A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases



Abstract
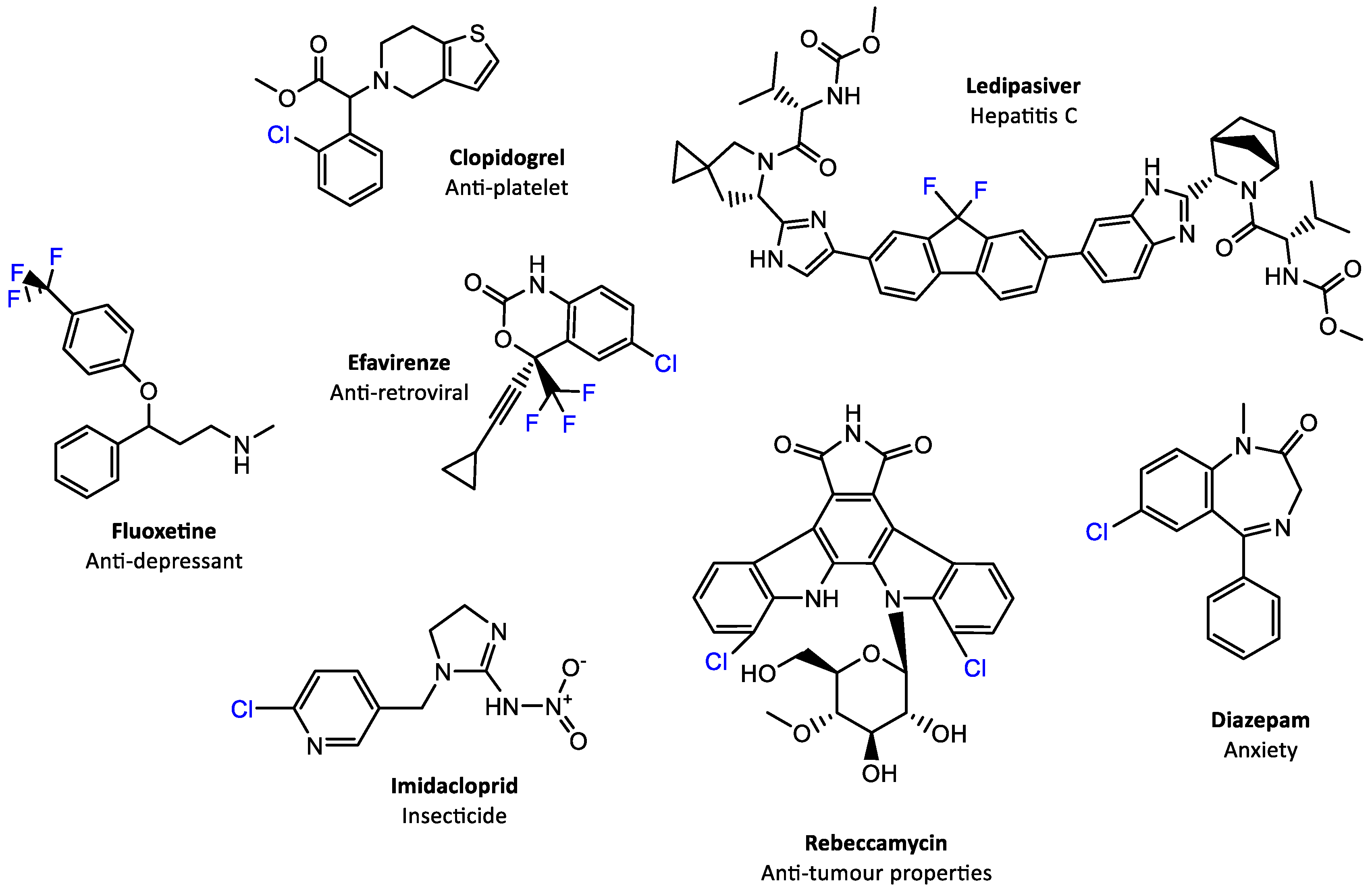
1. Introduction
1.1. Classification of Halogenases
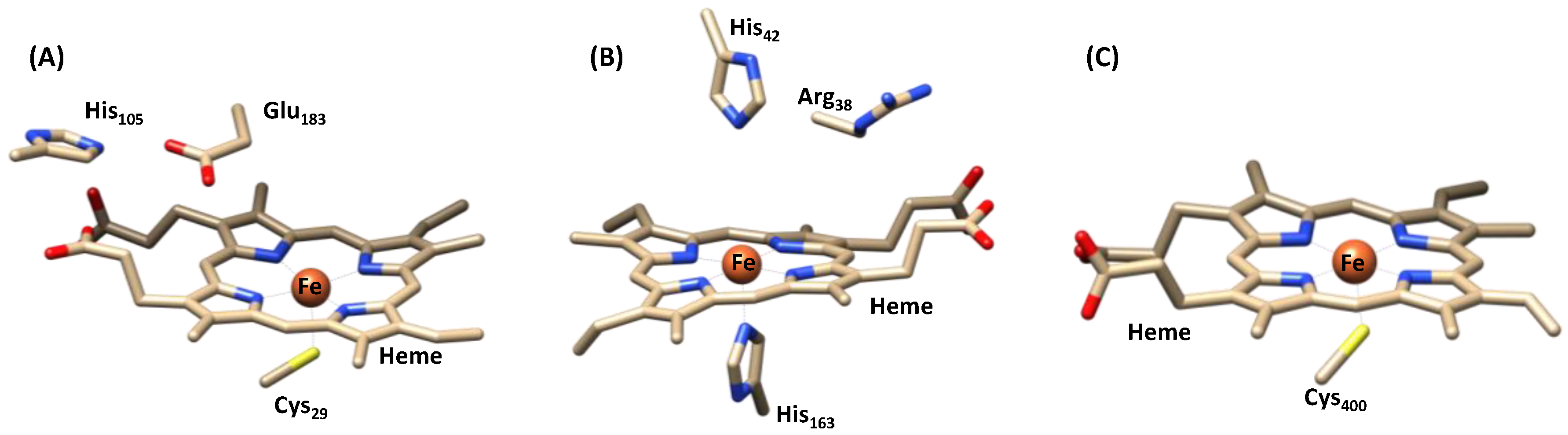
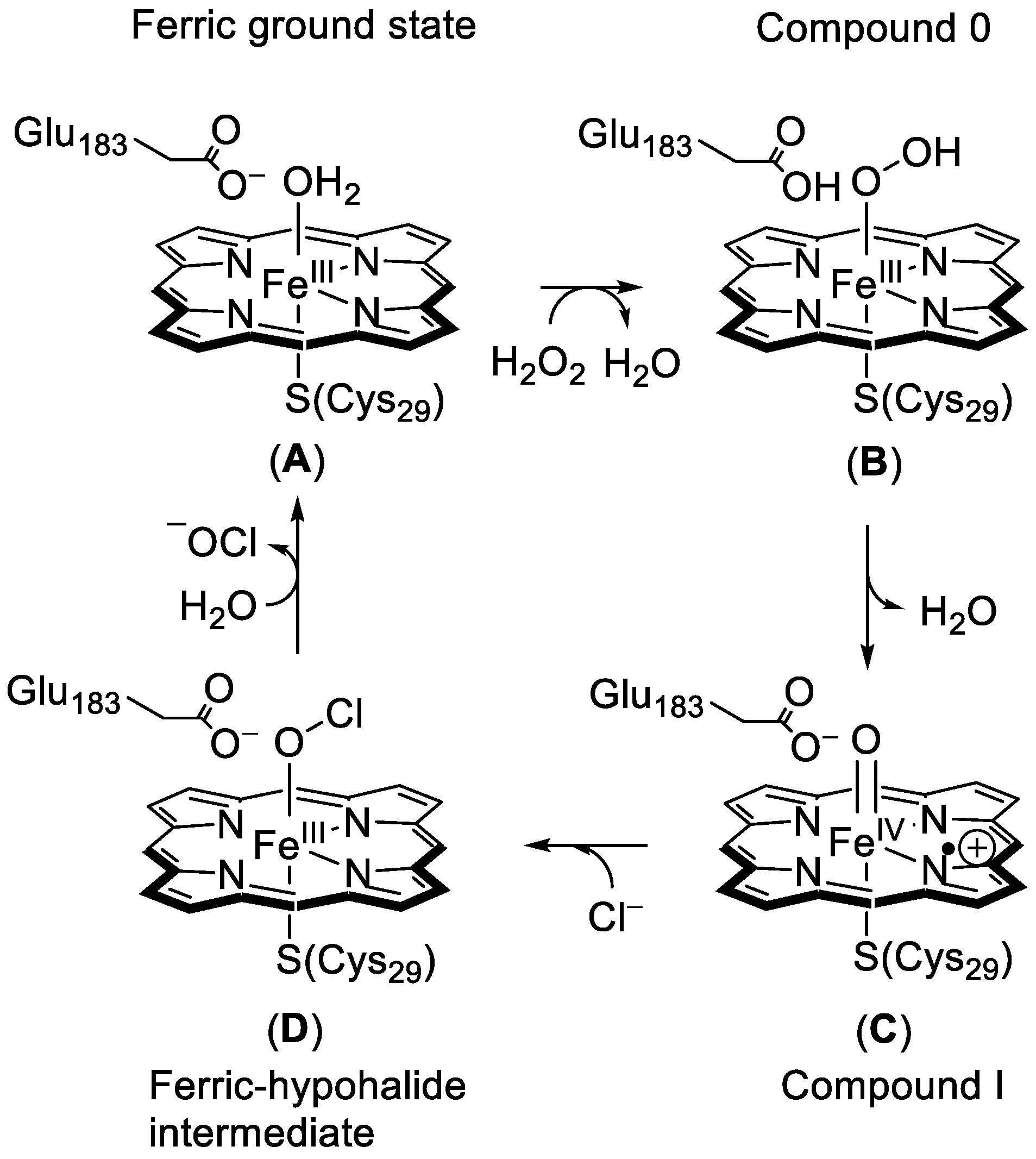
1.1.1. Heme Haloperoxidases
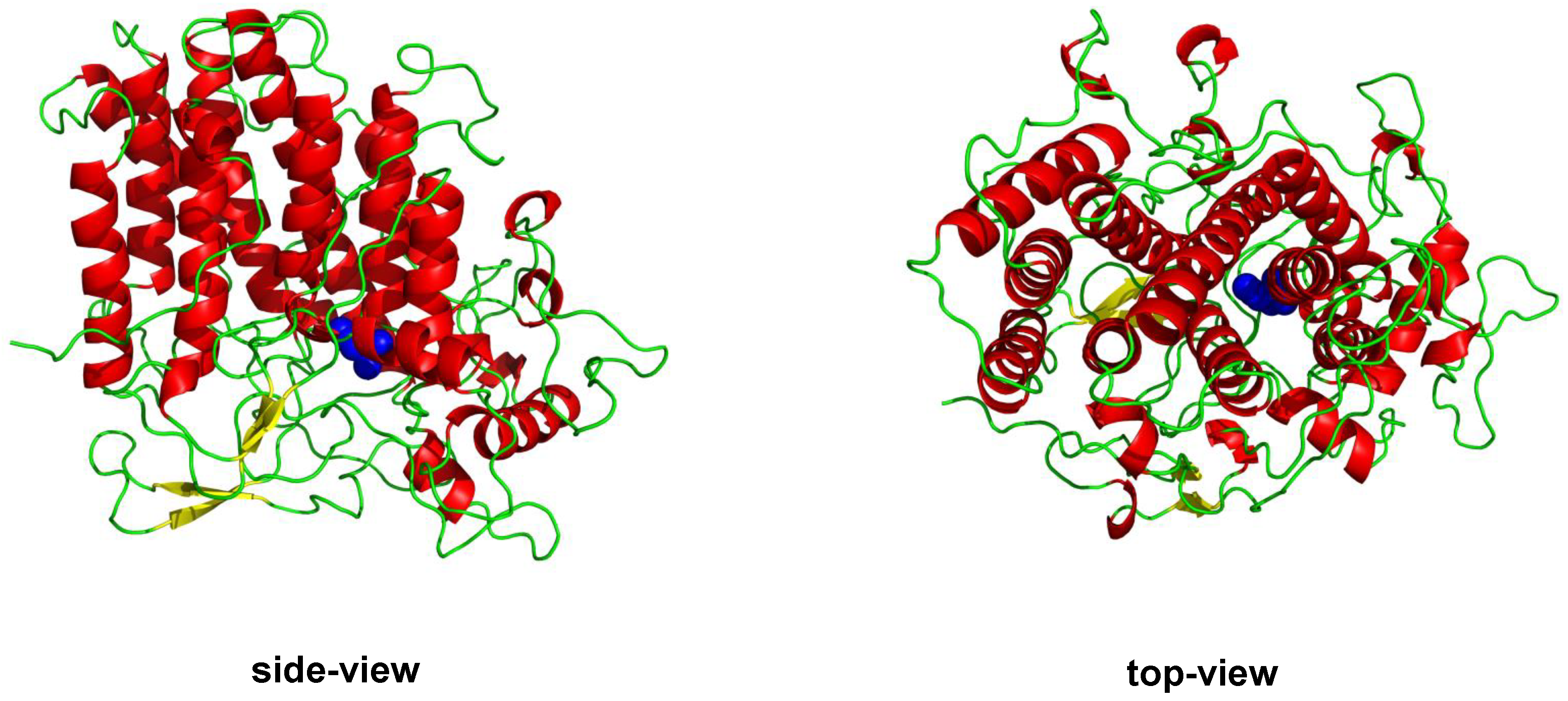
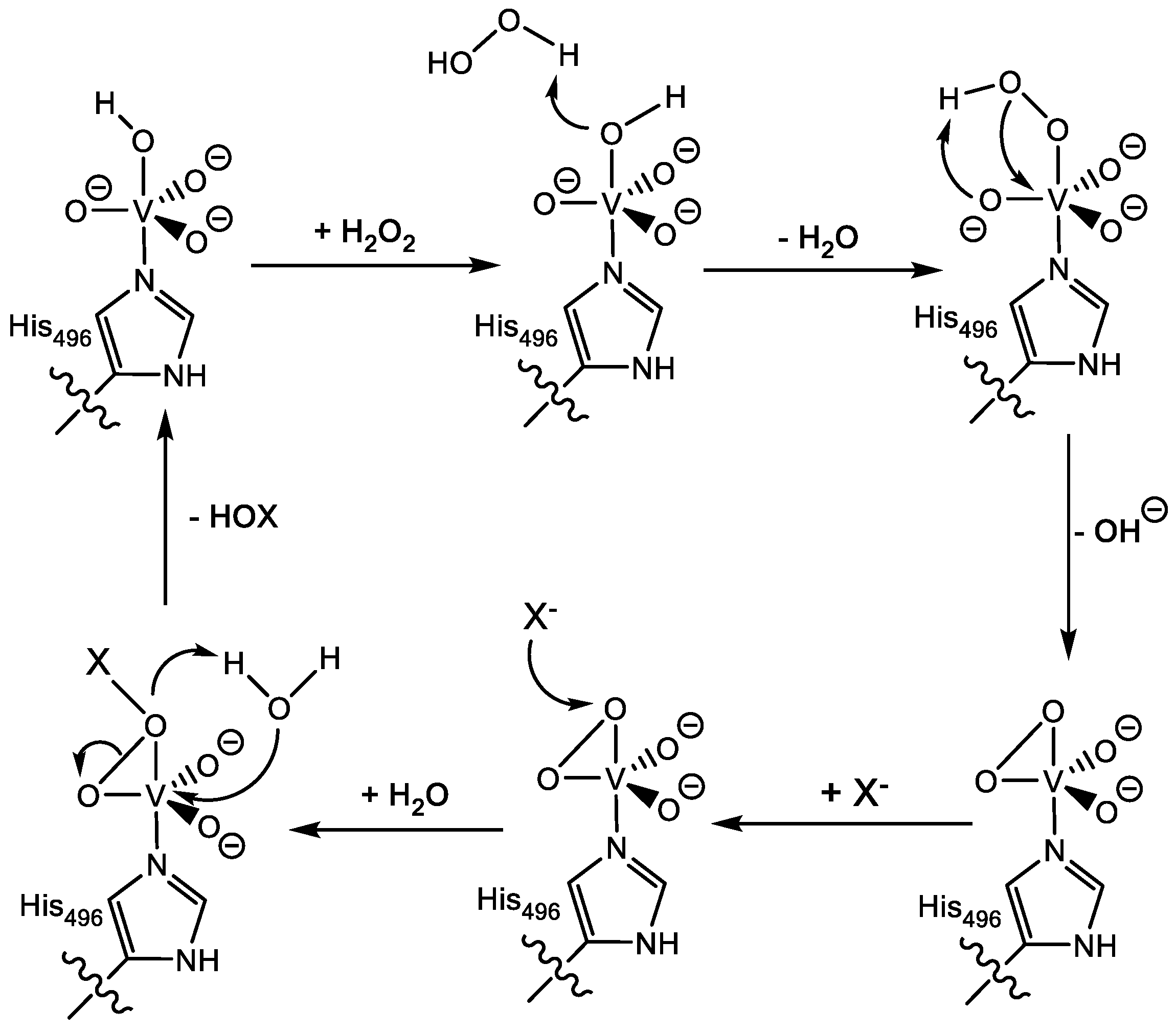
1.1.2. Vanadium Haloperoxidases
1.1.3. Flavin Adenine Dinucleotide Haloperoxidases
1.1.4. S-Adenosyl-l-Methionine Fluorinase
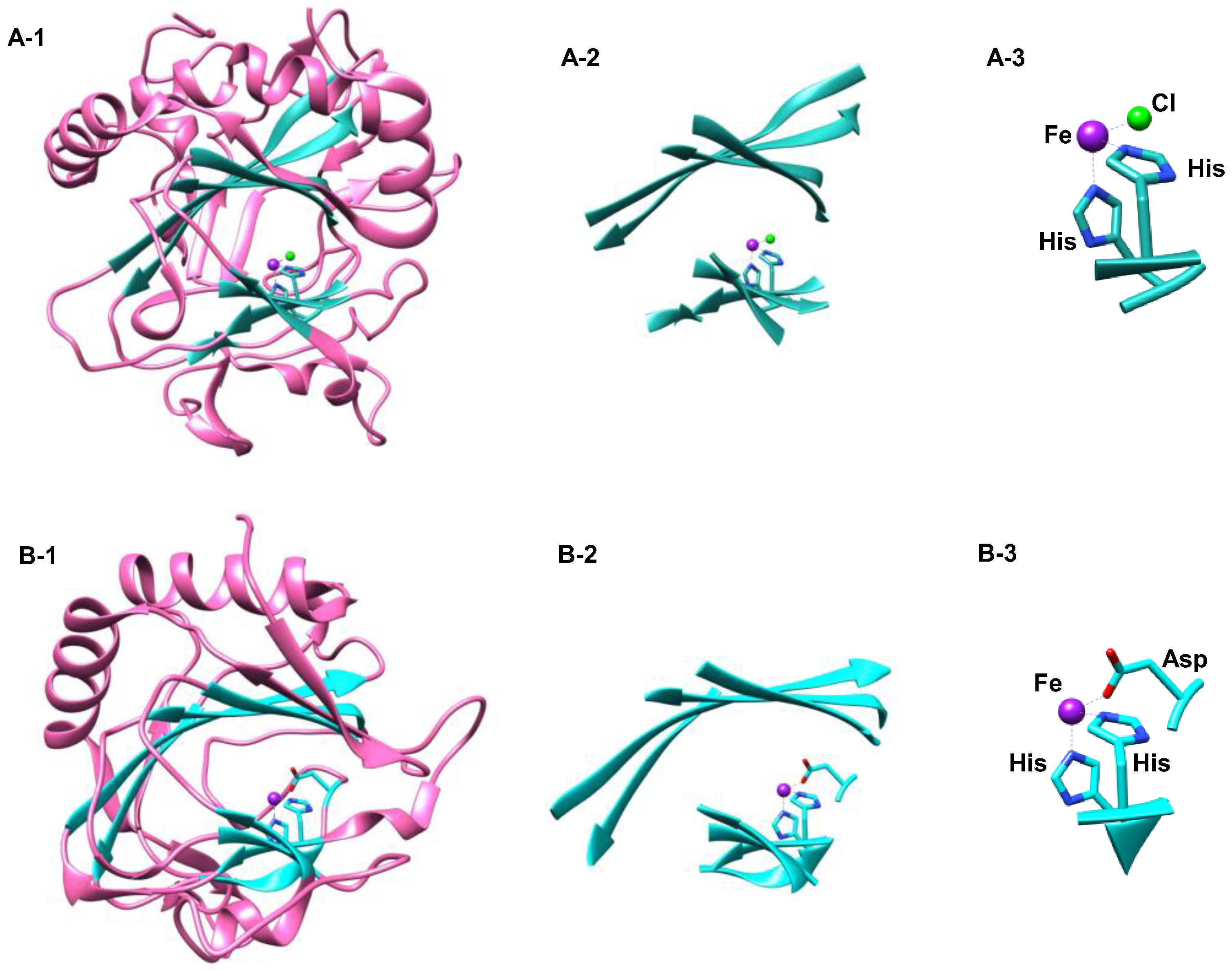
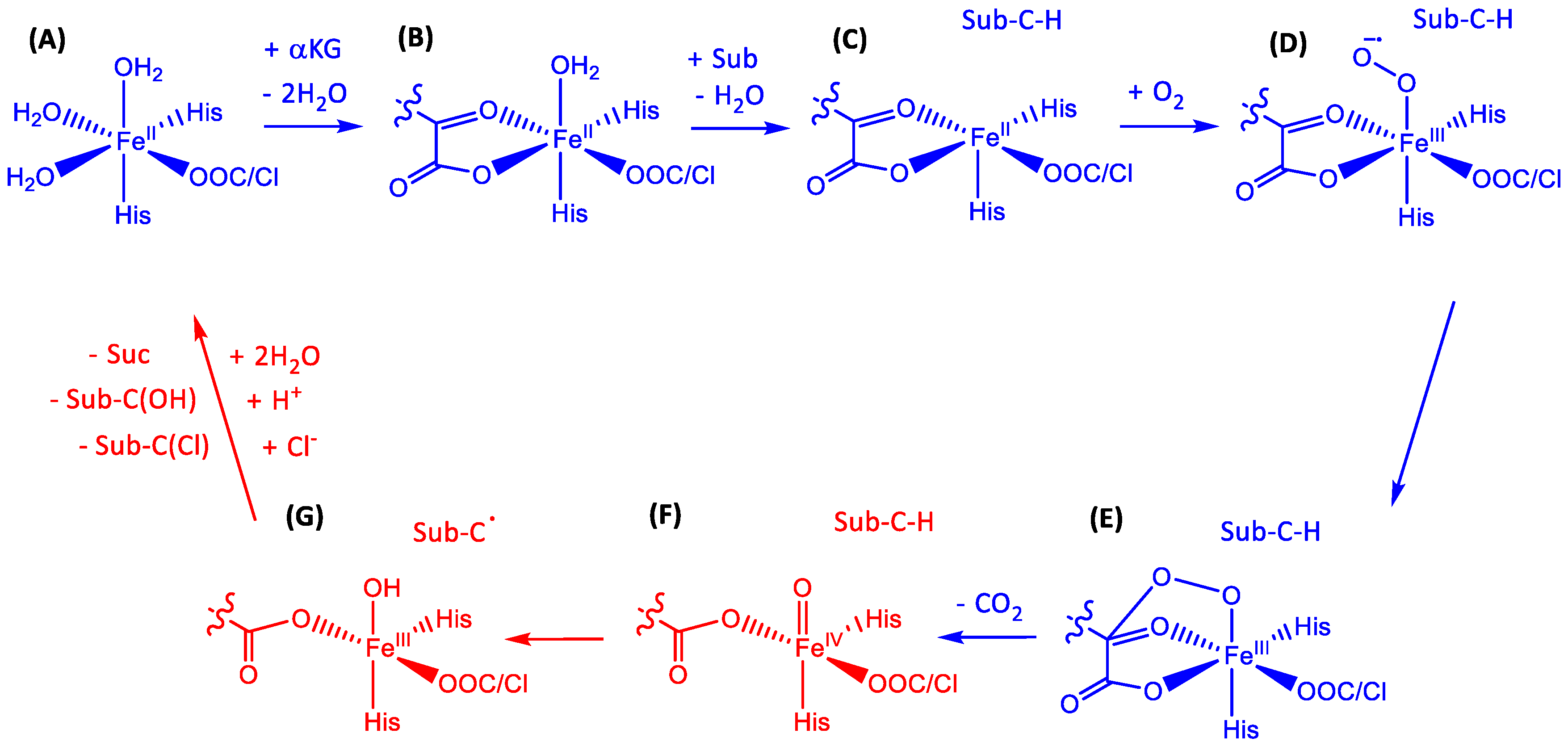
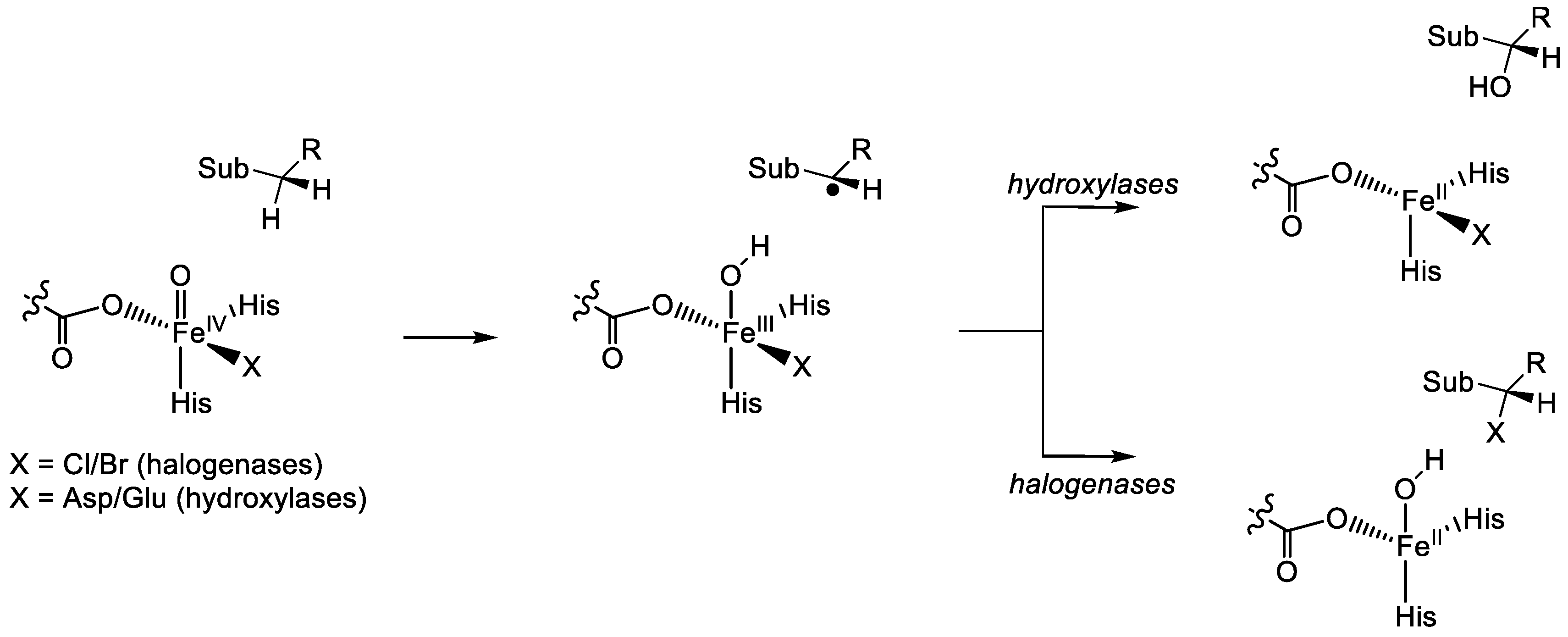
1.1.5. Nonheme Iron/α-Ketoglutarate Halogenases
2. Computational Studies on Nonheme Iron Hydroxylases and Halogenases
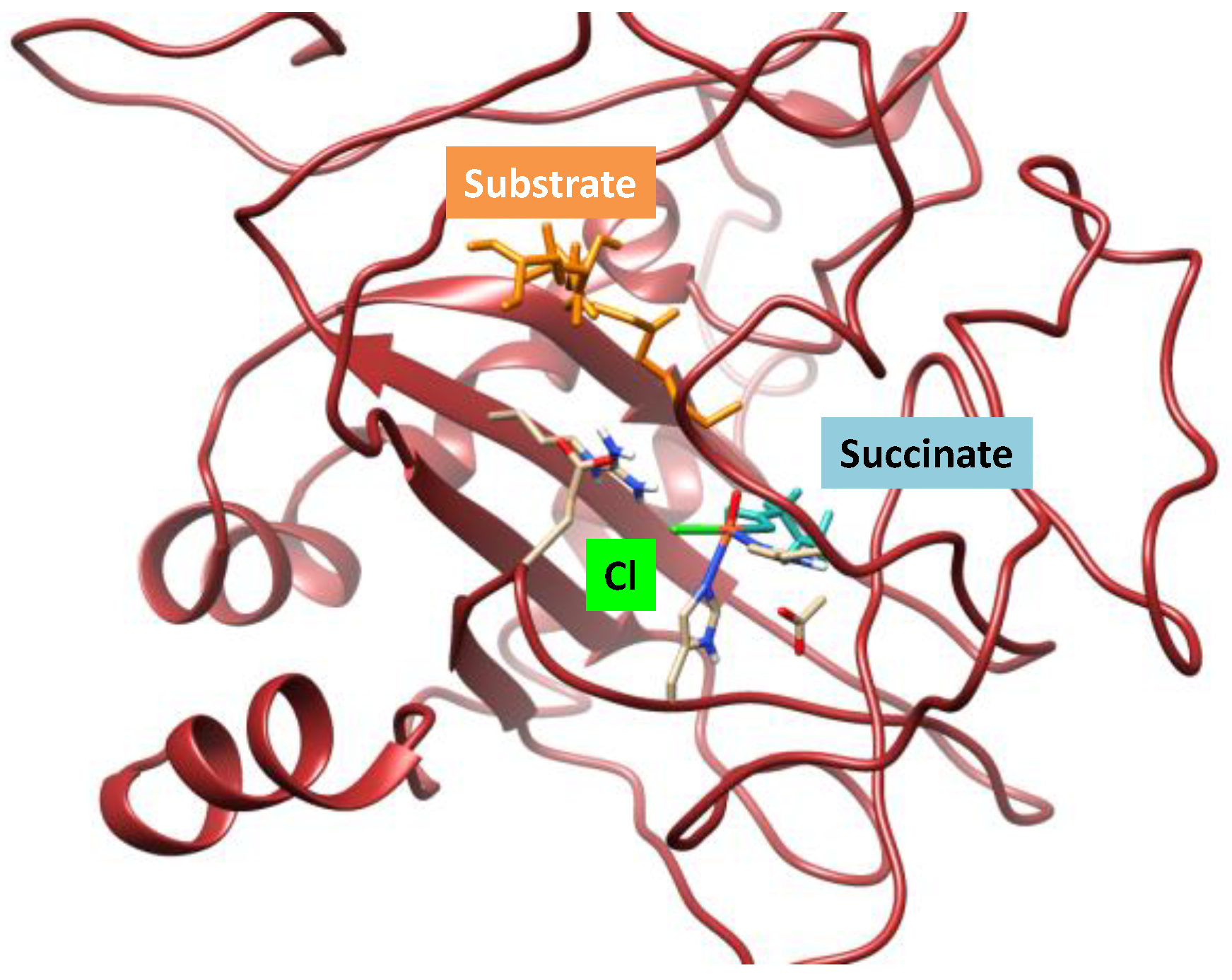
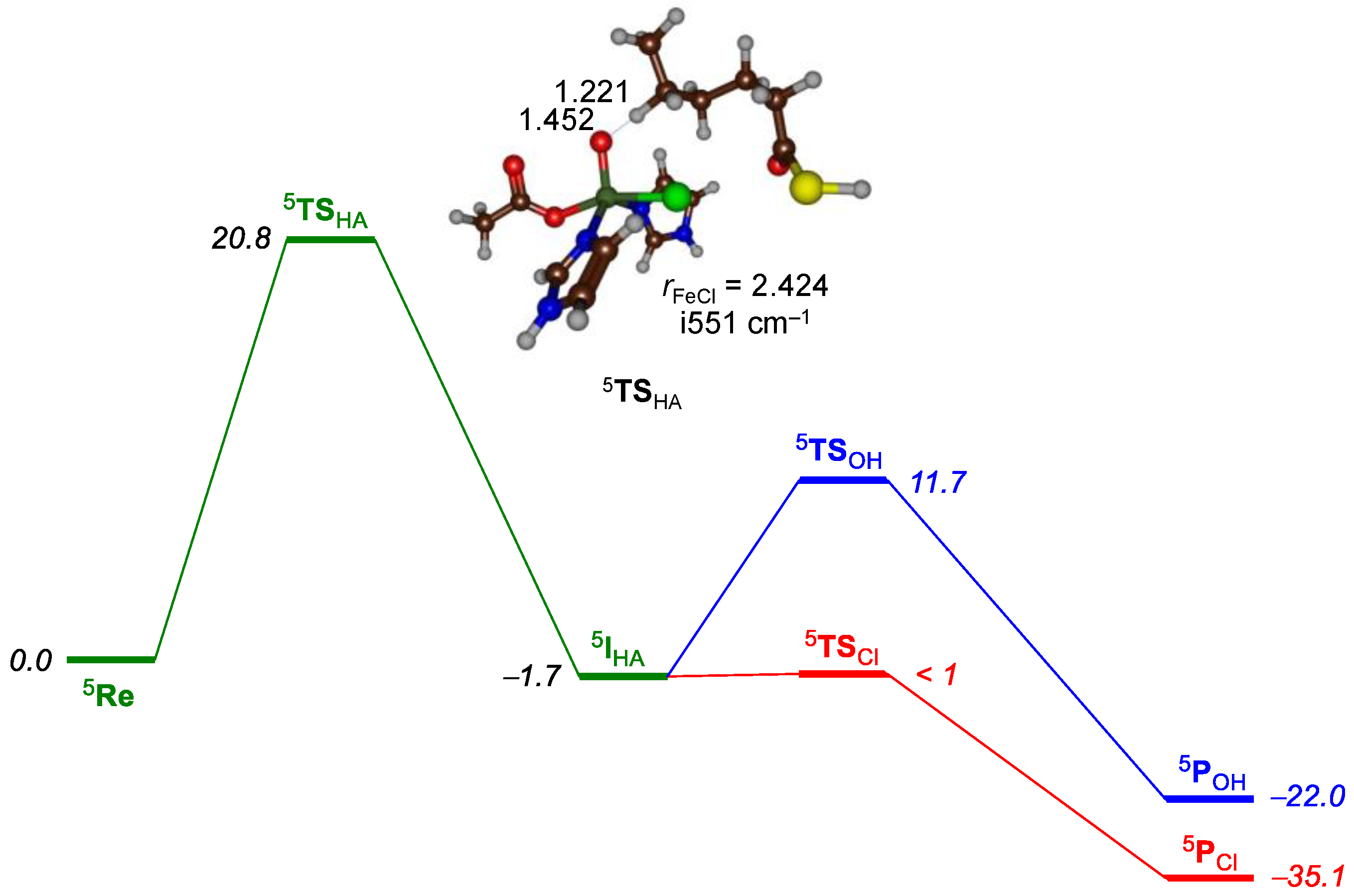
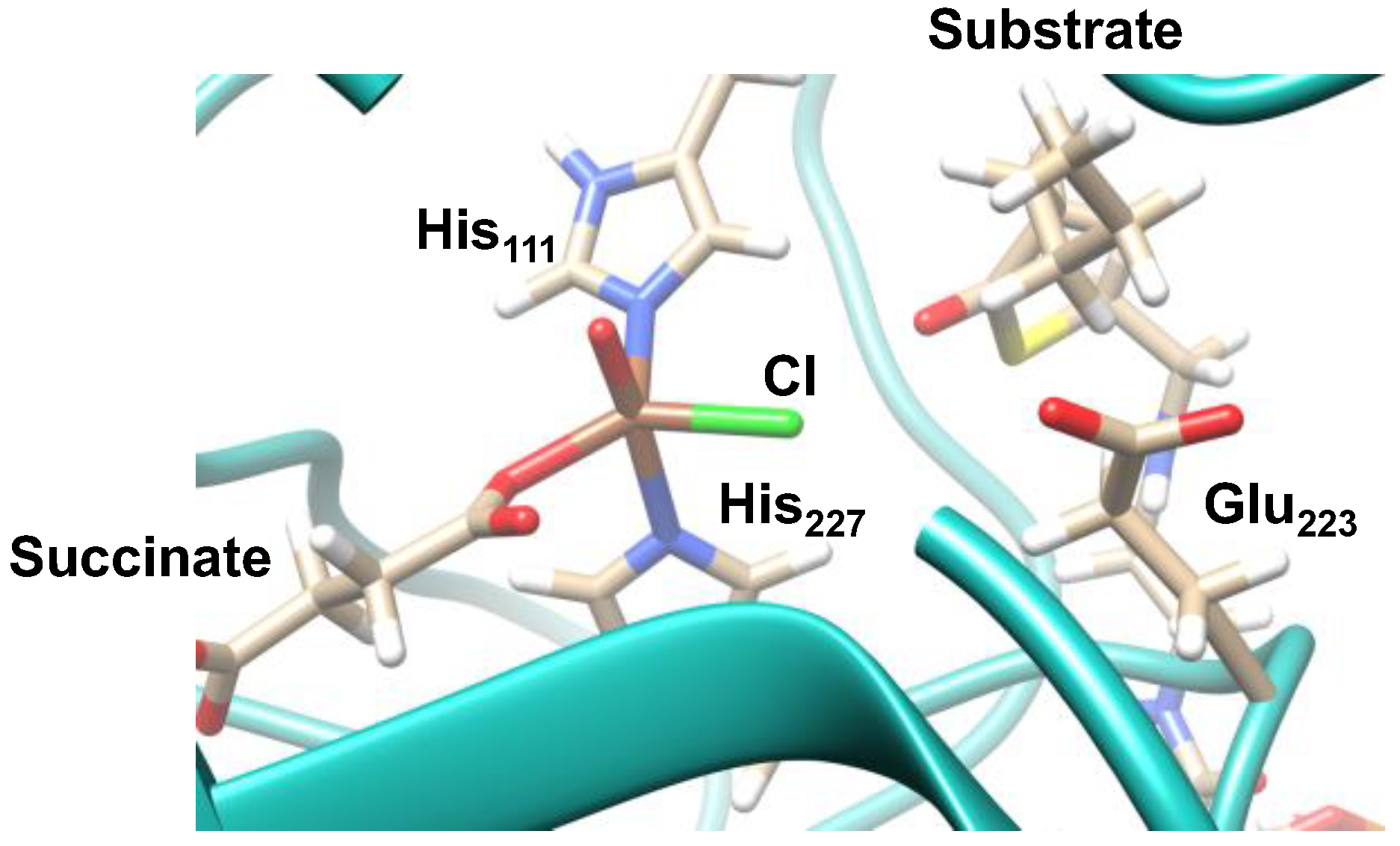
2.1. Hectochlorin Biosynthesis Enzyme
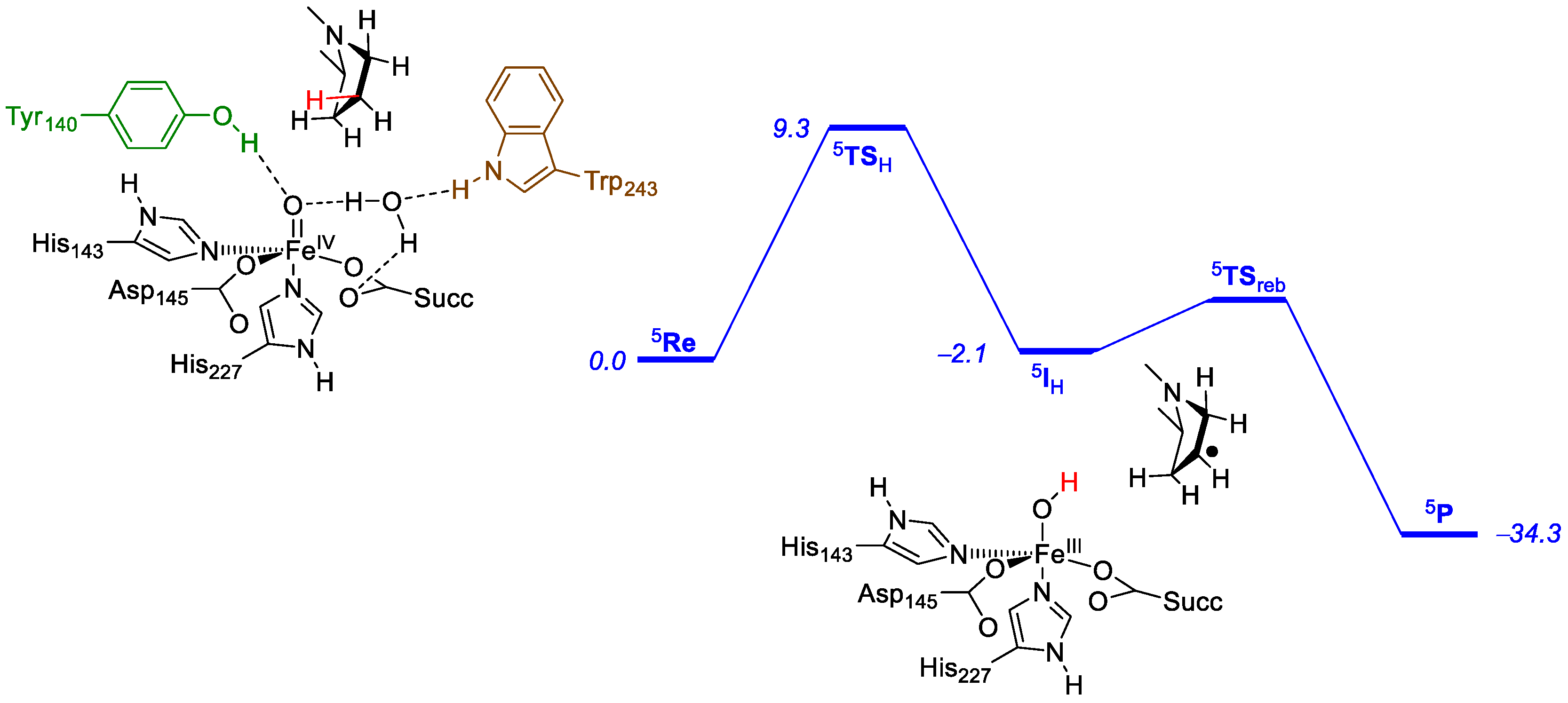
2.2. Prolyl-4-Hydroxylase
3. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gribble, G.W. The diversity of naturally produced organohalogens. Chemosphere 2003, 52, 289–297. [Google Scholar] [CrossRef]
- Vaillancourt, F.H.; Yeh, E.; Vosburg, D.A.; Garneaur-Tsodikova, S.; Walsh, C.T. Nature’s inventory of halogenation catalysts: Oxidative strategies predominate. Chem. Rev. 2006, 106, 3364–3378. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, M.T.; Vale, C.; Rauter, A.P. Halogenated compounds from marine algae. Mar. Drugs 2010, 8, 2301–2317. [Google Scholar] [CrossRef] [PubMed]
- Van Pee, K.-H. Biosynthesis of halogenated metabolited by bacteria. Annu. Rev. Microbiol. 1996, 50, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Laus, G. Biological activities of natural halogen compounds. Stud. Nat. Prod. Chem. 2001, 25, 757–809. [Google Scholar] [CrossRef]
- Stonik, V.A.; Fedorov, S.N. Marine low molecular weight natural products as potential cancer preventative compounds. Mar. Drugs 2014, 12, 636–671. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Hu, W.P.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2009, 26, 170–244. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Nakama, Y.; Yoshida, O.; Yoda, M.; Araki, K.; Sawada, Y.; Nakamura, J.; Xu, S.; Miura, K.; Maki, H.; Arimoto, H. Discovery of a novel series of semisynthetic vancomycin derivatives effective against vancomycin-resistant bacteria. J. Med. Chem. 2010, 53, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Matrangolo, P.; Milani, R.; Pilati, T.; Primagi, A.; Resnati, G.; Tarraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Lantham, J.; Brandenburger, E.; Shepherd, S.A.; Menon, B.R.K.; Micklefield, J. Development of halogenase enzymes for use in synthesis. Chem. Rev. 2017, 111, 232–269. [Google Scholar] [CrossRef]
- Tang, M.L.; Bao, Z. Halogenated materials as organic semiconductors. Chem. Mater. 2011, 23, 446–455. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen bonding in polymer science: From crystal engineering to functional supramolecular polymers and materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Amanchukwu, C.V.; Harding, J.R.; Shao-Horn, Y.; Hammond, P.T. Understanding the chemical stability of polymers for lithium-air batteries. Chem. Mater. 2015, 27, 550–561. [Google Scholar] [CrossRef]
- Sundaramoorthy, M.; Terner, J.; Poulus, T.L. The crystal structure of chloroperoxidase: A heme-peroxidase-cytochrome P450 functional hybrid. Structure 1995, 3, 1367–1377. [Google Scholar] [CrossRef]
- Badyal, S.K.; Joyce, M.G.; Sharp, K.H.; Seward, H.E.; Mewies, M.; Basranl, J.; Macdonald, I.K.; Moody, P.C.E.; Raven, E.L. Conformational mobility in the active site of the heme peroxidase. J. Biol. Chem. 2006, 281, 24512–24520. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Poulos, T.L. The structure of the cytochrome P450M-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat. Struct. Mol. Biol. 1997, 4, 140–146. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Nam, W. High-valent iron-oxo porphyrins in oxygenation reactions. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010; Chapter 44; pp. 85–140, ISBN-13 978-981-4307-23-9. [Google Scholar]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The experimentally elusive oxidant of cytochrome P450: A theoretical “trapping” defining more closely the “real” species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef]
- De Visser, S.P.; Ogliaro, F.; Gross, Z.; Shaik, S. What is the difference between the manganese porphyrin and corrole analogs of cytochrome P450’s Compound I? Chem. Eur. J. 2001, 7, 4954–4960. [Google Scholar] [CrossRef]
- De Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; de Visser, S.P.; Kumar, D. External electric field will control the selectivity of enzymatic-like bond activations. J. Am. Chem. Soc. 2004, 126, 11746–11749. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. What affects the quartet-doublet energy splitting in peroxidase enzymes? J. Phys. Chem. A 2005, 109, 11050–11057. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Derat, E.; Shaik, S. The intrinsic axial ligand effect on propene oxidation by horseradish peroxidase versus cytochrome P450 enzymes. J. Biol. Inorg. Chem. 2005, 10, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Hirao, H.; Shaik, S. Sulfoxidation mechanisms catalyzed by cytochrome P450 and horseradish peroxidase models: Spin selection induced by the ligand. Biochemistry 2005, 44, 8148–8158. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. The axial ligand effect of oxo-iron porphyrin catalysts. How does chloride compare to thiolate? J. Biol. Inorg. Chem. 2006, 11, 168–178. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Can the replacement of a single atom in the enzyme horseradish peroxidase convert it into a monoxygenase? A density functional study. J. Phys. Chem. B 2006, 110, 20759–20761. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; de Visser, S.P. How does the push/pull effect of the axial ligand influence the catalytic properties of Compound I of catalase and cytochrome P450? J. Inorg. Biochem. 2007, 101, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef] [PubMed]
