Reactions involving moisture-sensitive reagents were carried out under an argon atmosphere using standard vacuum line techniques. All glassware used was flame-dried and cooled under a vacuum. All solvents were dried using an Innovative Technologies PureSolv Solvent Purification System (INERT) and degassed via three freeze–pump–thaw cycles. All other commercial reagents were used as supplied without further purification, unless stated otherwise. The crude compounds were purified by a Combiflash Rf chromatography system (Teledyne Technologies, Inc., Thousand Oaks, CA, USA) unless specified otherwise. Analytical thin-layer chromatography was performed on pre-coated aluminum plates (Kieselgel 60 F254 silica). TLC visualization was carried out with ultraviolet light (254 nm), followed by staining with a 1% aqueous KMnO4 solution. NMR spectra were recorded on Bruker AMX 400 and 700 (Bruker, Karlruhe, Germany) spectrometers and referenced to the solvent residual peak. Elemental analyses were performed on a Vario MACRO CHN analyzer (Elementar Analysensysteme GmbH, Langenselbold, Germany). Optical rotations ([α]D) were measured on a PolAAr 3000 (Optical Activity Ltd., Cambridgeshire, UK) polarimeter. IR spectra were recorded on a Bruker Alfa spectrometer and are reported in terms of frequency of absorption cm−1. Mass spectra were collected on a Shimadzu HPLC Chromatograph/Mass Spectrometer LCMS-8030 (Shimadzu, Kyoto, Japan), (ESI, operating in positive mode). Enantiomeric excesses were determined by HPLC analysis on chiral stationary phase using 4.6 mm × 250 mm Phenomenex Lux Cellulose-1 and Luz Amylose-1 with n-hexane, 2-propanol as eluent.
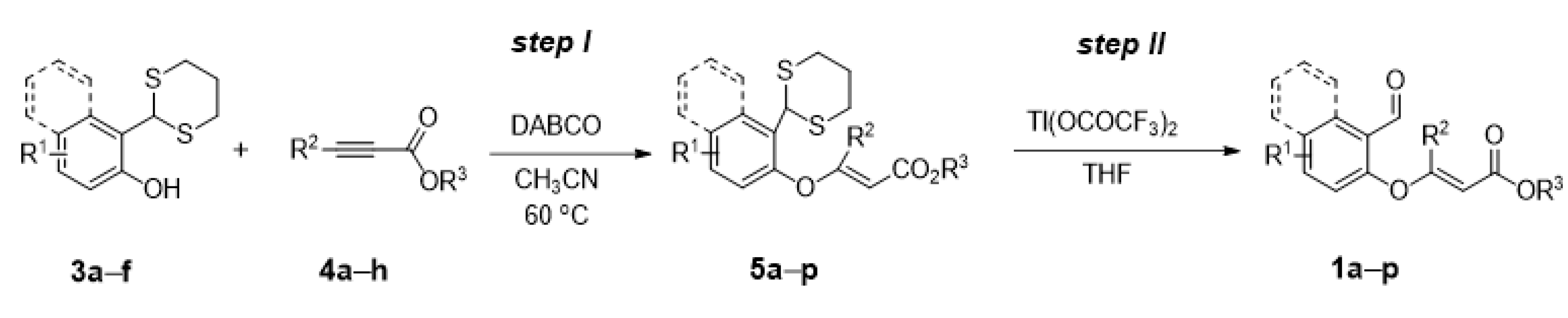
3.1. General Procedure for the Preparation of Compounds 1a–p
Step I: To a solution of dithiane [
52] (5.2 mmol) in acetonitrile (52 mL), DABCO (0.58 g, 5.2 mmol) was added. After stirring for 10 min, alkyne derivative (10.4 mmol, 2 equiv.) was added and the mixture was stirred at 60 °C overnight. After completion (monitored by TLC), most of the acetonitrile was evaporated, then water was added to the solution and the mixture was extracted with ethyl acetate. The combined ethyl acetate extract was washed with brine, dried over anhydrous MgSO
4 and then concentrated under reduced pressure. The crude product was purified by flash chromatography.
Step II: To a solution of dithane (3.0 mmol) in tetrahydrofuran (25 mL), thallium trifluoroacetate (3.3 g, 6.0 mmol, 2 equiv.) was added and the resulting cloudy solution was stirred until reaction was judged to be complete by TLC. The reaction mixture was then filtered through a Cellite pad and the resulting solution was concentrated in vacuo. The desired product was purified by decanting from a solution of petroleum ether, or a mixture of pentane and diethyl ether.
Methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5a). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and methyl hex-2-ynoate (4a). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5a (4.60 g, 64%) as a white solid. M.p. = 63–65 °C. 1H NMR (400 MHz, CDCl3) δ: 1.13 (t, J = 8.0 Hz, 3H), 1.81–1.90 (m, 2H), 1.92–1.99 (m, 1H), 2.15–2.20 (m, 1H), 2.88–2.90 (m, 1H), 2.92–2.94 (m, 1H), 3.00–3.07 (m, 4H), 3.63 (s, 3H), 4.85 (s, 1H), 5.30 (s, 1H), 6.97 (dd, J = 8.0, 4.0 Hz, 1H), 7.20 (dt, J = 7.2, 1.6 Hz, 1H), 7.33 (dt, J = 8.0, 2.0 Hz, 1H), 7.71 (dd, J = 7.6, 1.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 13.9, 21.0, 25.1, 32.3 (2C), 33.0, 44.2, 50.8, 96.1, 122.1, 126.4, 129.7, 129.9, 131.6, 149.6, 167.6, 175.7. IR (ATR) ν (cm−1): 2958, 2933, 2896, 1710, 1632, 1484, 1432, 1373, 1276, 1246, 1129, 1088, 1041, 938, 830, 822, 750, 670. LRMS (ESI): Mass calcd. for [M + Na]+ C17H22O3S2Na: 361.1; found 361.2. Anal. Calcd. for C17H22O3S2: C, 60.32; H, 6.55; found: C, 60.37; H, 6.63.
Benzyl (E)-3-(2-(1,3-dithian-2-yl)-5-phenoxy)but-2-enoate (5b). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and benzyl hex-2-ynoate (4b). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5b (4.67 g, 53%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 1.16 (t, J = 8.0 Hz, 3H), 1.86–1.95 (m, 3H), 2.12–2.18 (m, 1H), 2.90 (dt, J = 13.6, 4.0 Hz, 2H), 2.99–3.09 (m, 4H), 4. 97 (s, 1H), 5.12 (s, 2H), 5.34 (s, 1H), 7.00 (dd, J = 8.0, 1.2 Hz, 1H), 7.26 (dt, J = 6.4, 1.2 Hz, 1H), 7.29–7.36 (m, 6H), 7.74 (dd, J = 7.6, 1.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 14.0, 21.0, 25.2, 32.4 (2C), 33.2, 44.3, 65.5, 96.3, 122.1, 126.5, 128.0, 128.2 (2C), 128.5 (2C), 129.8, 130.0, 131.6, 136.4, 149.6, 167.1, 175.9. IR (ATR) ν (cm−1): 2959, 2932, 1709, 1629, 1484, 1451, 1383, 1258, 1223, 1121, 1090, 1023, 833, 732, 696. LRMS (ESI): Mass calcd. for [M + Na]+ C23H26O3S2Na: 437.1; found 437.2. Anal. Calcd. for C23H26O3S2: C, 66.63; H, 6.32; found: C, 66.71; H, 6.42.
Ethyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)pent-2-enoate (5d). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and ethyl pent-2-ynoate (4d). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5d (5.53 g, 77%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 1.22 (t, J = 7.2 Hz, 3H), 1.38 (t, J = 7.6 Hz, 3H), 1.88–1.98 (m, 1H), 2.14–2.19 (m, 1H), 2.88–2.93 (m, 2H), 3.00–3.07 (m, 4H), 4.10 (q, J = 7.2 Hz, 2H), 4.84 (s, 1H), 5.31 (s, 1H), 6.99 (dd, J = 8.4, 1.6 Hz, 1H), 7.25 (dt, J = 7.6, 1.2 Hz, 1H), 7.32 (dt, J = 7.6, 1.6 Hz, 1H), 7.71 (dd, J = 8.0, 1.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 12.1, 14.3, 24.8, 25.1, 32.3 (2C), 44.3, 59.6, 95.8, 122.1, 126.4, 129.7, 129.9, 131.6, 149.7, 167.2, 176.7. IR (ATR) ν (cm−1): 2975, 2936, 1706, 1629, 1448, 1378, 1275, 1227, 1211, 1127, 1086, 1041, 1003, 832. LRMS (ESI): Mass calcd. for [M + Na]+ C17H22O3S2Na: 361.1; found 361.3. Anal. Calcd. for C17H22O3S2: C, 60.32; H, 6.55; found: C, 60.41; H, 6.50.
Methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)oct-2-enoate (5e). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and methyl oct-2-ynoate (4e). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5e (3.96 g, 51%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 0.97 (t, J = 8.0 Hz, 3H), 1.41–1.52 (m, 4H), 1.78–1.86 (m, 2H), 1.88–1.98 (m, 1H), 2.14–2.19 (m, 1H), 2.87–2.93 (m, 2H), 2.99–3.06 (m, 4H), 3.62 (s, 3H), 4.84 (s, 1H), 5.30 (s, 1H), 6.97 (dd, J = 8.0, 4.0 Hz, 1H), 7.25 (dt, J = 8.0, 1.6 Hz, 1H), 7.31 (dt, J = 7.6, 1.6 Hz, 1H), 7.70 (dd, J = 7.6, 1.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 14.0, 22.6, 25.1, 27.4, 31.2, 31.6, 32.3 (2C), 44.2, 50.9, 95.6, 122.1, 126.4, 129.7, 129.9, 131.6, 149.7, 167.7, 176.0. IR (ATR) ν (cm−1): 2953, 2928, 2854, 1711, 1634, 1428, 1367, 1244, 1213, 1134, 1093, 825. LRMS (ESI): Mass calcd. for [M + Na]+ C19H26O3S2Na: 389.1; found 389.3. Anal. Calcd. for C19H26O3S2: C, 58.59; H, 6.73; found: C, 58.66; H, 6.82.
Methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)non-2-enoate (5f). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and methyl non-2-ynoate (4f). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5f (3.53 g, 41%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 0.93 (t, J = 8.0 Hz, 3H), 1.37–1.41 (m, 4H), 1.48–1.55 (m, 2H), 1.77–1.85 (m, 2H), 1.92–1.98 (m, 1H), 2.14–2.20 (m, 1H), 2.88–2.93 (m, 2H), 2.99–3.06 (m, 4H), 3.62 (s, 3H), 4.83 (s, 1H), 5.29 (s, 1H), 6.96 (dd, J = 8.0, 4.0 Hz, 1H), 7.25 (dt, J = 8.0, 1.6 Hz, 1H), 7.31 (dt, J = 7.6, 1.6 Hz, 1H), 7.70 (dd, J = 7.6, 1.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 14.0, 22.6, 25.1, 27.4, 31.2, 31.6, 32.3 (2C), 44.2, 50.9, 95.6, 122.1, 126.4, 129.7, 129.9, 131.6, 149.7, 167.7, 176.0. IR (ATR) ν (cm−1): 2950, 2924, 2850, 1709, 1636, 1427, 1366, 1240, 1212, 1134, 1093, 825. LRMS (ESI): Mass calcd. for [M + Na]+ C20H28O3S2Na: 403.1; found 403.2. Anal. Calcd. for C20H28O3S2: C, 63.12; H, 7.42; found: C, 63.27; H, 7.58.
Methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)-4-cyclohexylbut-2-enoate (5g). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and methyl 4-cyclohexylbut-2-ynoate (4g). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5g (3.53 g, 41%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 1.19–1.36 (m, 5H), 1.68–1.74 (m, 2H), 1.78–1.84 (m, 2H), 1.89–1.99 (m, 3H), 2.15–2.21 (m, 1H), 2.88–3.06 (m, 6H), 3.62 (s, 3H), 4.89 (s, 1H), 5.31 (s, 1H), 6.97 (dd, J = 8.0, 1.6 Hz, 1H), 7.26 (dt, J = 7.6, 1.6 Hz, 1H), 7.30 (dt, J = 7.6, 1.6 Hz, 1H), 7.72 (dd, J = 7.6, 1.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 25.1, 26.4, 26.4 (2C), 32.3 (2C), 33.0 (2C), 36.7, 38.4, 44.1, 50.8, 96.4, 122.1, 126.4, 129.8, 130.0, 131.6, 149.6, 167.7, 174.7. IR (ATR) ν (cm−1): 2920, 2850, 1713, 1630, 1447, 1430, 1373, 1274, 1258, 1194, 1170, 1134, 1104, 1040, 830. LRMS (ESI): Mass calcd. for [M + Na]+ C21H28O3S2Na: 415.1; found 415.3. Anal. Calcd. for C21H28O3S2: C, 64.25; H, 7.19; found: C, 64.31; H, 7.27.
Methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)-5-phenylpent-2-enoate (5h). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)phenol (3a) (5.0 g, 21.2 mmol) and methyl 5-phenylpent-2-ynoate (4h). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5h (3.65 g, 43%) as a yellow solid. M.p. = 97–99 °C. 1H NMR (700 MHz, CDCl3) δ: 1.90 (m, 1H), 2.13 (dspt, J = 14.2, 2.4 Hz, 1H), 2.87 (ddd, J = 14.6, 3.2, 0.9 Hz, 2H), 2.97 (ddd, J = 14.7, 12.5, 2.5 Hz, 2H), 3.10–3.14 (m, 2H), 3.28–3.40 (m, 2H), 3.63 (s, 3H), 4.86 (s, 1H), 5.24 (s, 1H), 6.87 (dd, J = 8.0, 1.3 Hz, 1H), 7.22–7.26 (m, 2H), 7.28–7.31 (m, 1 H), 7.32–7.35 (m, 2H), 7.36–7.39 (m, 2H), 7.70 (dd, J = 7.7, 1.7 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ: 24.68, 31.86, 32.59, 33.18, 43.75, 50.60, 96.11, 121.64, 125.82, 126.14, 128.09, 128.19, 129.40, 129.60, 131.21, 140.49, 149.22, 167.16, 174.33. IR (ATR) ν (cm−1): 2920, 2850, 1713, 1630, 1447, 1430, 1373, 1274, 1258, 1194, 1170, 1134, 1104, 1040, 830. LRMS (ESI): Mass calcd. for [M + Na]+ C22H24O3S2Na: 423.1; found 423.0. Anal. Calcd. for C22H24O3S2: C, 65.97; H, 6.04; found: C, 66.01 H, 6.11.
Methyl (E)-3-(2-(1,3-dithian-2-yl)-5-methylphenoxy)but-2-enoate (5i). The title compound was prepared according to the general procedure (Step I) using 2-(1,3-dithian-2-yl)-5-methylphenol (3b) (5.0 g, 21.2 mmol) and methyl but-2-ynoate (4c). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5i (6.17 g, 85%) as a yellow solid. M.p. = 108–109 °C. 1H NMR (400 MHz, CDCl3) δ: 1.88–1.99 (m, 1H), 2.15–2.22 (m, 1H), 2.36 (s, 3H), 2.55 (s, 3H), 2.88–2.93 (m, 2H), 3.02–3.10 (m, 2H), 3.64 (s, 3H), 4.96 (s, 1H), 5.24 (s, 1H), 6.86 (d, J = 8.4 Hz, 1H), 7.10–7.12 (m, 1H), 7.50 (d, J = 2.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 18.2, 20.8, 25.2, 32.4 (2C), 44.3, 50.9, 96.6, 121.8, 130.3, 130.4, 131.0, 136.3, 147.4, 168.0, 172.4. IR (ATR) ν (cm−1): 2949, 2927, 2822, 1706, 1629, 1493, 1434, 1383, 1340, 1257, 1122, 1094, 1041, 1004, 933, 876, 822. LRMS (ESI): Mass calcd. for [M + Na]+ C16H20O3S2Na: 347.1; found 347.2. Anal. calcd. for C16H20O3S2: C, 59.23; H, 6.21; found: C, 59.31; H, 6.34.
Methyl (E)-3-(4-chloro-2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5j). The title compound was prepared according to the general procedure (Step I) using 4-chloro-2-(1,3-dithian-2-yl)phenol (3c) (5.0 g, 21.2 mmol) and methyl hex-2-ynoate (4a). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5j (6.17 g, 85%) as a white solid. M.p. 103–104 °C. 1H NMR (400 MHz, CDCl3) δ: 1.11 (t, J = 7.2 Hz, 3H), 1.78–1.87 (m, 2H), 1.91–1.99 (m, 1H), 2.14–2.20 (m, 1H), 2.86–2.94 (m, 2H), 2.97–3.05 (m, 4H), 3.65 (s, 3H), 4.84 (s, 1H), 5.23 (s, 1H), 6.92 (d, J = 8.8 Hz, 1H), 7.28 (dd, J = 8.4, 2.4 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 13.8, 21.0, 25.0, 32.2 (2C), 32.9, 43.7, 51.0, 96.5, 123.4, 129.9, 130.0, 131.8, 133.4, 148.2, 167.4, 175.4. IR (ATR) ν (cm−1): 2954, 2888, 1711, 1625, 1479, 1436, 1370, 1226, 1210, 1126, 1102, 1080, 1037, 936, 828. LRMS (ESI): Mass calcd. for [M + Na]+ C17H21ClO3S2Na: 395.0; found 395.1. Anal. Calcd. for C17H21ClO3S2: C, 54.75; H, 5.68; found: C, 54.88; H, 5.72.
Methyl (E)-3-(4-bromo-2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5k). The title compound was prepared according to the general procedure (Step I) using 4-bromo-2-(1,3-dithian-2-yl)phenol (3d) (5.0 g, 21.2 mmol) and methyl hex-2-ynoate (4a). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5k (6.19 g, 70%) as a white solid. M.p. 87–89 °C. 1H NMR (700 MHz, CDCl3) δ: 1.12 (t, J = 7.7 Hz, 3H), 1.82–1.97 (m, 1H), 2.18–2.20 (m, 1H), 2.92–2.95 (m, 2H), 3.00–3.05 (m, 4H), 3.65 (s, 3H), 4.85 (s, 1H), 5.24 (s, 1H), 6.87 (d, J = 8.4 Hz, 1H), 7.44–7.46 (m, 1H), 7.85 (d, J = 2.1 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ: 14.9, 22.0, 26.0, 33.2 (2C), 33.9, 44.6, 52.0, 97.6, 120.4, 124.8, 133.8, 134.0, 134.8, 149.8, 168.4, 176.4. IR (ATR) ν (cm−1): 2957, 2933, 1710, 1626, 1477, 1436, 1369, 1226, 1209, 1125, 1102, 1079, 1035, 934, 829. LRMS (ESI): Mass calcd. for [M + Na]+ C17H21BrO3S2Na: 439.0; found 439.1. Anal. Calcd. for C17H21BrO3S2: C, 49.92; H, 5.07; found: C, 50.02; H, 5.17.
