Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules





Abstract
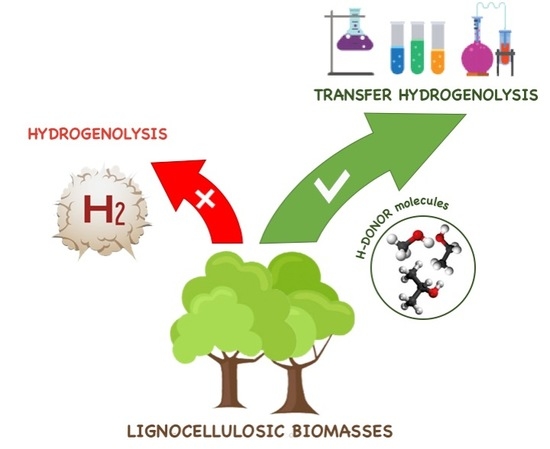
1. Introduction
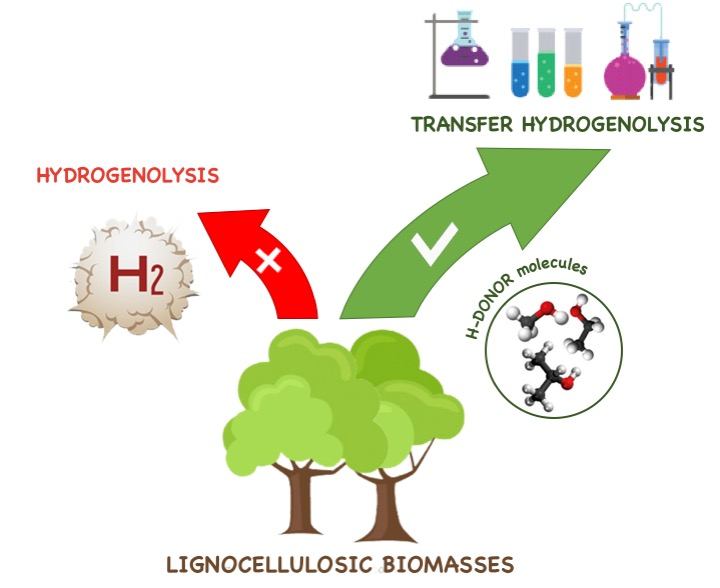
2. Catalytic Transfer Hydrogenolysis Applied to Cellulose and to Cellulose Derivable Molecules
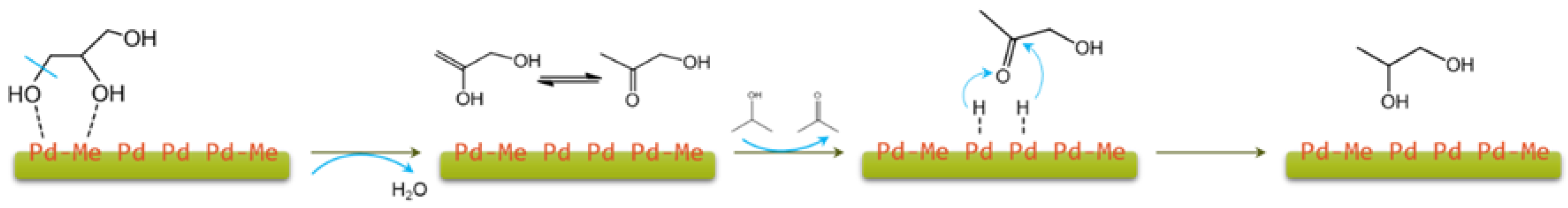
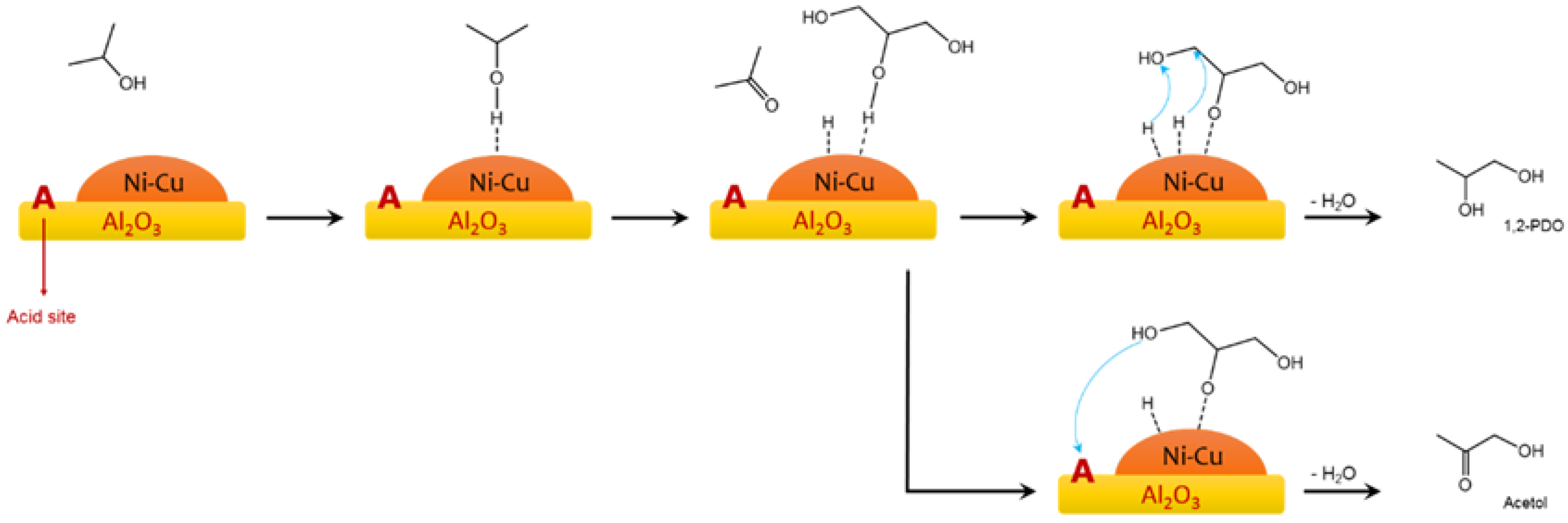
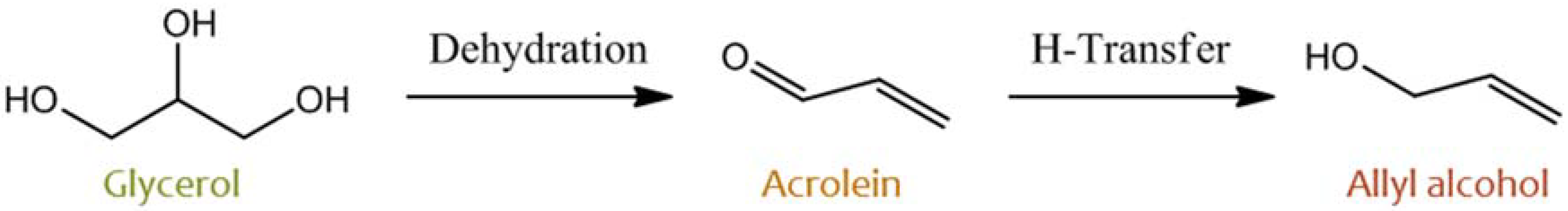
2.1. Glycerol and Other Polyols
2.2. Glucose and Carbohydrates
2.3. Cellulose