- Silaghi-Dumitrescu, R. Halide activation by heme peroxidases: Theoretical predictions on putative adducts of halides with Compound I. Eur. J. Inorg. Chem. 2008, 2008, 5404–5407. [Google Scholar] [CrossRef]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Sharma, P.K.; Shaik, S. Searching for the second oxidant in the catalytic cycle of cytochrome P450: A theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J. Am. Chem. Soc. 2002, 124, 2806–2817. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; Kevorkiants, R.; de Visser, S.P.; Kumar, D.; Shaik, S. Porphyrin trap its own terminator! Concerted and stepwise porphyrin degradation mechanisms induced by heme-oxygenase vs. cytochrome P450. Angew. Chem. Int. Ed. 2004, 43, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. Theory favors a stepwise mechanism of porphyrin degradation by a ferric hydroperoxide model of the active species of heme oxygenase. J. Am. Chem. Soc. 2005, 127, 8204–8213. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.S.; Sutcliffe, M.J.; de Visser, S.P. Quantum mechanics/molecular mechanics studies on the sulfoxidation of dimethyl sulfide by Compound I and Compound 0 of Cytochrome P450: Which is the better oxidant? J. Phys. Chem. A 2009, 113, 11635–11642. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; Sastri, C.V.; Banse, F.; de Visser, S.P. Differences and comparisons of the properties and reactivities of iron(III)-hydroperoxo complexes with saturated coordination sphere. Chem. Eur. J. 2015, 21, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Banse, F.; de Visser, S.P. Arene activation by a nonheme iron(III)-hydroperoxo complex: Pathways leading to phenol and ketone products. J. Biol. Inorg. Chem. 2016, 21, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Conesa, A.; Punt, P.J.; Hager, L.P. Examining the role of glutamic acid 183 in chloroperoxidase catalysis. J. Biol. Chem. 2003, 278, 13855–13859. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, H.-A.; Woggon, W.-D. Identification of intermediates in the catalytic cycle of chloroperoxidase. Chem. Biol. 1997, 4, 367–372. [Google Scholar] [CrossRef]
- Stone, K.L.; Behan, R.K.; Green, M.T. X-ray absorption spectroscopy of chloroperoxidase compound I: Insight into the reactive intermediate of P450 chemistry. Proc. Natl. Acad. Sci. USA 2005, 102, 16563–16565. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Perera, R.; Hager, L.P.; Dawson, J.H.; Hoffman, B.M. Rapid freeze-quench ENDOR study of chloroperoxidase Compound I: The site of the radical. J. Am. Chem. Soc. 2006, 128, 5598–5599. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T.; Dawson, J.H.; Gray, H.B. Oxoiron(IV) in chloroperoxidase compound II is basic: Implications for P450 chemistry. Science 2004, 304, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Shaik, S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Ogliaro, F.; Cohen, S.; de Visser, S.P.; Shaik, S. Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: What kind of a radical is it really? J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar] [CrossRef]
- Sharma, P.K.; de Visser, S.P.; Ogliaro, F.; Shaik, S. Is the ruthenium analogue of Compound I of cytochrome P450 an efficient oxidant? A theoretical investigation of the methane hydroxylation reaction. J. Am. Chem. Soc. 2003, 125, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Tan, T.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Porro, C.S.; Quesne, M.G.; Sainna, M.A.; Munro, A.W. Overview on recent theoretical studies discriminating the two-oxidant versus two-state-reactivity models for substrate monoxygenation by cytochrome P450 enzymes. Curr. Top. Med. Chem. 2013, 13, 2218–2232. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Biodegradation of cosmetics products: A computational study of cytochrome P450 metabolism of phthalates. Inorganics 2017, 5, 77. [Google Scholar] [CrossRef]
- Palcic, M.M.; Rutter, R.; Araiso, T.; Hager, L.P.; Dunford, H.B. Spectrum of chloroperoxidase compound I. Biochem. Biophys. Res. Commun. 1980, 94, 1123–1127. [Google Scholar] [CrossRef]
- Egawa, D.; Proshlyakov, T.; Miki, H.; Makino, R.; Ogura, K.; Ishimura, Y. Effects of a thiolate axial ligands on the pi-->pi* electronic states of oxoferryl porphyrins: A study of the optical and resonance Raman spectra of compounds I and II of chloroperoxidase. J. Biol. Inorg. Chem. 2001, 6, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Rutter, R.; Hager, L.P.; Dhonau, H.; Hendrich, M.; Valentine, J.; Debrunner, P. Chloroperoxidase compound I: Electron paramagnetic resonance and Mössbauer studies. Biochemistry 1984, 23, 6809–6816. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.M. Electron-nuclear double resonance spectroscopy (and electron spin-echo envelope modulation spectroscopy) in bioinorganic chemistry. Proc. Natl. Acad. Sci. USA 2003, 100, 3575–3578. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T. Evidence for sulfur-based radicals in thiolate compound I intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Anthony, J.; Grodzicki, M.; Trautwein, A.X. Local density functional study of oxoiron porphyrin complexes and their one-electron oxidised derivatives. Axial ligand effects. J. Phys. Chem. 1997, 101, 2692–2701. [Google Scholar] [CrossRef]
