-diones (Scheme 30). Three papers appeared in the literature starting from o-bromo- or o-iodoanilines [196,197,198]. All research groups carried out some control experiments to elucidate the reaction mechanism and all were in agreement to propose the mechanism depicted in Scheme 30. After the best reaction conditions and the scope of the reaction (Scheme 30, Equation (1)), the first paper reported also some post-functionalization, giving rise to N1-subsituted quinazolinediones, and 2,4-dichloro-6,7-dimethoxyquinazoline a key intermediate of some drugs. Moreover, by using the cheap 13CO2, labelled quinazolinediones were also synthesized. The reaction was scaled up to 5 mmol [196]. Almost simultaneously, another synthesis was developed starting from o-iodoanilines (Scheme 30, Equation (2)). It is worth to note that the reaction worked at atmospheric pressure of CO2, while the other worked at overpressure. The reaction was also carried out at a half gram scale and afforded product in 77% yield. Under these reaction conditions, o-bromo- or o-chloro-aniline, strong electron-withdrawing substituted o-iodoanilines, 4-nitrophenylisonitrile, and 2,6-dimethylphenylisonitrile did not react [197].
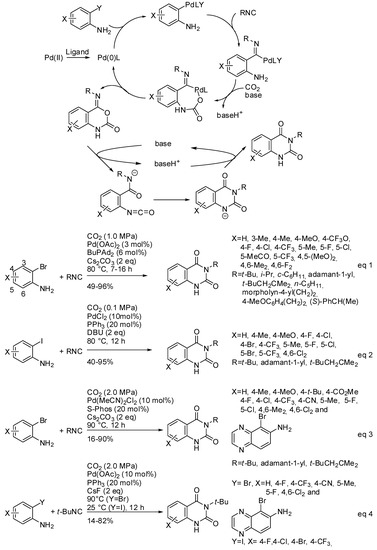
Scheme 30.
Three-component synthesis of quinazoline-2,4-(1H,3H)-diones.
Later, Zhang and coworkers used 2-dicyclohexylphosphino2′,6′-dimethoxybiphenyl, (SPhos) as the palladium ligand. Under these conditions, o-bromoanilines with electron-donating substituents gave satisfactory yield, but electron-withdrawing substituents afforded low yields (Scheme 30, Equation (3)). It should be noted that electron-deficient o-bromo- or o-iodoanilines gave good yields with PPh3 as the ligand, but with CsF as the base, conversely from Equation 2 (Scheme 30, Equation (4)) [198].
The direct carboxylation of diamines with carbon dioxide is another attractive manufacture of cyclic ureas. In the past years some catalysts have been introduced for this reaction and have been discussed in the reviews cited at the top of this section [188,189]. However, in the time range covered by us other interesting papers appeared in the literature. For instance, Lee, Kim and co-workers found that, in the presence of carbonate or bicarbonate as the bases, the synthesis of cyclic ureas is greatly enhanced by the presence of some amounts of imidazolidin-2-one. In particular, the reaction of ethylenediamine showed a classical autocatalytic rate, while 1,2- and 1,3-propanediamine yields increased from about 50% to 75%, if 10 mol% of imidazolidin-2-one was added at 200 °C, after 2 h and 5.0 MPa of CO2. The reactions went to completion in 4 h [199].
Since the carboxylation of amines by CO2 is known to proceed with almost no activation barrier, the role of imidazolidin-2-one cannot favor an increase of the nucleophilicity of the diamine, but rather to assist the catalyst in the protonation and deprotonation steps of the catalytic cycle, very likely via its enolic form.
Also the solvent play an important role, because it should be polar enough to stabilize anionic species, but it must not interact strongly with the base catalyst via hydrogen bonding. Authors performed a theoretical calculation to support these findings. On these bases, authors introduced 2-pyrrolidone as the best solvent for this reaction [200]. In fact, it can give rise to a keto-enol tautomerism and it is polar enough to stabilize the anionic species. The cyclic ureas from ethylenediamine, 1,2- and 1,3-propanediamine were recovered in 83–95% yields at 200 °C, after 2 h and 5.0 MPa of CO2 even in the absence of a base.
Benzimidazolones have been obtained from the reaction of o-phenylenediamines with CO2 in the presence of tributylamine as the base catalyst (Scheme 31) [187]. The catalyst can be recovered from the reaction mixture at 210–214 °C for 1 h and then reused without loss of activity, but slight reduction in yield was observed owing to the incomplete recovery. Electron-withdrawing substituents decrease nucleophilicity of the o-phenylenediamine, thus leading to less efficient reactions. Reaction with N-phenyl and N-methylphenylenediamine gave the expected product in 80 and 84% yields, respectively.
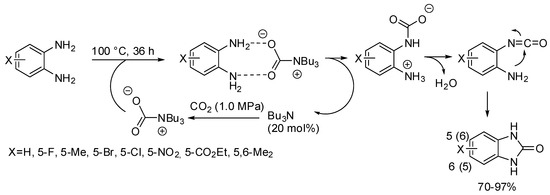
Scheme 31.
Synthesis of 2-benzimidazolones.
We already reported that [13C]-labeled quinazolinediones have been synthesized with the method described in Scheme 30, Equation (1) [196], but in the time range covered by this review, o-azidoamines and radiolabeled [11C and 14C] as well as labelled [13C] CO2 were used to prepare pharmaceutically important radiolabeled cyclic ureas [201]. The reaction proceeded in the presence of a phosphine, which reacted with the azide to give an iminophosphorane, which in turn underwent an aza-Wittig reaction to give an isocyanate. Finally the isocyanate is intramolecularly attacked by the amino group to cyclize (Scheme 32). During the experiments for the synthesis of N-alkylated heterocycles in a continuous flow reactor on a bed of γ-Al2O3 in supercritical CO2, authors found that under particular reaction conditions diethanolamine (1 M, at 250 °C, 15.0 MPa, and 0.2 mL/min) afforded 3-(2-hydroxyethyl)oxazolidin-2-one with a 56% of conversion and 73% selectivity [202]. At lower pressure and flow rate the oxazolidinone is converted in the expected 2,2′-(piperazine-1,4-diyl)diethanol. Owing to the higher nucleophilicity of amino groups the reaction was then carried out with N-(2-aminoethyl)ethanolamine. Actually at 250 °C, 1-(2-hydroxyethyl)imidazolidin-2-one was recovered with 85% selectivity and 70% yield. In a saturated CO2 atmosphere, but without supercritical CO2 as the solvent, the imidazolidinone was formed in 62% selectivity, 15% yield from 24% conversion. Conversely from oxazolidinone, imidazolidinone was stable in all the tested reaction conditions. Further studies are necessary to better understand this reaction.
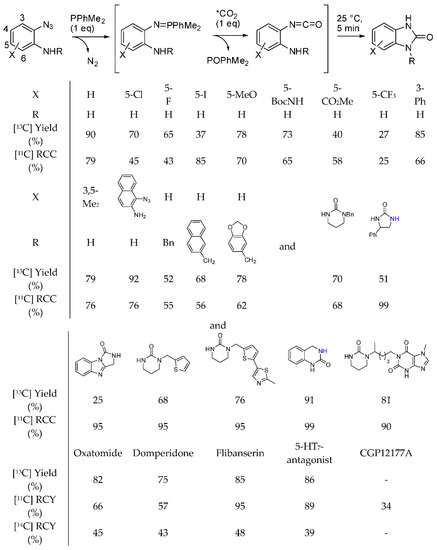
Scheme 32.
Synthesis of labelled pharmaceutically relevant cyclic ureas.
Finally, our research group was able to setup an easy access to imidazolidin-2-ones from the three-component reaction of propargylamines, primary amine and CO2 with 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as the catalyst under solvent-free conditions (Scheme 33) [203]. The most interesting features of this reaction were: (i) secondary alkyl amines as well as allylamine gave worse yields even increasing temperature to 120 °C. (ii) The higher stability of carbamate arising from benzylamine led to very low yields (<5%). However, changing the base to 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene and increasing temperature to 120 °C yield increased to 80%. (iii) Anilines reacted despite their low nucleophilicity, but yields greatly depended from electronic properties of the substituents, ranging from 81% for p-methoxyaniline to 22% for p-bromoaniline. (iv) Propargylamine led to 1H-imidazol-2(3H)-one, that is the most stable endocyclic double bond. (v) Also internal triple bond gave the product but in a 1:1 mixture of E/Z diastereoisomers. (vi) Only N-methylprop-2-yn-1-amine afforded 57% yield of cyclic urea, while other secondary propargylamines afforded only oxazolidinone. The reaction could be carried out in a gram scale (87% yield).
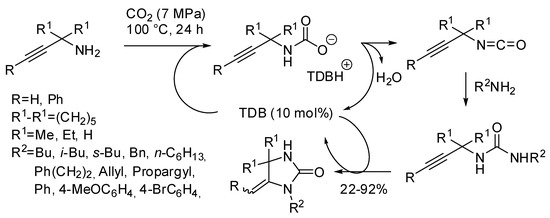
Scheme 33.
Urea derivatives from carbon dioxide and amines.
5. Other Heterocycles
In this last section, we report the synthesis of other heterocycles, which can be obtained by capture of carbon dioxide. For instance, a polythiazolium-based polymer was able to catalyze the cyclization of 2-aminobenzenethiols to benzothiazole in the presence of DBU and CO2 and phenylsilane as the reductant [204]. Authors found that primary amides used as the solvent reacted with 2-aminobenzenethiol in the presence of silane, so they used a cyclic amide, N-methyl-2-pyrrolidone (NMP). Moreover, temperatures >70 °C also reduced the yield. Four-substituted benzothiazoles were recovered in low yields, very likely for steric effects, but authors claimed that this was the first example of the use of three-substituted-2-aminobenzenethiols in the synthesis of four-substituted benzothiazoles. The polymer precatalyst was recovered adding excess HI and precipitation from methanol and reused for 7 times without losing its activity at 12 mol% of catalyst loading. Regarding the mechanism, authors proposed that DBU generated in situ a free carbene from the polymer precatalyst, which bound CO2. The following steps are reported in Scheme 34.