3. Catalytic Transfer Hydrogenolysis (CTH) Reactions of Hemicellulose Derived Molecules
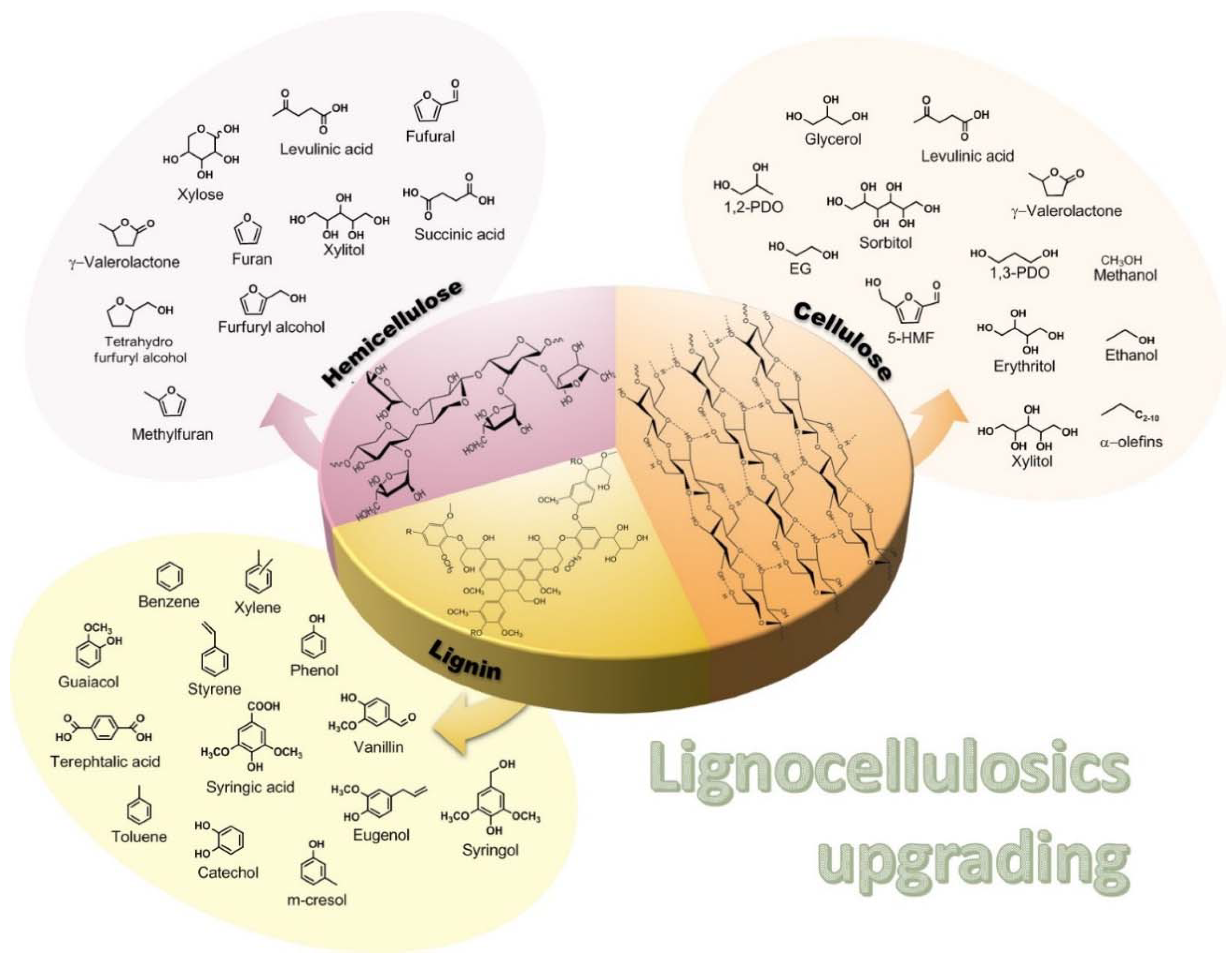
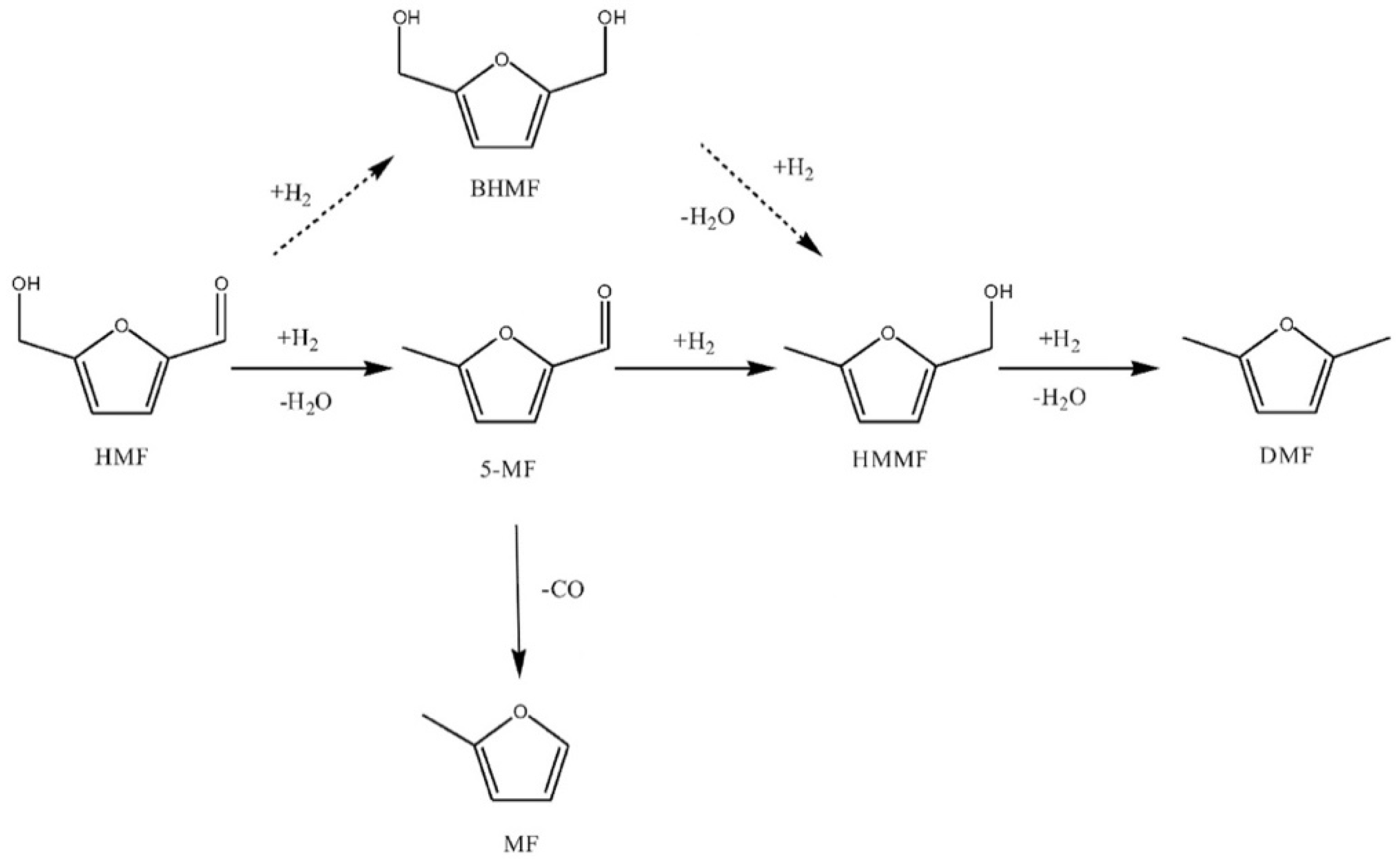
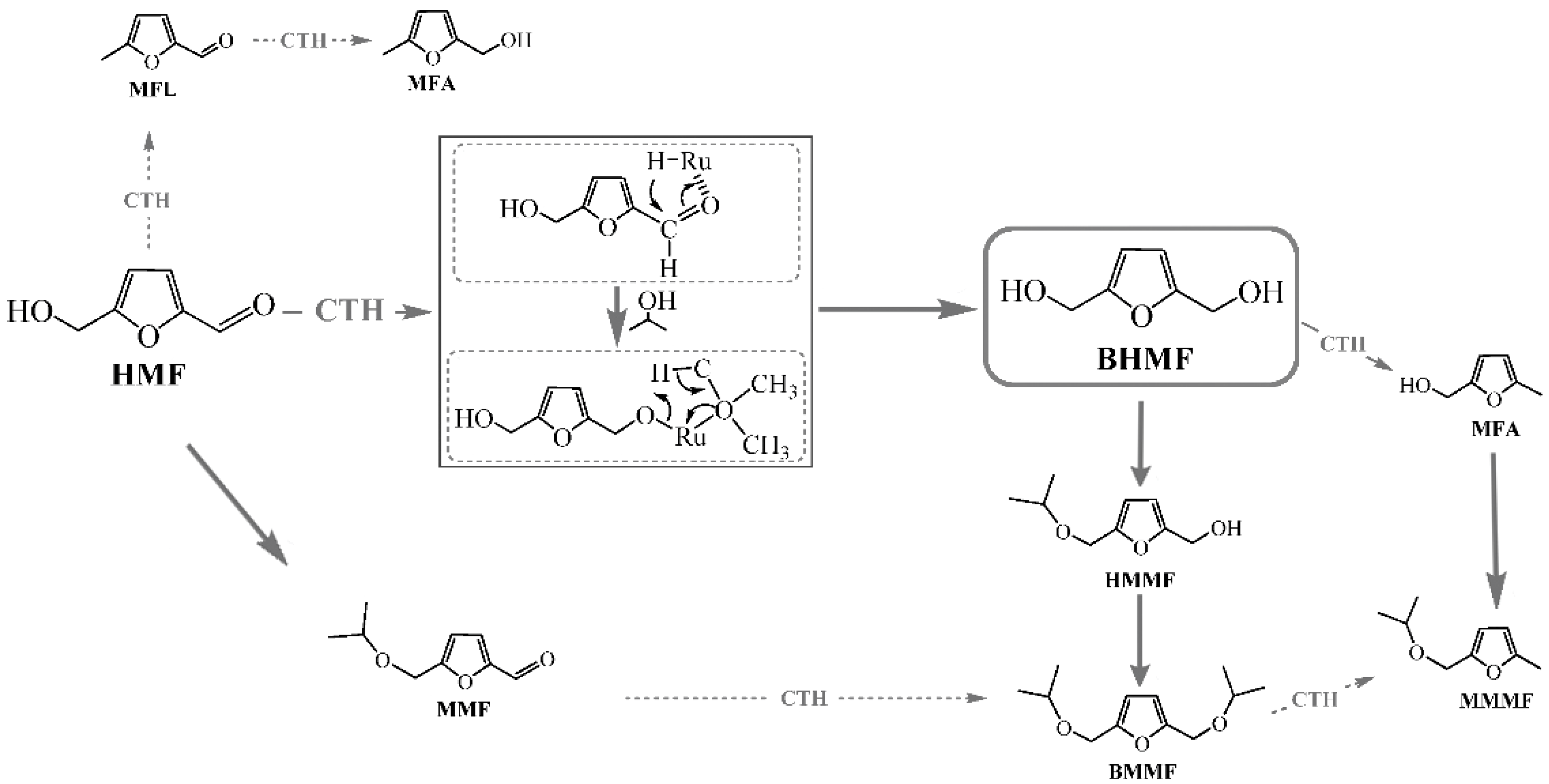
3.1. Furfural Derivatives
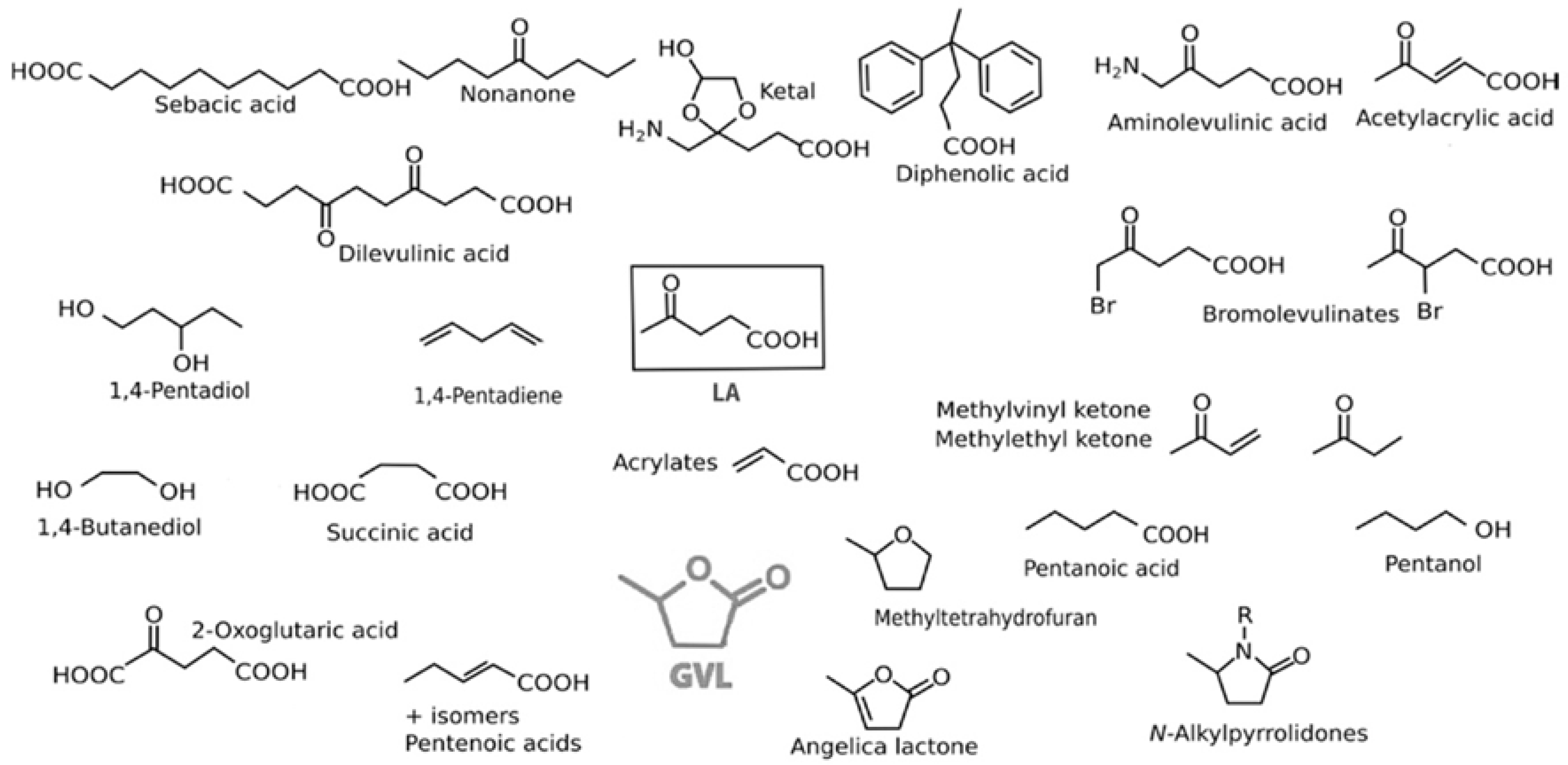
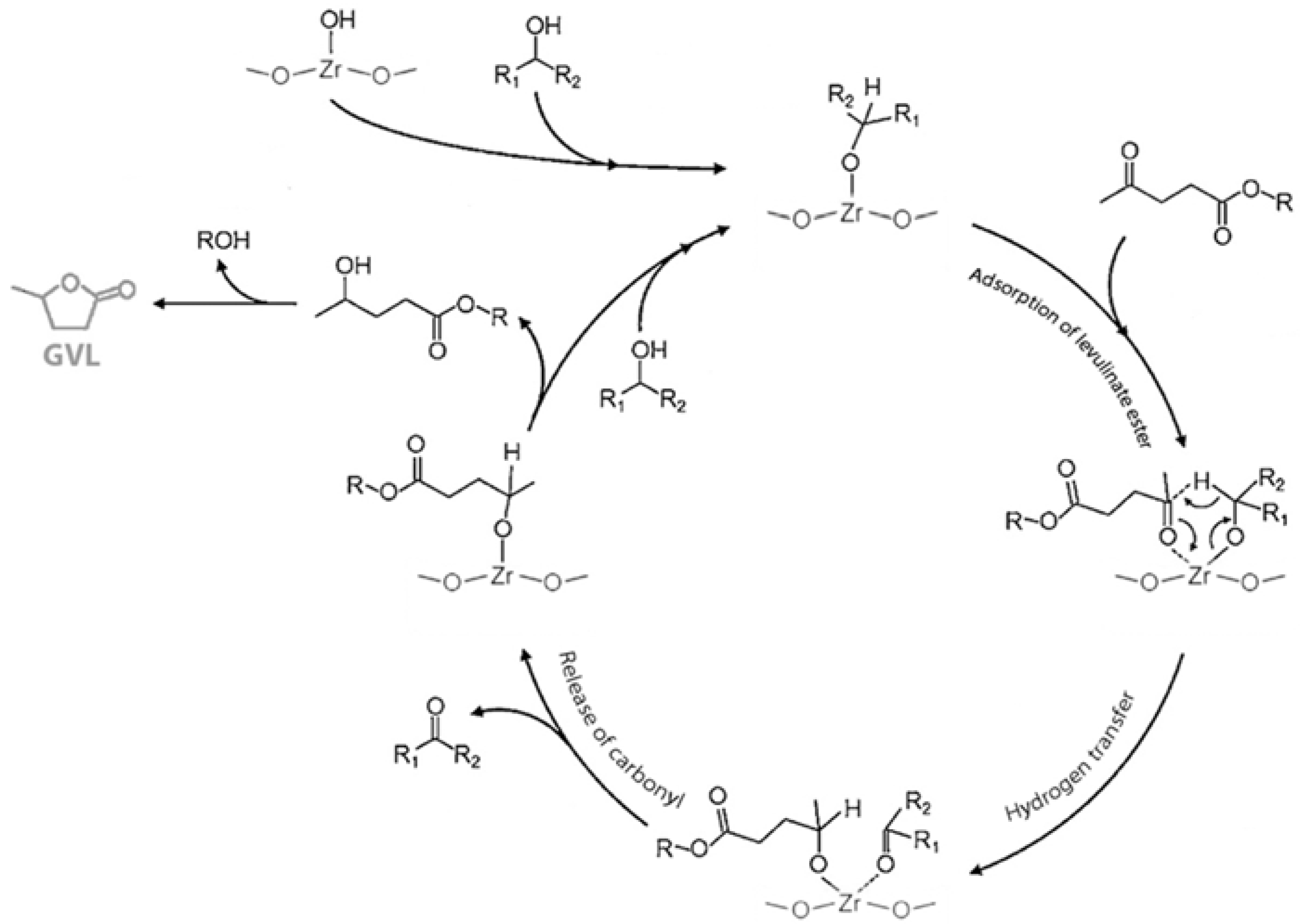
3.2. Levulinic Acid
4. Catalytic Transfer Hydrogenolysis (CTH) of Lignin and Its Derived Molecules
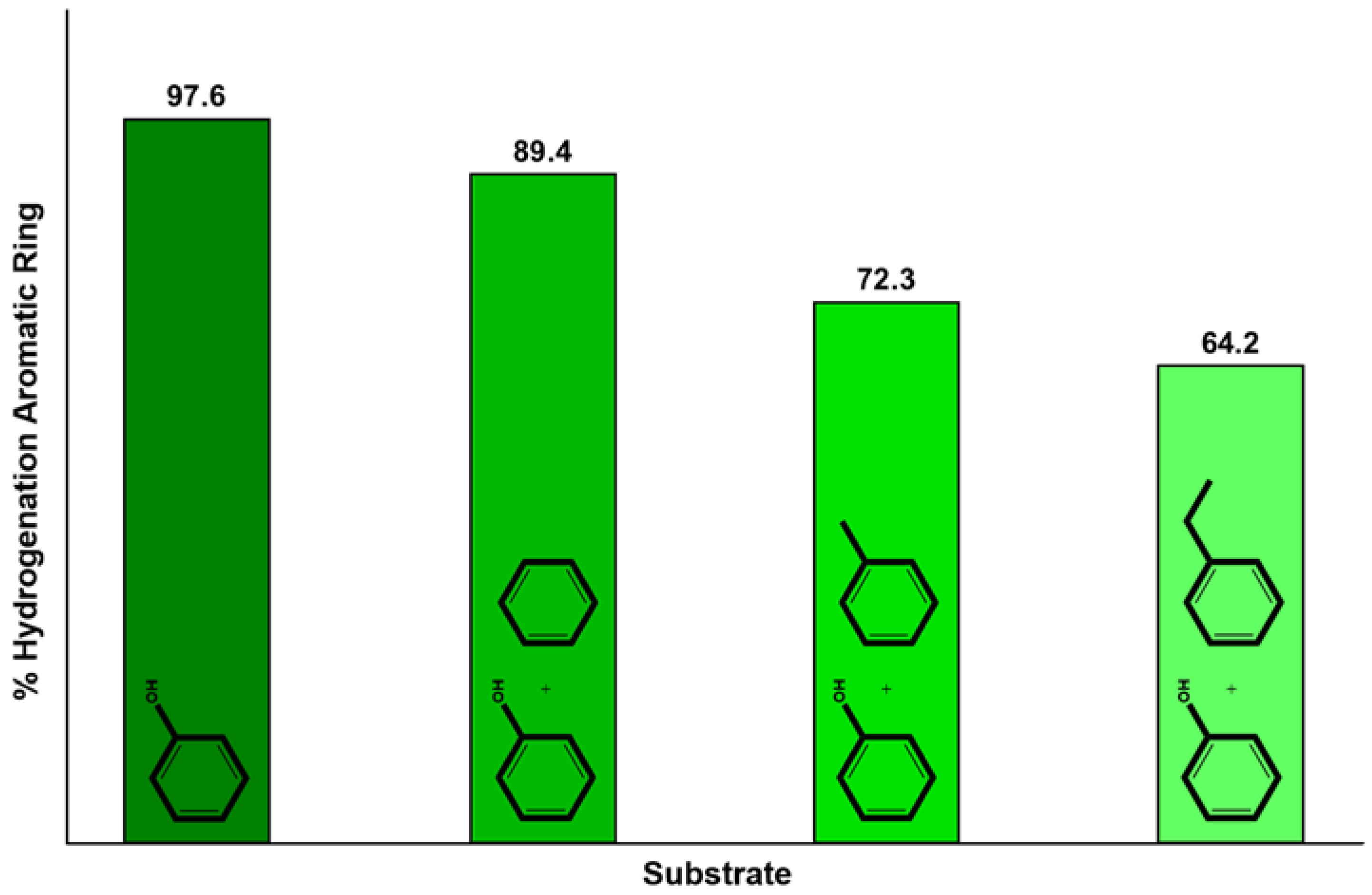
4.1. CTH of Lignin Derived Molecules
4.2. CTH of Lignin
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klass, D.L. Biomass for Renewable Energy, Fuels, and Chemicals; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- The White House-Washington. National Bio-economy Blueprint; The White House: Washington, DC, USA, 2012; pp. 1–43. Available online: https://obamawhitehouse.archives.gov/sites/default/files/microsites/ostp/national_bioeconomy_blueprint_april_2012.pdf (accessed on 30 June 2018).