Methyl (E)-3-(2-(1,3-dithian-2-yl)-4-(trifluoromethoxy)phenoxy)hex-2-enoate (5l). The title compound was prepared according to the general procedure (Step I) using 4-trifluoromethoxy-2-(1,3-dithian-2-yl)phenol (3e) (5.0 g, 21.2 mmol) and methyl hex-2-ynoate (4a). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5l (3.77 g, 42%) as a white solid. M.p. 57–60 °C. 1H NMR (700 MHz, CDCl3) δ: 1.13 (t, J = 7.7 Hz, 3H), 1.82–1.88 (m, 2H), 1.92–1.96 (m, 1H), 2.17–2.20 (m, 1H), 2.91–2.94 (m, 2H), 3.00–3.06 (m, 4H), 3.65 (s, 3H), 4.86 (s, 1H), 5.27 (s, 1H), 7.01 (d, J = 8.4 Hz, 1H), 7.18–7.20 (m, 1H), 7.59 (d, J = 2.1 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ: 14.8, 22.0, 25.9, 33.1 (2C), 33.9, 44.6, 52.0, 97.6, 121.0 (q, J = 258 Hz), 123.2, 123.7, 124.3, 134.6, 147.8, 148.9, 168.4, 176.4. IR (ATR) ν (cm−1): 2959, 2934, 2901, 1714, 1634, 1490, 1434, 1372, 1255, 1208, 1164, 1128, 1093, 1038, 937, 883, 834. LRMS (ESI): Mass calcd. for [M + Na]+ C18H21F3O4S2Na: 445.1; found 445.1. Anal. Calcd. for C18H21F3O4S2: C, 51.17; H, 5.01; found: C, 51.27; H, 5.10.
Methyl (E)-3-(4-fluoro-2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5m). The title compound was prepared according to the general procedure (Step I) using 4-fluoro-2-(1,3-dithian-2-yl)phenol (3g) (5.0 g, 21.1 mmol) and methyl hex-2-ynoate (4a). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5m (3.56 g, 47%) as a white solid. M.p. 75–78 °C. 1H NMR (700 MHz, CDCl3) δ: 1.11 (t, J = 7.2 Hz, 3H), 1.78–1.87 (m, 2H), 1.91–1.99 (m, 1H), 2.14–2.20 (m, 1H), 2.86–2.94 (m, 2H), 2.97–3.05 (m, 4H), 3.65 (s, 3H), 4.84 (s, 1H), 5.23 (s, 1H), 6.92 (d, J = 8.8 Hz, 1H), 7.28 (dd, J = 8.4, 2.4 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 13.8, 21.0, 25.0, 32.2 (2C), 32.9, 43.7, 51.0, 96.5, 123.4, 129.9, 130.0, 131.8, 133.4, 148.2, 167.4, 175.4. IR (ATR) ν (cm−1): 2956, 2915, 1714, 1622, 1490, 1432, 1221, 1206, 1163, 1125, 1085, 1071, 1036, 1002, 965, 933, 874, 831. LRMS (ESI): Mass calcd. for [M + Na]+ C17H21FO3S2Na: 379.1; found 379.2. Anal. Calcd. for C17H21FO3S2: C, 57.28; H, 5.94; found: C, 57.40; H, 5.99.
Methyl (E)-3-((1-(1,3-dithian-2-yl)naphthalen-2-yl)oxy)-5-phenylpent-2-enoate (5n). The title compound was prepared according to the general procedure (Step I) using 1-(1,3-dithian-2-yl)naphthalen-2-ol (3f) (5.0 g, 19.1 mmol) and methyl 5-phenylpent-2-ynoate (4h). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5n (3.93 g, 53%) as a white solid. M.p. 140–144 °C. 1H NMR (400 MHz, CDCl3) δ: 2.05–2.10 (m, 1H), 2.21–2.25 (m, 1H), 2.97 (t, J = 2.0 Hz, 1H), 2.99 (t, J = 2.0 Hz, 1H), 3.05–3.08 (m, 2H), 3.23–3.25 (m, 2H), 3.44–3.46 (m, 2H), 3.65 (s, 3H), 4.90 (s, 1H), 5.96 (s, 1H), 7.03 (d, J = 5.3 Hz, 1H), 7.30 (t, J = 4.4 Hz, 1H), 7.39–7.41 (m, 2H), 7.46 (m, 2H), 7.53–7.55 (m, 1H), 7.63–7.65 (m, 1H), 7.82 (d, J = 5.2 Hz, 1H), 7.85 (d, J = 4.4 Hz, 1H), 9.24 (d, J = 4.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 25.7, 33.0, 33.2 (2C), 33.8, 45.4, 51.0, 97.4, 120.7, 125.7, 126.0, 126.1, 126.3, 127.4, 128.4, 128.5 (2C), 128.6 (2C), 130.9, 132.4, 132.5, 140.8, 147.0, 167.5, 174.2. IR (ATR) ν (cm−1): 3026, 2980, 2899, 1710, 1632, 1432, 1365, 1232, 1215, 1185, 1161, 1150, 1113, 1046, 1008, 931, 838, 828. LRMS (ESI): Mass calcd. for [M + Na]+ C26H26O3S2Na: 473.1; found 473.3. Anal. Calcd. for C26H26O3S2: C, 69.30; H, 5.82; found: C, 69.41; H, 5.87.
Methyl (E)-3-((1-(1,3-dithian-2-yl)naphthalen-2-yl)oxy)hex-2-enoate (5o). The title compound was prepared according to the general procedure (Step I) using 1-(1,3-dithian-2-yl)naphthalen-2-ol (3f) (5.0 g, 19.1 mmol) and methyl hex-2-ynoate (4a). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5o (2.37 g, 32%) as a yellow solid. M.p. 108–109 °C. 1H NMR (700 MHz, CDCl3) δ: 1.19 (t, J = 7.7 Hz, 3H), 1.95–1.97 (m, 2H), 2.08–2.12 (m, 1H), 2.26–2.28 (m, 1H), 2.98–3.02 (m, 2H) 3.08–3.13 (m, 4H), 3.62 (s, 3H), 4.86 (s, 1H), 5.97 (s, 1H), 7.11 (d, J = 9.1 Hz, 1H), 7.51–7.54 (m, 1H), 7.61–7.63 (m, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 9.20 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 15.0, 22.2, 26.7, 34.0, 34.2 (2C), 46.4, 51.9, 98.0, 121.8, 126.7, 127.0, 127.1, 128.4, 129.4, 131.9, 133.4, 133.5, 148.0, 168.6, 176.2. IR (ATR) ν (cm−1): 2967, 2939, 1710, 1625, 1509, 1431, 1368, 1273, 1257, 1129, 1080, 830. LRMS (ESI): Mass calcd. for [M + Na]+ C21H24O3S2Na: 411.1; found 411.2. Anal. Calcd. for C21H24O3S2: C, 64.92; H, 6.23; found: C, 65.01; H, 6.37.
Methyl (E)-3-((3-(1,3-dithian-2-yl)naphthalen-2-yl)oxy)but-2-enoate (5p). The title compound was prepared according to the general procedure (Step I) using 1-(1,3-dithian-2-yl)naphthalen-2-ol (3f) (5.0 g, 19.1 mmol) and methyl but-2-ynoate (4c). The product was purified by flash chromatography (petroleum ether/EtOAc 8:2) to give 5p (3.02 g, 44%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ: 2.03–2.13 (m, 1H), 2.21–2.29 (m, 1H), 2.62 (s, 3H), 2.96–3.02 (m, 2H), 3.10–3.16 (m, 2H), 3.63 (s, 3H), 4.96 (s, 1H), 5.94 (s, 1H), 7.12 (d, J = 8.8 Hz, 1H), 7.49–7.53 (m, 1H), 7.59–7.63 (m, 1H), 7.80–7.84 (m, 2H), 9.21 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 18.2, 25.7, 33.3 (2C), 45.5, 50.9, 97.7, 120.8, 125.6, 125.9, 126.0, 127.4, 128.4, 130.8, 132.3, 132.4, 147.2, 167.9, 171.6. IR (ATR) ν (cm−1): 2951, 2908, 1706, 1638, 1620, 1508, 1345, 1248, 1213, 1144, 1128, 1016, 930 856, 831. LRMS (ESI): Mass calcd. for [M + Na]+ C19H20O3S2Na: 383.1; found 383.2. Anal. Calcd. for C19H20O3S2: C, 63.31; H, 5.59; found: C, 63.40; H, 5.70.
Methyl (E)-3-(2-formylphenoxy)hex-2-enoate (1a). The title compound was prepared according to the general procedure (Step II) using methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5a) (1.0 g, 3.0 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 94% yield (0.70 g). 1H NMR (400 MHz, CDCl3) δ: 1.09 (t, J = 8.0 Hz, 3H), 1.77–1.86 (m, 2H), 2.99–3.03 (m, 2H), 3.62 (s, 3H), 4.81 (s, 1H), 7.10 (dd, J = 8.0, 0.8 Hz, 1H), 7.35–7.39 (m, 1H), 7.65 (dddd, J = 15.6, 9.2, 7.6, 2.0 Hz, 1H), 7.95 (dd, J = 8.0, 2.0 Hz, 1H), 10.16 (d, J = 0.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 13.9, 20.8, 33.0, 51.0, 97.3, 122.8, 126.2, 128.4, 129.0, 135.9, 155.7, 167.0, 176.4, 188.1. IR (ATR) ν (cm−1): 2951, 2929, 1715, 1696, 1629, 1600, 1455, 1434, 1371, 1236, 1187, 1097, 1042, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C14H16O4Na: 271.1; found 271.2. Anal. Calcd. for C14H16O4: C, 67.73; H, 6.50; found: C, 67.82; H, 6.66.
