Abstract
Benzoic acid (C6H5COOH) is selected as coal-based model compound with Co compounds (Co3O4, CoO and Co) as the catalysts, and the influence of the valence state change of the catalyst for pyrolysis process is investigated using density functional theory (DFT). DFT results shows that the highest energy barrier of C6H5COOH pyrolysis is in the following order: Ea(CoO) <Ea(Co3O4) <Ea(no catalyst) <Ea(Co). In general, Co3O4 catalyst accelerates C6H5COOH pyrolysis. Then, the catalytic activity further increases when Co3O4 is reduced to CoO. Finally, Co shows no activity for C6H5COOH pyrolysis due to the reduction of CoO to metallic Co.
1. Introduction
Coal pyrolysis is an essential step for coal combustion, gasification, carbonization, liquefaction and so forth. In other words, to achieve the effective utilization of coal, pyrolysis is a significant method in the condition of inert gas [1,2,3]. With the addition of catalysts, energy consumption is reduced, the production rate is less volatile and the quality of products are improved [1,4]. Therefore, some researchers have studied coal catalytic pyrolysis. Wang conducted experiments and found that K2CO3 exhibits a catalytic effect on coal pyrolysis [5]. Fu proposed that metal chlorides KCl, alkali metal CaO, and transition metal Fe2O3 promote coal pyrolysis [6]. Some researchers also find that Co compounds improve the catalytic activity, e.g. coal depolymerization and hydrogenation, tar yield and so on [7,8,9,10].
The structure of coal is complex and it has undefined molecular compositions [11,12]. Furthermore, the separation of coal and catalysts is difficult after the catalytic pyrolysis reaction, and the effect of the catalyst for coal catalytic pyrolysis is hard to investigate via experiment. Hence, to study the coal catalytic pyrolysis mechanism, some organic compounds were selected as model compounds. It is well known that carboxyl (COOH) is the most common functional groups in coal [13], and aromatic compounds such as benzene (the simplest aromatic) are also common [11]. Therefore, the behavior of COOH group in coal during pyrolysis by using the compounds C6H5COOH is modeled by experiment and calculation [12,14,15,16,17]. However, those theoretical studies focus on the coal pyrolysis [18,19], and the role of catalysts are not considered. Recently, the role of some catalysts (ZnO, γ-Al2O3, CaO, and MgO) for coal catalytic pyrolysis is studied by using DFT, which C6H5COOH, C6H5CHO and C6H5OCH3 are selected as the model compounds [20,21,22]. It is found that the catalysts alter the energy barrier and reaction pathway. For example, ZnO, CaO, and MgO catalysts promote C6H5COOH decomposition, but γ-Al2O3 catalyst causes no catalytic effect on C6H5COOH decomposition. However, all the catalysts alter the reaction pathway. In fact, reducing gases are formed such as H2 and CO during the process of coal pyrolysis [6]. In the case, some unstable catalysts are possibly reduced. For example, it is found that the reduction process of Co3O4 under reducing gases is in this order: Co3O4→CoO→Co [23,24]. What is the influence of the catalyst valence state change for coal catalytic pyrolysis?
To answer the question, benzoic acid (C6H5COOH) is selected as a coal-based model compound, and Co compounds (Co3O4, CoO and Co) are selected as the unstable catalysts under coal catalytic pyrolysis condition. Then, the effect of the valence state change of the Co-based catalyst for coal catalytic pyrolysis is investigated using DFT.
2. Results and Discussion
2.1. C6H5COOH Pyrolysis
As shown in Figure 1, three possible pathways are studied for C6H5COOH pyrolysis: C6H5COOH →CO2 + C6H6 (Ea = 2.89 eV, ΔH = -0.05 eV), C6H5COOH →C6H6COO (Ea = 2.78 eV, ΔH = 2.70 eV), and C6H5COOH →C6H5COO + H (Ea = 5.93 eV, ΔH = 5.90 eV). The energy barriers of pathways 1 and 2 are similar, which are far lower than that of pathway 3. Therefore, pathways 1 and 2 for C6H5COOH pyrolysis are possible, which is similar with our previous study [20]. The potential energy diagrams of C6H5COOH pyrolysis and the corresponding initial states (IS), transition states (TS) and final states (FS) are shown in Figure 2. As shown in Figure 2, the pyrolysis pathways of C6H5COOH are C6H5COOH →CO2 + C6H6 and C6H5COOH →C6H6COO →CO2 + C6H6, which are in accordance with the experimental results [25,26,27].