- European Commission. Innovating for Sustainable Growth: A Bioeconomy for Europe; European Commission: Brussels, Belgium, 2012; pp. 1–9. Available online: http://ec.europa.eu/research/bioeconomy/pdf/official-strategy_en.pdf (accessed on 30 June 2018).
- European Commission. A Roadmap for Moving to a Competitive Low Carbon Economy in 2050; European Commission: Brussels, Belgium, 2011; Available online: http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:52011DC0112 (accessed on 30 December 2016).
- Lee, D.-H. Bio-based economies in Asia: Economic analysis of development of bio-based industry in China, India, Japan, Korea, Malaysia and Taiwan. Int. J. Hydrogen Energy 2016, 41, 4333–4346. [Google Scholar] [CrossRef]
- Dey, S. Asian bioeconomy and biobusiness: Current scenario and future prospects. New Biotechnol. 2014, 31, S34. [Google Scholar] [CrossRef]
- Ignaciuk, A.; Vöhringer, F.; Ruijs, A.; Van Ierland, E.C. Competition between biomass and food production in the presence of energy policies: A partial equilibrium analysis. Energy Policy 2006, 34, 1127–1138. [Google Scholar] [CrossRef]
- Somerville, C.; Youngs, H.; Taylor, C.; Davis, S.C.; Long, S.P. Feedstocks for lignocellulosic biofuels. Science 2010, 329, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Tuck, C.O.; Pérez, E.; Horváth, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Yang, Y.; Chai, J.; Lu, Y. Catalytic reactions of gamma-valerolactone: A platform to fuels and value-added chemicals. Appl. Catal. B Environ. 2015, 179, 292–304. [Google Scholar] [CrossRef]
- Hu, L.; Lin, L.; Wu, Z.; Zhou, S.; Liu, S. Chemocatalytic hydrolysis of cellulose into glucose over solid acid catalysts. Appl. Catal. B Environ. 2015, 174–175, 225–243. [Google Scholar] [CrossRef]
- Negahdar, L.; Delidovich, I.; Palkovits, R. Aqueous-phase hydrolysis of cellulose and hemicelluloses over molecular acidic catalysts: Insights into the kinetics and reaction mechanism. Appl. Catal. B Environ. 2016, 184, 285–298. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, J.; Han, B. Catalytic transformation of lignocellulose into chemicals and fuel products in ionic liquids. Chem. Rev. 2016, 117, 6834–6880. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.H.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.P. Lignocellulose conversion: An introduction to chemistry, process and economics. Biofuels Bioprod. Biorefin. 2007, 1, 39–48. [Google Scholar] [CrossRef]
- Stöcker, M. Biofuels and biomass-to-liquid fuels in the biorefinery: Catalytic conversion of lignocellulosic biomass using porous materials. Angew. Chem. Int. Ed. 2008, 47, 9200–9211. [Google Scholar] [CrossRef] [PubMed]
- Espro, C.; Gumina, B.; Paone, E.; Mauriello, F. Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts. Catalysts 2017, 7, 78. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; De Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.-F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Global Bio-Based Chemical Market Forecast 2018–2026. Available online: https://www.reportlinker.com/p05001382/Global-Bio-Based-Chemicals-Market-Forecast.html (accessed on 30 June 2018).
- Dornburg, V.; Hermann, B.G.; Patel, M.K. Scenario Projections for Future Market Potentials of Biobased Bulk Chemicals. Environ. Sci. Technol. 2008, 42, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. Chemical composition and structure of natural lignocellulose. In Biotechnology of Lignocellulose: Theory and Practice; Springer Science + Business Media B.V.: Dordrecht, The Netherlands, 2014; pp. 25–71. [Google Scholar]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Fukuoka, A. Synthesis and utilisation of sugar compounds derived from lignocellulosic biomass. Green Chem. 2013, 15, 1740–1763. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic Biomass: A sustainable platform for production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.B.; Raines, R.T. Simple Chemical Transformation of Lignocellulosic Biomass into Furans for Fuels and Chemicals. J. Am. Chem. Soc. 2009, 131, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef] [PubMed]
- Deuss, P.J.; Barta, K. From models to lignin: Transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016, 306, 510–532. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Samec, J.S.M. Lignin Valorization through Catalytic Lignocellulose Fractionation: A Fundamental Platform for the Future Biorefinery. ChemSusChem 2016, 9, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Bridgwater, A.V.; Meierb, D.; Radlein, D. An overview of fast pyrolysis of biomass. Org. Geochem. 1999, 30, 1479–1493. [Google Scholar] [CrossRef]
- De, S.; Saha, B.; Luque, R. Hydrodeoxygenation processes: Advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels. Bioresour. Technol. 2015, 178, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, A.M.; Weinberg, K.; Palkovits, R. Hydrogenolysis goes bio: From carbohydrates and sugar alcohols to platform chemicals. Angew. Chem. Int. Ed. 2012, 51, 2564–2601. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, W.; Zheng, M.; Zhang, T. General Reaction Mechanisms in Hydrogenation and Hydrogenolysis for Biorefining. In Catalytic Hydrogenation for Biomass Valorization; Rinaldi, R., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 22–50. [Google Scholar]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Gilkey, M.J.; Xu, B. Heterogeneous Catalytic Transfer Hydrogenation as an Effective Pathway in Biomass Upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Muzart, J. Pd-Catalyzed Hydrogen-Transfer Reactions from Alcohols to C=C, C=O and C=N Bonds. Eur. J. Org. Chem. 2015, 2015, 5693–5707. [Google Scholar] [CrossRef]
- Barta, K.; Ford, P.C. Catalytic Conversion of Nonfood Woody Biomass Solids to Organic Liquids. Acc. Chem. Res. 2014, 47, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material–development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef] [PubMed]
- Loges, B.; Boddien, A.; Gärtner, F.; Junge, H.; Beller, M. Catalytic Generation of Hydrogen from Formic acid and its Derivatives: Useful Hydrogen Storage Materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G.; Hirose, T.; Raspail, P.; Liu, S.L.; Wu, Y.P.; Guo, Q.-X.; Ludwig, R.; Beller, M. Formic acid as a hydrogen source–recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Musolino, M.G.; Scarpino, L.A.; Mauriello, F.; Pietropaolo, R. Selective transfer hydrogenolysis of glycerol promoted by palladium catalysts in absence of hydrogen. Green Chem. 2009, 11, 1511–1513. [Google Scholar] [CrossRef]
- Mauriello, F.; Ariga, H.; Musolino, M.G.; Pietropaolo, R.; Takakusagi, S.; Asakura, K. Exploring the catalytic properties of supported palladium catalysts in the transfer hydrogenolysis of glycerol. Appl. Catal. B Environ. 2015, 166–167, 121–131. [Google Scholar] [CrossRef]
- Gandarias, I.; Arias, P.L.; Requies, J.; El Doukkali, M.; Güemez, M.B. Liquid-phase glycerol hydrogenolysis to 1,2-propanediol under nitrogen pressure using 2-propanol as hydrogen source. J. Catal. 2011, 282, 237–247. [Google Scholar] [CrossRef]
- Gandarias, I.; Arias, P.L.; Fernández, S.G.; Requies, J.; El Doukkali, M.; Güemez, M.B. Hydrogenolysis through catalytic transfer hydrogenation: Glycerol conversion to 1,2-propanediol. Catal. Today 2012, 195, 22–31. [Google Scholar] [CrossRef]
- Gandarias, I.; Requies, J.; Arias, P.L.; Armbruster, U.; Martin, A. Liquid-phase glycerol hydrogenolysis by formic acid over Ni–Cu/Al2O3 catalysts. J. Catal. 2012, 290, 79–89. [Google Scholar] [CrossRef]