- Schöneboom, J.C.; Lin, H.; Reuter, N.; Thiel, W.; Cohen, S.; Ogliaro, F.; Shaik, S. The elusive oxidant species of cytochrome P450 enzymes: Characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 2002, 124, 8142–8151. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 2011, 47, 11044–11046. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Mechanistic insight into halide oxidation by non-heme iron complexes. Haloperoxidase versus halogenase activity. Chem. Commun. 2013, 49, 10926–10928. [Google Scholar] [CrossRef] [PubMed]
- Van Pee, K.-H.; Dong, C.; Flecks, S.; Naismith, J.; Patallo, E.P.; Wage, T. Biological halogenation has moved far beyond haloperoxidases. Adv. Appl. Microbiol. 2006, 59, 127–157. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; de Visser, S.P. Enzymatic halogenases and haloperoxidases: Computational studies on mechanism and function. Adv. Protein Chem. Struct. Biol. 2015, 100, 113–151. [Google Scholar] [CrossRef] [PubMed]
- Spreti, N.; Germani, R.; Icani, A.; Savelli, G. Stabilization of chloroperoxidase by polyethylene glycols in aqueous media: Kinetic studies and synthetic applications. Biotechnol. Prog. 2004, 20, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; de Visser, S.P. Regioselectivity of substrate hydroxylation versus halogenation by a non-heme iron(IV)-oxo complex: Possibility of rearrangement pathways. J. Biol. Inorg. Chem. 2012, 17, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Quesne, M.G.; Davies, C.G.; Dürr, M.; Ivanović-Burmazović, I.; Siegler, M.A.; Jameson, G.N.L.; de Visser, S.P.; Goldberg, D.P. Direct observation of a non-heme iron(IV)-oxo complex that mediates aromatic C-F hydroxylation. J. Am. Chem. Soc. 2014, 136, 13542–13545. [Google Scholar] [CrossRef] [PubMed]
- Draksharapu, A.; Angelone, D.; Quesne, M.G.; Padamati, S.K.; Gómez, L.; Hage, R.; Costas, M.; Browne, W.R.; de Visser, S.P. Identification and spectroscopic characterization of nonheme iron(III) hypochlorite intermediates. Angew. Chem. Int. Ed. 2015, 54, 4357–4361. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.; Faponle, A.S.; Vardhaman, A.K.; Angelone, D.; Löhr, A.-M.; Browne, W.R.; Comba, P.; Sastri, C.V.; de Visser, S.P. Influence of ligand architecture in tuning reaction bifurcation pathways for chlorite oxidation by nonheme iron complexes. Inorg. Chem. 2016, 55, 10170–10181. [Google Scholar] [CrossRef] [PubMed]
- Rehder, D. The bioinorganic chemistry of vanadium. Angew. Chem. Int. Ed. Engl. 1991, 30, 148–167. [Google Scholar] [CrossRef]
- Butler, A.; Carter, J.N.; Simpson, M.T. Handbook of Metalloproteins; Marcel Dekker Inc.: New York, NY, USA, 2001; pp. 153–179. ISBN 9780470869819. [Google Scholar]
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Moore, B.S. Exploring the chemistry and biology of vanadium-dependent haloperoxidases. J. Biol. Chem. 2009, 284, 18577–18581. [Google Scholar] [CrossRef] [PubMed]
- De Boer, E.; Wever, R. The reaction mechanism of the novel vanadium-bromoperoxidase. A steady-state kinetic analysis. J. Biol. Chem. 1988, 263, 12326–12332. [Google Scholar] [PubMed]
- Van Schijndel, J.W.P.M.; Barnett, P.; Roelse, J.; Vollenbroed, E.G.M.; Wever, R. The stability and steady state kinetics of vanadium chloroperoxidase from the fungus Curvularia inequalis. Eur. J. Biochem. 1994, 225, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, A.; Wever, R. X-ray structure of a vanadium-containing enzyme: Chloroperoxidase from the fungus Curvularia inaequalis. Proc. Natl. Acad. Sci. USA 1996, 93, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.S.; Carroll, G.L.; Tschirret-Guth, R.A.; Altenhoff, G.; Little, D.R.; Butler, A. On the regiospecificity of vanadium bromoperoxidase. J. Am. Chem. Soc. 2001, 123, 3289–3294. [Google Scholar] [CrossRef] [PubMed]
- Carter-Franklin, J.N.; Butler, A. Vanadium bromoperoxidase-catalyzed biosynthesis of halogenated marine natural products. J. Am. Chem. Soc. 2004, 126, 15060–15066. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.K.; Jensen, P.R.; Fenical, W. Neomangicols: Structures and absolute stereochemistries of unprecedented halogenated sesterterpenes from a marine fungus of the genus Fusarium. J. Org. Chem. 1998, 63, 8346–8354. [Google Scholar] [CrossRef]
- Borchardt, S.A.; Allain, E.J.; Michels, J.J.; Stearns, G.W.; Kelly, R.F.; McCoy, W.F. Reaction of acylated homoserine lactone bacterial signalling molecules with oxidized halogen antimicrobials. Appl. Environ. Microbiol. 2001, 67, 3174–3179. [Google Scholar] [CrossRef] [PubMed]
- Wever, R.; Hemrika, W. Handbook of Metalloproteins; Messershmidt, A., Huber, R., Wieghardt, K., Poulos, T.L., Eds.; Wiley & Sons: Chichester, UK, 2001; Volume 1, ISBN 0-471-62743-7. [Google Scholar]
- Vilter, H. Peroxidases from Phaeophyceae. Bot. Mar. 1983, 26, 429–435. [Google Scholar] [CrossRef]
- Vilter, H. Peroxidases from Phaeophycea: A vanadium(V)-dependent peroxidase from Ascophyllum nodosum. Phytochemistry 1984, 23, 1387–1390. [Google Scholar] [CrossRef]
- Butler, A.; Sandy, M. Mechanistic considerations of halogenating enzymes. Nature 2009, 460, 848–854. [Google Scholar] [CrossRef] [PubMed]