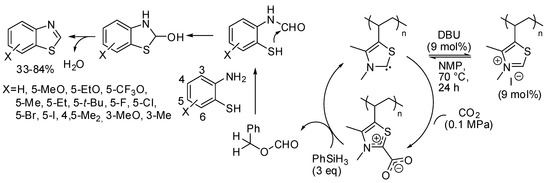
Scheme 34.
Cyclization of 2-aminobenzenethiols to benzothiazole.
In addition, developing the reaction already cited in Section 2 (Scheme 11) [127], 2-alkynylanilines, aryl iodides, and CO2 provided a series of 3,3-diaryl 2,4-quinolinediones (Scheme 35) [205]. It should be noted that this palladium catalyst allowed the incorporation of CO2, conversely from the palladium catalyst described in Section 3 (Scheme 15) [134]. The reaction was scaled up to a 1 mmol scale and product was recovered in 75% yield. In addition, bromobenzene afforded 3,3-diphenyl-2,4-quinolinedione in 72% yield. Some control experiments allowed authors the formulation of the mechanism depicted in Scheme 35.
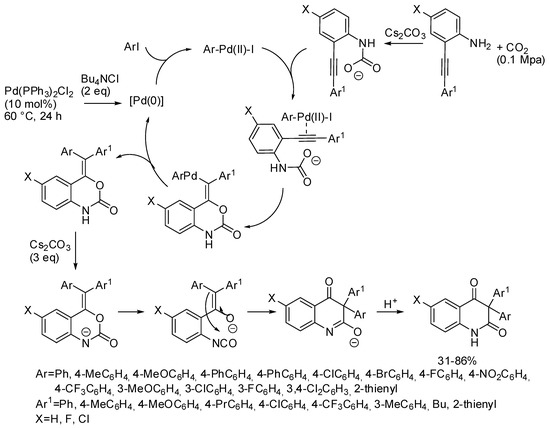
Scheme 35.
Multicomponent synthesis of 3,3-diaryl 2,4-quinolinediones.
The reaction of o-allylanilines with CO2 in the presence of a strong base led to 2-quinolinones. The detailed mechanism has been recently studied by density functional theory calculations. The calculated minimum energy reaction pathway is depicted in Scheme 36 [206]. Moreover, the base was found to play a significant role in reducing the energy barriers. The presence of urea and Boc-protected aniline among the by-products was explained by the competitive addition of ter-buatanol or aniline to the isocyanate intermediate. Calculation also predicted that weaker bases such as Na2CO3 or NaHCO3 might promote the reaction as well.
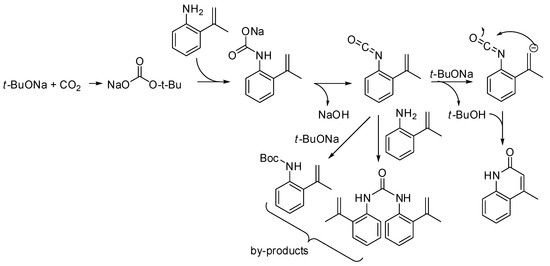
Scheme 36.
Lactamization of sp2 C–H bonds with CO2.
6. Conclusions
The importance of CO2 as C1 synthon in chemical reactions is greatly increasing in recent years. In fact, human activity in its industrial processes produces as by products about 3.3 1010 metric tons of CO2 increasing the deleterious greenhouse effect. Thus, since sequestration of some of the circulating carbon dioxide is possible, its use as feedstock in chemical process creates added value to a waste material and positive effect on the environment. Among the many transformations of carbon dioxide into valuable chemicals, the synthesis of heterocycles undoubtedly plays an indisputable role, for the widespread presence of these moieties in new drugs and as building blocks for multistep synthesis [207,208]. The next future should address this research field towards sustainable methods such as the use of recoverable organocatalysts instead of precious metal ones, and towards asymmetric reaction [209], since most of the organic active pharmaceuticals are chiral molecules.
Author Contributions
Conceptualization, B.G. and R.D.; Resources, and blibliographic research R.M.; Data Curation, R.M.; Writing-Original Draft Preparation, R.D.; Writing-Review & Editing, R.D.; Visualization, B.G., N.D.C.; Supervision, N.D.C.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Solomon, S.; Plattner, G.-K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [Google Scholar] [CrossRef]
- Zhou, Z.; Xia, S.; He, L. Green Catalysis for Three-Component Reaction of Carbon Dioxide, Propargylic Alcohols and Nucleophiles. Acta Phys. Chim. Sin. 2018, 34, 838–844. [Google Scholar] [CrossRef]
- Cantat, T.; He, L.-N. (Eds.) CO2 Capture and Chemistry 2017. Curr. Opin. Green Sustain. Chem. 2017, 3, 1–66. Available online: https://www.sciencedirect.com/journal/current-opinion-in-green-and-sustainable-chemistry/vol/3/suppl/C (accessed on 5 June 2019).
- Song, Q.; Zhou, Z.; He, L. Efficient: Selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- ChemSusChem: Hot Topic: Carbon Dioxide First published: 1 April 2017, Last updated: 23 April 2018. Available online: https://onlinelibrary.wiley.com/doi/toc/10.1002/(ISSN)1864-564X.hottopic-co2 (accessed on 5 June 2019).
- Lu, X.B. Carbon Dioxide and Organometallics. In Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2015; Volume 53. [Google Scholar]
- Fiorani, G.; Guo, W.; Kleij, A.W. Sustainable conversion of carbon dioxide: The advent of organocatalysis. Green Chem. 2015, 17, 1375–1389. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, X. Lewis base-CO2 adducts as organocatalysts for CO2 transformation. Sci. China Chem. 2017, 60, 904–911. [Google Scholar] [CrossRef]
- Tortajada, A.; Juliá-Hernández, F.; Börjesson, M.; Moragas, T.; Martin, R. Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem. Int. Ed. 2018, 57, 15948–15982. [Google Scholar] [CrossRef]
- Poliakoff, M.; Leitner, W.; Streng, E.S. The Twelve Principles of CO2 Chemistry. Faraday Discuss. 2015, 183, 9–17. [Google Scholar] [CrossRef]
- Kumar, M.; Sundaram, S.; Gnansounou, E.; Larroche, C.; Thakura, I.S. Carbon dioxide capture, storage and production of biofuel and biomaterials by bacteria: A review. Bioresour. Technol. 2018, 247, 1059–1068. [Google Scholar] [CrossRef]
- Muthuramalingam, S.; Velusamy, M.; Mayilmurugan, R. Fixation and sequestration of carbon dioxide by copper(II) complexes. J. Chem. Sci. 2018, 130, 78. [Google Scholar] [CrossRef]
- Speight, J.G. Carbonylation. In Environmental Organic Chemistry for Engineers; Elsevier: Oxford, UK, 2017. [Google Scholar]
- Qiao, C.; Cao, Y.; He, L.-N. Transition Metal-Catalyzed Carboxylation of Terminal Alkynes with CO2. Mini-Rev. Org. Chem. 2018, 15, 283–290. [Google Scholar] [CrossRef]
- Zou, B.; Hu, C. Halogen-free processes for organic carbonate synthesis from CO2. Curr. Opin. Green Sustain. Chem. 2017, 3, 11–16. [Google Scholar] [CrossRef]
- Vessally, E.; Hosseinian, A.; Babazadeh, M.; Edjlali, L.; Hosseinzadeh-Khanmiri, R. Metal Catalyzed Carboxylative Coupling of Terminal Alkynes, Organohalides and Carbon Dioxide: A Novel and Promising Synthetic Strategy Toward 2-Alkynoates (A Review). Curr. Org. Chem. 2018, 22, 315–322. [Google Scholar] [CrossRef]
- Buttner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent Developments in the Synthesis of Cyclic Carbonates from Epoxides and CO2. Top. Curr. Chem. 2017, 375, 50. [Google Scholar] [CrossRef]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef]
- Cotton, C.A.R.; Edlich-Muth, C.; Bar-Even, A. Reinforcing carbon fixation: CO2 reduction replacing and supporting carboxylation. Curr. Opin. Biotechnol. 2018, 49, 49–56. [Google Scholar] [CrossRef]
- Pander, J.E., III; Ren, D.; Huang, Y.; Loo, N.W.X.; Hong, S.H.L.; Yeo, B.S. Understanding the Heterogeneous Electrocatalytic Reduction of Carbon Dioxide on Oxide-Derived Catalysts. ChemElectroChem 2018, 5, 219–237. [Google Scholar] [CrossRef]
- Voiry, D.; Shin, H.S.; Loh, K.P.; Chhowalla, M. Low-dimensional catalysts for hydrogen evolution and CO2 reduction. Nat. Rev. Chem. 2018, 2, 0105. [Google Scholar] [CrossRef]
- Pagliaro, M.; Fidalgo, A.; Palmisano, L.; Ilharco, L.M.; Parrino, F.; Ciriminna, R. Polymers of limonene oxide and carbon dioxide: Polycarbonates of the solar economy. Preprints 2018, 2018, 040067. [Google Scholar] [CrossRef]
- Muthuraj, R.; Mekonnen, T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: Co-polymers and polymer blends. Polymer 2018, 145, 348–373. [Google Scholar] [CrossRef]
- Kamphuis, A.J.; Picchioni, F.; Pescarmona, P.P. CO2-fixation into cyclic and polymeric carbonates: Principles and applications. Green Chem. 2019, 21, 406–448. [Google Scholar] [CrossRef]
- Tomishige, K.; Tamura, M.; Nakagawa, Y. CO2 Conversion with Alcohols and Amines into Carbonates, Ureas, and Carbamates over CeO2 Catalyst in the Presence and Absence of 2-Cyanopyridine. Chem. Rec. 2018, 18, 1–26. [Google Scholar] [CrossRef]