- Mane, R.B.; Rode, C.V. Continuous Dehydration and Hydrogenolysis of Glycerol over Non-Chromium Copper Catalyst: Laboratory-Scale Process Studies. Org. Process Res. Dev. 2012, 16, 1043–1052. [Google Scholar] [CrossRef]
- Yuan, J.; Li, S.; Yu, L.; Liu, Y.; Cao, Y. Efficient catalytic hydrogenolysis of glycerol using formic acid as hydrogen source. Chin. J. Catal. 2013, 34, 2066–2074. [Google Scholar] [CrossRef]
- Xia, S.; Yuan, Z.; Wang, L.; Chen, P.; Hou, Z. Hydrogenolysis of glycerol on bimetallic Pd-Cu/solid-base catalysts prepared via layered double hydroxides precursors. Appl. Catal. A Gen. 2011, 403, 173–182. [Google Scholar] [CrossRef]
- Bienholz, A.; Hofmann, H.; Claus, P. Selective hydrogenolysis of glycerol over copper catalysts both in liquid and vapour phase: Correlation between the copper surface area and the catalyst’s activity. Appl. Catal. A Gen. 2011, 391, 153–157. [Google Scholar] [CrossRef]
- Xia, S.; Yuan, Z.; Wang, L.; Chen, P.; Hou, Z. Catalytic production of 1,2-propanediol from glycerol in bio-ethanol solvent. Bioresour. Technol. 2012, 104, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Musolino, M.G.; Scarpino, L.A.; Mauriello, F.; Pietropaolo, R. Glycerol hydrogenolysis promoted by supported palladium catalysts. ChemSusChem 2011, 4, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Lü, Y.; Ding, Y.; Lin, R.; Li, J.; Dong, W.; Wang, T.; Chen, W. Solvent Effect on Selective Dehydroxylation of Glycerol to 1,3-Propanediol over a Pt/WO3/ZrO2 Catalyst. Chin. J. Catal. 2009, 30, 1189–1191. [Google Scholar] [CrossRef]
- Liu, Y.; Tüysü, H.; Jia, C.-J.; Schwickardi, M.; Rinaldi, R.; Lu, A.-H.; Schmidt, W.; Schüt, F. From glycerol to allyl alcohol: Iron oxide catalyzed dehydration and consecutive hydrogen transfer. Chem. Commun. 2010, 46, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Konaka, A.; Tago, T.; Yoshikawa, T.; Nakamura, A.; Masuda, T. Conversion of glycerol into allyl alcohol over potassium-supported zirconia–iron oxide catalyst. Appl. Catal. B Environ. 2014, 146, 267–273. [Google Scholar] [CrossRef]
- Arceo, E.; Marsden, P.; Bergman, R.G.; Ellman, J.A. An efficient didehydroxylation method for the biomass-derived polyols glycerol and erythritol. Mechanistic studies of a formic acid-mediated deoxygenation. Chem. Commun. 2009, 23, 3357–3359. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, J.R.; Lupp, D.; Teshome, A.; Nielsen, L.B.; Fristrup, P. Molybdenum-catalyzed conversion of diols and biomass-derived polyols to alkenes using isopropyl alcohol as reductant and solvent. ACS Catal. 2015, 5, 3638–3647. [Google Scholar] [CrossRef]
- Deng, L.; Li, J.; Lai, D.-M.; Fu, Y.; Guo, Q.-X. Catalytic conversion of biomass-derived carbohydrates into γ-valerolactone without using an external H2 supply. Angew. Chem. Int. Ed. 2009, 48, 6529–6532. [Google Scholar] [CrossRef] [PubMed]
- Heeres, H.; Handana, R.; Chunai, D.; Rasrendra, C.B.; Girisuta, B.; Heeres, H.J. Combined dehydration/(transfer)-hydrogenation of C6-sugars (d-glucose and d-fructose) to γ-valerolactone using ruthenium catalysts. Green Chem. 2009, 11, 1247–1255. [Google Scholar] [CrossRef]
- Du, X.-L.; He, L.; Zhao, S.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Hydrogen-independent reductive transformation of carbohydrate biomass into γ-valerolactone and pyrrolidone derivatives with supported gold catalysts. Angew. Chem. 2011, 123, 7961–7965. [Google Scholar] [CrossRef]
- Scholz, D.; Aellig, C.; Mondelli, C.; Pèrez-Ramìrez, J. Continuous transfer hydrogenation of sugars to alditols with bioderived donors over Cu–Ni–Al catalysts. ChemCatChem 2015, 7, 1551–1558. [Google Scholar] [CrossRef]
- Van Hengstum, A.J.; Kieboom, A.P.G.; van Bekkum, H. Catalytic transfer hydrogenation of glucose-fructose syrups in alkaline solution. Starch 1984, 36, 317–320. [Google Scholar] [CrossRef]
- Kobayashi, H.; Matsuhashi, H.; Komanoya, T.; Hara, K.; Fukuoka, A. Transfer hydrogenation of cellulose to sugar alcohols over supported ruthenium catalysts. Chem. Commun. 2011, 47, 2366–2368. [Google Scholar] [CrossRef] [PubMed]
- Shrotri, A.; Kobayashi, H.; Tanksale, A.; Fukuoka, A.; Beltramini, J. Transfer Hydrogenation of Cellulose-based Oligomers over Carbon-supported Ruthenium Catalyst in a Fixed-bed Reactor. ChemCatChem 2014, 6, 1349–1356. [Google Scholar] [CrossRef]
- Dalvand, K.; Rubin, J.; Gunukula, S.; Clayton Wheeler, M.; Hunt, G. Economics of biofuels: Market potential of furfural and its derivatives. Biomass Bioenergy 2018, 115, 56–63. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Li, X.; Jia, P.; Wang, T. Furfural: A Promising Platform Compound for Sustainable Production of C4 and C5 Chemicals. ACS Catal. 2016, 6, 7621–7640. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Zhou, H.; Fu, Y. Catalytic Transfer Hydrogenation of Furfural to Furfuryl Alcohol over Nitrogen-Doped Carbon-Supported Iron Catalysts. ChemSusChem 2016, 9, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wei, J.; Ding, N.; Sun, Y.; Zeng, X.; Hu, L.; Liu, S.; Lei, T.; Lin, L. Chemoselective hydrogenation of biomass derived 5-hydroxymethylfurfural to diols: Key intermediates for sustainable chemicals, materials and fuels. Renew. Sustain. Energy Rev. 2017, 77, 287–296. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Martin, N.; Vlachos, D.G. Effect of hydrogen donor on liquid phase catalytic transfer hydrogenation of furfural over a Ru/RuO2/C catalyst. J. Mol. Catal. A Chem. 2014, 392, 223–228. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Panagiotopoulou, P.; Mironenko, A.V.; Jenness, G.R.; Vlachos, D.G.; Xu, B. Mechanistic Insights into Metal Lewis Acid-Mediated Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran. ACS Catal. 2015, 5, 3988–3994. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Vlachos, D.G. Liquid phase catalytic transfer hydrogenation of furfural over a Ru/C catalyst. Appl. Catal. A Gen. 2014, 480, 17–24. [Google Scholar] [CrossRef]
- Wang, B.; Li, C.; He, B.; Qi, J.; Liang, C. Highly stable and selective Ru/NiFe2O4 catalysts for transfer hydrogenation of biomass-derived furfural to 2-methylfuran. J. Energy Chem. 2017, 26, 799–807. [Google Scholar] [CrossRef]
- Zhang, Z.; Pei, Z.; Chen, H.; Chen, K.; Hou, Z.; Lu, X.; Ouyang, P.; Fu, J. Catalytic in-Situ Hydrogenation of Furfural over Bimetallic Cu-Ni Alloy Catalysts in Isopropanol. Ind. Eng. Chem. Res. 2018, 57, 4225–4230. [Google Scholar] [CrossRef]
- Gong, W.; Chen, C.; Fan, R.; Zhang, H.; Wang, G.; Zhao, H. Transfer-hydrogenation of furfural and levulinic acid over supported copper catalyst. Fuel 2018, 231, 165–171. [Google Scholar] [CrossRef]
- Chang, X.; Liu, A.-F.; Cai, B.; Luo, J.-Y.; Pan, H.; Huang, Y.-B. Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran and 2-Methyltetrahydrofuran over Bimetallic Copper-Palladium Catalysts. ChemSusChem 2016, 9, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J. Selective Transfer Hydrogenation of Biomass-Based Furfural and 5-Hydroxymethylfurfural over Hydrotalcite-Derived Copper Catalysts Using Methanol as a Hydrogen Donor. ACS Sustain. Chem. Eng. 2017, 5, 5982–5993. [Google Scholar] [CrossRef]
- Scholz, D.; Aellig, C.; Hermans, I. Catalytic Transfer Hydrogenation/Hydrogenolysis for Reductive Upgrading of Furfural and 5-(Hydroxymethyl)furfural. ChemSusChem 2014, 7, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Xia, Q.; Liu, X.; Wang, Y. Catalytic transfer hydrogenation/hydrogenolysis of 5-hydroxymethylfurfural to 2,5-dimethylfuran over Ni-Co/C catalyst. Fuel 2017, 187, 159–166. [Google Scholar] [CrossRef]