Benzyl (E)-3-(2-formylphenoxy)hex-2-enoate (1b). The title compound was prepared according to the general procedure (Step II) using benzyl (E)-3-(2-(1,3-dithian-2-yl)-5-phenoxy)but-2-enoate (5b) (1.2 g, 3.0 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 90% yield (0.70 g). 1H NMR (700 MHz, CDCl3) δ: 1.08 (t, J = 7.4 Hz, 4H), 1.81 (sxtt, J = 7.6, 1.7 Hz, 2H), 2.99–3.03 (m, 2H), 4.84 (s, 1H), 5.07 (s, 2H), 7.08 (dd, J = 8.1, 1.0 Hz, 1H), 7.29–7.34 (m, 6H), 7.63 (ddd, J = 8.2, 7.3, 1.7 Hz, 1H), 7.94 (ddd, J = 7.7, 1.8, 0.3 Hz, 1H), 10.15 (d, J = 0.8 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ: 13.6, 20.5, 32.8, 65.4, 96.9, 122.5, 125.9, 127.8, 128.0, 128.2, 128.7, 135.6, 135.7, 155.2, 166.1, 176.4, 187.6, 187.7. IR (ATR) ν (cm−1): 2946, 2932, 1711, 1692, 1633, 1598, 1455, 1430, 1373, 1235, 1189, 1100, 1042, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C20H20O4Na: 347.1; found 347.2. Anal. Calcd. for C20H20O4: C, 74.06; H, 6.22; found: C, 74.15; H, 6.34.
Methyl (E)-3-(2-formylphenoxy)but-2-enoate (1c). The title compound was prepared according to the general procedure (Step II) using methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)but-2-enoate (5c) (0.93 g, 3.0 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 95% yield (0.63 g). 1H NMR (700 MHz, CDCl3) δ: 2.57 (d, J = 0.6 Hz, 3H), 3.63 (s, 3H), 4.84 (d, J = 0.6 Hz, 1H), 7.11 (dd, J = 8.2, 1.0 Hz, 1H), 7.36–7.39 (m, 1H), 7.65 (ddd, J = 8.1, 7.4, 1.7 Hz, 1H), 7.94 (dd, J = 7.7, 1.8 Hz, 1H), 10.12–10.14 (m, 1H). 13C NMR (176 MHz, CDCl3) δ: 17.7, 50.7, 97.2, 122.4, 126.0, 127.8, 128.9, 135.5, 155.0, 166.9, 172.5, 187.7, 187.8. IR (ATR) ν (cm−1): 2946, 1713, 1694, 1636, 1602, 1457, 1428, 1375, 1232, 1189, 1100, 1045, 834. LRMS (ESI): Mass calcd. for [M + Na]+ C12H12O4Na: 243.1; found 243.2. Anal. Calcd. for C12H12O4: C, 65.45; H, 6.49; found: C, 65.54; H, 6.48.
Ethyl (E)-3-(2-formylphenoxy)pent-2-enoate (1d). The title compound was prepared according to the general procedure (Step II) using ethyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)pent-2-enoate (5d) (1.0 g, 3.0 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 92% yield (0.68 g). 1H NMR (700 MHz, CDCl3) δ: 1.19–1.22 (m, 3H), 1.31–1.35 (m, 3H), 3.03 (q, J = 7.5 Hz, 2H), 4.08 (q, J = 7.1 Hz, 2H), 4.74 (s, 1H), 7.10 (dd, J = 8.2, 0.9 Hz, 1H), 7.35–7.39 (m, 1H), 7.65 (ddd, J = 8.2, 7.3, 1.7 Hz, 1H), 7.95 (dd, J = 7.7, 1.7 Hz, 1H), 10.16 (d, J = 0.9 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ: 11.4, 13.8, 24.4, 59.4, 96.6, 122.4, 125.9, 128.0, 128.5, 135.6, 155.4, 166.2, 177.1, 187.8. IR (ATR) ν (cm−1): 2940, 1710, 1691, 1629, 1600, 1453, 1428, 1373, 1232, 1189, 1110, 1045, 834. LRMS (ESI): Mass calcd. for [M + Na]+ C14H16O4Na: 271.1; found 271.1. Anal. Calcd. for C14H16O4: C, 67.73; H, 6.50; found: C, 67.70; H, 6.59.
Methyl (E)-3-(2-formylphenoxy)oct-2-enoate (1e). The title compound was prepared according to the general procedure (Step II) using methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)oct-2-enoate (5e) (1.0 g, 2.72 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 85% yield (0.64 g). 1H NMR (700 MHz, CDCl3) δ: 0.93 (t, J = 7.2 Hz, 3H), 1.37–1.47 (m, 4H), 1.74–1.79 (m, 2H), 2.98–3.02 (m, 2H), 3.61 (s, 3H), 4.78 (s, 1H), 7.08 (dd, J = 8.2, 1.0 Hz, 1H), 7.35–7.38 (m, 1H), 7.64 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.95 (dd, J = 7.8, 1.8 Hz, 1H), 10.15 (d, J = 0.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 22.4, 27.1, 31.2, 31.6, 51.0, 97.1, 122.8, 126.3, 128.4, 128.9, 135.9, 155.8, 167.0, 176.8, 188.1. IR (ATR) ν (cm−1): 2951, 2929, 2857, 1715, 1696, 1629, 1600, 1455, 1434, 1371, 1272, 1236, 1187, 1110, 1045, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C16H20O4Na: 299.1; found 299.1. Anal. Calcd. for C14H16O4: C, 69.55; H, 7.30; found: C, 69.60; H, 7.42.
Methyl (E)-3-(2-formylphenoxy)non-2-enoate (1f). The title compound was prepared according to the general procedure (Step II) using methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)non-2-enoate (5f) (1.0 g, 2.63 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 82% yield (0.63 g). 1H NMR (400 MHz, CDCl3) δ: 0.93 (t, J = 7.1 Hz, 3H), 1.35–1.40 (m, 4H), 1.43–1.53 (m, 2H), 1.73–1.82 (m, 2H), 2.99–3.05 (m, 2H), 3.63 (s, 3H), 4.80 (s, 1H), 7.10 (dd, J = 8.1, 1.0 Hz, 1H), 7.35–7.40 (m, 1H), 7.66 (ddd, J = 8.2, 7.3, 1.8 Hz, 1H), 7.96 (dd, J = 7.8, 1.7 Hz, 1H), 10.17 (d, J = 0.7 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 22.6, 27.4, 29.1, 31.2, 31.6, 51.0, 97.2, 122.8, 126.2, 128.4, 129.0, 135.9, 155.8, 167.0, 176.8, 188.1. IR (ATR) ν (cm−1): 2950, 2929, 2855, 1717, 1694, 1632, 1602, 1455, 1434, 1374, 1272, 1236, 1187, 1113, 1045, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C17H22O4Na: 313.1; found 313.2. Anal. Calcd. for C17H22O4: C, 70.32; H, 7.64; found: C, 70.28; H, 7.74.
Methyl (E)-4-cyclohexyl-3-(2-formylphenoxy)but-2-enoate (1g). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)-4-cyclohexylbut-2-enoate (5g) (1.0 g, 2.55 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 87% yield (0.67 g). 1H NMR (700 MHz, CDCl3) δ: 0.90 (t, J = 7.0 Hz, 3H), 1.13–1.30 (m, 5H), 1.65–1.89 (m, 5H), 2.93 (d, J = 7.0 Hz, 2H), 3.60 (s, 3H), 4.85 (s, 1H), 7.07 (d, J = 7.0, 1H), 7.34 (m, 1H), 7.63 (m, 1H), 7.93 (dd, J = 14.0, 7.0 Hz, 1H), 10.01 (s, 1H). 13C NMR (176 MHz, CDCl3) δ: 25.8 (2C), 25.9, 32.7 (2C), 36.2, 37.8, 50.6, 97.8, 122.3, 125.8, 127.9, 128.5, 135.5, 155.4, 166.7, 175.0, 187.8. IR (ATR) ν (cm−1): 2950, 2929, 1710, 1691, 1632, 1602, 1457, 1435, 1372, 1274, 1236, 1187, 1113, 1047, 830. LRMS (ESI): Mass calcd. for [M + Na]+ C18H22O4Na: 325.1; found 325.3. Anal. Calcd. for C18H22O4: C, 71.50; H, 7.33; found: C, 71.56; H, 7.45.
Methyl (E)-3-(2-formylphenoxy)-5-phenylpent-2-enoate (1h). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(2-(1,3-dithian-2-yl)phenoxy)-5-phenylpent-2-enoate (5h) (1.0 g, 2.50 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 93% yield (0.72 g). 1H NMR (400 MHz, CDCl3) δ: 3.11 (dd, J = 9.0, 6.6 Hz, 8H), 3.36 (dd, J = 9.8, 6.6 Hz, 8H), 3.65 (s, 3H), 4.81 (s, 1H), 6.97 (dd, J = 8.1, 1.0 Hz, 1H), 7.23–7.28 (m, 1H), 7.33–7.40 (m, 5H), 7.64 (ddd, J = 8.2, 7.3, 1.8 Hz, 1H), 7.95 (dd, J = 7.8, 1.7 Hz, 1H), 10.00 (d, J = 0.7 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 33.0, 33.4, 51.1, 97.6, 122.8, 126.3, 126.4, 128.5, 128.6, 129.1, 136.0, 140.5, 155.5, 166.9, 175.5, 188.1. IR (ATR) ν (cm−1): 2954, 2932, 1714, 1689, 1634, 1600, 1455, 1433, 1373, 1272, 1236, 1187, 1111, 1044, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C19H18O4Na: 333.1; found 333.1. Anal. Calcd. for C19H18O4: C, 73.53; H, 5.85; found: C, 73.61; H, 5.89.