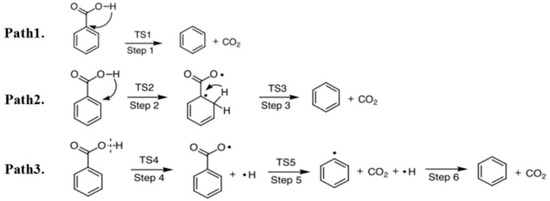
Figure 1.
Three pathways for the pyrolysis of C6H5COOH.
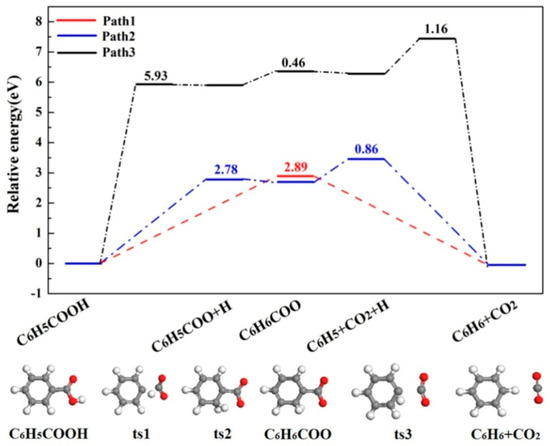
Figure 2.
The potential energy diagrams and corresponding IS, TS and FS of C6H5COOH pyrolysis.
2.2. Catalytic Pyrolysis
The adsorption structures of possible intermediates of C6H5COOH catalytic pyrolysis on Co3O4(110)-B, CoO(100) and Co(111) surfaces are shown in Figure 3. The corresponding adsorption energies and geometrical parameters are shown in Table 1.
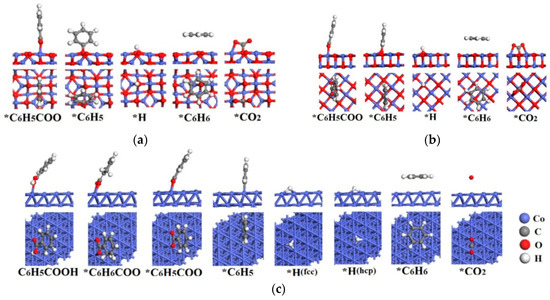
Figure 3.
Structures of the possible intermediates of C6H5COOH pyrolysis on (a) Co3O4(110)-B, (b) CoO(100) and (c) Co(111) surfaces.

Table 1.
Adsorption energies and geometrical parameters of relevant species on Co3O4(110)-B, CoO(100) and Co(111) surfaces.
2.2.1. Co3O4(110)-B Surface
The O-H bond scission of *C6H5COOH after optimization indicates that C6H5COOH is dissociative adsorption on Co3O4(110)-B, which is the same as HCOOH adsorption on ZnO surface [28,29], and C6H5COOH adsorption on ZnO, MgO, CaO and γ-Al2O3 surfaces [20]. *C6H5COO adsorbs on Cooct site through one O, the other O points to the adjacent Cooct site with the adsorption energy of −1.91 eV. *H tends to bond with O2f site, which the adsorption energy is −3.59 eV. For *C6H5COO, the further reaction has two pathways: one is *C6H5COO dissociation directly without the assistance of *H (*C6H5COO + *H→*C6H5 + *CO2 + *H →*C6H6 + *CO2), and the other is *C6H5COO dissociation with *H assistance (*C6H5COO + *H→*C6H6 + *CO2).
For *C6H5COO →*C6H5 + *CO2, the C-C bond length gradually extends from 1.521 Å in the IS to 3.205 Å in the TS. In the FS, *C6H5 adsorbs on O2f site and *CO2 bonds with O2f and Cooct sites, which the adsorption energies of *C6H5 and *CO2 are −3.37 and −0.64 eV, respectively. The reaction needs to overcome an energy barrier of 1.88 eV with reaction energy −1.63 eV. Finally, *C6H6 is formed through *C6H5 hydrogenation (*C6H5+ *H →*C6H6), for which the energy barrier and reaction energy are 2.28 and 1.47 eV. *C6H6 parallels adsorption on the surface with the adsorption energy of −0.01 eV, which is in accordance with the result calculated by Yildirim [30]. For *C6H5COO + *H → *CO2 + *C6H6, *H bonds with O2f site and *C6H5COO adsorbs on Cooct site in the IS. In the TS, the C-C bond length gradually increases to 2.873 Å from 1.517 Å in the IS. The reaction needs to overcome an energy barrier of 2.69 eV with an exothermicity of 0.16 eV.