- Liang, J.; Huang, J.-B.; Cao, R. Metal-organic frameworks and porous organic polymers for sustainable fixation of carbon dioxide into cyclic carbonates. Coord. Chem. Rev. 2019, 378, 32–65. [Google Scholar] [CrossRef]
- Li, J.-Y.; Song, Q.-W.; Zhang, K.; Liu, P. Catalytic Conversion of Carbon Dioxide through C-N Bond Formation. Molecules 2019, 24, 182. [Google Scholar] [CrossRef]
- Farshbaf, S.; Fekri, L.Z.; Nikpassand, M.; Mohammadi, R.; Vessally, E. Dehydrative condensation of β-aminoalcohols with CO2: An environmentally benign access to 2-oxazolidinone derivatives. J. CO2 Util. 2018, 25, 194–204. [Google Scholar] [CrossRef]
- Didehbana, K.; Vessally, E.; Salary, M.; Edjlalic, L.; Babazadeh, M. Synthesis of a variety of key medicinal heterocyclic compounds via chemical fixation of CO2 onto o-alkynylaniline derivatives. J. CO2 Util. 2018, 23, 42–50. [Google Scholar] [CrossRef]
- Vessally, E.; Babazadeh, M.; Hosseinian, A.; Arshadi, S.; Edjlali, L. Nanocatalysts for chemical transformation of carbon dioxide. J. CO2 Util. 2017, 21, 491–502. [Google Scholar] [CrossRef]
- Vessally, E.; Didehban, K.; Babazadeh, M.; Hosseinian, A.; Edjlali, L. Chemical fixation of CO2 with aniline derivatives: A new avenue to the synthesis of functionalized azole compounds (A review). J. CO2 Util. 2017, 21, 480–490. [Google Scholar] [CrossRef]
- Vessally, E.; Soleimani-Amiri, S.; Hosseinian, A.; Edjlali, L.; Babazadeh, M. Chemical fixation of CO2 to 2-aminobenzonitriles: A straightforward route to quinazoline-2,4(1H,3H)-diones with green and sustainable chemistryperspectives. J. CO2 Util. 2017, 21, 342–352. [Google Scholar] [CrossRef]
- Arshadi, S.; Vessally, E.; Sobati, M.; Hosseinian, A.; Bekhradnia, A. Chemical fixation of CO2 to N-propargylamines: A straightforward route to 2-oxazolidinones. J. CO2 Util. 2017, 19, 120–129. [Google Scholar] [CrossRef]
- Hosseinian, A.; Ahmadi, S.; Mohammadi, R.; Monfared, A.; Rahmani, Z. Three-component reaction of amines, epoxides, and carbon dioxide: A straightforward route to organic carbamates. J. CO2 Util. 2018, 27, 381–389. [Google Scholar] [CrossRef]
- Liu, X.-F.; Wang, M.-Y.; He, L.-N. Heterogeneous Catalysis for Oxazolidinone Synthesis from Aziridines and CO2. Curr. Org. Chem. 2017, 21, 698–707. [Google Scholar] [CrossRef]
- Arshadi, S.; Vessally, E.; Hosseinian, A.; Soleimani-Amiri, S.; Edjlali, L. Three component coupling of CO2, propargyl alcohols, and amines: An environmentally benign access to cyclic and acyclic carbamates (A Review). J. CO2 Util. 2017, 21, 108–118. [Google Scholar] [CrossRef]
- Yu, B.; He, L.-N. Upgrading Carbon Dioxide by Incorporation into Heterocycles. ChemSusChem 2015, 8, 52–62. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef]
- Poland, S.J.; Darensbourg, D.J. A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes. Green Chem. 2017, 19, 4990–5011. [Google Scholar] [CrossRef]
- Zhang, S.S. A review on electrolyte additives for lithium-ion batteries. J. Power Sources 2006, 162, 1379–1394. [Google Scholar] [CrossRef]
- He, Q.; O’Brien, J.W.; Kitselman, K.A.; Tompkins, L.E.; Curtis, G.C.T.; Kerton, F.M. Synthesis of cyclic carbonates from CO2 and epoxides using ionic liquids and related catalysts including choline chloride–metal halide mixtures. Catal. Sci. Technol. 2014, 4, 1513–1528. [Google Scholar] [CrossRef]
- Lan, D.-H.; Fan, N.; Wang, Y.; Gao, X.; Zhang, P.; Chen, L.; Au, C.-T.; Yin, S.-F. Recent advances in metal-free catalysts for the synthesis of cyclic carbonates from CO2 and epoxides. Chin. J. Catal. 2016, 37, 826–845. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Dyson, P.J. Synthesis of carbonates and related compounds incorporating CO2 using ionic liquid-type catalysts: State-of-the-art and beyond. J. Catal. 2016, 343, 52–61. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: Catalyst design and mechanistic studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal. 2018, 8, 419–450. [Google Scholar] [CrossRef]
- Kim, H.-U.; Babu, R.; Roshan, R.; Park, D.-W. Catalytic performance of metal azolate frameworks in the solventless synthesis of cyclic carbonates from CO2 and epoxides. Appl. Catal. A Gen. 2017, 538, 59–65. [Google Scholar] [CrossRef]
- Elkurtehi, A.I.; Kerton, F.M. Coupling Reactions of Carbon Dioxide with Epoxides Catalyzed by Vanadium Aminophenolate Complexes. ChemSusChem 2017, 10, 1249–1254. [Google Scholar] [CrossRef]
- Chen, F.; Liu, N.; Dai, B. Iron(II) Bis-CNN Pincer Complex-Catalyzed Cyclic Carbonate Synthesis at Room Temperature. ACS Sustain. Chem. Eng. 2017, 5, 9065–9075. [Google Scholar] [CrossRef]
- Wu, X.; North, M. A Bimetallic Aluminium(Salphen) Complex for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 74–78. [Google Scholar] [CrossRef]
- Luo, R.; Yang, Z.; Zhang, W.; Zhou, X.; Ji, H. Recyclable bifunctional aluminum salen catalyst for CO2 fixation: The efficient formation of five-membered heterocyclic compounds. Sci. China Chem. 2017, 60, 979–989. [Google Scholar] [CrossRef]
- Zhou, F.; Xie, S.-L.; Gao, X.-T.; Zhang, R.; Wang, C.-H.; Yin, G.-Q.; Zhou, J. Activation of (salen)CoI complex by phosphorane for carbon dioxide transformation at ambient temperature and pressure. Green Chem. 2017, 19, 3908–3915. [Google Scholar] [CrossRef]
- Kaneko, S.; Shirakawa, S. Potassium Iodide−Tetraethylene Glycol Complex as a Practical Catalyst for CO2 Fixation Reactions with Epoxides under Mild Conditions. ACS Sustain. Chem. Eng. 2017, 5, 2836–2840. [Google Scholar] [CrossRef]
- Steinbauer, J.; Werner, T. Poly(ethylene glycol)s as Ligands in Calcium-Catalyzed Cyclic Carbonate Synthesis. ChemSusChem 2017, 10, 3025–3029. [Google Scholar] [CrossRef]
- Steinbauer, J.; Spannenberg, A.; Werner, T. An in situ formed Ca2+–crown ether complex and its use in CO2-fixation reactions with terminal and internal epoxides. Green Chem. 2017, 19, 3769–3779. [Google Scholar] [CrossRef]
- Longwitz, L.; Steinbauer, J.; Spannenberg, A.; Werner, T. Calcium-Based Catalytic System for the Synthesis of Bio-Derived Cyclic Carbonates under Mild Conditions. ACS Catal. 2018, 8, 665–672. [Google Scholar] [CrossRef]
- Zhao, T.-X.; Zhang, Y.-Y.; Liang, J.; Li, P.; Hu, X.-B.; Wu, Y.-T. Multisite activation of epoxides by recyclable CaI2/N-methyldiethanolamine catalyst for CO2 fixation: A facile access to cyclic carbonates under mild conditions. Mol. Catal. 2018, 450, 87–94. [Google Scholar] [CrossRef]
- Chowdhury, A.H.; Bhanja, P.; Salam, N.; Bhaumik, A.; Islam, S.M. Magnesium oxide as an efficient catalyst for CO2 fixation and N-formylation reactions under ambient conditions. Mol. Catal. 2018, 450, 46–54. [Google Scholar] [CrossRef]
- Kilic, A.; Durgun, M.; Aytar, E.; Yavuz, R. Synthesis and characterization of novel positively charged organocobaloximes as catalysts for the fixation of CO2 to cyclic carbonates. J. Organomet. Chem. 2018, 858, 78–88. [Google Scholar] [CrossRef]
- Della Monica, F.; Buonerba, A.; Paradiso, V.; Milione, S.; Grassi, A.; Capacchione, C. [OSSO]-Type Fe(III) Metallate as Single-Component Catalyst for the CO2 Cycloaddition to Epoxides. Adv. Synth. Catal. 2018, 361, 283–288. [Google Scholar] [CrossRef]