Methyl (E)-3-(2-formyl-5-methoxyphenoxy)but-2-enoate (1i). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(2-(1,3-dithian-2-yl)-5-methoxyphenoxy)but-2-enoate (5i) (1.0 g, 2.94 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 95% yield (0.70 g). 1H NMR (400 MHz, CDCl3) δ: 2.42 (s, 3H), 2.57 (d, J = 0.5 Hz, 3H), 3.64 (s, 3H), 4.84 (d, J = 0.5 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 7.41–7.50 (m, 1H), 7.74 (d, J = 2.2 Hz, 1H), 10.10 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 18.1, 20.7, 51.0, 97.3, 122.7, 127.8, 129.3, 136.4, 136.7, 153.3, 167.4, 173.2, 188.4. IR (ATR) ν (cm−1): 2955, 2931, 1711, 1691, 1632, 1601, 1456, 1433, 1373, 1269, 1234, 1187, 1113, 1045, 834. LRMS (ESI): Mass calcd. for [M + Na]+ C13H14O5Na: 273.1; found 273.1. Anal. Calcd. for C13H14O5: C, 62.39; H, 5.64; found: C, 62.47; H, 5.72.
Methyl (E)-3-(4-chloro-2-formylphenoxy)hex-2-enoate (1j). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(4-chloro-2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5j) (1.0 g, 2.68 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 91% yield (0.69 g). 1H NMR (400 MHz, CDCl3) δ: 1.08 (t, J = 7.0 Hz, 3H), 1.77 (m, 2H), 2.97–2.99 (m, 2H), 3.62 (s, 3H), 4.80 (s, 1H), 7.03 (d, J = 7.0 Hz, 1H), 7.58 (dd, J = 9.1, 3.5 Hz, 1H), 7.89 (d, J = 2.1 Hz, 1H), 10.07 (s, 1H). 13C NMR (176 MHz, CDCl3) δ: 13.4, 20.4, 32.5, 50.7, 97.4, 123.9, 128.2, 128.9, 131.9, 131.8, 135.4, 153.7, 166.4, 175.8, 186.4. IR (ATR) ν (cm−1): 2953, 2930, 1705, 1690, 1632, 1600, 1456, 1433, 1373, 1265, 1230, 1187, 1113, 1045, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C14H15ClO4Na: 305.1; found 305.2. Anal. Calcd. for C14H15ClO4: C, 59.48; H, 5.35; found: C, 59.56; H, 5.41.
Methyl (E)-3-(4-bromo-2-formylphenoxy)hex-2-enoate (1k). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(4-bromo-2-(1,3-dithian-2-yl)phenoxy)hex-2-enoate (5k) (1.0 g, 2.40 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 90% yield (0.70 g). 1H NMR (400 MHz, CDCl3) δ: 1.08 (t, J = 7.3 Hz, 3H), 1.80 (dq, J = 15.0, 7.5 Hz, 1H), 2.97–3.03 (m, 2H), 4.83 (s, 1H), 3.65 (s, 3H), 7.01 (d, J = 8.6 Hz, 1H), 7.75 (dd, J = 8.7, 2.6 Hz, 1H), 8.07 (d, J = 2.4 Hz, 1H),10.09 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 13.8, 20.8, 32.9, 51.1, 97.9, 119.7, 124.6, 129.6, 131.7, 138.7, 154.6, 166.8, 176.1, 186.7. IR (ATR) ν (cm−1): 2953, 1701, 1692, 1632, 1600, 1456, 1433, 1371, 1263, 1232, 1184, 1110, 1044, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C14H15BrO4Na: 349.0; found 349.1. Anal. Calcd. for C14H15BrO4: C, 51.40; H, 4.62; found: C, 51.48; H, 4.70.
Methyl (E)-3-(2-formyl-4-(trifluoromethoxy)phenoxy)hex-2-enoate (1l). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(2-(1,3-dithian-2-yl)-4-(trifluoromethoxy)phenoxy)hex-2-enoate (5l) (1.0 g, 2.37 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 85% yield (0.67 g). 1H NMR (400 MHz, CDCl3) δ: 1.10 (t, J = 7.3 Hz, 3H), 1.81 (sxt, J = 7.5 Hz, 1H), 2.97–3.04 (m, 2H), 3.66 (s, 3H), 4.85 (s, 1H), 7.17 (d, J = 8.8 Hz, 1H), 7.50 (ddd, J = 8.9, 3.1, 0.7 Hz, 1H), 7.80 (dd, J = 2.9, 1.0 Hz, 1H), 10.13 (s, 1H), 13C NMR (101 MHz, CDCl3) δ: 13.8, 20.8, 32.9, 51.1, 98.1, 120.7, 124.4, 128.3, 129.4, 146.7–146.8 (m), 153.8, 166.7, 176.0, 186.5. IR (ATR) ν (cm−1): 2950, 1704, 1690, 1635, 1603, 1458, 1431, 1370, 1262, 1231, 1184, 1110, 1044, 831. LRMS (ESI): Mass calcd. for [M + Na]+ C15H15F3O5Na: 355.1; found 355.1. Anal. Calcd. for C15H15F3O5: C, 54.22; H, 4.55; found: C, 54.30; H, 4.67.
Methyl (E)-3-(4-fluoro-2-formylphenoxy)hex-2-enoate (1m). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-(2-(1,3-dithian-2-yl)-4-fluorophenoxy)hex-2-enoate (5m) (1.0 g, 2.80 mmol) and thallium (III) trifluoroacetate. The product was obtained as a colorless oil in 94% yield (0.70 g). 1H NMR (400 MHz, CDCl3) δ: 1.09 (t, J = 8.0 Hz, 3H), 1.78–1.84 (m, 2H), 2.98–3.02 (m, 2H), 3.64 (s, 3H), 4.79 (s, 1H), 7.10 (dd, J = 12.0, 8.0 Hz, 1H), 7.34–7.39 (m, 1H), 7.62 (dd, J = 8.0, 4.0 H, 1H), 10.08 (d, J = 4.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 13.8, 20.8, 32.9, 51.1, 97.5, 114.7 (d, J = 23.1 Hz), 123.0 (d, J = 25.2 Hz), 124.7 (d, J = 8.0 Hz), 129.6 (d, J = 6.0 Hz), 151.6 (d, J = 2.0 Hz), 160.2 (d, J = 248.5 Hz), 166.8, 176.6, 186.9 (d, J = 2.0 Hz). IR (ATR) ν (cm−1): 2954, 1700, 1693, 1636, 1606, 1454, 1429, 1371, 1262, 1231, 1184, 1112, 1046, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C14H15FO4Na: 289.1; found 289.1. Anal. Calcd. for C14H15FO4: C, 63.15; H, 5.68; found: C, 63.21; H, 5.79.
Methyl (E)-3-((1-formylnaphthalen-2-yl)oxy)-5-phenylpent-2-enoate (1n). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-((1-(1,3-dithian-2-yl)naphthalen-2-yl)oxy)-5-phenylpent-2-enoate (5n) (1.0 g, 2.22 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 85% yield (0.68 g). 1H NMR (400 MHz, CDCl3) δ: 3.12–3.19 (m, 2H), 3.38–3.45 (m, 2H), 3.64 (s, 3H), 4.88 (s, 1H), 7.02 (d, J = 9.0 Hz, 1H), 7.23–7.33 (m, 1H), 7.34–7.44 (m, 4H), 7.59 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.71 (ddd, J = 8.6, 7.0, 1.3 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 9.27 (dd, J = 8.8, 0.7 Hz, 1H), 10.44 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 32.9, 33.4, 51.1, 98.8, 120.8, 121.7, 125.6, 126.4, 126.8, 128.4, 128.5, 128.6, 129.9, 131.0, 131.6, 137.3, 140.4, 157.9, 166.8, 175.2, 190.6. IR (ATR) ν (cm−1): 2951, 1703, 1690, 1635, 1604, 1456, 1429, 1371, 1262, 1231, 1184, 1110, 1048, 835. LRMS (ESI): Mass calcd. for [M + Na]+ C23H20O4Na: 383.1; found 383.2. Anal. Calcd. for C23H20O4: C, 76.65; H, 5.59; found: C, 76.76; H, 5.70.
Methyl (E)-3-((1-formylnaphthalen-2-yl)oxy)hex-2-enoate (1o). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-((1-(1,3-dithian-2-yl)naphthalen-2-yl)oxy)hex-2-enoate (5o) (1.0 g, 2.57 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 90% yield (0.69 g). 1H NMR (400 MHz, CDCl3) δ: 1.13 (t, J = 7.5 Hz, 3H), 1.86 (sxt, J = 7.5 Hz, 2H), 3.05 (ddd, J = 8.5, 6.1, 1.4 Hz, 2H), 3.63 (s, 3H), 4.88 (s, 1H), 7.19 (d, J = 8.8 Hz, 1H), 7.60 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 7.72 (ddd, J = 8.6, 7.0, 1.3 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 8.13 (d, J = 8.8 Hz, 1H), 9.28 (dd, J = 8.6, 1.0 Hz, 1H), 10.57 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 13.9, 20.8, 33.0, 51.1, 98.6, 120.8, 125.6, 126.8, 128.4, 130.0, 131.0, 131.5, 137.4, 158.2, 167.1, 176.2, 190.7. IR (ATR) ν (cm−1): 2947, 1715, 1694, 1638, 1600, 1456, 1429, 1374, 1263, 1231, 1184, 1111, 1045, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C18H18O4Na: 321.1; found 321.2. Anal. Calcd. for C18H18O4: C, 72.47; H, 6.08; found: C, 72.55; H, 6.18.