The potential energy diagrams and corresponding IS, TS and FS geometrical structures of C6H5COOH catalytic pyrolysis on Co3O4(110)-B surface are shown in Figure 4. It is demonstrated that the energetically preferred pathway of C6H5COOH catalytic pyrolysis on Co3O4(110)-B is C6H5COOH(g) →*C6H5COO + *H →*C6H5 + *CO2 +*H→*CO2+*C6H6 →CO2(g) + C6H6(g), which is similar with C6H5COOH pyrolysis on MgO, CaO and γ-Al2O3 surfaces [20]. The highest energy barrier (2.28 eV) on Co3O4(110)-B surface is smaller than that of C6H5COOH pyrolysis without a catalyst (2.78 and 2.89 eV). The above results showed that Co3O4(110)-B have catalytic effect on C6H5COOH pyrolysis.
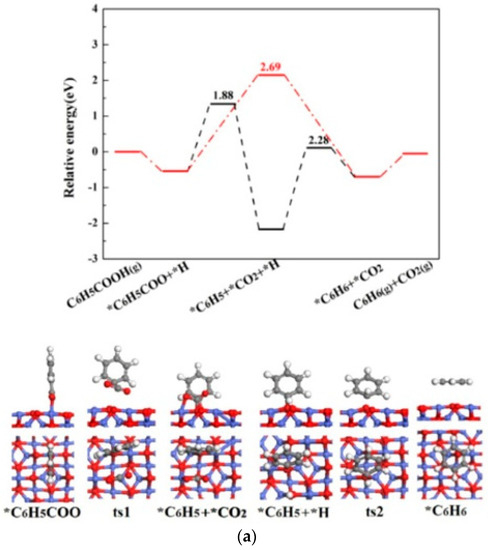
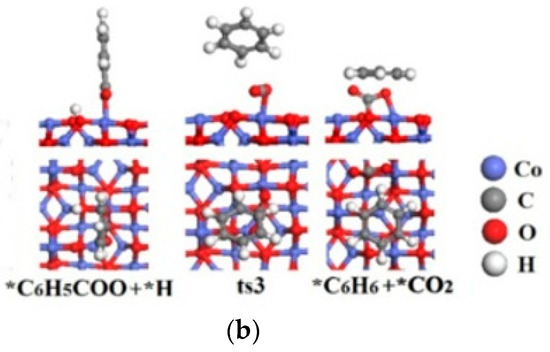
Figure 4.
The potential energy diagrams (Red line: C6H5COOH(g) → *C6H5COO + *H → *CO2 + *C6H6→CO2(g) + C6H6(g); Black line: C6H5COOH(g) → *C6H5COO + *H →*C6H5 + *CO2 + *H→*CO2 + *C6H6→CO2(g) + C6H6(g)) and corresponding IS, TS and FS geometrical structures of C6H5COOH catalytic pyrolysis on Co3O4(110)-B surface(a) related to black line; (b) related to red line.
2.2.2. CoO(100) Surface
*C6H5COOH is also dissociative adsorption on CoO(100) surface after optimization [31]. *C6H5COO binds to Cobri site through two O atoms with the binding energy of −3.06 eV. *H prefers to adsorb on Otop site with the adsorption energy of −2.59 eV.
For *C6H5COO dissociation, the C-C bond length gradually increases from 1.497 Å in the IS to 2.798 Å in the TS. The reaction of *C6H5COO →*C6H5 + *CO2 overcomes an energy barrier of 1.70 eV with reaction energy 1.21 eV. *C6H5 tends to bond with Otop site via ipso-C, which the adsorption energy is −2.03 eV. *CO2 bonds with Otop and Cotop sites with the binding energy of −0.40 eV. The energy barrier and reaction energy of *C6H5 hydrogenation (*C6H5+ *H →*C6H6) are 2.01 and −1.12 eV. *C6H6 is the parallel adsorption on the surface with the adsorption energy of −0.26 eV. With H assistance, the process of *C6H5COO + *H → *CO2 + *C6H6 needs to overcome the energy barrier of 0.82 eV with the reaction energy of 0.09 eV.