- Qi, L.; Mui, Y.F.; Lo, S.W.; Lui, M.Y.; Akien, G.R.; Horváth, I.T. Catalytic conversion of fructose, glucose, and sucrose to 5-(hydroxymethyl)furfural and levulinic and formic acids in γ-valerolactone as a green solvent. ACS Catal. 2014, 4, 1470–1477. [Google Scholar] [CrossRef]
- Jae, J.; Zheng, W.; Lobo, R.F.; Vlachos, D.G. Production of dimethylfuran from hydroxymethylfurfural through catalytic transfer hydrogenation with ruthenium supported on carbon. ChemSusChem 2013, 6, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, J.; Xie, W.; Tang, Y.; Guo, D.; Ni, Y. Catalytic Transfer Hydrogenation of Biobased HMF to 2,5-Bis-(Hydroxymethyl)Furan over Ru/Co3O4. Catalysts 2017, 7, 92. [Google Scholar] [CrossRef]
- Aellig, C.; Jenny, F.; Scholz, D.; Wolf, P.; Giovinazzo, I.; Kollhoff, F.; Hermans, I. Combined 1,4-butanediol lactonization and transfer hydrogenation/hydrogenolysis of furfural-derivatives under continuous flow conditions. Catal. Sci. Technol. 2014, 4, 2326–2331. [Google Scholar] [CrossRef]
- Hansen, T.S.; Barta, K.; Anastas, P.T.; Ford, P.C.; Riisager, A. One-pot reduction of 5-hydroxymethylfurfural via hydrogen transfer from supercritical methanol. Green Chem. 2012, 14, 2457–2461. [Google Scholar] [CrossRef]
- Jae, J.; Mahmoud, E.; Lobo, R.F.; Vlachos, D.G. Cascade of liquid-phase catalytic transfer hydrogenation and etherification of 5-hydroxymethylfurfural to potential biodiesel components over Lewis acid zeolites. ChemCatChem 2014, 6, 508–513. [Google Scholar] [CrossRef]
- Hao, W.; Li, W.; Tang, X.; Zeng, X.; Sun, Y.; Liu, S.; Lin, L. Catalytic transfer hydrogenation of biomass-derived 5-hydroxymethyl furfural to the building block 2,5-bishydroxymethyl furan. Green Chem. 2016, 18, 1080–1088. [Google Scholar] [CrossRef]
- Hu, L.; Yang, M.; Xu, N.; Xu, J.; Zhou, S.; Chu, X.; Zhao, Y. Selective transformation of biomass-derived 5-hydroxymethylfurfural into 2,5-dihydroxymethylfuran via catalytic transfer hydrogenation over magnetic zirconium hydroxides. Korean J. Chem. Eng. 2018, 35, 99–109. [Google Scholar] [CrossRef]
- Pasini, T.; Lolli, A.; Albonetti, S.; Cavani, F.; Mella, M. Methanol as a clean and efficient H-transfer reactant for carbonyl reduction: Scope, limitations, and reaction mechanism. J. Catal. 2014, 317, 206–219. [Google Scholar] [CrossRef]
- Thananatthanachon, T.; Rauchfuss, T.B. Efficient production of the liquid fuel 2,5-dimethylfuran from fructose using formic acid as a reagent. Angew. Chem. Int. Ed. 2010, 49, 6616–6618. [Google Scholar] [CrossRef] [PubMed]
- Thananatthanachon, T.; Rauchfuss, T.B. Efficient route to hydroxymethylfurans from sugars via transfer hydrogenation. ChemSusChem 2010, 3, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, J.; Choudhary, H.; Nishimura, S.; Ebitani, K. Direct Synthesis of 1,6-Hexanediol from HMF over a Heterogeneous Pd/ZrP Catalyst using Formic Acid as Hydrogen Source. ChemSusChem 2014, 7, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, C.; Fan, G.; Yang, L.; Li, F. Nitrogen-doped carbon-decorated copper catalyst for highly efficient transfer hydrogenolysis of 5-hydroxymethylfurfural to convertibly produce 2,5-dimethylfuran or 2,5-dimethyltetrahydrofuran. Appl. Catal. B Environ. 2018, 226, 523–533. [Google Scholar] [CrossRef]
- Bozell, J.J.; Moens, L.; Elliott, D.; Wang, Y.; Neuenscwander, G.; Fitzpatrick, S.; Bilski, R.; Jarnefeld, J. Production of levulinic acid and use as a platform chemical for derived products. Resour. Conserv. Recycl. 2000, 28, 227–239. [Google Scholar] [CrossRef]
- Antonetti, C.; Licursi, D.; Fulignati, S.; Valentini, G.; Raspolli Galletti, A. New Frontiers in the Catalytic Synthesis of Levulinic Acid: From Sugars to Raw and Waste Biomass as Starting Feedstock. Catalysts 2016, 6, 196. [Google Scholar] [CrossRef]
- Hengne, A.M.; Kadu, B.S.; Biradar, N.S.; Chikate, R.C.; Rode, C.V. Transfer hydrogenation of biomass-derived levulinic acid to γ-valerolactone over supported Ni catalysts. RSC Adv. 2016, 6, 59753–59761. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, Y.B.; Guo, Q.X.; Fu, Y. RANEY® Ni catalyzed transfer hydrogenation of levulinate esters to γ-valerolactone at room temperature. Chem. Commun. 2013, 49, 5328–5330. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wu, L.; Zhou, B.; Zhou, H.; Fan, H.; Yang, Y.; Meng, Q.; Han, B. A new porous Zr-containing catalyst with a phenate group: An efficient catalyst for the catalytic transfer hydrogenation of ethyl levulinate to γ-valerolactone. Green Chem. 2015, 17, 1626–1632. [Google Scholar] [CrossRef]
- Tang, X.; Hu, L.; Sun, Y.; Zhao, G.; Hao, W.; Lin, L. Conversion of biomass-derived ethyl levulinate into γ-valerolactone via hydrogen transfer from supercritical ethanol over a ZrO2 catalyst. RSC Adv. 2013, 3, 10277–10284. [Google Scholar] [CrossRef]
- Tang, X.; Chen, H.; Hu, L.; Hao, W.; Sun, Y.; Zeng, X.; Lin, L.; Liu, S. Conversion of biomass to γ-valerolactone by catalytic transfer hydrogenation of ethyl levulinate over metal hydroxides. Appl. Catal. B Environ. 2014, 147, 827–834. [Google Scholar] [CrossRef]
- Li, F.; France, L.J.; Cai, Z.; Li, Y.; Liu, S.; Lou, H.; Long, J.; Li, X. Catalytic transfer hydrogenation of butyl levulinate to Γ-valerolactone over zirconium phosphates with adjustable Lewis and Brønsted acid sites. Appl. Catal. B Environ. 2017, 214, 67–77. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Kaburagi, W.; Osada, Y.; Fujitani, T.; Yamashita, H. Catalytic transfer hydrogenation of biomass-derived levulinic acid and its esters to γ-valerolactone over ZrO2 catalyst supported on SBA-15 silica. Catal. Today 2017, 281, 418–428. [Google Scholar] [CrossRef]
- Wang, J.; Jaenicke, S.; Chuah, G.-K. Zirconium–Beta zeolite as a robust catalyst for the transformation of levulinic acid to γ-valerolactone via Meerwein–Ponndorf–Verley reduction. RSC Adv. 2014, 4, 13481–13489. [Google Scholar] [CrossRef]
- Chia, M.; Dumesic, J.A. Liquid-phase catalytic transfer hydrogenation and cyclization of levulinic acid and its esters to γ-valerolactone over metal oxide catalysts. Chem. Commun. 2011, 47, 12233–12235. [Google Scholar] [CrossRef] [PubMed]
- Bui, L.; Luo, H.; Gunther, W.R.; Román-Leshkov, Y. Domino Reaction Catalyzed by Zeolites with Brønsted and Lewis Acid Sites for the Production of γ-Valerolactone from Furfural. Angew. Chem. Int. Ed. 2013, 52, 8022–8025. [Google Scholar] [CrossRef] [PubMed]
- Valekar, A.H.; Cho, K.H.; Chitale, S.K.; Hong, D.Y.; Cha, G.Y.; Lee, U.H.; Hwang, D.W.; Serre, C.; Chang, J.S.; Hwang, Y.K. Catalytic transfer hydrogenation of ethyl levulinate to γ-valerolactone over zirconium-based metal-organic frameworks. Green Chem. 2016, 18, 4542–4552. [Google Scholar] [CrossRef]
- Tang, X.; Zeng, X.; Li, Z.; Hu, L.; Sun, Y.; Liu, S.; Lei, T.; Lin, L. Production of γ-valerolactone from lignocellulosic biomass for sustainable fuels and chemicals supply. Renew. Sustain. Energy Rev. 2014, 40, 608–620. [Google Scholar] [CrossRef]
- Ortiz-Cervantes, C.; García, J.J. Hydrogenation of levulinic acid to γ-valerolactone using ruthenium nanoparticles. Inorg. Chim. Acta 2013, 397, 124–128. [Google Scholar] [CrossRef]
- Deng, L.; Zhao, Y.; Li, J.; Fu, Y.; Liao, B.; Guo, Q.-X. Conversion of Levulinic Acid and Formic Acid into γ-Valerolactone over Heterogeneous Catalysts. ChemSusChem 2010, 3, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Son, P.A.; Nishimura, S.; Ebitani, K. Production of γ-valerolactone from biomass-derived compounds using formic acid as a hydrogen source over supported metal catalysts in water solvent. RSC Adv. 2014, 4, 10525–10530. [Google Scholar] [CrossRef]