- De Boer, E.; Plat, H.; Tromp, M.G.; Wever, R.; Franssen, M.C.; van der Plas, H.C.; Meijer, E.M.; Shoemaker, H.E. Vanadium containing bromoperoxidase: An example of an oxidoreductase with high operational stability in aqueous and organic media. Biotechnol. Bioeng. 1987, 30, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Coupe, E.E.; Smyth, M.G.; Fosberry, A.P.; Hall, R.M.; Littlechild, J.A. The dodecameric vanadium-dependent haloperoxidase from the marine algae Corallina officinalis: Cloning, expression, and refolding of the recombinant enzyme. Protein Expr. Purif. 2007, 52, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Willetts, A.; Allenmark, S. Asymmetric sulfoxidation catalyzed by a vanadium-containing bromoperoxidase. J. Org. Chem. 1997, 62, 8455–8458. [Google Scholar] [CrossRef] [PubMed]
- Ten Brink, H.B.; Dekker, H.L.; Schoemaker, H.E.; Wever, R. Oxidation reactions catalyzed by vanadium chloroperoxidase from Curvularia inaequalis. J. Inorg. Biochem. 2000, 80, 91–98. [Google Scholar] [CrossRef]
- Bernhardt, P.; Okino, T.; Winter, J.M.; Miyanaga, A.; Moore, B.S. A stereoselective vanadium-dependent chloroperoxidase in bacterial antibiotic biosynthesis. J. Am. Chem. Soc. 2011, 133, 4268–4270. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, A.; Prade, L.; Wever, R. Implications for the catalytic mechanism of the vanadium-containing enzyme chloroperoxidase from the fungus Curvularia inaequalis by X-ray structures of the native and peroxide form. J. Biol. Chem. 1997, 378, 309–315. [Google Scholar] [CrossRef]
- Macedo-Ribeiro, S.; Hemrika, W.; Renirie, R.; Wever, R.; Messerschmidt, A. X-ray crystal structures of active site mutants of the vanadium-containing chloroperoxidase from the fungus Curvularia inaequalis. J. Biol. Inorg. Chem. 1999, 4, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Weyand, M.; Hecht, H.; Kiess, M.; Liaud, M.; Vitler, H.; Schomburg, D. X-ray structure determination of a vanadium-dependent haloperoxidase from Ascophyllum nodosum at 2.0 Å resolution. J. Mol. Biol. 1999, 293, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Isupov, M.N.; Dalby, A.R.; Brindley, A.A.; Izumi, Y.; Tanabe, T.; Murshudov, G.N.; Littlechild, J.A. Crystal structure of dodecameric vanadium-dependent bromoperoxidase from the red algae Corallina officinalis. J. Mol. Biol. 2000, 299, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Carter-Franklin, J.N. The role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products. Nat. Prod. Rep. 2004, 21, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Hemrika, W.; Renirie, R.; Macedo-Ribeiro, S.; Messerschmidt, A.; Wever, R. Heterologous Expression of the Vanadium-containing Chloroperoxidase from Curvularia inaequalis in Saccharomyces cerevisiae and Site-directed Mutagenesis of the Active Site Residues His496, Lys353, Arg360, and Arg490. J. Biol. Chem. 1999, 274, 23820–23827. [Google Scholar] [CrossRef] [PubMed]
- Renirie, R.; Hemrika, W.; Wever, R. Peroxidase and Phosphatase Activity of Active-site Mutants of Vanadium Chloroperoxidase from the Fungus Curvularia inaequalis. J. Biol. Chem. 2000, 275, 11650–11657. [Google Scholar] [CrossRef] [PubMed]
- Zampella, G.; Fantucci, P.; Pecoraro, V.L.; De Gioia, L. Reactivity of peroxo forms of the vanadium haloperoxidase cofactor. A DFT investigation. J. Am. Chem. Soc. 2005, 127, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Zampella, G.; Fantucci, P.; Pecoraro, V.L.; De Gioia, L. Insight into the catalytic mechanism of vanadium haloperoxidases. DFT investigation of vanadium cofactor reactivity. Inorg. Chem. 2006, 45, 7133–7143. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.J.; Zampella, G.; Greco, C.; Pecoraro, V.L.; De Gioia, L. Mechanistic analysis of nucleophilic substrates oxidation by functional models of vanadium-dependent haloperoxidases: A density functional theory study. Eur. J. Inorg. Chem. 2007, 2007, 515–523. [Google Scholar] [CrossRef]
- Waller, M.P.; Bühl, M.; Geethalakshmi, K.R.; Wang, D.; Thiel, W. 51V NMR chemical shifts calculated from QM/MM models of vanadium chloroperoxidase. Chem. Eur. J. 2007, 13, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Waller, M.P.; Geethalakshmi, K.R.; Bühl, M. 51V NMR chemical shifts from quantum-mechanical/molecular-mechanical models of vanadium bromoperoxidase. J. Phys. Chem. B 2008, 112, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Geethalakshmi, K.R.; Waller, M.P.; Thiel, W.; Bühl, M. 51V NMR chemical shifts calculated from QM/MM models of peroxo forms of vanadium haloperoxidases. J. Phys. Chem. B 2009, 113, 4456–4465. [Google Scholar] [CrossRef] [PubMed]
- Rehder, D.; Casny, M.; Grosse, R. A vanadium-51 NMR study of the binding of vanadate and peroxovanadate to proteins. Magn. Reson. Chem. 2004, 42, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Pacios, L.F.; Gálvez, O. Active site, catalytic cycle, and iodination reactions of vanadium iodoperoxidase: A computational study. J. Chem. Theory Comput. 2010, 6, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Conte, V.; Coletti, A.; Floris, B.; Licini, G.; Zonta, C. Mechanistic aspects of vanadium catalysed oxidations with peroxides. Coord. Chem. Rev. 2011, 255, 2165–2177. [Google Scholar] [CrossRef]
- Butler, A. Mechanistic considerations of the vanadium haloperoxidases. Coord. Chem. Rev. 1999, 187, 17–35. [Google Scholar] [CrossRef]
- Van Schijndel, J.W.; Vollenbroek, E.G.; Wever, R. The chloroperoxidase from the fungus Curvularia inaequalis; a novel vanadium enzyme. Biochim. Biophys. Acta 1993, 1161, 249–256. [Google Scholar] [CrossRef]