The potential energy diagrams and corresponding IS, TS and FS geometrical structures of C6H5COOH catalytic pyrolysis on CoO(100) surface are shown in Figure 5. As shown in Figure 5, the energetically preferred pathway of C6H5COOH catalytic pyrolysis on CoO(100) is C6H5COOH(g) →*C6H5COO + *H→*CO2 + *C6H6 →CO2(g) + C6H6(g). The highest energy barrier (2.01 eV) on CoO(100) surface is also smaller than that of C6H5COOH pyrolysis (2.78 and 2.89 eV), showing that CoO(100) surface exhibits a catalytic effect on C6H5COOH decomposition. C6H5COOH catalytic pyrolysis pathway on CoO(100) surface is same as on ZnO [20], but is different on Co3O4(110)-B surface.
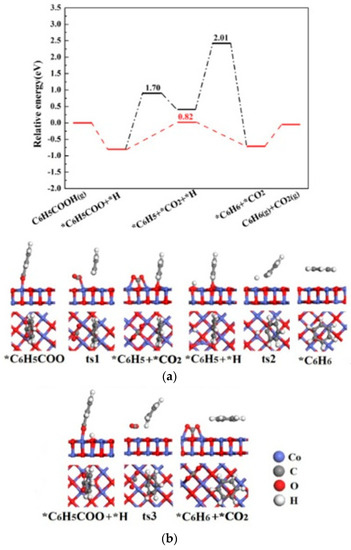
Figure 5.
The potential energy diagrams (Red line: C6H5COOH(g)→ *C6H5COO + *H → *CO2 + *C6H6→CO2(g) + C6H6(g); Black line:C6H5COOH(g)→ *C6H5COO + *H →*C6H5 + *CO2 + *H→*CO2 + *C6H6→CO2(g) + C6H6(g)) and corresponding IS, TS, and FS geometrical structures of C6H5COOH catalytic pyrolysis on CoO(100) surface (a) related to black line; (b) related to red line.
2.2.3. Co(111) Surface
On Co(111) surface, *C6H5COOH binds to top site via O atom, and the adsorption energy is −0.18 eV. The result shows that *C6H5COOH on Co(111) surface uses nondissociation adsorption, which is different from *C6H5COOH adsorption on CoO(100) and Co3O4(110)-B surface. There are three possible pathways for *C6H5COOH further reaction: the first is *C6H6 and *CO2 formation (*C6H5COOH →*C6H6 + *CO2), the second is the formation of *C6H6COO (*C6H5COOH →*C6H6COO) and the third is *C6H5COOH dehydrogenation (*C6H5COOH →*C6H5COO + *H).
The energy barrier and reaction energy of *C6H5COOH →*C6H6 + *CO2 are 6.10 and −0.16 eV. *C6H6 and CO2 are parallel adsorption on the surface, and the corresponding adsorption energies are −0.01 and −0.28 eV. For the reaction of *C6H5COOH → *C6H6COO, the process overcomes an energy barrier of 8.68 eV with an endothermicity of 0.71 eV. *C6H6COO prefers to bind at a bridge site through O atoms with an adsorption energy of −2.18 eV. However, the energy barrier of *C6H5COOH →*C6H5COO + *H is 4.99 eV, which is far smaller than that of *C6H5COOH →*C6H6 + *CO2 and *C6H5COOH →*C6H6COO. The result shows that *C6H5COOH dehydrogenation is the most favorable pathway for *C6H5COOH further reaction. The adsorption sites of *H are fcc and hcp sites, which the adsorption energies are −2.76 and −2.73 eV, respectively. The calculation result is consistent with the results of Chen et al. [32]. C6H5COO adsorbs strongly at the bridge site, for which the adsorption energy is −3.08 eV.
Similarity of C6H5COO dissociation on Co3O4(110)-B and CoO(100) surfaces, there are also two possible pathways on Co(111): one is the formation of *C6H5 and *CO2 without *H assistance, for which the energy barrier and reaction energy are 2.77 and 0.99 eV. *C6H5 adsorbs on the top site via ipso-C with the adsorption energy of −2.29 eV. Then, *C6H6 is formed by *C6H5 hydrogenation (Ea = 3.84 eV, ΔH = −0.45 eV). The other is *C6H6 and *CO2 formation with *H assistance, for which the energy barrier and reaction energy are 2.39 and 0.54 eV.
Figure 6 shows the potential energy diagrams and corresponding IS, TS and FS geometrical structures of C6H5COOH catalytic pyrolysis on Co(111) surface. It is demonstrated that the energetically preferred pathway of C6H5COOH catalytic pyrolysis on Co(111) surface is C6H5COOH(g) → *C6H5COOH → *C6H5COO + *H → *CO2 + *C6H6→CO2(g) + C6H6(g). The highest energy barrier (4.99 eV) of C6H5COOH catalytic pyrolysis on Co(111) surface is greater than that without catalysts (2.78 and 2.89 eV), indicating that Co(111) surface does not show catalytic effect on C6H5COOH pyrolysis.