- Lomate, S.; Sultana, A.; Fujitani, T. Vapor Phase Catalytic Transfer Hydrogenation (CTH) of Levulinic Acid to γ-Valerolactone over Copper Supported Catalysts Using Formic Acid as Hydrogen Source. Catal. Lett. 2018, 148, 348–358. [Google Scholar] [CrossRef]
- Yuan, J.; Li, S.-S.; Yu, L.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Copper-based catalysts for the efficient conversion of carbohydrate biomass into γ-valerolactone in the absence of externally added hydrogen. Energy Environ. Sci. 2013, 6, 3308–3313. [Google Scholar] [CrossRef]
- Hengne, A.M.; Malawadkar, A.V.; Biradar, N.S.; Rode, C.V. Surface synergism of an Ag–Ni/ZrO2 nanocomposite for the catalytic transfer hydrogenation of bio-derived platform molecules. RSC Adv. 2014, 4, 9730–9736. [Google Scholar] [CrossRef]
- Mascala, S.M.; Matson, T.D.; Johnson, C.L.; Lewis, R.S.; Iretskii, A.V.; Ford, P.C. Hydrogen Transfer from Supercritical Methanol over a Solid Base Catalyst: A Model for Lignin Depolymerization. ChemSusChem 2009, 2, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Besse, X.; Schuurman, Y.; Guilhame, N. Reactivity of lignin model compounds through hydrogen transfer catalysis in ethanol/water mixtures. Appl. Catal. B 2017, 209, 265–272. [Google Scholar] [CrossRef]
- Wu, H.; Song, J.; Xie, C.; Wu, C.; Chen, C.; Han, B. Efficient and Mild Transfer Hydrogenolytic Cleavage of Aromatic Ether Bonds in Lignin-Derived Compounds over Ru/C. ACS Sustain. Chem. Eng. 2018, 6, 2872–2877. [Google Scholar] [CrossRef]
- Galkin, M.V.; Sawadjoon, S.; Rohde, V.; Dawange, M.; Samec, J.S.M. Mild Heterogeneous Palladium-Catalyzed Cleavage of β-O-4′-Ether Linkages of Lignin Model Compounds and Native Lignin in Air. ChemCatChem 2014, 6, 179–184. [Google Scholar] [CrossRef]
- Sawadjoon, S.; Lundstedt, A.; Samec, J.S.M. Pd-Catalyzed Transfer Hydrogenolysis of Primary, Secondary, and Tertiary Benzylic Alcohols by Formic Acid: A Mechanistic Study. ACS Catal. 2013, 3, 635–642. [Google Scholar] [CrossRef]
- Zhang, D.; Ye, F.; Xue, T.; Guan, Y.; Wang, Y.M. Transfer hydrogenation of phenol on supported Pd catalysts using formic acid as an alternative hydrogen source. Catal. Today 2014, 234, 133–138. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. Exploiting H-transfer reactions with RANEY® Ni for upgrade of phenolic and aromatic biorefinery feeds under unusual, low-severity conditions. Energy Environ. Sci. 2012, 5, 8244–8260. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. A Route for Lignin and Bio-Oil Conversion: Dehydroxylation of Phenols into Arenes by Catalytic Tandem Reactions. Angew. Chem. Int. Ed. 2013, 52, 11499–11503. [Google Scholar] [CrossRef] [PubMed]
- Kennema, M.; de Castro, I.B.D.; Meemken, F.; Rinaldi, R. Liquid-Phase H-Transfer from 2-Propanol to Phenol on Raney Ni: Surface Processes and Inhibition. ACS Catal. 2017, 7, 2437–2445. [Google Scholar] [CrossRef]
- Mauriello, F.; Paone, E.; Pietropaolo, R.; Balu, A.M.; Luque, R. Catalytic transfer hydrogenolysis of lignin derived aromatic ethers promoted by bimetallic Pd/Ni systems. ACS Sustain. Chem. Eng. 2018, 6, 9269–9276. [Google Scholar] [CrossRef]
- Cozzula, D.; Vinci, A.; Mauriello, F.; Pietropaolo, R.; Müller, T.E. Directing the Cleavage of Ester C–O Bonds by Controlling the Hydrogen Availability on the Surface of Coprecipitated Pd/Fe3O4. ChemCatChem 2016, 8, 1515–1522. [Google Scholar] [CrossRef]
- Paone, E.; Espro, C.; Pietropaolo, R.; Mauriello, F. Selective arene production from transfer hydrogenolysis of benzyl phenyl ether promoted by a co-precipitated Pd/Fe3O4 catalyst. Catal. Sci. Technol. 2016, 6, 7937–7941. [Google Scholar] [CrossRef]
- Kim, M.; Ha, J.-M.; Lee, K.-Y.; Jae, J. Catalytic transfer hydrogenation/hydrogenolysis of guaiacol to cyclohexane over bimetallic RuRe/C catalysts. Catal. Commun. 2016, 86, 113–118. [Google Scholar] [CrossRef]
- Guo, T.; Xia, Q.; Shao, Y.; Liu, X.; Wang, Y. Direct deoxygenation of lignin model compounds into aromatic hydrocarbons through hydrogen transfer reaction. Appl. Catal. A 2017, 547, 30–36. [Google Scholar] [CrossRef]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640–1647. [Google Scholar] [CrossRef]
- Deuss, P.J.; Scott, M.; Tran, F.; Westwood, N.J.; de Vries, J.G.; Barta, K. Aromatic Monomers by in Situ Conversion of Reactive Intermediates in the Acid-Catalyzed Depolymerization of Lignin. J. Am. Chem. Soc. 2015, 137, 7456–7467. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Samec, J.S.M. Selective Route to 2-Propenyl Aryls Directly from Wood by a Tandem Organosolv and Palladium-Catalysed Transfer Hydrogenolysis. ChemSusChem 2014, 7, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Smit, A.T.; Subbotina, E.; Artemenko, K.A.; Bergquist, J.; Huijgen, W.J.; Samec, J.S.M. Hydrogen-free catalytic fractionation of woody biomass. ChemSusChem 2016, 9, 3280–3287. [Google Scholar] [CrossRef] [PubMed]
- Kumaniaev, I.; Samec, J.S.M. Valorization of Quercus suber Bark toward Hydrocarbon Bio-Oil and 4-Ethylguaiacol. ACS Sustain. Chem. Eng. 2018, 6, 5737–5742. [Google Scholar] [CrossRef]
- Kumaniaev, I.; Subbotina, E.; Savmarker, J.; Larhed, M.; Galkin, M.V.; Samec, J.S.M. Lignin depolymerization to monophenolic compounds in a flow-through system. Green Chem. 2017, 19, 5767–5771. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Schutyser, W.; Koelewijn, S.-F.; Renders, T.; Courtin, C.M.; Sels, B.F. Tuning the lignin oil OH-content with Ru and Pd catalysts during lignin hydrogenolysis on birch wood. Chem. Commun. 2015, 51, 13158–13161. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Simmons, B.A.; Singh, S. Catalytic transfer hydrogenolysis of ionic liquid processed biorefinery lignin to phenolic compounds. Green Chem. 2017, 19, 215–224. [Google Scholar] [CrossRef]
- Song, Q.; Wang, F.; Cai, J.; Wang, Y.; Zhang, J.; Yu, W.; Xu, J. Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process. Energy Environ. Sci. 2013, 6, 994–1007. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Pineda, A.; Romero, A.A.; Luque, R.; Labidi, J. Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: Catalyst screening. Appl. Catal. B. 2014, 145, 43–55. [Google Scholar] [CrossRef]
- Ferrini, P.; Rezende, C.A.; Rinaldi, R. Catalytic Upstream Biorefining through Hydrogen Transfer Reactions: Understanding the Process from the Pulp Perspective. ChemSusChem 2016, 9, 3171–3180. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, P.; Rinaldi, R. Catalytic Biorefining of Plant Biomass to Non-Pyrolytic Lignin Bio-Oil and Carbohydrates through Hydrogen Transfer Reactions. Angew. Chem. Int. Ed. 2014, 53, 8634–8639. [Google Scholar] [CrossRef] [PubMed]
- Molinari, V.; Clavel, G.; Graglia, M.; Antonietti, M.; Esposito, D. Mild Continuous Hydrogenolysis of Kraft Lignin over Titanium Nitride–Nickel Catalyst. ACS Catal. 2016, 6, 1663–1670. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Lu, G.-P.; Cai, C. Self-hydrogen transfer hydrogenolysis of β-O-4 linkages in lignin catalyzed by MIL-100(Fe) supported Pd–Ni BMNPs. Green Chem. 2017, 19, 4538–4543. [Google Scholar] [CrossRef]
- Regmi, Y.N.; Mann, J.K.; McBride, J.R.; Tao, J.; Barnes, C.E.; Labbè, N.; Chmely, S.C. Catalytic transfer hydrogenolysis of organosolv lignin using B-containing FeNi alloyed catalysts. Catal. Today 2018, 302, 190–195. [Google Scholar] [CrossRef]