- Dairi, T.; Nakano, T.; Aisaka, K.; Katsumata, R.; Hasegawa, M. Cloning and nucleotide sequence of the gene responsible for chlorination of tetracycline. Biosci. Biotechnol. Biochem. 1995, 59, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.T.; Hubbard, B.K.; Shah, A.N.; Eide, J.; Fredenburg, R.A.; Walsh, C.T.; Khosla, C. Molecular cloning and sequence analysis of the complestatin biosynthetic gene cluster. Proc. Natl. Acad. Sci. USA 2001, 98, 8548–8553. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Flecks, S.; Unversucht, S.; Haupt, C.; van Pée, K.H.; Naismith, J.H. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Eustaquio, A.S.; Gust, B.; Luft, T.; Chater, K.F.; Heide, L. Clorobiocin biosynthesis in Streptomyces: Identification of the halogenase and generation of structural analogs. Chem. Biol. 2003, 10, 279–288. [Google Scholar] [CrossRef]
- Hammer, P.E.; Hill, D.S.; Lam, S.T.; van Pee, K.H.; Ligon, J.M. Four genes from Pseudomonas fluorescens that encode the biosynthesis of pyrrolnitrin. Appl. Environ. Microbiol. 1997, 63, 2147–2154. [Google Scholar] [PubMed]
- Hohaus, K.; Altmann, A.; Burd, W.; Fischer, I.; Hammer, P.E.; Hill, D.S.; Ligon, J.M.; van Pee, K.-H. NADH-dependent halogenases are more likely to be involved in halometabolite biosynthesis than haloperoxidases. Angew. Chem. Int. Ed. Engl. 1997, 36, 2012–2013. [Google Scholar] [CrossRef]
- Keller, S.; Wage, T.; Hohaus, K.; Hölzer, M.; Eichhorn, E.; van Pée, K.H. Purification and partial characterization of tryptophan 7-halogenase(PrnA) from Pseudomonas fluorescens. Angew. Chem. Int. Ed. Engl. 2000, 39, 2300–2302. [Google Scholar] [CrossRef]
- Nowak-Thompson, B.; Chaney, N.; Wing, J.S.; Gould, S.J.; Loper, J.E. Characterization of the pyoluteorin biosynthetic gene cluster of Pseudomonas fluorescens Pf-5. J. Bacteriol. 1999, 181, 2166–2174. [Google Scholar] [PubMed]
- Puk, O.; Huber, P.; Bischoff, D.; Recktenwald, J.; Jung, G.; Süßmuth, R.D.; van Pee, K.-H.; Wohlleben, W.; Pelzer, S. Glycoprotein biosynthesis in Amycalotopsis mediterranei DSM5908: Function of a halogenase and a haloperoxidase/perhydrolase. Chem. Biol. 2002, 9, 225–235. [Google Scholar] [CrossRef]
- Sánchez, C.; Butovich, I.A.; Brana, A.F.; Rohr, J.; Méndez, C.; Salas, J.A. The biosynthetic gene cluster for the antitumor rebeccamycin. Characterization and generation of indolocarbazole derivatives. Chem. Biol. 2002, 9, 519–531. [Google Scholar] [CrossRef]
- Seibold, C.; Schnerr, H.; Rumpf, J.; Kunzendorf, A.; Hatscher, C.; Wage, T.; Ernyei, A.J. A flavin-dependent tryptophan 6-halogenase and its use in modification of pyrrolnitrin biosynthesis. Biocatal. Biotransform. 2006, 24, 401–408. [Google Scholar] [CrossRef]
- Trefzer, A.; Pelzer, S.; Schimana, J.; Stockert, S.; Bihlmaier, C.; Fiedler, H.P.; Welzel, K.; Vente, A.; Bechthold, A. Biosynthetic gene cluster of simocyclinone, a natural multihybrid antibiotic. Antimicrob. Agents Chem. 2002, 46, 1174–1182. [Google Scholar] [CrossRef]
- Weitnauer, G.; Mühlenweg, A.; Trefzer, A.; Hoffmeister, D.; Süßmuth, R.D.; Jung, G.; Welzel, K.; Vente, A.; Girreser, U.; Bechthold, A. Biosynthesis of the orthosomycin antibiotic avilamycin A: Deductions from the molecular analysis of the avi biosynthetic gene cluster of Streptomyces viridochromogenes Tü57 and production of new antibiotics. Chem. Biol. 2001, 8, 569–581. [Google Scholar] [CrossRef]
- Wijnands, I.; van Pee, K.H. A novel halogenase gene from the pentachloropseudilin producer Actinoplanes sp. ATCC 33002 and detection of in vitro halogenase activity. FEMS Microbiol. Lett. 2004, 237, 363–367. [Google Scholar] [CrossRef]
- Yeh, E.; Cole, L.J.; Barr, E.W.; Bollinger, J.M., Jr.; Ballou, D.P.; Walsh, C.T. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry 2006, 45, 7904–7912. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Kotzsch, A.; Dorward, M.; van Pee, K.H.; Naismith, J.H. Crystallization and X-ray diffraction of a halogenating enzyme, tryptophan 7-halogenase, from Pseudomonas fluorescens. Acta Crystallogr. D 2004, 60, 1438–1440. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Schaffrath, C.; Cobb, S.L.; Hamilton, J.T.G.; Murphy, C.D. Biochemistry: Biosynthesis of an organofluorine molecule. Nature 2002, 416, 279. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Deng, H. Enzymatic fluorination and biotechnological developments of the fluorinase. Chem. Rev. 2015, 115, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Ma, L. Identification of fluorinases from Streptomyces sp MA37, Norcardia brasiliensis, and Actinoplanes sp N902-109 by genome mining. Chembiochem 2014, 15, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Senn, H.M. Insights into enzymatic halogenation from computational studies. Front. Chem. 2014, 2, 98. [Google Scholar] [CrossRef] [PubMed]
- Senn, H.M.; O’Hagan, D.; Thiel, W. Insight into enzymatic C–F bond formation from QM and QM/MM calculations. J. Am. Chem. Soc. 2005, 127, 13643–13655. [Google Scholar] [CrossRef] [PubMed]