- Kuznetsova, S.A.; Rulev, Y.A.; Larionov, V.A.; Smol’yakov, A.F.; Zubavichus, Y.V.; Maleev, V.I.; Li, H.; North, M.; Saghyan, A.S.; Belokon, Y.N. Self-Assembled Ionic Composites of Negatively Charged Zn(salen) Complexes and Triphenylmethane Derived Polycations as Recyclable Catalysts for the Addition of Carbon Dioxide to Epoxides. ChemCatChem 2019, 11, 511–519. [Google Scholar] [CrossRef]
- Milani, J.L.S.; Oliveira, I.S.; Dos Santos, P.A.; Valdo, A.K.S.M.; Martins, F.T.; Cangussu, D.; Das Chagas, R.P. Chemical fixation of carbon dioxide to cyclic carbonates catalyzed by zinc(II) complex bearing 1,2-disubstituted benzimidazole ligand. Chin. J. Catal. 2018, 39, 245–249. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Yang, Z.; Zhou, X.; Ji, H. Imidazolium-based ionic liquid decorated zinc porphyrin catalyst for converting CO2 into five-membered heterocyclic molecules. Sustain. Energy Fuels 2018, 2, 125–132. [Google Scholar] [CrossRef]
- Sogukomerogullari, H.G.; Aytar, E.; Ulusoy, M.; Demir, S.; Dege, N.; Richeson, D.S.; Sönmez, M. Synthesis of complexes Fe, Co and Cu supported by “SNS” pincer ligands and their ability to catalytically form cyclic carbonates. Inorg. Chim. Acta 2018, 471, 290–296. [Google Scholar] [CrossRef]
- Bresciani, G.; Marco Bortoluzzi, M.; Marchetti, F.; Pampaloni, G. Iron(III) N,N-Dialkylcarbamate-Catalyzed Formation of Cyclic Carbonates from CO2 and Epoxides under Ambient Conditions by Dynamic CO2 Trapping as Carbamato Ligands. ChemSusChem 2018, 11, 2737–2743. [Google Scholar] [CrossRef]
- Arayachukiat, S.; Kongtes, C.; Barthel, A.; Vummaleti, S.V.C.; Poater, A.; Wannakao, S.; Cavallo, L.; D’Elia, V. Ascorbic Acid as a Bifunctional Hydrogen Bond Donor for the Synthesis of Cyclic Carbonates from CO2 under Ambient Conditions. ACS Sustain. Chem. Eng. 2017, 5, 6392–6397. [Google Scholar] [CrossRef]
- Sopeña, S.; Martin, E.; Escudero-Adán, E.C.; Kleij, A.W. Pushing the Limits with Squaramide-Based Organocatalysts in Cyclic Carbonate Synthesis. ACS Catal. 2017, 7, 3532–3539. [Google Scholar] [CrossRef]
- Kumatabara, Y.; Okada, M.; Shirakawa, S. Triethylamine Hydroiodide as a Simple Yet Effective Bifunctional Catalyst for CO2 Fixation Reactions with Epoxides under Mild Conditions. ACS Sustain. Chem. Eng. 2017, 5, 7295–7301. [Google Scholar] [CrossRef]
- Bettner, H.; Steinbauer, J.; Wulf, C.; Dindaroglu, M.; Schmalz, H.-G.; Werner, T. Organocatalyzed Synthesis of Oleochemical Carbonates from CO2 and Renewables. ChemSusChem 2017, 10, 1076–1079. [Google Scholar] [CrossRef]
- Saptal, V.B.; Bhanage, B.M. Bifunctional Ionic Liquids Derived from Biorenewable Sources as Sustainable Catalysts for Fixation of Carbon Dioxide. ChemSusChem 2017, 10, 1145–1151. [Google Scholar] [CrossRef]
- Ma, R.; He, L.-N.; Liu, X.-F.; Liu, X.; Wang, M.-I. DBU as activator for the N-iodosuccinimide promoted chemical fixation of carbon dioxide with epoxides. J. CO2 Util. 2017, 19, 28–32. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, F.; Xing, H.; Yang, Q.; Bao, Z.; Ren, Q. Efficient Synthesis of Cyclic Carbonates from Atmospheric CO2 Using a Positive Charge Delocalized Ionic Liquid Catalyst. ACS Sustain. Chem. Eng. 2017, 5, 2841–2846. [Google Scholar] [CrossRef]
- Hirose, T.; Qu, S.; Kodama, K.; Wang, X. Organocatalyst system for disubstituted carbonates from cycloaddition between CO2 and internal epoxides. J. CO2 Util. 2018, 24, 261–265. [Google Scholar] [CrossRef]
- Wang, T.; Zheng, D.; Ma, Y.; Guo, J.; He, Z.; Ma, B.; Liu, L.; Ren, T.; Wang, L.; Zhang, J. Benzyl substituted imidazolium ionic liquids as efficient solvent-free catalysts for the cycloaddition of CO2 with epoxides: Experimental and Theoretic study. J. CO2 Util. 2017, 22, 44–52. [Google Scholar] [CrossRef]
- Yue, S.; Hao, X.J.; Wang, P.-P.; Li, J. Amino acid-based ionic liquids for CO2 conversion to form cyclic carbonate under solvent-free conditions. Mol. Catal. 2017, 433, 420–429. [Google Scholar] [CrossRef]
- Jose, T.; Cañellas, S.; Pericàs, M.A.; Kleij, A.W. Polystyrene-supported bifunctional resorcinarenes as cheap, metal-free and recyclable catalysts for epoxide/CO2 coupling reactions. Green Chem. 2017, 19, 5488–5493. [Google Scholar] [CrossRef]
- Li, K.; Wu, X.; Gu, Q.; Zhao, X.; Yuan, M.; Ma, W.; Ni, W.; Hou, Z. Inclusion complexes of organic salts with β-cyclodextrin as organocatalysts for CO2 cycloaddition with epoxides. RSC Adv. 2017, 7, 14721–14732. [Google Scholar] [CrossRef]
- Hu, J.; Ma, J.; Liu, H.; Qian, Q.; Xiea, C.; Han, B. Dual-ionic liquid system: An efficient catalyst for chemical fixation of CO2 to cyclic carbonates under mild conditions. Green Chem. 2018, 20, 2990–2994. [Google Scholar] [CrossRef]
- Peng, J.; Wang, S.; Yang, H.-J.; Ban, B.; Wei, Z.; Wang, L.; Lei, B. Highly efficient fixation of carbon dioxide to cyclic carbonates with new multi-hydroxyl bis-(quaternary ammonium) ionic liquids as metal-free catalysts under mild conditions. Fuel 2018, 224, 481–488. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Zhao, Y.; Kodama, K.; Hirose, T. Efficient and practical organocatalytic system for the synthesis of cyclic carbonates from carbon dioxide and epoxides: 3-hydroxypyridine /tetra-n-butylammonium iodide. Tetrahedron 2017, 73, 1190–1195. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Martínez, J.; de la Cruz-Martínez, F.; Caballero, M.P.; Fernández-Baeza, J.; Rodríguez-López, J.; Otero, A.; Lara-Sánchez, A.; Tejeda, J. Development of hydroxy-containing imidazole organocatalysts for CO2 fixation into cyclic carbonates. Catal. Sci. Technol. 2018, 8, 1981–1987. [Google Scholar] [CrossRef]
- Liu, A.-H.; Dang, Y.-L.; Zhou, H.; Zhang, J.-J.; Lu, X.-B. CO2 Adducts of Carbodicarbenes: Robust and Versatile Organocatalysts for Chemical Transformation of Carbon Dioxide into Heterocyclic Compounds. ChemCatChem 2018, 10, 2686–2692. [Google Scholar] [CrossRef]
- Maeda, C.; Sasaki, S.; Takaishi, K.; Ema, T. Calix[4]pyrroles as macrocyclic organocatalysts for the synthesis of cyclic carbonates from epoxides and carbon dioxide. Catal. Sci. Technol. 2018, 8, 4193–4198. [Google Scholar] [CrossRef]
- Hong, M.; Kim, Y.; Kim, H.; Cho, H.J.; Baik, M.-H.; Kim, Y. Scorpionate Catalysts for Coupling CO2 and Epoxides to Cyclic Carbonates: A Rational Design Approach for Organocatalysts. J. Org. Chem. 2018, 83, 9370–9380. [Google Scholar] [CrossRef]
- Liu, N.; Xie, Y.-F.; Wang, C.; Li, S.-J.; Wei, D.; Li, M.; Dai, B. Cooperative Multifunctional Organocatalysts for Ambient Conversion of Carbon Dioxide into Cyclic Carbonates. ACS Catal. 2018, 8, 9945–9957. [Google Scholar] [CrossRef]
- Mosteirin, N.F.; Jehanno, C.; Ruiperez, F.; Sardon, H.; Dove, A.P. Rational study of DBU salts for the CO2 insertion into epoxides for the synthesis of cyclic carbonates. ACS Sustain. Chem. Eng. 2019. [Google Scholar] [CrossRef]
- Wang, S.; Song, K.; Zhang, C.; Shu, Y.; Li, T.; Tan, B. A novel metalporphyrin-based microporous organic polymer with high CO2 uptake and efficient chemical conversion of CO2 under ambient conditions. J. Mater. Chem. A 2017, 5, 1509–1515. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Xu, Q.; Zhang, W.; Zhou, X.; Ji, H. State-of-the-Art Aluminum Porphyrin-based Heterogeneous Catalysts for the Chemical Fixation of CO2 into Cyclic Carbonates at Ambient Conditions. ChemCatChem 2017, 9, 767–773. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Xu, Q.; Jiang, J.; Zhou, X.; Ji, H. Charged Metalloporphyrin Polymers for Cooperative Synthesis of Cyclic Carbonates from CO2 under Ambient Conditions. ChemSusChem 2017, 10, 2534–2541. [Google Scholar] [CrossRef]