Methyl (E)-3-((1-formylnaphthalen-2-yl)oxy)but-2-enoate (1p). The title compound was prepared according to the general procedure (Step II) methyl (E)-3-((1-(1,3-dithian-2-yl)naphthalen-2-yl)oxy)hex-2-enoate (5p) (1.0 g, 2.77 mmol) and thallium (III) trifluoroacetate. The product was obtained as a yellow oil in 93% yield (0.70 g). 1H NMR (700 MHz, CDCl3) δ: 2.61 (s, 3H), 3.61 (s, 3H), 4.90 (s, 1H), 7.19 (d, J = 7.0 Hz, 1H), 7.56–7.59 (m, 1H), 7.68–7.71 (m, 1H), 7.87 (dd, J = 7.7, 1.4 Hz, 1H), 8.1 (d, J = 9.1 Hz, 1H), 9.24 (m, 1H), 10.56 (s, 1H). 13C NMR (101 MHz, CDCl3) δ: 18.1, 51.1, 98.8, 120.9, 121.6, 125.6, 126.8, 128.4, 129.9, 131.1, 131.6, 137.3, 157.8, 167.2, 172.5, 190.5. IR (ATR) ν (cm−1): 2942, 1713, 1690, 1631, 1604, 1456, 1429, 1374, 1263, 1231, 1184, 1112, 1044, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C16H14O4Na: 293.1; found 293.2. Anal. Calcd. for C16H14O4: C, 71.10; H, 5.22; found: C, 71.17; H, 5.31.
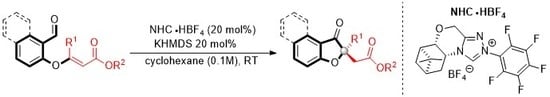
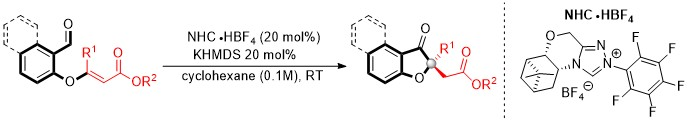
3.2. General Procedure for Enantioselective Intramolecular Stetter Reaction Using Triazolium Salts O and L
A flame dried round bottom flask was charged with triazolium salt O (19.0 mg, 20 mol%) and 1.0 mL of cyclohexane. To this solution, KHMDS (0.5M in toluene 80 μL, 0.04 mmol, 20 mol%) was added via syringe and the solution was allowed to stir at ambient temperature for 20 min. A solution of the substrate (0.20 mmol) in 1.0 mL of cyclohexane was added. The resulting solution was allowed to stir at ambient temperature and monitored by TLC. The reaction mixture was placed directly onto a silica gel column and eluted with a suitable solution of hexane and ethyl acetate (80:20). Evaporation of solvent allowed analytically pure product.
A flame dried round bottom flask was charged with triazolium salt L (28.8 mg, 20 mol%) and 1.0 mL of cyclohexane. To this solution, potassium tert-butoxide (1.0 M in THF 40 μL, 0.04 mmol, 20 mol%)was added via syringe and the solution was allowed to stir at ambient temperature for 20 min. A solution of the substrate (0.20 mmol) in 1.0 mL of cyclohexane was added. The resulting solution was allowed to stir at ambient temperature and monitored by TLC. The reaction mixture was placed directly onto a silica gel column and eluted with a suitable solution of hexane and ethyl acetate (80:20). Evaporation of solvent allowed analytically pure product.
Methyl (R)-2-(3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2a). Yellow oil (98% yield, 97% ee). HPLC (Phenomenex Lux Celluose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 14.0 min, t (minor) = 12.6 min. [α] = −19.2 (c 1.0, CH2Cl2).1H NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 8.0 Hz, 3H), 1.14–1.23 (m, 1H), 1.30–1.37 (m, 1H), 1.84 (m, 2H), 2.96 (d, J = 16.0 Hz, 1H), 3.03 (d, J = 16.0 Hz, 1H), 3.53 (s, 3H), 7.09–7.11 (m, 2H), 7.59–7.64 (m, 1H), 7.69–7.72 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 16.2, 38.7, 40.6, 51.7, 88.8, 112.9, 121.8, 124.1, 137.7, 169.0, 171.5, 202.7. IR (ATR) ν (cm−1): 2961, 2936, 2875, 1720, 1602, 1462, 1436, 1268, 1211, 1171, 1120, 993, 820. LRMS (ESI): Mass calcd. for [M + Na]+ C14H16O4Na: 271.1; found 271.1. Anal. Calcd. for C14H16O4: C, 67.73; H, 6.50; found: C, 67.80; H, 6.61.
Methyl (S)-2-(3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2a’). Yellow oil (98% yield, 97% ee). HPLC (Phenomenex Lux Celluose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 12.7 min, t (minor) = 14.2 min. [α] = +19.4 (c 1.0, CH2Cl2).
Benzyl (R)-2-(3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2b). Yellow oil (95% yield, 96% ee). HPLC (Phenomenex Lux Celluose-1, hexanes/2-propanol 95:5 v = 0.7 mL/min, λ = 254 nm) t (major) = 19.1 min, t (minor) = 21.1 min. [α] = −1.4 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.86 (t, J = 8.0 Hz, 1H), 1.18–1.20 (m, 1H), 1.31–1.35 (m, 1H), 1.80–1.85 (m, 2H), 3.01 (d, J = 16.0 Hz, 1H), 3.09 (d, J = 16.0 Hz, 1H), 4.96 (s, 1H), 7.03–7.08 (m, 2H), 7.15–7.18 (m, 2H), 7.28–7.31 (m, 3H), 7.56–7.60 (m, 1H), 7.63 (dd, J = 8.0, 4.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 16.2, 38.9, 40.8, 66.7, 88.8, 112.9, 121.7, 121.8, 124.2, 128.2, 128.3 (2C), 128.5 (2C), 135.2, 137.7, 168.5, 171.5, 202.7. IR (ATR) ν (cm−1): 2953, 2935, 2874, 1726, 1482, 1437, 1250, 1213, 1163, 1123, 1010, 836. LRMS (ESI): Mass calcd. for [M + Na]+ C20H20O4Na: 347.1; found 347.2. Anal. Calcd. for C20H20O4: C, 74.06; H, 6.22; found: C, 74.12; H, 6.34.
Benzyl (S)-2-(3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2b’). Yellow oil (98% yield, 97% ee). HPLC (Phenomenex Lux Celluose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 14.7 min, t (minor) = 13.2 min. [α] = +1.5 (c 1.0, CH2Cl2).
Methyl (R)-2-(2-methyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2c). Yellow oil (96% yield, 96% ee). HPLC (Phenomenex Lux Celluose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 15.6 min, t (minor) = 14.0 min. [α] = −12.6 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 2.96 (d, J = 16.0 Hz, 1H), 3.05 (d, J = 16.0 Hz, 1H), 3.56 (s, 3H), 7.09–7.13 (m, 2H), 7.61–7.65 (m, 1H), 7.71–7.74 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 22.3, 41.2, 51.8, 86.3, 113.3, 120.4, 121.9, 124.6, 137.8, 169.1, 170.9, 202.6. IR (ATR) ν (cm−1): 2958, 2940, 2870, 1722, 1601, 1473, 1434, 1262, 1210, 1163, 1123, 1011, 836. LRMS (ESI): Mass calcd. for [M + Na]+ C12H12O4Na: 243.1; found 243.1. Anal. Calcd. for C12H12O4: C, 65.45; H, 5.49; found: C, 65.52; H, 5.56.
Methyl (S)-2-(2-methyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2c’). Yellow oil (98% yield, <99% ee). HPLC (Phenomenex Lux Celluose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 14.2 min, t (minor) = 15.6 min. [α] = +13.4 (c 1.0, CH2Cl2).
Ethyl (R)-2-(2-ethyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2d). Yellow oil (96% yield, 96% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 98:2, v = 0.7 mL/min, λ = 254 nm) t (major) = 23.5 min, t (minor) = 26.0 min. [α] = −19.5 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.85 (t, J = 7.5 Hz, 3H), 0.97 (t, J = 7.2 Hz, 3H), 1.89 (q, J = 7.3 Hz, 2H), 2.99 (dd, J = 16.1, 16.1 Hz, 2H), 3.95 (m, 2H), 7.04–7.14 (m, 2H), 7.61 (ddd, J = 8.4, 7.2, 1.5 Hz, 1H), 7.69 (ddd, J = 7.6, 1.5, 0.7 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 7.3, 13.7, 29.9, 40.7, 60.8, 89.1, 112.9, 121.7, 122.0, 124.1, 137.7, 168.6, 171.7, 202.8. IR (ATR) ν (cm−1): 2952, 2930, 2874, 1720, 1482, 1434, 1256, 1211, 1168, 1120, 1012, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C14H16O4Na: 271.1; found 271.1. Anal. Calcd. for C14H16O4: C, 67.73; H, 6.50; found: C, 67.80; H, 6.61.
Ethyl (S)-2-(2-ethyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2d’). Yellow oil (98% yield, 91% ee). HPLC (Phenomenex Lux Amylose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 10.0 min, t (minor) = 9.5 min. [α] = +18.8 (c 1.0, CH2Cl2).