- Aluri, S.; de Visser, S.P. The mechanism of cysteine oxygenation by cysteine dioxygenase enzymes. J. Am. Chem. Soc. 2007, 129, 14846–14847. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Straganz, G.D. Why do cysteine dioxygenase enzymes contain a 3-His ligand motif rather than a 2His/1Asp motif like most nonheme dioxygenases? J. Phys. Chem. A 2009, 113, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; Goldberg, D.P.; de Visser, S.P. Mechanism of S-oxygenation by a cysteine dioxygenase model complex. J. Phys. Chem. A 2012, 116, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Fellner, M.; Siakkou, E.; Faponle, A.S.; Tchesnokov, E.P.; de Visser, S.P.; Wilbanks, S.M.; Jameson, G.N.L. Influence of cysteine 164 on active site structure in rat cysteine dioxygenase. J. Biol. Inorg. Chem. 2016, 21, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Tchesnokov, E.P.; Faponle, A.S.; Davies, C.G.; Quesne, M.G.; Turner, R.; Fellner, M.; Souness, R.J.; Wilbanks, S.M.; de Visser, S.P.; Jameson, G.N.L. An iron-oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase. Chem. Commun. 2016, 52, 8814–8817. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Seebeck, F.P.; de Visser, S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 2017, 139, 9259–9270. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.L.; Chapman, S.K. Molecular mechanisms of enzyme-catalysed halogenation. Mol. BioSyst. 2006, 2, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.M.; Gruschow, S.; Goss, R.J.M. Scope and potential of halogenases in biosynthetic applications. Curr. Opin. Chem. Biol. 2013, 17, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Omari, M.E.; Konig, G.M. Biohalogenation: Nature’s way to synthesize halogenated metabolites. J. Nat. Prod. 2009, 72, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; Saint-André, M.; de Visser, S.P. Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C-H bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; de Visser, S.P. How are substrate binding and catalysis affected by mutating Glu127 and Arg161 in prolyl-4-hydroxylase? A QM/MM and MD study. Front. Chem. 2017, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Hillwig, M.L.; Fuhrman, H.A.; Ittiamornkul, K.; Sevco, T.J.; Kwak, D.H.; Liu, X. Identification and characterization of a welwitindolinone alkaloid biosynthetic gene cluster in the Stigonematalean cyanobacterium Hapalosiphon welwitschii. ChemBioChem 2014, 15, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Hillwig, M.L.; Liu, X. A new family of iron-dependent halogenases acts on freestanding substrates. Nat. Chem. Biol. 2014, 10, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Zhu, Q.; Maggiolo, A.O.; Ananth, N.R.; Hillwig, M.L.; Liu, X.; Boal, A.K. Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat. Chem. Biol. 2016, 12, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, J.M., Jr.; Price, J.C.; Hoffart, L.M.; Barr, E.W.; Krebs, C. Mechanism of taurine: α-Ketoglutarate dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem. 2005, 2005, 4245–4254. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; van Koten, G.; Klein Gebbink, R.J.M. Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: Recent developments in enzymology and modeling studies. Chem. Soc. Rev. 2008, 37, 2716–2744. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Elucidating enzyme mechanism and intrinsic chemical properties of short-lived intermediates in the catalytic cycles of cysteine dioxygenase and taurine/α-ketoglutarate dioxygenase. Coord. Chem. Rev. 2009, 253, 754–768. [Google Scholar] [CrossRef]
- Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reaction in Nature; de Visser, S.P., Kumar, D., Eds.; RSC Publishing: Cambridge, UK, 2011. [Google Scholar]
- Buongiorno, D.; Straganz, G.D. Structure and function of atypically coordinated enzymatic mononuclear non-heme-Fe(II) centers. Coord. Chem. Rev. 2013, 257, 541–563. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Bagherzadeh, M.; de Visser, S.P. Origin of the correlation of the rate constant of substrate hydroxylation by nonheme iron(IV)-oxo complexes with the bond-dissociation energy of the C–H bond of the substrate. Chem. Eur. J. 2009, 15, 6651–6662. [Google Scholar] [CrossRef] [PubMed]
- Karamzadeh, B.; Kumar, D.; Sastry, G.N.; de Visser, S.P. Steric factors override thermodynamic driving force in regioselectivity of proline hydroxylation by prolyl-4-hydroxylase enzymes. J. Phys. Chem. A 2010, 114, 13234–13243. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Latifi, R.; Tahsini, L.; Nam, W. The axial ligand effect on aliphatic and aromatic hydroxylation by nonheme iron(IV)-oxo biomimetic complexes. Chem. Asian J. 2011, 6, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Konstantinovics, C.; DiGiuro, C.L.M.; Leitner, E.; Kumar, D.; de Visser, S.P.; Grogan, G.; Straganz, G.D. Inversion of enantio-selectivity of a mononuclear non-heme iron(II)-dependent hydroxylase by tuning the interplay of metal-center geometry and protein structure. Angew. Chem. Int. Ed. 2013, 52, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Jastrzebski, R.; Quesne, M.G.; Weckhuysen, B.M.; de Visser, S.P.; Bruijnincx, P.C.A. Experimental and computational evidence for the mechanism of intradiol catechol dioxygenation by non-heme iron(III) complexes. Chem. Eur. J. 2014, 20, 15686–15691. [Google Scholar] [CrossRef] [PubMed]
- Sallmann, M.; Kumar, S.; Chernev, P.; Nehrkorn, J.; Schnegg, A.; Kumar, D.; Dau, H.; Limberg, C.; de Visser, S.P. Structure and mechanism leading to formation of the cysteine sulfinate product complex of a biomimetic cysteine dioxygenase model. Chem. Eur. J. 2015, 21, 7470–7479. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, M.A.; Malhotra, A.; Balan, G.A.; Timmins, A.; de Visser, S.P. Nitrogen reduction to ammonia on a biomimetic mononuclear iron center: Insights into the nitrogenase enzyme. Chem. Eur. J. 2018, 24, 5293–5302. [Google Scholar] [CrossRef] [PubMed]
- Price, J.C.; Barr, E.W.; Glass, T.E.; Krebs, C.; Bollinger, J.M., Jr. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. J. Am. Chem. Soc. 2003, 125, 13008–13009. [Google Scholar] [CrossRef] [PubMed]