- Luo, L.; Yang, J.; Yao, G.; Jin, F. Controlling the selectivity to chemicals from catalytic depolymerization of kraft lignin with in-situ H2. Bioresour. Technol. 2018, 264, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atay, C.; Korànyi, T.I.; Boot, M.D.; Hensen, E.J.M. Role of Cu–Mg–Al Mixed Oxide Catalysts in Lignin Depolymerization in Supercritical Ethanol. ACS Catal. 2015, 5, 7359–7370. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
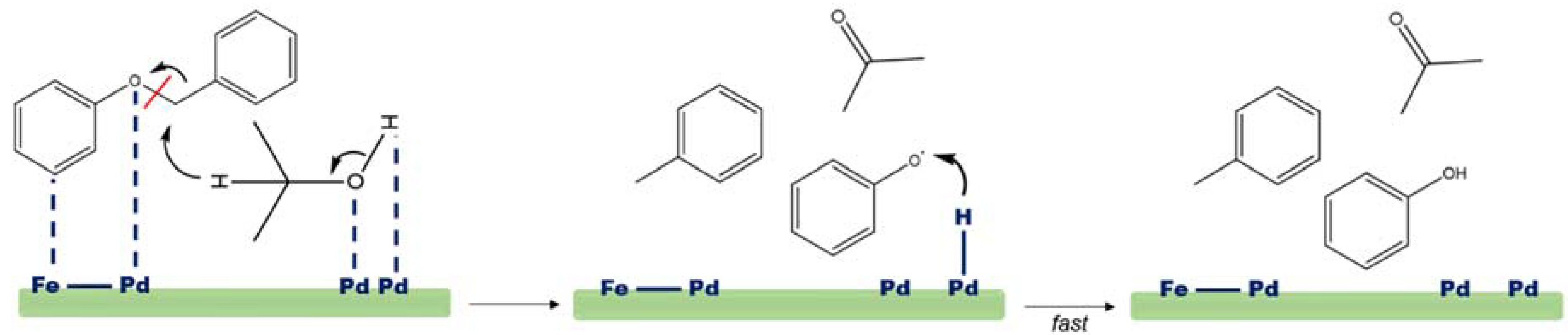{kind=link}
| Entry | Catalyst | H-Donor 1 | Cat/Gly 2 | Temp (°C) | Time (h) | Conv. (%) | 1,2-PDO Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | PdO/Fe2O3 | EtOH | 0.237 | 180 | 24 | 100 | 90 | [45] |
| 2 | PdO/Fe2O3 | 2-PO | 0.237 | 180 | 24 | 100 | 94 | [45] |
| 3 | PdO/Fe2O3 | 2-PO | 0.237 | 180 | 8 | 96 | 87 | [45] |
| 4 | Pd/Fe3O4 | 2-PO | 0.237 | 180 | 8 | 100 | 84 | [45] |
| 5 | Pd/Fe3O4 | 2-PO | 0.207 | 180 | 24 | 100 | 56 | [46] |
| 6 | Pd/Co3O4 | 2-PO | 0.207 | 180 | 24 | 100 | 64 | [46] |
| 7 | Ni-Cu/Al2O3 | - | 0.166 | 220 | 24 | 70.5 | 66.9 | [47] |
| 8 | Ni-Cu/Al2O3 | 2-PO | 0.166 | 220 | 24 | 60.4 | 64.6 | [47] |
| 9 | Ni-Cu/Al2O3 | 2-PO | 0.166 | 220 | 10 | 28.2 | 77.4 | [48] |
| 10 | Ni-Cu/Al2O3 | MeOH | 0.120 | 220 | 10 | 26.2 | 51.2 | [48] |
| 11 | Ni-Cu/Al2O3 | FA | 0.120 | 220 | 10 | 33.5 | 85.9 | [48] |
| 12 | Ni-Cu/Al2O3 | FA | 0.498 | 220 | 24 | 90 | 82 | [49] |
| 13 | 70Cu30Al | 2-PO | - | 220 | 5 | 69 | 90 | [50] |
| 14 | 20Cu/ZrO2 | FA | - | 220 | 18 | 97 | 95 | [51] |
| Entry | Catalyst | Solvent 1 | Cat/Gly 2 | Temp (°C) | Gas (bar) | Time (h) | Conv. (%) | Desired Prod. Select. (%) 3 | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Pd0.04Cu0.4/Mg5.5Al2O8.5 | MeOH | 0.125 | 180 | H2 (20) | 10 | 89.5 | 1,2-PDO (98) | [52] |
| 2 | Pd0.04Cu0.4/Mg5.5Al2O8.5 | EtOH | 0.125 | 180 | H2 (20) | 10 | 88.0 | 1,2-PDO (99) | [52] |
| 3 | Rh0.02Cu0.4/Mg5.6Al1.9O8.6 | EtOH | 0.167 | 180 | H2 (20) | 10 | 91.0 | 1,2-PDO (99) | [54] |
| 4 | Pd/Fe3O4 | 2-PO | 0.237 | 180 | H2 (5) | 24 | 100 | 1,2-PDO (71) | [55] |
| 5 | 2Pt/20WO3/ZrO2 | EtOH | 0.250 | 170 | H2 (55) | 12 | 45.7 | 1,3-PDO (21) | [56] |
| Entry | Substrate 1 | Catalyst | H-Donor 2 | Temp (°C) | Time (h) | Conv. (%) | Yield Sorbitol (%) | Yield Mannitol (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MC | Ru/C-Q10 | 2-PO | 190 | 18 | 80.2 | 36.8 | 9.0 | [66] |
| 2 | MC | Ru/CMK-3 | 2-PO | 190 | 18 | 81.2 | 35.7 | 9.3 | [66] |
| 3 | MC | Ru/AC(N) | 2-PO | 190 | 18 | 74.4 | 33.5 | 9.0 | [66] |
| 4 | Glucose | Ru/AC(N) | 2-PO | 180 | 0.33 | 82 | 77.0 | 2.7 | [67] |
| 5 | ACO | Ru/AC(N) | 2-PO | 180 | 0.33 | 100 | 32.2 | 3.1 | [67] |
| Entry | Catalyst | H-Donor 1 | Reaction Conditions 2 (Temperature, Time, Solvent) | Conv. (%) | Main Product 3 | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Ru/RuO2/C | 2-PE, 2-BU | 180 °C, 10 h, 2-PE, 2-BU | 100.0 | MF | 76.0 | [73] |
| 2 | Ru/RuOx/C | 2-BU | 180 °C, 10 h, TU | 100.0 | MF | 76.0 | [74] |
| 3 | Ru/C | 2-PO | 180 °C, 10 h, 2-PO | 100.0 | MF | 61.0 | [75] |
| 4 | Ru/NiFe2O4 | 2-PO | 180 °C, 10 h, 2-PO | >97.0 | MF | 83.0 | [76] |
| 5 | Cu-Ni/Al2O3 | 2-PO | 230 °C, 4 h, 2-PO | >97.0 | MF, MTHF | 82.5 | [77] |
| 6 | Cu/C | 2-PO | 200 °C, 5 h, 2-PO | 96.3 | MF | 84.0 | [78] |
| 7 | Cu-Pd/C | 2-PO | 200 °C, 4 h, 2-PO | 100.0 | MF, MTHF | 83.9 | [79] |
| 8 | Cu3Al-A | MeOH | 240 °C, 1.5 h, MeOH | >97.7 | MF | 88.2 | [80] |
| 9 | Pd/Fe2O3 | 2-PO | 180 °C, 7.5 h, 2-PO | 95.0 | MF, MTHF | 62.0 | [81] |
| Entry | Substrate 1 | Catalyst 2 | H-Donor 3 | Reaction Conditions (Temperature, Time, Solvent) | Conv. (%) | GVL Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | LA | Ni/MMT | 2-PO | 200 °C, 1 h, 2-PO | 99.0 | 99.0 | [98] |
| 2 | EL | Raney® Ni | 2-PO | 25 °C, 9 h, 2-PO | - | 99.0 | [99] |
| 3 | EL | Zr-HBA | 2-BU | 150 °C, 4 h, 2-BU | 100.0 | 95.9 | [100] |
| 4 | EL | ZrO2 | EtOH | 250 °C, 3 h, EtOH | 95.5 | 81.5 | [101] |
| 5 | EL | ZrO(OH)2 | 2-PO | 200 °C, 1 h, 2-PO | 93.6 | 94.5 | [102] |
| 6 | BL | ZrPO-1.00 | 2-PO | 210 °C, 2 h, 2-PO | 98.1 | 95.7 | [103] |
| 7 | ML | ZrO2/SBA-15 | 2-PO | 150 °C, 6 h, 2-PO | 99.9 | 95.0 | [104] |
| 8 | LA | Zr-Beta | 2-PO | 250 °C, vap. phase, 2-PO | 100.0 | >99.0 | [105] |
| 9 | LA | ZrO2 | 2-BU | 150 °C, 16 h, 2-BU | >99.9 | 84.7 | [106] |
| 10 | FU | Zr-Beta + Al-MFI-ns | 2-BU | 120 °C, 48 h, 2-BU | - | 78.0 | [107] |
| 11 | EL | UiO66(Zr) | 2-PO | 200 °C, 2 h, 2-PO | >98.0 | 92.7 | [108] |
| Entry | Catalyst | Reaction Conditions (Temperature, Time, Solvent) 1 | Conv. (%) | GVL Yield (%) | Ref. |
|---|---|---|---|---|---|
| 1 | Ru NPs | 130 °C, 42 h, FA + triethylamine + water | 100.0 | 100.0 | [110] |
| 2 | Ru-P/SiO2 + Ru/TiO2 | 150 °C, 6 h, LA | 100.0 | 30.0 | [111] |
| 3 | Ru/C | 150 °C, 5 h, water | 100.0 | 90.0 | [112] |
| 4 | Cu/SiO2 | 250 °C, vap. phase, -, FA + water | 48.0 | 90.0 | [113] |
| 5 | Cu/ZrO2 | 200 °C, 5 h, water | 100.0 | 100.0 | [114] |
| 6 | Ag-Ni/ZrO2 | 220 °C, 5 h, water | 100.0 | 99.0 | [115] |
| Entry | Lignin Type 1 | Catalyst | H-Source | Temp. [°C] | Time [h] | Conversion [%] | Main Products | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | OL | Cu-MPO | MeOH | 300 | 24 | 100 | Cycloexyl derivates | [130] |
| 2 | DWL | Trifilic acid | 1,4-dioxane | 140 | 4 | 100 | C2-aldehyde fragments | [131] |
| 3 | PS | Pd/C | Formic acid | 195 | 1 | 100 | Aryl propene monomers | [132] |
| 4 | OL | Pd/C | Hemicellulose | 210 | 15 | 100 | Phenols and propylphenols | [133] |
| 5 | OL | Pd/C | Carbohydrate fractions | 200 | 2 | 100 | 4-ethylguaiacol | [134] |
| 6 | OL | Pd/C | Hemicellulose | 160–220 | 3–6 | 100 | Monophenolic products | [135] |
| 7 | OL | Pd/C | MeOH | 250 | 3 | 90 | 4-n-propanolguaiacol and 4-n-propanolsyringol | [136] |
| 8 | OL | Ru/C | MeOH | 250 | 3 | 85 | para-propyl phenolics | [137] |
| 9 | OL | Ru/C | 2-PO | 300 | 1-3 | 100 | 4-ethyl phenol, 2-methoxy phenol and phenol | [137] |
| 10 | BVL | Ni/C | Aliphatic alcohols | 200 | 6 | 50 | 4-propylguaiacol and 4-propylsyringol | [138] |
| 11 | OL | Al-SBA-15 | Tetraline/FA | 140 | 1/2 | 100 | Mesitol and syrangaldehyde | [139] |
| 12 | OL | RANEY® Ni | Hemicellulose | 160–220 | 3 | 100 | Monocyclic products | [140] |
| 13 | OL | RANEY® Ni | 2-PO/H2O | 160–220 | 18 | 100 | Alkenes and arenes | [141] |
| 14 | KL | TiN-Ni and TiO2-Ni | MeOH, EtOH, 2-PO, THF | 150 | 4.5 min | 100 | Guaiacol products | [142] |
| 15 | OL | Pd1Ni4/MIL-100(Fe) | H2O | 180 | 6 | 100 | Phenol and guaiacol derivates | [143] |
| 16 | OL | FeB, NiB and FeNiB | EtOH | 320 | 2 | 100 | 21 depolymerization products | [144] |
| 17 | KL | Fe on Rh/La2O3/CeO2-ZrO2 | 2-PO/H2O | 373 | 2 | 100 | C12–26 aliphatic, C6–16 aromatic and C7–10 hydrogenated cycles compounds | [145] |
| 18 | OL | Cu-Mg-Al oxides | EtOH | 340 | 4 | 100 | C6–12 aromatics, C3–8 alcohols, C3–12 esters | [146] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espro, C.; Gumina, B.; Szumelda, T.; Paone, E.; Mauriello, F. Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules. Catalysts 2018, 8, 313. https://doi.org/10.3390/catal8080313
Espro C, Gumina B, Szumelda T, Paone E, Mauriello F. Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules. Catalysts. 2018; 8(8):313. https://doi.org/10.3390/catal8080313
Chicago/Turabian StyleEspro, Claudia, Bianca Gumina, Tomasz Szumelda, Emilia Paone, and Francesco Mauriello. 2018. "Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules" Catalysts 8, no. 8: 313. https://doi.org/10.3390/catal8080313
APA StyleEspro, C., Gumina, B., Szumelda, T., Paone, E., & Mauriello, F. (2018). Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules. Catalysts, 8(8), 313. https://doi.org/10.3390/catal8080313