- Zhou, K.; Mousavi, B.; Luo, Z.; Phatanasri, S.; Chaemchuen, S.; Verpoort, F. Characterization and properties of Zn/Co zeolitic imidazolate frameworks vs. ZIF-8 and ZIF-67. J. Mater. Chem. A 2017, 5, 952–957. [Google Scholar] [CrossRef]
- Zanon, A.; Chaemchuen, S.; Mousavi, B.; Verpoort, F. 1 Zn-doped ZIF-67 as catalyst for the CO2 fixation into cyclic carbonates. J. CO2 Util. 2017, 20, 282–291. [Google Scholar] [CrossRef]
- Buyukcakir, O.; Je, S.H.; Talapaneni, S.N.; Kim, D.; Coskun, A. Charged Covalent Triazine Frameworks for CO2 Capture and Conversion. ACS Appl. Mater. Interfaces 2017, 9, 7209–7216. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, Q.; Shi, Y.; Li, J.; Yang, X.; Hou, W.; Zhou, Y.; Wang, J. Tethering Dual Hydroxyls into Mesoporous Poly(ionic liquid)s for Chemical Fixation of CO2 at Ambient Conditions: A Combined Experimental and Theoretical Study. ACS Catal. 2017, 7, 6770–6780. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Phang, H.S.; Li, Y.; Zhao, Y. Highly Effective Carbon Fixation via Catalytic Conversion of CO2 by an Acylamide-Containing Metal−Organic Framework. Chem. Mater. 2017, 29, 9256–9261. [Google Scholar] [CrossRef]
- Lu, B.-B.; Yang, J.; Liu, Y.-Y.; Ma, J.F. A Polyoxovanadate−Resorcin[4]arene-Based Porous Metal−Organic Framework as an Efficient Multifunctional Catalyst for the Cycloaddition of CO2 with Epoxides and the Selective Oxidation of Sulfides. Inorg. Chem. 2017, 56, 11710–11720. [Google Scholar] [CrossRef]
- Mousavi, B.; Chaemchuen, S.; Moosavi, B.; Zhou, K.; Yusubov, M.; Verpoort, F. CO2 Cycloaddition to Epoxides by using M-DABCO Metal-Organic Frameworks and the Influence of the Synthetic Method on Catalytic Reactivity. ChemistryOpen 2017, 6, 674–680. [Google Scholar] [CrossRef]
- Ding, M.; Chen, S.; Liu, X.-Q.; Sun, L.-B.; Lu, J.; Jiang, H.-L. Metal–Organic Framework-Templated Catalyst: Synergy in Multiple Sites for Catalytic CO2 Fixation. ChemSusChem 2017, 10, 1898–1903. [Google Scholar] [CrossRef]
- Song, L.; Zhang, X.; Chen, C.; Liu, X.; Zhang, N. UTSA-16 as an efficient microporous catalyst for CO2 conversion to cyclic carbonates. Microporous Mesoporous Mater. 2017, 241, 36–42. [Google Scholar] [CrossRef]
- Liu, M.; Lan, J.; Liang, L.; Sun, J.; Arai, M. Heterogeneous catalytic conversion of CO2 and epoxides to cyclic carbonates over multifunctional tri-s-triazine terminal-linked ionic liquids. J. Catal. 2017, 347, 138–147. [Google Scholar] [CrossRef]
- Zhong, W.; Bobbink, F.D.; Fei, Z.; Dyson, P.J. Polyimidazolium Salts: Robust Catalysts for the Cycloaddition of Carbon Dioxide into Carbonates in Solvent-Free Conditions. ChemSusChem 2017, 10, 2728–2735. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Van Muyden, A.P.; Gopakumar, A.; Fei, Z.; Dyson, P.J. Synthesis of Cross-linked Ionic Poly(styrenes) and their Application as Catalysts for the Synthesis of Carbonates from CO2 and Epoxides. ChemPlusChem 2017, 82, 144–151. [Google Scholar] [CrossRef]
- Zhang, X.; Su, D.; Xiao, L.; Wu, W. Immobilized protic ionic liquids: Efficient catalysts for CO2 fixation with epoxides. J. CO2 Util. 2017, 17, 37–42. [Google Scholar] [CrossRef]
- Ravi, S.; Puthiaraj, P.; Ahn, W.-S. Cyclic carbonate synthesis from CO2 and epoxides over diamine functionalized porous organic frameworks. J. CO2 Util. 2017, 21, 450–458. [Google Scholar] [CrossRef]
- Steinbauer, J.; Longwitz, L.; Frank, M.; Epping, J.; Kragld, U.; Werner, T. Immobilized bifunctional phosphonium salts as recyclable organocatalysts in the cycloaddition of CO2 and epoxides. Green. Chem. 2017, 19, 4435–4445. [Google Scholar] [CrossRef]
- Zhi, Y.; Shao, P.; Feng, X.; Xia, H.; Zhang, Y.; Shi, Z.; Mua, Y.; Liu, X. Covalent organic frameworks: Efficient, metal-free, heterogeneous organocatalysts for chemical fixation of CO2 under mild conditions. J. Mater. Chem. A 2018, 6, 374–382. [Google Scholar] [CrossRef]
- Maya, M.M.; Rangel-Rangel, E.; Díaz, U.; Iglesias, M. Efficient cycloaddition of CO2 to epoxides using novel heterogeneous organocatalysts based on tetramethylguanidine-functionalized porous polyphenylenes. J. CO2 Util. 2018, 25, 170–179. [Google Scholar] [CrossRef]
- Song, X.; Wu, Y.; Pan, D.; Wei, R.; Gao, L.; Zhang, J.; Xiao, G. Melem based multifunctional catalyst for chemical fixation of carbon dioxide into cyclic carbonate. J. CO2 Util. 2018, 24, 287–297. [Google Scholar] [CrossRef]
- Subramanian, S.; Park, J.; Byun, J.; Jung, Y.; Yavuz, C.T. Highly Efficient Catalytic Cyclic Carbonate Formation by Pyridyl Salicylimines. ACS Appl. Mater. Interfaces 2018, 10, 9478–9484. [Google Scholar] [CrossRef]
- Wang, X.; Liu, M.S.; Yang, L.; Lan, J.W.; Chen, Y.L.; Sun, J.M. Synthesis of Zn Modified Carbon Nitrides Heterogeneous Catalyst for the Cycloaddition of CO2 to Epoxides. ChemistrySelect 2018, 3, 4101–4109. [Google Scholar] [CrossRef]
- Liu, D.; Li, G.; Liu, H. Functionalized MIL-101 with imidazolium-based ionic liquids for the cycloaddition of CO2 and epoxides under mild condition. Appl. Surf. Sci. 2018, 428, 218–225. [Google Scholar] [CrossRef]
- Shi, X.-L.; Chen, Y.; Duan, P.; Zhang, W.; Hu, Q. Conversion of CO2 into Organic Carbonates over a Fiber-Supported Ionic Liquid Catalyst in Impellers of the Agitation System. ACS Sustain. Chem. Eng. 2018, 6, 7119–7127. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Tao, L.; Li, C.; Liu, L.; Chen, J.; Yang, Q. Cationic Zn-Porphyrin Immobilized in Mesoporous Silicas as Bifunctional Catalyst for CO2 Cycloaddition Reaction under Cocatalyst Free Conditions. ACS Sustain. Chem. Eng. 2018, 6, 9237–9245. [Google Scholar] [CrossRef]
- Jayakumar, S.; Li, H.; Chen, J.; Yang, Q. Cationic Zn−Porphyrin Polymer Coated onto CNTs as a Cooperative Catalyst for the Synthesis of Cyclic Carbonates. ACS Appl. Mater. Interfaces 2018, 10, 2546–2555. [Google Scholar] [CrossRef]
- Tiffner, M.; Häring, M.; Díaz Díaz, D.; Waser, M. Cationic Polymers Bearing Quaternary Ammonium Groups-Catalyzed CO2 Fixation with Epoxides. Top. Catal. 2018, 61, 1545–1550. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, S.; Wu, Q.; Wang, X.; Wu, H.; Jiang, Z. Heterobimetallic Metal-Organic Framework Nanocages as Highly Efficient Catalysts for CO2 Conversion under Mild Condition. J. Mater. Chem. A 2018, 6, 2964–2973. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Wang, H.-H.; Yang, H.-Y.; Hou, L.; Wang, Y.-Y.; Zhu, Z. An Uncommon Carboxyl-Decorated Metal–Organic Framework with Selective Gas Adsorption and Catalytic Conversion of CO2. Chem. Eur. J. 2018, 24, 865–871. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, H.; Wu, H.; Qian, Y.; Chen, L.; Chen, J. Chemical Fixation of CO2 by Using Carbon Material-Grafted N-Heterocyclic Carbene Silver and Copper Complexes. ACS Appl. Nano Mater. 2018, 1, 6463–6476. [Google Scholar] [CrossRef]
- Calabrese, C.; Liotta, L.F.; Giacalone, F.; Gruttadauria, M.; Aprile, C. Supported Polyhedral Oligomeric Silsesquioxane-Based (POSS) Materials as Highly Active Organocatalysts for the Conversion of CO2. ChemCatChem 2019, 11, 560–567. [Google Scholar] [CrossRef]
- Rintjema, J.; Kleij, A.W. Aluminum-mediated formation of cyclic carbonates: Benchmarking catalytic performance metrics. ChemSusChem 2017, 10, 1274–1282. [Google Scholar] [CrossRef]