- Grzyska, P.K.; Ryle, M.J.; Monterosso, G.R.; Liu, J.; Ballou, D.P.; Hausinger, R.P. Steady-state and transient kinetic analysis of taurine/α-ketoglutarate dioxygenase: Effects of oxygen concentration, alternative sulfonates, and active-site variants on the FeIV-oxo intermediate. Biochemistry 2005, 44, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Propene activation by the oxo-iron active species of taurine/α-ketoglutarate dioxygenase (TauD) enzyme. How does the catalysis compare to heme-enzymes? J. Am. Chem. Soc. 2006, 128, 9813–9824. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Borowski, T.; Bassan, A.; Siegbahn, P.E.M. 4-Hydroxyphenylpyruvate dioxygenase: A hybrid density functional study of the catalytic reaction mechanism. Biochemistry 2004, 43, 12331–12342. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Can the peroxosuccinate complex in the catalytic cycle of taurine/α-ketoglutarate dioxygenase (TauD) act as an alternative oxidant? Chem. Commun. 2007, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.; Porro, C.S.; de Visser, S.P. Comparative quantum mechanics/molecular mechanics (QM/MM) and density functional theory calculations on the oxo-iron species of taurine/α-ketoglutarate dioxygenase. J. Phys. Chem. A 2008, 112, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Latifi, R.; Gonzalez-Ovalle, L.E.; Kumar, D.; de Visser, S.P. Quantum mechanics/molecular mechanics study on the oxygen binding and substrate hydroxylation step in AlkB repair enzymes. Chem. Eur. J. 2014, 20, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Diebold, A.R.; Brown-Marshall, C.D.; Neidig, M.L.; Brownlee, J.M.; Moran, G.R.; Solomon, E.I. Activation of α-keto acid-dependent dioxygenases: Application of an {FeNO}7/{FeO2}8 methodology for characterizing the initial steps of O2 activation. J. Am. Chem. Soc. 2011, 133, 18148–18160. [Google Scholar] [CrossRef] [PubMed]
- Borowski, T.; Bassan, A.; Siegbahn, P.E.M. Mechanism of dioxygen activation in 2-oxoglutarate-dependent enzymes. A hybrid DFT study. Chem. Eur. J. 2004, 10, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Topol, I.A.; Nemukhin, A.V.; Salnikow, K.; Cachau, R.E.; Abashkin, Y.G.; Kasprzak, K.S.; Burt, S.K. Quantum chemical modeling of reaction mechanism for 2-glutarate dependent enzymes: Effect of substitution of iron by nickel and cobalt. J. Phys. Chem. A 2006, 110, 4223–4228. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Riplinger, C.; Hansen, A.; Krebs, C.; Bollinger, J.M., Jr.; Neese, F. Electronic structure analysis of the oxygen-activation mechanism by FeII- and α-ketoglutarate (αKG)-dependent dioxygenases. Chem. Eur. J. 2012, 18, 6555–6567. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.L.; Krest, C.M.; Barr, E.W.; Vaillancourt, F.H.; Walsh, C.T.; Green, M.T.; Krebs, C.; Bollinger, J.M., Jr. Substrate-triggered formation and remarkable stability of the C-H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. [Google Scholar] [CrossRef] [PubMed]
- Matthews, M.L.; Neumann, C.S.; Miles, L.A.; Grove, T.L.; Booker, S.J.; Krebs, C.; Walsh, C.T.; Bollinger, J.M., Jr. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. USA 2009, 106, 17723–17728. [Google Scholar] [CrossRef] [PubMed]
- Kulik, H.J.; Drennan, C.L. Substrate placement influences reactivity in non-heme Fe(II) halogenases and hyroxylases. J. Biol. Chem. 2013, 288, 11233–11241. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.D.; Srnec, M.; Matthews, M.L.; Liu, L.V.; Kwak, Y.; Park, K.; Bell, C.B.; Alp, E.; Zhao, J.; Yoda, Y.; et al. Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 2013, 499, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Pandian, S.; Vincent, M.A.; Hillier, I.H.; Burton, N.A. Why does the enzyme SyrB2 chlorinate, but does not hydroxylate, saturated hydrocarbons? A density functional theory (DFT) study. Dalton Trans. 2009, 31, 6201–6207. [Google Scholar] [CrossRef] [PubMed]
- Kulik, H.J.; Blasiak, L.C.; Marzari, N.; Drennan, C.L. First-principles study of non-heme Fe(II) halogenase SyrB2 reactivity. J. Am. Chem. Soc. 2009, 131, 14426–14433. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Latifi, R. Carbon dioxide: A waste product in the catalytic cycle of alpha-ketoglutarate dependent halogenases prevents the formation of hydroxylated by-products. J. Phys. Chem. B 2009, 113, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Leising, R.A.; Yan, S.; Que, L., Jr. Alkane functionalization at nonheme iron centers. Stoichiometric transfer of metal-bound ligands to alkane. J. Am. Chem. Soc. 1993, 115, 11328–11335. [Google Scholar] [CrossRef]
- Borowski, T.; Noack, H.; Radon, M.; Zych, K.; Siegbahn, P.E.M. Mechanism of selective halogenation by SyrB2: A computational study. J. Am. Chem. Soc. 2010, 132, 12887–12898. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.; Wang, B.; Sharon, D.A.; Wu, W.; Shaik, S. Selective chlorination of substrates by the halogenase SyrB2 is controlled by the protein according to a combined quantum mechanics/molecular mechanics and molecular dynamics study. ACS Catal. 2016, 6, 2694–2704. [Google Scholar] [CrossRef]
- Timmins, A.; Fowler, N.J.; Warwicker, J.; Straganz, G.D.; de Visser, S.P. Does substrate positioning affect the selectivity and reactivity in the hectochlorin biosynthesis halogenase? 2018; Submitted for publication. [Google Scholar]