Methyl (R)-2-(3-oxo-2-pentyl-2,3-dihydrobenzofuran-2-yl)acetate (2e). Yellow oil (95% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 14.0 min, t (minor) = 11.6 min. [α] = −27.8 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.83 (t, J = 7.1 Hz, 3H), 0.88–1.03 (m, 2H), 1.19–1.25 (m, 4H), 1.80–1.87 (m, 2H), 2.97 (d, J = 16.4 Hz, 1H), 3.00 (d, J = 16.4 Hz, 1H), 3.52 (s, 3H), 7.06–7.11 (m, 2H), 7.61 (ddd, J = 8.4, 7.2, 1.5 Hz, 1H), 7.69 (dd, J = 7.9, 1.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 13.9, 22.2, 22.4, 31.7, 36.5, 40.6, 51.8, 88.8, 112.9, 121.8, 124.2, 137.7, 169.1, 171.6, 202.8. IR (ATR) ν (cm−1): 2950, 2934, 2870, 1725, 1480, 1438, 1257, 1209, 1171, 1122, 1012, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C16H20O4Na: 299.1; found 299.2. Anal. Calcd. for C16H20O4: C, 69.55; H, 7.30; found: C, 69.64; H, 7.38.
Methyl (S)-2-(3-oxo-2-pentyl-2,3-dihydrobenzofuran-2-yl)acetate (2e’). Yellow oil (95% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 11.6 min, t (minor) = 14.6 min. [α] = +27.2 (c 1.0, CH2Cl2).
Methyl (R)-2-(2-hexyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2f). Yellow oil (93% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 13.7 min, t (minor) = 10.9 min. [α] = −28.3 (c 1.0, CH2Cl2). 1H NMR (700 MHz, CDCl3) δ: 0.86 (t, J = 7.0 Hz, 3H), 1.17–1.26 (m, 7H), 1.27–1.32 (m, 2H), 1.82–1.90 (m, 2H), 2.97 (d, J = 14.0 Hz, 1H), 3.02 (d, J = 14.0 Hz, 1H), 3.54 (s, 3H), 7.11 (m, 2H), 7.62–7.64 (m, 1H), 7.69 (dd, J = 7.0, 2.0 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ: 14.9, 23.4, 23.6, 30.2, 32.4, 37.6, 41.6, 52.7, 89.8, 113.9, 122.7, 122.8, 125.2, 138.7, 170.1, 172.6, 203.8. IR (ATR) ν (cm−1): 2958, 2940, 2870, 1722, 1601, 1473, 1434, 1262, 1210, 1163, 1123, 1011, 836. LRMS (ESI): Mass calcd. for [M + Na]+ C17H22O4Na: 313.1; found 313.2. Anal. Calcd. for C17H22O4: C, 70.32; H, 7.64; found: C, 70.39; H, 7.72.
Methyl (S)-2-(2-hexyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2f’). Yellow oil (93% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 10.6 min, t (minor) = 13.6 min. [α] = +28.3 (c 1.0, CH2Cl2).
Methyl (R)-2-(2-(cyclohexylmethyl)-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2g). Yellow oil (91% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 15.3 min, t (minor) = 12.2 min. [α] = −19.2 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.87–1.01 (m, 3H), 1.04–1.17 (m, 4H), 1.24–1.36 (m, 3H), 1.67 (dd, J = 14.7, 6.8 Hz, 3H), 1.79 (dd, J = 14.7, 5.1 Hz, 1H), 3.00 (s, 2H), 3.50 (s, 3H), 7.07–7.12 (m, 2H), 7.61 (ddd, J = 8.4, 7.2, 1.5 Hz, 1H), 7.71 (dd, J = 8.1, 1.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 26.0, 26.1, 33.1, 34.2, 34.7, 40.8, 43.6, 51.7, 89.1, 113.0, 121.8, 124.2, 137.7, 169.1, 171.3, 203.0. IR (ATR) ν (cm−1): 2952, 2944, 2876, 1709, 1607, 1466, 1430, 1267, 1212, 1155, 1123, 1006, 834. LRMS (ESI): Mass calcd. for [M + Na]+ C18H22O4Na: 325.1; found 325.2. Anal. Calcd. for C18H22O4: C, 66.45; H, 6.82; found: C, 66.54; H, 6.88.
Methyl (S)-2-(2-(cyclohexylmethyl)-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2g’). Yellow oil (95% yield, <99% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 12.0 min, t (minor) = 14.6 min. [α] = +20.2 (c 1.0, CH2Cl2).
Methyl (R)-2-(3-oxo-2-phenethyl-2,3-dihydrobenzofuran-2-yl)acetate (2h). Yellow oil (92% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 18.4 min, t (minor) = 15.2 min. [α] = −38.0 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 2.19 (ddd, J = 11.3, 6.1, 2.9 Hz, 2H), 2.47 (ddd, J = 13.4, 11.0, 6.4 Hz, 1H), 2.63 (ddd, J = 13.4, 11.0, 6.3 Hz, 1H), 2.98–3.10 (m, 2H), 3.55 (s, 3H), 7.09–7.21 (m, 5H), 7.22–7.28 (m, 2H), 7.64 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 7.73 (ddd, J = 7.6, 1.5, 0.7 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 29.2, 38.3, 40.7, 51.8, 88.4, 113.1, 121.7, 122.0, 124.2, 126.2, 128.3 (2C), 128.4 (2C), 137.9, 140.6, 169.0, 171.6, 202.3. IR (ATR) ν (cm−1): 2953, 2939, 2870, 1701, 1604, 1459, 1433, 1271, 1215, 1148, 1119, 1012, 834. LRMS (ESI): Mass calcd. for [M + Na]+ C19H18O4Na: 333.1; found 333.1. Anal. Calcd. for C19H18O4: C, 73.53; H, 5.85; found: C, 73.60; H, 5.94.
Methyl (S)-2-(3-oxo-2-phenethyl-2,3-dihydrobenzofuran-2-yl)acetate (2h’). Yellow oil (92% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 15.6 min, t (minor) = 18.6 min. [α] = +38.4 (c 1.0, CH2Cl2).
Methyl (R)-2-(6-methyl-2-methyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2i). Yellow oil (94% yield, 94% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 23.3 min, t (minor) = 20.3 min. [α] = −1.6 (c 1.0, CH2Cl2). 1H NMR (700 MHz, CDCl3) δ: 1.44 (s, 3H), 2.34 (s, 3H), 2.90 (d, J = 14.0 Hz, 1H), 3.00 (d, J = 14.0 Hz, 1H), 3.53 (s, 3H), 6.96 (d, J = 7.0 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.47 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 20.2, 22.0, 40.8, 51.4, 86.0, 112.5, 119.9, 123.6, 131.1, 138.7, 168.8, 169.0, 202.3. IR (ATR) ν (cm−1): 2956, 2941, 2869, 1715, 1609, 1462, 1433, 1270, 1212, 1155, 1123, 1006, 833. LRMS (ESI): Mass calcd. for [M + Na]+ C13H14O5Na: 273.1; found 273.2. Anal. Calcd. for C13H14O5: C, 62.39; H, 5.64; found: C, 62.47; H, 5.70.
Methyl (S)-2-(6-methyl-2-methyl-3-oxo-2,3-dihydrobenzofuran-2-yl)acetate (2i’). Yellow oil (96% yield, 93% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 20.2 min, t (minor) = 23.5 min. [α] = +1.7 (c 1.0, CH2Cl2).
Methyl (R)-2-(5-chloro-3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2j). Yellow oil (95% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 9.5 min, t (minor) = 7.6 min. [α] = −24.8 (c 1.0, CH2Cl2). 1H NMR (700 MHz, CDCl3) δ: 0.89 (t, J = 7.0 Hz, 3H), 1.20 (m, 1H), 1.34 (m, 1H), 1.81 (dtd, J = 17.6, 13.9, 5.6 Hz, 2H), 3.00 (d, J = 14.0 Hz, 1H), 3.06 (d, J = 14.0 Hz, 1H) 3.56 (s, 3H), 7.02 (d, J = 7.0 Hz, 1H), 7.56 (dd, J = 8.4, 2.1 Hz, 1H), 7.67 (d, J = 1.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 15.0, 17.2, 39.8, 41.6, 52.8, 90.9, 115.2, 124.0, 124.5, 128.2, 138.4, 170.0, 170.8, 202.6. IR (ATR) ν (cm−1): 2961, 2878, 1722, 1620, 1482, 1247, 1210, 1155, 1123, 1009, 829. LRMS (ESI): Mass calcd. for [M + Na]+ C14H15ClO4Na: 305.1; found 305.2. Anal. Calcd. for C14H15ClO4: C, 59.48; H, 5.35; found: C, 59.55; H, 5.43.
Methyl (S)-2-(5-chloro-3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2j’). Yellow oil (94% yield, 98% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 7.6 min, t (minor) = 9.6 min. [α] = +25.6 (c 1.0, CH2Cl2).