- Yingcharoen, P.; Kongtes, C.; Arayachukiat, S.; Suvarnapunya, K.; Vummaleti, S.V.C.; Wannakao, S.; Cavallo, L.; Poater, A.; D’Elia, V. Assessing the pKa-Dependent Activity of Hydroxyl Hydrogen Bond Donors in the Organocatalyzed Cycloaddition of Carbon Dioxide to Epoxides: Experimental and Theoretical Study. Adv. Synth. Catal. 2019, 361, 366–373. [Google Scholar] [CrossRef]
- Laserna, V.; Martin, E.; Escudero-Adán, E.C.; Kleij, A.W. Substrate-Triggered Stereoselective Preparation of Highly Substituted Organic Carbonates. ACS Catal. 2017, 7, 5478–5482. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, J.; Kim, H. Stereocontrolled, Divergent, Al(lll)-Catalyzed Coupling of Chiral N-Aryl Epoxy Amines and CO2. Org. Lett. 2018, 20, 5036–5039. [Google Scholar] [CrossRef]
- Jawad, A.; Rezaei, F.; Rownaghi, A.A. Porous polymeric hollow fibers as bifunctional catalysts for CO2 conversion to cyclic carbonates. J. CO2 Util. 2017, 21, 589–596. [Google Scholar] [CrossRef]
- Liu, F.; Huang, K.; Wu, Q.; Dai, S. Solvent-Free Self-Assembly to the Synthesis of Nitrogen-Doped Ordered Mesoporous Polymers for Highly Selective Capture and Conversion of CO2. Adv. Mater. 2017, 29, 1700445. [Google Scholar] [CrossRef]
- Kim, K.; Kim, S.; Talapaneni, S.N.; Buyukcakir, O.; Almutawa, A.M.I.; Polychronopoulou, K.; Coskun, A. Transition metal complex directed synthesis of porous cationic polymers for efficient CO2 capture and conversion. Polymer 2017, 126, 296–302. [Google Scholar] [CrossRef]
- Mirabaud, A.; Mulatier, J.-C.; Martinez, A.; Dutasta, J.-P.; Dufaud, V. Merging host-guest chemistry and organocatalysis for the chemical valorization of CO2. Catal. Today 2017, 281, 387–391. [Google Scholar] [CrossRef]
- Bresciani, G.; Marchetti, F.; Rizzi, G.; Gabbani, A.; Pineider, F.; Pampaloni, G. Metal N,N-dialkylcarbamates as easily available catalytic precursors for the carbon dioxide/propylene oxide coupling under ambient conditions. J. CO2 Util. 2018, 28, 168–173. [Google Scholar] [CrossRef]
- Samanta, S.; Srivastava, R. A novel method to introduce acidic and basic bifunctional sites in graphitic carbon nitride for sustainable catalysis: Cycloaddition, esterification, and transesterification reactions. Sustain. Energy Fuels 2017, 1, 1390–1404. [Google Scholar] [CrossRef]
- Kim, H.G.; Limb, C.S.; Kim, D.-W.; Cho, D.H.; Lee, D.K.; Chung, J.S. Multifunctional alkanolamine as a catalyst for CO2 and propylene oxide cycloaddition. Mol. Catal. 2017, 438, 121–129. [Google Scholar] [CrossRef]
- Onyenkeadi, V.; Kellici, S.; Saha, B. Greener synthesis of 1,2-butylene carbonate from CO2 using graphene-inorganic nanocomposite catalyst. Energy 2018, 165A, 867–876. [Google Scholar] [CrossRef]
- Liu, W.; Lu, G.; Xiao, B.; Xie, C. Potassium iodide–polyethylene glycol catalyzed cycloaddition reaction of epoxidized soybean oil fatty acid methyl esters with CO2. RSC Adv. 2018, 8, 30860–30867. [Google Scholar] [CrossRef]
- Liu, S.; Suematsu, N.; Maruoka, K.; Shirakawa, S. Design of bifunctional quaternary phosphonium salt catalysts for CO2 fixation reaction with epoxides under mild conditions. Green Chem. 2016, 18, 4611–4615. [Google Scholar] [CrossRef]
- Ema, T.; Yokoyama, M.; Watanabe, S.; Sasaki, S.; Ota, H.; Takaishi, K. Chiral Macrocyclic Organocatalysts for Kinetic Resolution of Disubstituted Epoxides with Carbon Dioxide. Org. Lett. 2017, 19, 4070–4073. [Google Scholar] [CrossRef]
- Maeda, C.; Mitsuzane, M.; Ema, T. Chiral Bifunctional Metalloporphyrin Catalysts for Kinetic Resolution of Epoxides with Carbon Dioxide. Org. Lett. 2019, 21, 1397–1401. [Google Scholar] [CrossRef]
- Qin, J.; Larionov, V.A.; Harms, K.; Meggers, E. Kinetic Resolution of Epoxides with CO2 Catalyzed by a Chiral-at-Iridium Complex. ChemSusChem 2019, 12, 320–325. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Boyaval, A.; Méreau, R.; De Winter, J.; Gerbaux, P.; Detrembleur, C.; Tassaing, T.; Jérôme, C. Organocatalytic Coupling of CO2 with Oxetane. ChemSusChem 2017, 10, 1128–1138. [Google Scholar] [CrossRef]
- Huang, J.; de Winter, J.; Dove, A.P.; Coulembier, O. Metal-free synthesis of poly(trimethylene carbonate) by efficient valorization of carbon dioxide. Green Chem. 2019, 21, 472–477. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, G.-X.; Lu, X.-B. CO2 Adducts of α-Carbon Alkylated N-Heterocyclic Olefins: Highly Active Organocatalysts for CO2 Chemical Transformation. Asian. J. Org. Chem. 2017, 6, 1264–1269. [Google Scholar] [CrossRef]
- Qiu, J.; Zhao, Y.; Li, Z.; Wang, H.; Fan, M.; Wang, J. Efficient Ionic-Liquid-Promoted Chemical Fixation of CO2 into α-Alkylidene Cyclic Carbonates. ChemSusChem 2017, 10, 1120–1127. [Google Scholar] [CrossRef]
- Panwar, V.; Jain, S.L. Zinc grafted to magnetic nanostarch for cyclic carbonate synthesis from propargylic alcohols and CO2 at room temperature. J. CO2 Util. 2018, 24, 306–314. [Google Scholar] [CrossRef]
- Grignard, B.; Ngassamtounzoua, C.; Gennen, S.; Gilbert, B.; Méreau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Boosting the Catalytic Performance of Organic Salts for the Fast and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Propargylic Alcohols. ChemCatChem 2018, 10, 2584–2592. [Google Scholar] [CrossRef]
- Jung, M.E.; Piizi, G. gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev. 2005, 105, 1735–1766. [Google Scholar] [CrossRef]
- Boyaval, A.; Méreau, R.; Grignard, B.; Detrembleur, C.; Jerome, C.; Tassaing, T. Organocatalytic Coupling of CO2 with a Propargylic Alcohol: A Comprehensive Mechanistic Study. ChemSusChem 2017, 10, 1241–1248. [Google Scholar] [CrossRef]
- Della Ca’, N.; Gabriele, B.; Ruffolo, G.; Veltri, L.; Zanetta, T.; Costa, M. Effective Guanidine-Catalyzed Synthesis of Carbonate and Carbamate Derivatives from Propargyl Alcohols in Supercritical Carbon Dioxide. Adv. Synth. Catal. 2011, 353, 133–146. [Google Scholar] [CrossRef]
- Méreau, R.; Grignard, B.; Boyaval, A.; Detrembleur, C.; Jerome, C.; Tassaing, T. Tetrabutylammonium Salts: Cheap Catalysts for the Facile and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Alkynols. ChemCatChem 2018, 10, 956–960. [Google Scholar] [CrossRef]
- Sun, S.; Wang, B.; Gu, N.; Yu, J.-T.; Cheng, J. Palladium-Catalyzed Arylcarboxylation of Propargylic Alcohols with CO2 and Aryl Halides: Access to Functionalized α-Alkylidene Cyclic Carbonates. Org. Lett. 2017, 19, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.L.; Ulmann, M.; Buchard, A. Synthesis of 6-membered cyclic carbonates from 1,3-diols and low CO2 pressure: A novel mild strategy to replace phosgene reagents. RSC Adv. 2015, 5, 39404–39408. [Google Scholar] [CrossRef]
- McGuire, T.M.; López-Vidal, E.M.; Gregory, G.L.; Buchard, A. Synthesis of 5- to 8-membered cyclic carbonates from diols and CO2: A onestep, atmospheric pressure and ambient temperature procedure. J. CO2 Util. 2018, 27, 283–288. [Google Scholar] [CrossRef]
- Pati, D.; Chen, Z.; Feng, X.; Hadjichristidis, N.; Gnanou, Y. Synthesis of polyglycocarbonates through polycondensation of glucopyranosides with CO2. Polym. Chem. 2017, 8, 2640–2646. [Google Scholar] [CrossRef]
- Pati, D.; Feng, X.; Hadjichristidis, N.; Gnanou, Y. CO2 as versatile carbonation agent of glycosides: Synthesis of 5- and 6-membered cyclic glycocarbonates and investigation of their ring-opening. J. CO2 Util. 2018, 24, 564–571. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, C.; Zhao, B.; Wang, Q.; Yao, Y. Cycloaddition of Aziridine with CO2/CS2 Catalyzed by Amidato Divalent Lanthanide Complexes. J. Org. Chem. 2019, 84, 1951–1958. [Google Scholar] [CrossRef]