- Srnec, M.; Solomon, E.I. Frontier molecular orbital contributions to chlorination versus hydroxylation selectivity in the non-heme iron halogenase SyrB2. J. Am. Chem. Soc. 2017, 139, 2396–2407. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Light, K.M.; Solomon, E.I.; Straganz, G.D. The role of chloride in the mechanism of O2 activation at the mononuclear nonheme Fe(II) center of the halogenase HctB. J. Am. Chem. Soc. 2014, 136, 9385–9395. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Ivkovic, J.; Birner-Gruienberger, R.; Breinbauer, R.; Zangger, K.; Stranganz, G.D. More than just a halogenase: Modification of fatty acyl moieties by a trifunctional metal enzyme. ChemBioChem 2014, 15, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modelling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Thiel, W.; de Visser, S.P. Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J. Am. Chem. Soc. 2011, 133, 3869–3882. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heuts, D.P.H.M.; Steiner, R.A.; Heyes, D.J.; de Visser, S.P.; Scrutton, N.S. Origin of the proton-transfer step in the cofactor-free 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase: Effect of the basicity of an active site His residue. J. Biol. Chem. 2014, 289, 8620–8632. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heyes, D.J.; Steiner, R.A.; Scrutton, N.S.; de Visser, S.P. Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates. J. Am. Chem. Soc. 2015, 137, 7474–7487. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production: What Drives the Reaction to Biofuel Production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef] [PubMed]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef] [PubMed]
- Sainna, M.A.; Sil, D.; Sahoo, D.; Martin, B.; Rath, S.P.; Comba, P.; de Visser, S.P. Spin state ordering in hydroxo-bridged diiron(III)bisporphyrin complexes. Inorg. Chem. 2015, 54, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Quesne, M.G.; Neu, H.M.; Cantú Reinhard, F.G.; Goldberg, D.P.; de Visser, S.P. Singlet versus triplet reactivity in an Mn(V)-Oxo species: Testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate sulfoxidation by an iron(IV)-oxo complex: Benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.J.; Blanford, C.F.; Warwicker, J.; de Visser, S.P. Prediction of reduction potentials of copper proteins with continuum electrostatics and density functional theory. Chem. Eur. J. 2017, 23, 15436–15445. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. Hydrogen atom versus hydride transfer in cytochrome P450 oxidations: A combined mass spectrometry and computational study. Eur. J. Inorg. Chem. 2018, 2018, 1854–1865. [Google Scholar] [CrossRef]
- Swart, M. Accurate spin-state energies for iron complexes. J. Chem. Theory Comput. 2008, 4, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Kumar, D.; Neumann, R.; Shaik, S. Computer-generated high-valent iron-oxo and manganese-oxo species with polyoxometalate ligands: How do they compare with the iron-oxo active species of heme enzymes? Angew. Chem. Int. Ed. 2004, 43, 5661–5665. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Differences in and comparison of the catalytic properties of heme and non-heme enzymes with a central oxo-iron group. Angew. Chem. Int. Ed. 2006, 45, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. What factors influence the ratio of C–H hydroxylation versus C=C epoxidation by a nonheme cytochrome P450 biomimetic? J. Am. Chem. Soc. 2006, 128, 15809–15818. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Faponle, A.S.; Barman, P.; Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Long-range electron transfer triggers mechanistic differences between iron(IV)-oxo and iron(IV)-imido oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.; Upadhyay, P.; Faponle, A.S.; Kumar, J.; Nag, S.S.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Deformylation reaction by a nonheme manganese(III)-peroxo complex via initial hydrogen atom abstraction. Angew. Chem. Int. Ed. 2016, 55, 11091–11095. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: How does the reactivity compare to Cytochrome P450 Compound? Chem. Eur. J. 2017, 23, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Barman, P.; Mukherjee, G.; Kumar, J.; Kumar, D.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Keto-enol tautomerization triggers an electrophilic aldehyde deformylation reaction by a nonheme manganese(III)-peroxo complex. J. Am. Chem. Soc. 2017, 139, 18328–18338. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Heyes, D.J.; Sakuma, M.; de Visser, S.P.; Scrutton, N.S. Nuclear quantum tunneling in the light-activated enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 2009, 284, 3762–3767. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Comparison of the reactivity of nonheme iron(IV)-oxo versus iron(IV)-imido complexes: Which is the better oxidant? Angew. Chem. Int. Ed. 2013, 52, 12288–12292. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of µ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrophilic | Nucleophilic | Radical |
|---|---|---|
| Heme-dependent | SAM fluorinases | Nonheme iron/αKG-dependent halogenase |
| Vanadium-dependent | ||
| Flavin-dependent |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmins, A.; De Visser, S.P. A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases. Catalysts 2018, 8, 314. https://doi.org/10.3390/catal8080314
Timmins A, De Visser SP. A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases. Catalysts. 2018; 8(8):314. https://doi.org/10.3390/catal8080314
Chicago/Turabian StyleTimmins, Amy, and Sam P. De Visser. 2018. "A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases" Catalysts 8, no. 8: 314. https://doi.org/10.3390/catal8080314
APA StyleTimmins, A., & De Visser, S. P. (2018). A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases. Catalysts, 8(8), 314. https://doi.org/10.3390/catal8080314