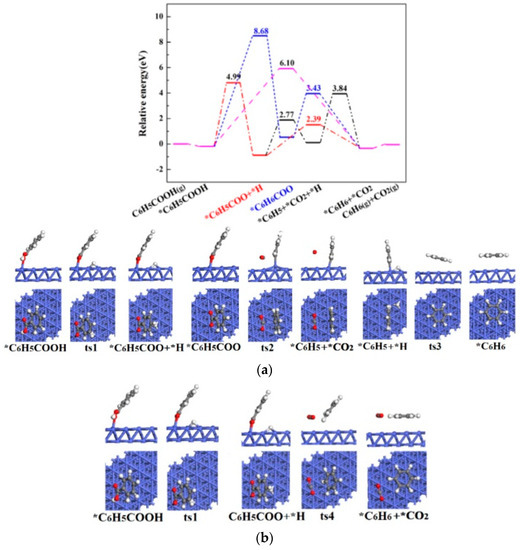
Figure 6.
The potential energy diagrams (Purple line: C6H5COOH(g) →*C6H5COOH →*C6H6 + *CO2→C6H6(g)+ CO2(g); Blue line: C6H5COOH(g) → *C6H5COOH →*C6H6COO →*C6H6 + *CO2→C6H6(g)+ CO2(g); Red line: C6H5COOH(g) →*C6H5COOH → *C6H5COO + *H →*C6H6+ *CO2→C6H6(g)+ CO2(g); Black line: C6H5COOH(g) → *C6H5COOH → *C6H5COO + *H →*C6H5 + *CO2 + *H→*C6H6 + *CO2→C6H6(g) + CO2(g)) and corresponding IS, TS and FS geometrical structures of C6H5COOH catalytic pyrolysis on Co(111) surface (a) related to black line; (b) related to red line.
2.3. d-band Center Analyses
The calculation of d-band center is based on all cobalt atoms to each surface. The corresponding d-band centers of Co3O4(110)-B, CoO(100) and Co(111) are −4.32, −2.35 and −1.42 eV relative to the Fermi level, respectively. The highest energy barriers of C6H5COOH catalytic pyrolysis on Co3O4(110)-B, CoO(100) and Co(111) are 2.28, 0.82, and 4.99 eV, respectively. It is found that there is a volcano-like relationship between d-band center and the highest energy barrier [33].
3. Computational Methods and Models
DFT calculations were carried out with the Vienna ab initio simulation package (VASP) [34,35]. The projector-augmented-wave (PAW) potentials were employed to calculate the interaction between ion core and valence electron [36]. The Perdew−Burke−Ernzerh (PBE) [37] generalized gradient approximation (GGA) [38] functional was performed to describe electronic structure [37]. The climbing-image nudged-elastic-band method (CI-NEB) was applied to derive the transition states (TS) [39], which was confirmed by one imaginary frequency. 3 × 3 × 1 k-points and the cutoff energy with 415 eV were used.
Co3O4(110)-B, CoO(100) and Co(111) surfaces were modeled with (2 × 1), (3 × 4) and (4 × 6) supercells separated by a 15 Å vacuum. The bottom two layers were fixed while other layers and the adsorbates were relaxed during the geometry optimization. Co2+ and Co3+ of Co3O4(110)-B both are antimagnetic [40], which the magnetic moments were set to 2.53 and 2.44 μB, respectively. For CoO(100), the magnetic moment of Co2+ was set to 2.64 μB [41]. The DFT+U method [42,43,44] was performed to Co3O4(110)-B and CoO(100) surfaces because of the error caused by 3d-oribit spin in the Co atoms. The Ueff=U-J values of Co3O4(110)-B and CoO(100) were set to 2.0 and 3.3 eV, respectively. The surface energies of the three surfaces with different numbers of layers are shown in Table 2. According to Table 2, a four-layer slab, a five-layer slab and a four-layer slab for Co3O4(110)-B, CoO(100) and Co(111) surfaces were used. Top and side views of three surfaces are shown in Figure 7.
Table 2.
The surface energies of Co3O4(110)-B, CoO(100) and Co(111) surfaces.