- Phung, C.; Tantillo, D.J.; Hein, J.E.; Pinhas, A.R. The mechanism of the reaction between an aziridine and carbon dioxide with no added catalyst. J. Phys. Org. Chem. 2018, 31, e3735. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, J.-H.; Wu, D.-S.; Zhou, Y.-Q.; Yu, D.-G. Synthesis of Oxazolidin-2-ones from Unsaturated Amines with CO2 by Using Homogeneous Catalysis. Chem. Asian. J. 2018, 13, 2292–2306. [Google Scholar] [CrossRef]
- Yuan, R.; Wei, B.; Fu, G. How the Coordinated Structures of Ag(I) Catalysts Affect the Outcomes of Carbon Dioxide Incorporation into Propargylic Amine: A DFT Study. J. Org. Chem. 2017, 82, 3639–3647. [Google Scholar] [CrossRef]
- Brunel, P.; Monot, J.; Kefalidis, C.E.; Maron, L.; Martin-Vaca, B.; Bourissou, D. Valorization of CO2: Preparation of 2-Oxazolidinones by Metal-Ligand Cooperative Catalysis with SCS Indenediide Pd Complexes. ACS Catal. 2017, 7, 2652–2660. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, X.-H.; Zhu, C.; Kang, Y.-S.; Wang, P.; Shi, Z.; Lu, Y.; Sun, W.-Y. Efficient and Reusable Metal–Organic Framework Catalysts for Carboxylative Cyclization of Propargylamines with Carbon Dioxide. ChemCatChem 2017, 9, 4598–4606. [Google Scholar] [CrossRef]
- Yu, B.; Kim, D.; Kim, S.; Hong, S.H. Cyanuric Acid-Based Organocatalyst for Utilization of Carbon Dioxide at Atmospheric Pressure. ChemSusChem 2017, 10, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, M.-Y.; Wang, S.-Y.; Wang, Q.; He, L.-N. In Situ Generated Zinc(II) Catalyst for Incorporation of CO2 into 2-Oxazolidinones with Propargylic Amines at Atmospheric Pressure. ChemSusChem 2017, 10, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiu, J.; Li, Z.; Wang, H.; Fan, M.; Wang, J. An Experimental and Theoretical Study on the Unexpected Catalytic Activity of Triethanolamine for the Carboxylative Cyclization of Propargylic Amines with CO2. ChemSusChem 2017, 10, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Choi, J.-C.; Fujita, K.-i. Quaternary ammonium salt-catalyzed carboxylative cyclization of propargylic amines with CO2. Tetrahedron Lett. 2017, 58, 4483–4486. [Google Scholar] [CrossRef]
- Fujii, A.; Matsuo, H.; Choi, J.-C.; Fujitani, T.; Fujita, K.-i. Efficient synthesis of 2-oxazolidinones and quinazoline-2,4(1H,3H)-diones from CO2 catalyzed by tetrabutylammonium fluoride. Tetrahedron 2018, 74, 2914–2920. [Google Scholar] [CrossRef]
- Inagaki, F.; Maeda, K.; Nakazawa, K.; Mukai, C. Construction of the Oxazolidinone Framework from Propargylamine and CO2 in Air at Ambient Temperature: Catalytic Effect of a Gold Complex Featuring an L2/Z-Type Ligand. Eur. J. Org. Chem. 2018, 2972–2976. [Google Scholar] [CrossRef]
- Sadeghzadeh, S.M.; Zhiani, R.; Emrani, S. Ni@Pd nanoparticles supported on ionic liquid-functionalized KCC-1 as robust and recyclable nanocatalysts for cycloaddition of propargylic amines and CO2. Appl. Organometal. Chem. 2018, 32, e3941. [Google Scholar] [CrossRef]
- Yoshida, M.; Mizuguchi, T.; Shishido, K. Synthesis of Oxazolidinones by Efficient Fixation of Atmospheric CO2 with Propargylic Amines by using a Silver/1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) Dual-Catalyst System. Chem. Eur. J. 2012, 18, 15578–15581. [Google Scholar] [CrossRef]
- Inagaki, F.; Okada, Y.; Matsumoto, C.; Yamada, M.; Nakazawa, K.; Mukai, C. Energyless CO2 Absorption, Generation, and Fixation Using Atmospheric CO2. Chem. Pharm. Bull. 2016, 64, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-T.; Gan, C.-C.; Liu, S.-I.; Zhou, F.; Wu, H.-H.; Zhou, J. Utilization of CO2 as a C1 Building Block in a Tandem Asymmetric A3 Coupling-Carboxylative Cyclization Sequence to 2-Oxazolidinones. ACS Catal. 2017, 7, 8588–8593. [Google Scholar] [CrossRef]
- Kim, N.-K.; Sogawa, H.; Yamamoto, K.; Hayashi, Y.; Kawauchi, S.; Takata, T. DBU-mediated Highly Efficient CO2-fixation to Propargylamines and Polypropargylamine. Chem. Lett. 2018, 47, 1063–1066. [Google Scholar] [CrossRef]
- Kim, N.-K.; Sogawa, H.; Felicia, M.D.; Takata, T. DBU-catalyzed CO2 fixation in polypropargylamines under solvent-free conditions. Polym. J. 2019, 51, 351–357. [Google Scholar] [CrossRef]
- Sugiyama, N.; Ohseki, M.; Kobayashi, R.; Sekine, K.; Saito, K.; Yamada, T. Silver-catalyzed Three-component Reaction of Propargylic Amines, Carbon Dioxide, and N-Bromosuccinimide for Stereoselective Preparation of (E)-Bromovinyloxazolidinones. Chem. Lett. 2017, 46, 1323–1326. [Google Scholar] [CrossRef]
- Yin, Z.-B.; Ye, J.-H.; Zhou, W.-J.; Zhang, Y.-H.; Ding, L.; Gui, Y.-Y.; Yan, S.-S.; Li, J.; Yu, D.-G. Oxy-Difluoroalkylation of Allylamines with CO2 via Visible-Light Photoredox Catalysis. Org. Lett. 2018, 20, 190–193. [Google Scholar] [CrossRef]
- Sun, L.; Ye, J.-H.; Zhou, W.-J.; Zeng, X.; Yu, D.-G. Oxy-Alkylation of Allylamines with Unactivated Alkyl Bromides and CO2 via Visible-Light-Driven Palladium Catalysis. Org. Lett. 2018, 20, 3049–3052. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Li, S.-H. Photoinduced radical-initiated carboxylative cyclization of allyl amines with carbon dioxide. Green Chem. 2017, 19, 1240–1244. [Google Scholar] [CrossRef]
- Seo, U.R.; Chung, Y.K. Potassium phosphate-catalyzed one-pot synthesis of 3-aryl-2-oxazolidinones from epoxides, amines, and atmospheric carbon dioxide. Green Chem. 2017, 19, 803–808. [Google Scholar] [CrossRef]
- Lv, M.; Wang, P.; Yuan, D.; Yao, Y. Conversion of Carbon Dioxide into Oxazolidinones Mediated by Quaternary Ammonium Salts and DBU. ChemCatChem 2017, 9, 4451–4455. [Google Scholar] [CrossRef]
- Chen, F.; Li, M.; Wang, J.; Dai, B.; Liu, N. Fe(II) complexes: Reservoirs for Lewis acids and carbenes and their utility in the conversion of CO2 to oxazolidinones. J. CO2 Util. 2018, 28, 181–188. [Google Scholar] [CrossRef]
- Sadeghzadeh, S.M.; Zhiani, R.; Moradi, M. KCC-1 Supported Cu(II)-β-Cyclodextrin Complex as a Reusable Catalyst for the Synthesis of 3-Aryl-2-oxazolidinones from Carbon Dioxide, Epoxide, Anilines. ChemistrySelect 2018, 3, 3516–3522. [Google Scholar] [CrossRef]
- Mancuso, R.; Raut, D.S.; Della Ca’, N.; Fini, F.; Carfagna, C.; Gabriele, B. Catalytic oxidative carbonylation of amino moieties to ureas, oxamides, 2-oxazolidinones, and benzoxazolones. ChemSusChem 2015, 8, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Song, Q.-W.; Lang, X.-D.; Chang, Y.; He, L.-N. AgI/TMG-Promoted Cascade Reaction of Propargyl Alcohols, Carbon Dioxide, and 2-Aminoethanols to 2-Oxazolidinones. ChemPhysChem 2017, 18, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Cao, Y.; Ma, R.; He, L.-N. Thermodynamically favorable protocol for the synthesis of 2-oxazolidinones via Cu(I)-catalyzed three-component reaction of propargylic alcohols, CO2 and 2-aminoethanols. J. CO2 Util. 2018, 25, 338–345. [Google Scholar] [CrossRef]
- Xia, S.; Song, Y.; Li, X.; Li, H.; He, L.-N. Ionic Liquid-Promoted Three-Component Domino Reaction of Propargyl Alcohols, Carbon Dioxide and 2-Aminoethanols: A Thermodynamically Favorable Synthesis of 2-Oxazolidinones. Molecules 2018, 23, 3033. [Google Scholar] [CrossRef] [PubMed]