Methyl (R)-2-(5-bromo-3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2k). Yellow oil (90% yield, 93% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 13.0 min, t (minor) = 9.5 min. [α] = −28.3 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 7.3 Hz, 3H), 1.10–1.24 (m, 1H), 1.26–1.38 (m, 1H), 1.81 (dtd, J = 17.6, 13.9, 5.6 Hz, 2H), 2.99 (d, J = 16.6 Hz, 1H), 3.04 (d, J = 16.6 Hz, 1H), 3.54 (s, 3H), 7.00 (d, J = 8.8 Hz, 1H), 7.68 (dd, J = 8.8, 2.2 Hz, 1H), 7.81 (dd, J = 2.2, 0.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.01, 16.22, 38.80, 40.63, 51.89, 89.80, 114.30, 114.65, 123.63, 126.69, 140.15, 168.99, 170.24, 201.38. IR (ATR) ν (cm−1): 2955, 2870, 1715, 1622, 1478, 1244, 1211, 1156, 1123, 1009, 831. LRMS (ESI): Mass calcd. for [M + Na]+ C14H15BrO4Na: 349.0; found 349.1. Anal. Calcd. for C14H15BrO4: C, 51.40; H, 4.62; found: C, 51.49; H, 4.70.
Methyl (S)-2-(5-bromo-3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2k’). Yellow oil (90% yield, 94% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 9.5 min, t (minor) = 12.7 min. [α] = +28.7 (c 1.0, CH2Cl2).
Methyl (R)-2-(3-oxo-2-propyl-5-(trifluoromethoxy)-2,3-dihydrobenzofuran-2-yl)acetate (2l). Yellow oil (94% yield, 97% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 9.5 min, t (minor) = 10.7 min. [α] = −17.3 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.88 (t, J = 7.2 Hz, 1H), 1.17–1.24 (m, 1H), 1.31–1.39 (m, 1H), 1.78–1.87 (m, 2H), 2.99 (d, J = 16.8 Hz, 1H), 3.07 (d, J = 16.8 Hz, 1H), 3.55 (s, 3H), 7.11 (d, J = 8.8 Hz, 1H), 7.46 (dd, J = 9.2, 2.8 Hz, 1H), 7.54 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 16.2, 38.7, 40.5, 51.8, 90.3, 114.0, 116.3, 120.5 (d, J = 257.6 Hz), 122.4, 130.9, 143.5 (d, J = 2.0 Hz), 169.2 (d, J = 52.3 Hz), 201.8. IR (ATR) ν (cm−1): 2961, 2878, 1722, 1620, 1482, 1247, 1210, 1195, 1155, 1116, 1009, 829. LRMS (ESI): Mass calcd. for [M + Na]+ C15H15F3O5Na: 355.1; found 355.2. Anal. Calcd. for C15H15F3O5: C, 54.22; H, 4.55; found: C, 54.31; H, 4.65.
Methyl (S)-2-(3-oxo-2-propyl-5-(trifluoromethoxy)-2,3-dihydrobenzofuran-2-yl)acetate (2l’). Yellow oil (92% yield, 85% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 10.7 min, t (minor) = 9.6 min. [α] = +15.9 (c 1.0, CH2Cl2).
Methyl (R)-2-(5-fluoro-3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2m). Yellow oil (97% yield, 86% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 12.2 min, t (minor) = 9.5 min. [α] = −33.9 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 7.3 Hz, 3H), 1.11–1.26 (m, 1H), 1.28–1.38 (m, 1 H), 1.81 (dtd, J = 17.3, 13.0, 5.6 Hz, 2H), 2.99 (d, J = 16.6 Hz, 1H), 3.03 (d, J = 16.6 Hz, 1H), 3.54 (s, 3H), 7.05 (dd, J = 9.7, 3.5 Hz, 1H), 7.31–7.37 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 16.2, 38.8, 40.7, 51.8, 90.0, 109.2 (d, J = 23.1 Hz), 113.9 (d, J = 8.1 Hz), 122.3 (d, J = 7.0 Hz), 125.2 (d, J = 26.1 Hz), 156.4, 158.8, 168.2 (d, J = 134.8 Hz), 202.5. IR (ATR) ν (cm−1): 2953, 2935, 1726, 1482, 1437, 1250, 1213, 1163, 1123, 1084, 1009, 837. LRMS (ESI): Mass calcd. for [M + Na]+ C14H15FO4Na: 289.1; found 289.1. Anal. Calcd. for C14H15FO4: C, 58.13; H, 5.23; found: C, 58.20; H, 5.33.
Methyl (S)-2-(5-fluoro-3-oxo-2-propyl-2,3-dihydrobenzofuran-2-yl)acetate (2m’). Yellow oil (95% yield, 96% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 9.6 min, t (minor) = 12.0 min. [α] = +36.5 (c 1.0, CH2Cl2).
Methyl (R)-2-(1-oxo-2-phenethyl-1,2-dihydronaphtho [2,1-b]furan-2-yl)acetate (2n). Yellow oil (94% yield, 28% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 28.2 min, t (minor) = 28.0 min. [α] = −4.3 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 2.26–2.35 (m, 2H), 2.48–2.56 (m, 1H), 2.61–2.68 (m, 1H), 3.04 (d, J = 16.0 Hz, 1H), 3.10 (d, J = 16.0 Hz, 1H), 3.55 (s, 3H), 7.11–7.14 (m, 2H), 7.15–7.18 (m, 1H), 7.22–7.26 (m, 2H), 7.30 (d, J = 8.0 Hz, 1H), 7.51 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 7.70 (ddd, J = 8.4, 7.2, 1.6 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 8.8 Hz, 1H), 8.80–8.82 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 29.2, 38.2, 40.5, 51.8, 89.4, 113.6, 113.7, 123.2, 125.4, 126.1, 128.3 (2C), 128.4 (2C), 128.5, 129.2, 129.3, 129.8, 140.0, 140.6, 169.0, 174.3, 201.6. IR (ATR) ν (cm−1): 3081, 3060, 3023, 2948, 1713, 1683, 1585, 1509, 1430, 1366, 1234, 1207, 1163, 1151, 1117, 1073, 1061, 1044, 1002, 1482, 1437, 1250, 1213, 1163, 1123, 1084, 1009, 837. LRMS (ESI): Mass calcd. for [M + Na]+ C23H20O4Na: 383.1; found 383.2. Anal. Calcd. for C23H20O4: C, 76.65; H, 5.59; found: C, 76.77; H, 5.68.
Methyl (S)-2-(1-oxo-2-phenethyl-1,2-dihydronaphtho[2,1-b]furan-2-yl)acetate (2n’). Yellow oil (90% yield, 50% ee). HPLC (Phenomenex Lux Amylose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 19.2 min, t (minor) = 26.8 min. [α] = +9.2 (c 1.0, CH2Cl2).
Methyl (R)-2-(1-oxo-2-propyl-1,2-dihydronaphtho[2,1-b]furan-2-yl)acetate (2o). Yellow oil (94% yield, 50% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 22.0 min, t (minor) = 15.8 min. [α] = −7.7 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 0.85 (t, J = 7.0 Hz, 3H), 1.88–1.96 (m, 2H), 2.99 (d, J = 16.0 Hz, 1H), 3.04 (d, J = 16.0 Hz, 1H), 3.51 (s, 3H), 7.24 (d, J = 9.0 Hz, 1H), 7.47 (m, 1H), 7.66 (m, 1H), 7.81 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 8.75 (d, J = 8.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 14.0, 16.3, 22.7, 29.7, 38.6, 40.5, 51.8, 89.9, 113.6, 123.2, 125.2, 128.5, 129.2, 129.7, 139.7, 169.1, 174.2, 202.0. IR (ATR) ν (cm−1): 2949, 2928, 1734, 1688, 1630, 1578, 1515, 1437, 1359, 1288, 1211, 1163, 1151, 1123, 1076, 991, 912, 837. LRMS (ESI): Mass calcd. for [M + Na]+ C18H18O4Na: 321.1; found 321.2. Anal. Calcd. for C18H18O4: C, 72.47; H, 6.08; found: C, 72.55; H, 6.17.
Methyl (S)-2-(1-oxo-2-propyl-1,2-dihydronaphtho[2,1-b]furan-2-yl)acetate (2o’). Yellow oil (94% yield, 32% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 22.0 min, t (minor) = 15.8 min. [α] = +5.7 (c 1.0, CH2Cl2).
Methyl (R)-2-(2-methyl-1-oxo-1,2-dihydronaphtho[2,1-b]furan-2-yl)acetate (2p). Yellow oil (98% yield, 32% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 18.8 min, t (minor) = 24.0 min. [α] = −6.2 (c 1.0, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ: 1.59 (s, 3H), 2.99 (d, J = 16.0 Hz, 1H), 3.06 (d, J = 16.0 Hz, 1H), 3.57 (s, 3H), 7.25 (d, J = 9.2 Hz, 1H), 7.45 (m, 1H), 7.68 (m, 1H), 7.85 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 9.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 22.3, 41.0, 51.8, 87.3, 112.1, 113.9, 123.2, 125.3, 128.5, 129.3, 129.5, 129.8, 139.8, 169.1, 173.6, 202.0. IR (ATR) ν (cm−1): 2940, 2924, 1730, 1680, 1634, 1580, 1510, 1430, 1355, 1285, 1209, 1163, 1151, 1128, 1070, 991, 912, 837. LRMS (ESI): Mass calcd. for [M + Na]+ C16H14O4Na: 293.1; found 293.2. Anal. Calcd. for C16H14O4: C, 71.10; H, 5.22; found: C, 71.18; H, 5.31.
Methyl (S)-2-(2-methyl-1-oxo-1,2-dihydronaphtho[2,1-b]furan-2-yl)acetate (2p’). Yellow oil (98% yield, 3% ee). HPLC (Phenomenex Lux Cellulose-1, hexanes/2-propanol 90:10, v = 0.7 mL/min, λ = 254 nm) t (major) = 18.8 min, t (minor) = 24.0 min. [α] = +1.8 (c 1.0, CH2Cl2).






{kind=link}
{kind=link}
{kind=link}
{kind=link}