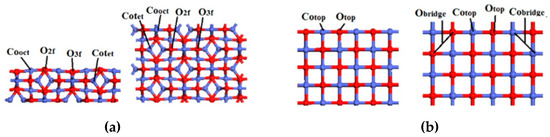
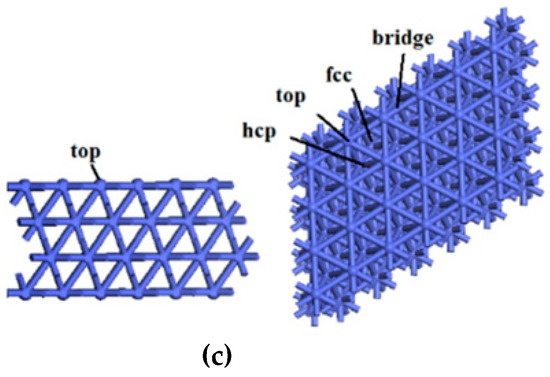
Figure 7.
The top and side views of (a) Co3O4(110)-B, (b) CoO(100) and (c) Co(111) surfaces.
4. Conclusions
In this study, during the pyrolysis of C6H5COOH, the effect of the valence state change of cobalt oxides is investigated with the DFT method. Based on the calculated results, there are two possible pathways for C6H5COOH pyrolysis without catalyst: one is C6H5COOH →CO2 + C6H6; the other is C6H5COOH →C6H6COO →CO2 + C6H6. There are three possibilities for C6H5COOH pyrolysis with catalyst. On Co3O4(110)-B surface, the energetically preferred pathway is C6H5COOH(g) →*C6H5COO + *H → *CO2 + *C6H5 + *H → *CO2 + *C6H6 →CO2(g) + C6H6(g). On CoO(100) surface, the energetically preferred pathway is C6H5COOH(g) →*C6H5COO + *H → *CO2 + *C6H6→CO2(g) + C6H6(g). On Co(111) surface, the energetically preferred pathway is C6H5COOH(g) →*C6H5COOH →*C6H5COO + *H → *CO2 + *C6H6→CO2(g) + C6H6(g). Compared with the highest energy barrier of C6H5COOH pyrolysis, it is found in the following order: Ea(CoO) <Ea(Co3O4) <Ea(no catalyst) <Ea(Co). The above results showed that the presence of catalysts could change the reaction pathway and energy barrier. Co3O4 and CoO can promote C6H5COOH pyrolysis, and the catalytic effect of CoO is much more effective. However, metallic Co does not show catalytic effect on the C6H5COOH pyrolysis. In short, cobalt oxides improve the catalytic activity of C6H5COOH pyrolysis, but catalysis does not occur when cobalt oxides are reduced as metallic Co.
Author Contributions
S.-M.F. and J.-T.L. carried out the research; Y.Z. wrote the paper; W.-S.L. and G.-S.L. prepared the paper; W.H. and Z.-J.Z. revised the paper.
Funding
This work was financially supported by the National Natural Science Foundation of China (21776197 and 21776195) and Shanxi Province Science Foundation for Youths (201701D211003).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, L.L.; Kumar, S.; Wang, Z.H.; He, Y.; Liu, J.Z.; Cen, K. Catalytic Effect of Metal Chlorides on Coal Pyrolysis and Gasification Part I. Combined TG-FTIR Study for Coal Pyrolysis. Thermochim. Acta 2017, 655, 331–336. [Google Scholar] [CrossRef]
- Hu, H.Q.; Zhou, Q.; Zhu, S.W.; Meyer, B.; Krzack, S.; Chen, G.H. Product distribution and sulfur behavior in coal pyrolysis. Fuel Process. Technol. 2004, 85, 849–861. [Google Scholar] [CrossRef]
- Solomon, P.R.; Serio, M.A.; Suuberg, E.M. Coal pyrolysis: Experiments, kinetic rates and mechanisms. Prog. Energy Combust. Sci. 1992, 18, 133–220. [Google Scholar] [CrossRef]
- Zarnegar, S. A review on catalytic pyrolysis of coal and biomass for value added fuel and chemicals. Energy Sources Part A Recover. Util. Environ. Eff. 2018, 40, 1427–1433. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhu, H.L.; Wang, X.M.; Liu, H.F.; Wang, F.C.; Yu, G.S. Transformation and Reactivity of a Potassium Catalyst during Coal–Steam Catalytic Pyrolysis and Gasification. Energy Technol. 2014, 2, 598–603. [Google Scholar] [CrossRef]
- Fu, Y.; Guo, Y.H.; Zhang, K.X. Effect of Three Different Catalysts (KCl, CaO, and Fe2O3) on the Reactivity and Mechanism of Low-Rank Coal Pyrolysis. Energy Fuels 2016, 30, 2428–2433. [Google Scholar] [CrossRef]