- Niemi, T.; Fernández, I.; Steadman, B.; Mannisto, J.K.; Repo, T. Carbon dioxide-based facile synthesis of cyclic carbamates from amino alcohols. Chem. Commun. 2018, 54, 3166–3169. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.; Zhao, Y.; Chen, Q.; Cao, C.; Pang, G.; Shi, Y. Synthesis of Oxazolidinones and Derivatives through Three-Component Fixation of Carbon Dioxide. ChemCatChem 2018, 10, 3057–3068. [Google Scholar] [CrossRef]
- Yousefi, R.; Struble, T.J.; Payne, J.L.; Vishe, M.; Schley, N.D.; Johnston, J.N. Catalytic, Enantioselective Synthesis of Cyclic Carbamates from Dialkyl Amines by CO2-Capture: Discovery, Development, and Mechanism. J. Am. Chem. Soc. 2019, 141, 618–625. [Google Scholar] [CrossRef]
- Nikpassand, M.; Fekri, L.Z.; Pourahmad, A. One-pot Synthesis of new azo-linked 4H-benzo[d][1,3]oxazine-2,4-diones from carbon dioxide using CuO@RHA/MCM-41 nanocomposite in green media. J. CO2 Util. 2018, 27, 320–325. [Google Scholar] [CrossRef]
- Guo, C.-X.; Zhang, W.-Z.; Zhang, N.; Lu, X.-B. 1,3-Dipolar Cycloaddition of Nitrile Imine with Carbon Dioxide: Access to 1,3,4-Oxadiazole-2(3H)-ones. J. Org. Chem. 2017, 82, 7637–7642. [Google Scholar] [CrossRef]
- Murillo, F.; Barroso, J.; de los Santos, M.G.; Ávila, G.; Pan, S.; Fernández-Herrera, M.A.; Merino, G. Revisiting the Formation Mechanism of 1,3,4-Oxadiazole-2(3H)-ones from Hydrazonyl Chloride and Carbon Dioxide. J. Org. Chem. 2018, 83, 13045–13050. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Zhang, H.; Yuan, G. KI-catalyzed reactions of aryl hydrazines with α-oxocarboxylic acids in the presence of CO2: Access to 1,3,4-oxadiazol-2(3H)-ones. Org. Chem. Front. 2019, 6, 532–536. [Google Scholar] [CrossRef]
- Brahmayya, M.; Dai, S.A.; Suen, S.-Y. Facile synthesis of 2-benzimidazolones via carbonylation of o-phenylenediamines with CO2. J. CO2 Util. 2017, 22, 135–142. [Google Scholar] [CrossRef]
- Wang, H.; Xin, Z.; Li, Y. Synthesis of Ureas from CO2. Top. Curr. Chem. 2017, 375, 49. [Google Scholar] [CrossRef]
- He, X.; Li, X.-Y.; Song, Y.; Xia, S.-M.; Lang, X.-D.; He, L.-N. Synthesis of Urea Derivatives using Carbon Dioxide as Carbonylation Reagent in Ionic Liquids. Curr. Organocat. 2017, 4, 112–121. [Google Scholar] [CrossRef]
- Hulla, M.; Chamam, S.M.A.; Laurenczy, G.; Das, S.; Dyson, P.J. Delineating the Mechanism of Ionic Liquids in the Synthesis of Quinazoline-2,4(1H,3H)-dione from 2-Aminobenzonitrile and CO2. Angew. Chem. Int. Ed. 2017, 56, 10559–10563. [Google Scholar] [CrossRef] [PubMed]
- Sabet-Sarvestani, H.; Eshghi, H.; Izadyar, M. A theoretical study on the efficiency and role of guanidines-based organic superbases on carbon dioxide utilization in quinazoline-2,4(1H, 3H)-diones synthesis. Struct. Chem. 2017, 28, 675–686. [Google Scholar] [CrossRef]
- Yan, C.; Ren, Y.; Jia, J.-F.; Wu, H.-S. Mechanism of the chemical fixation of carbon dioxide with 2-aminobenzonitrile catalyzed by cesium carbonate: A computational study. Mol. Catal. 2017, 432, 172–186. [Google Scholar] [CrossRef]
- Li, C.; Lu, X.; Yang, Y.; Yang, S.; Zhang, L. Ionic liquid promoted synthesis of heterocycle-fused pyrimidine-2,4-(1H,3H)-diones utilising CO2. Tetrahedron Lett. 2018, 59, 2463–2466. [Google Scholar] [CrossRef]
- Shi, G.; Chen, K.; Wang, Y.; Li, H.; Wang, C. Highly Efficient Synthesis of Quinazoline-2,4(1H,3H)-diones from CO2 by Hydroxyl Functionalized Aprotic Ionic Liquids. ACS Sustain. Chem. Eng. 2018, 6, 5760–5765. [Google Scholar] [CrossRef]
- Hu, J.; Chen, S.; Guo, Y.; Li, L.; Deng, T. Basic Salt-Lake Brine: An Efficient Catalyst for the Transformation of CO2 into Quinazoline-2,4(1H,3H)-diones. ChemSusChem 2018, 11, 4219–4225. [Google Scholar] [CrossRef] [PubMed]
- Mampuys, P.; Neumann, H.; Sergeyev, S.; Orru, R.V.A.; Jiao, H.; Spannenberg, A.; Maes, B.U.W.; Beller, M. Combining Isocyanides with Carbon Dioxide in Palladium-Catalyzed Heterocycle Synthesis: N3-Substituted Quinazoline-2,4-(1H,3H)-diones via a Three-Component Reaction. ACS Catal. 2017, 7, 5549–5556. [Google Scholar] [CrossRef]
- Xu, P.; Wang, F.; Wei, T.-Q.; Yin, L.; Wang, S.-Y.; Ji, S.J. Palladium-Catalyzed Incorporation of Two C1 Building Blocks: The Reaction of Atmospheric CO2 and Isocyanides with 2-Iodoanilines Leading to the Synthesis of Quinazoline-2,4(1H,3H)-diones. Org. Lett. 2017, 19, 4484–4487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Z.; Li, H.; Zeng, Y.; Tao, X.; Lu, X. Palladium-Catalyzed Cyclization Reaction of o-Haloanilines, CO2 and Isocyanides: Access to Quinazoline-2,4(1H,3H)-diones. Chin. J. Chem. 2018, 36, 112–118. [Google Scholar] [CrossRef]
- Jin, S.-J.; Khan, Y.; Maeng, J.-H.; Kim, Y.J.; Hwang, J.; Cheong, M.; Lee, J.S.; Kim, H.S. Efficient catalytic systems for the carboxylation of diamines to cyclic ureas using ethylene urea as a promoter. Appl. Catal. B Environ. 2017, 209, 139–145. [Google Scholar] [CrossRef]
- Hwang, J.; Han, D.; Oh, J.J.; Cheong, M.; Koo, H.-J.; Lee, J.S.; Kim, H.S. Efficient Non-Catalytic Carboxylation of Diamines to Cyclic Ureas Using 2-Pyrrolidone as a Solvent and a Promoter. Adv. Synth. Catal. 2019, 361, 297–306. [Google Scholar] [CrossRef]
- Del Vecchio, A.; Caillé, F.; Chevalier, A.; Loreau, O.; Horkka, K.; Halldin, C.; Schou, M.; Camus, N.; Kessler, P.; Kuhnast, B.; et al. Late-Stage Isotopic Carbon Labeling of Pharmaceutically Relevant Cyclic Ureas Directly from CO2. Angew. Chem. Int. Ed. 2018, 57, 9744–9748. [Google Scholar] [CrossRef] [PubMed]
- Streng, E.S.; Lee, D.S.; George, M.W.; Poliakoff, M. Continuous N-alkylation reactions of amino alcohols using γ-Al2O3 and supercritical CO2: Unexpected formation of cyclic ureas and urethanes by reaction with CO2. Beilstein J. Org. Chem. 2017, 13, 329–337. [Google Scholar] [CrossRef]
- Marchegiani, M.; Nodari, M.; Tansini, F.; Massera, C.; Mancuso, R.; Gabriele, B.; Costa, M.; Della Cà, N. Urea derivatives from carbon dioxide and amines by guanidine catalysis: Easy access to imidazolidin-2-ones under solvent-free conditions. J. CO2 Util. 2017, 21, 553–561. [Google Scholar] [CrossRef]
- Chun, S.; Yang, S.; Chung, Y.K. Synthesis of benzothiazoles from 2-aminobenzenethiols in the presence of a reusable polythiazolium precatalyst under atmospheric pressure of carbon dioxide. Tetrahedron 2017, 73, 3438–3442. [Google Scholar] [CrossRef]
- Wang, B.; Sun, S.; Yu, J.-T.; Jiang, Y.; Cheng, J. Palladium-Catalyzed Multicomponent Reactions of o-Alkynylanilines, Aryl Iodides, and CO2 toward 3,3-Diaryl 2,4-Quinolinediones. Org. Lett. 2017, 19, 4319–4322. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, C.; Lyu, Y. Lactamization of sp2 C–H bonds with CO2 under transition-metal-free and redox-neutral conditions: A computational mechanistic study. Org. Chem. Front. 2018, 5, 2189–2201. [Google Scholar] [CrossRef]
- Wang, S.; Xi, C. Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles. Chem. Soc. Rev. 2019, 48, 382–404. [Google Scholar] [CrossRef] [PubMed]
- Niemi, T.; Repo, T. Antibiotics from Carbon Dioxide: Sustainable Pathways to Pharmaceutically Relevant Cyclic Carbamates. Eur. J. Org. Chem. 2019, 1180–1188. [Google Scholar] [CrossRef]
- Vaitla, J.; Guttormsen, Y.; Mannisto, J.K.; Nova, A.; Repo, T.; Bayer, A.; Hopmann, K.H. Enantioselective Incorporation of CO2: Status and Potential. ACS Catal. 2017, 7, 7231–7244. [Google Scholar] [CrossRef]






















































































































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).