- Lei, Z.; Sha, X.L.; Lei, Z.; Wang, R.; Zhang, L.X.; Shu, X.Q. Influences of Different Preparation Conditions on Catalytic Activity of Ag2O-Co3O4/γ-Al2O3for Hydrogenation of Coal Pyrolysis. J. Spectrosc. 2014, 2014, 1–6. [Google Scholar]
- Yan, S.; Zhang, J.S.; Yan, X.Q.; Pan, D.F.; Ren, H.; Qu, X. Catalytic coal hydrogasification by cobalt-calcium catalyst in a pressurized fluidized bed: Role of hydropyrolysis and catalysis process. J. Anal. Appl. Pyrolysis 2018, 135, 251–259. [Google Scholar] [CrossRef]
- Liang, L.T.; Huai, J.T.; Zhang, Q.; Liu, J.W.; Huang, W.; Zhang, Z.L.; Hao, X.G.; Guan, G.Q. Catalytic depolymerization of a typical lignite for improving tar yield by Co and Zn catalyst. Sci. Rep. 2017, 7, 14433. [Google Scholar] [CrossRef]
- Takarada, T.; Onoyama, Y.; Takayama, K.; Sakashita, T. Hydropyrolysis of coal in a pressurized powder-particle fluidized bed using several catalysts. Catal. Today 1997, 39, 127–136. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Shi, L.; Jin, L.J.; Tang, Z.C.; Fan, H.J.; Hu, H.Q. Experimental and Theoretical Study on the Pyrolysis Mechanism of Three Coal-Based Model Compounds. Energy Fuels 2014, 28, 980–986. [Google Scholar] [CrossRef]
- Liu, S.Y.; Zhang, Z.Q.; Wang, H.F. Quantum chemical investigation of the thermal pyrolysis reactions of the carboxylic group in a brown coal model. J. Mol. Model. 2012, 18, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Hosokawa, M.; Kidena, K.; Nomura, M. Analysis ofoxygen-functional groups in brown coals. Fuel Process. Technol. 2000, 67, 231–243. [Google Scholar] [CrossRef]
- Li, L.; Fan, H.J.; Hu, H.Q. A theoretical study on bond dissociation enthalpies of coal based model compounds. Fuel 2015, 153, 70–77. [Google Scholar] [CrossRef]
- Li, J.; Zhang, F.; Fang, W.H. Probing Photophysical and Photochemical Processes of Benzoic Acid from ab Initio Calculations. J. Phys. Chem. A 2005, 109, 7718–7724. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Wang, J.; Liu, J.Z.; Yang, Y.M.; Cheng, J.; Wang, Z.H.; Zhou, J.H.; Cen, K.F. Moisture removal mechanism of low-rank coal by hydrothermal dewatering: Physicochemical property analysis and DFT calculation. Fuel 2017, 187, 242–249. [Google Scholar] [CrossRef]
- Li, B.; Liu, S.Y.; Guo, J.Y.; Zhang, L. Interaction between low rank coal and kaolinite particles: A DFT simulation. Appl. Surf. Sci. 2018, 456, 215–220. [Google Scholar] [CrossRef]
- Kong, L.H.; Li, G.; Jin, L.J.; Hu, H.Q. Pyrolysis behaviors of two coal-related model compounds on a fixed-bed reactor. Fuel Process. Technol. 2015, 129, 113–119. [Google Scholar] [CrossRef]
- Gao, M.J.; Li, X.X.; Guo, L. Pyrolysis simulations of Fugu coal by large-scale ReaxFF molecular dynamics. Fuel Process. Technol. 2018, 178, 197–205. [Google Scholar] [CrossRef]
- Wang, M.F.; Zuo, Z.J.; Ren, R.P.; Gao, Z.H.; Huang, W. Theoretical Study on Catalytic Pyrolysis of Benzoic Acid as a Coal-Based Model Compound. Energy Fuels 2016, 30, 2833–2840. [Google Scholar] [CrossRef]
- Cui, L.P.; Liu, J.T.; Liu, S.Z.; Wang, M.F.; Gao, Z.H.; Zuo, Z.J.; Huang, W. A DFT study of the catalytic pyrolysis of benzaldehyde on ZnO, γ-Al2O3. J. Mol. Model. 2018, 24, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Wang, M.F.; Gao, Z.H.; Zuo, Z.J.; Huang, W. The role of catalysts in the decomposition of phenoxy compounds in coal: A density functional theory study. Appl. Surf. Sci. 2018, 428, 541–548. [Google Scholar] [CrossRef]
- Wang, B.W.; Liu, S.H.; Hu, Z.Y.; Li, Z.H.; Ma, X.B. Active phase of highly active Co3O4 catalyst for synthetic natural gas production. RSC Adv. 2014, 4, 57185–57191. [Google Scholar] [CrossRef]
- Garces, L.J.; Hincapie, B.; Zerger, R.; Suib, S.L. The Effect of Temperature and Support on the Reduction of Cobalt Oxide: An in Situ X-ray Diffraction Study. J. Phys. Chem. C 2015, 119, 5484–5490. [Google Scholar] [CrossRef]
- Manion, J.A.; McMillen, D.F.; Malhotra, R. Decarboxylation and coupling reactions of aromatic acids under coal-liquefaction conditions. Energy Fuels 1996, 10, 776–788. [Google Scholar] [CrossRef]
- Eskay, T.P.; Britt, P.F.; Buchanan, A.C., III. Does Decarboxylation Lead to Cross-Linking in Low-Rank Coals? Energy Fuels 1996, 10, 1257–1261. [Google Scholar] [CrossRef]
- Eskay, T.P.; Britt, P.F.; Buchanan, A.C., III. Pyrolysis of Coal Model Compounds Containing Aromatic Carboxylic Acids: The Role of Carboxylic Acids in Cross-Linking Reactions in Low-Rank Coal; Oak Ridge National Lab.: Oak Ridge, TN, USA, 1997.
- Labat, F.; Ciofini, I.; Adamo, C. Modeling ZnO phases using a periodic approach: From bulk to surface and beyond. J. Chem. Phys. 2009, 131, 044708. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Li, Q.; Noei, H.; Nefedov, A.; Wang, Y.M.; Muhler, M.; Fink, K.; Wöll, C. The Interaction of Formic Acid with Zinc Oxide: A Combined Experimental and Theoretical Study on Single Crystal and Powder Samples. Top. Catal. 2015, 58, 174–183. [Google Scholar] [CrossRef]
- Yildirim, H.; Greber, T.; Kara, A. Trends in Adsorption Characteristics of Benzene on Transition Metal Surfaces: Role of Surface Chemistry and van der Waals Interactions. J. Phys. Chem. C 2013, 117, 20572–20583. [Google Scholar] [CrossRef]
- Schwarz, M.; Hohner, C.; Mohr, S.; Libuda, J. Dissociative Adsorption of Benzoic Acid on Well-Ordered Cobalt Oxide Surfaces: Role of the Protons. J. Phys. Chem. C 2017, 121, 28317–28327. [Google Scholar] [CrossRef]
- Chen, C.B.; Wang, Q.; Wang, G.R.; Hou, B.; Jia, L.T.; Li, D.B. Mechanistic Insight into the C2 Hydrocarbons Formation from Syngas on fcc-Co(111) Surface: A DFT Study. J. Phys. Chem. C 2016, 120, 9132–9147. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Choi, Y.; Hwang, S.M.; Park, G.G.; Yang, T.H.; Su, D.; Sasaki, K.; Liu, P.; Adzic, R.R. Enhancement of the oxygen reduction on nitride stabilized pt-M (M=Fe, Co, and Ni) core–shell nanoparticle electrocatalysts. Nano Energy 2015, 13, 442–449. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phy. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.; Xiao, P.H.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lan, J.; Guo, Y.; Cao, X.M.; Hu, P. Origin of Efficient Catalytic Combustion of Methane over Co3O4(110) Active Low-Coordination Lattice Oxygen and Cooperation of Multiple Active Sites. ACS Catal. 2016, 6, 5508–5519. [Google Scholar] [CrossRef]
- Shojaee, K.; Haynes, B.S.; Montoya, A. Molecular modelling of the decomposition of NH3 over CoO(100). Mater. Chem. Phys. 2015, 156, 141–149. [Google Scholar] [CrossRef]
- Selcuk, S.; Selloni, A. DFT+U Study of the Surface Structure and Stability of Co3O4(110): Dependence on U. J. Phys. Chem. C 2015, 119, 9973–9979. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).