Abstract
The aim of this study was to develop an effective carbonaceous solid acid for synthesizing green fuel additive through esterification of lignocellulose-derived levulinic acid (LA) and n-butanol. Two different sulfonated carbons were prepared from glucose-derived amorphous carbon (GC400) and commercial active carbon (AC400). They were contrastively studied by a series of characterizations (N2 adsorption, X-ray diffraction, elemental analysis, transmission electron microscopy, Fourier transform infrared spectroscopy and NH3 temperature programmed desorption). The results indicated that GC400 possessed stronger acidity and higher –SO3H density than AC400, and the amorphous structure qualified GC400 for good swelling capacity in the reaction solution. Assessment experiments showed that GC400 displayed remarkably higher catalytic efficiency than AC400 and other typical solid acids (HZSM-5, Hβ, Amberlyst-15 and Nafion-212 resin). Up to 90.5% conversion of LA and 100% selectivity of n-butyl levulinate could be obtained on GC400 under the optimal reaction conditions. The sulfonated carbon retained 92% of its original catalytic activity even after five cycles.
1. Introduction
In recent years, the foreseeable depletion of fossil energy and growing environmental concerns have drawn tremendous attention towards the conversion of renewable biomass into fuels [1,2,3] and fine chemicals [4,5]. Lignocellulose is the major component of agricultural waste and forest residues. Levulinic acid (LA) derived from the deconstruction of lignocellulose is a versatile platform chemical, and it has been highlighted by the United States Department of Energy as a promising building block for chemistry in 2004 and 2010 [6]. Among the downstream derivativities of LA, alkyl levulinates are of particular interest due to their extensive applications in the flavoring, solvent and plasticizer sectors [4,7]. Moreover, the low toxicity, high lubricity, stable flash point and moderate flow property make alkyl levulinates suitable additives for gasoline and diesel fuels [8].
Generally, alkyl levulinates are obtained by esterification of LA with alcohols using mineral acids, which lead to some difficulties in catalyst recycling, product separation and environmental protection. Substitution of homogeneous catalysts by heterogeneous catalysts is highly desirable to avoid handling corrosion and environmental problems. As far as we know, most studies were focused on the synthesis of ethyl levulinate (EL). For this purpose, various solid acid catalysts have been developed, including supported heteropolyacid [9,10], zeolites [11,12], sulfonated metal oxides [13] and silicas [14], sulfonated carbon nanotubes [15], Starbon® mesoporous carbon [16], and hybrid catalysts [17].
Comparatively, the potential of butyl levulinate (BL) has been left unexploited. It is worth noting that BL has been proved to be more promising than EL as a fuel additive [18,19]. They both could improve lubricity, conductivity and reduce particulate emissions in diesel blending. The esters displayed 25% (BL) and 31% (EL) less energy per volume than conventional diesel fuel. It is already an upgrading on bioethanol blends. On the other hand, BL has an evidently lower solubility in water (1.3 wt %) than EL (15.6 wt %). In diesel blends containing 20% (v/v) alkyl levulinate, EL was found to form a separate liquid phase from diesel at 283 K, while BL remained constant miscibility in diesel even at the diesel cloud point (247 K) [20].
Nowadays, the application of acetone-butanol-ethanol fermentation technology to lignocellulose makes bio-butanol (n-butanol) become an inexpensive platform chemical to produce BL. However, the literature on the synthesis of BL via the esterification of LA are sparse. Several types of solid acids such as supported montmorillonite [21], zeolites [22,23], metal–organic frameworks [24], Al-MCM-41 [25], ion-exchange resins [26,27], graphene oxide [28] and immobilized lipase [29] have been utilized in the esterification of LA with n-butanol. Surprisingly, the catalysis with amorphous carbon materials to produce BL has not been explored before. The amorphous carbon obtained by incomplete carbonization of carbohydrate is a kind of soft carbon possessing polycyclic aromatic carbon sheets in a three-dimensional sp3-bonded structure [30,31]. After sulfonation, this amorphous carbon could generate a stable solid acid catalyst with abundant sulfonic sites, which has been reported to effectively catalyze the esterification of fatty acids to produce biodiesel [32,33,34]. So far, there has been no report about its utilization in esterification of LA to produce fuel additives.
In this work, two different sulfonated carbons obtained from glucose-derived amorphous carbon and conventional active carbon were prepared and evaluated their catalytic performance in the esterification of LA with n-butanol. Both carbon materials were characterized by N2 adsorption, X-ray diffraction (XRD), CHNS elemental analysis, Fourier-transform infrared spectra (FT-IR), high-resolution transmission electron microscopy (HRTEM), and NH3 temperature programmed desorption (NH3-TPD). The relationship between the catalytic performance and the physicochemical properties of carbon materials were discussed. Moreover, different experimental parameters, such as reaction temperature, reaction time, molar ratio of reactants, catalyst loading, and cycle times were optimized over sulfonated glucose-derived carbon.
2. Results and Discussion
2.1. Catalyst Characterizations
Figure 1 illustrates the XRD patterns of two sulfonated carbon materials. GC400 exhibited a weak and broad carbon (002) diffraction peak at 10–30°, which was attributed to amorphous carbon composed of aromatic carbon sheets oriented in a considerably random fashion [31]. This indicated that the sample was comprised of a high content of non-graphitic carbon structure. For an AC400 sample, a sharp diffraction peak at 26.1° and a definitive carbon (101) diffraction peak at 40–50° both demonstrated the formation of graphite structure in the active carbon [35]. The XRD results indicated that AC400 was more carbonized than GC400 and possessed larger carbon sheets. The amorphous structure of GC400 was further confirmed by HRTEM images (see Figure S1), as no evident graphene sheets were observed, while the image of AC400 clearly revealed the existence of curved graphene sheets that were randomly distributed.
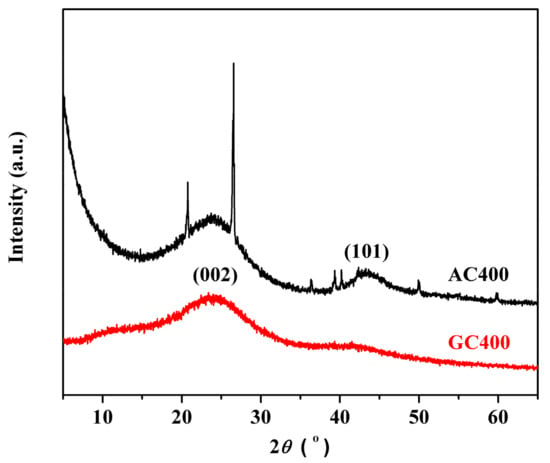
Figure 1.
XRD patterns of two sulfonated carbon materials.
According to the results of CHNS elemental analysis shown in Table 1, GC400 had a lower carbon content and a higher hydrogen content than AC400, indicating the incomplete carbonization of glucose at 673 K. In addition, the sulfur content of GC400 was remarkably higher than that of AC400. Since all the sulfur atoms have been proved to be existing as –SO3H groups [32], the –SO3H density of GC400 and AC400 were calculated to be 0.66 mmol·g−1 and 0.2 mmol·g−1, respectively. The higher –SO3H density of GC400 can be rationalized by its small non-graphitic carbon sheets that have more access to bind with –SO3H groups, as –SO3H groups can be only attached onto the edge of carbon sheets [31].

Table 1.
The results of CHNS elemental analysis and surface areas.
Despite having high –SO3H density, GC400 had a rather small specific surface area (2 m2·g−1, see Table 1), which was comparable to those reported in the literature [31,32]. It indicated most –SO3H groups were in the bulk structure, as opposed to on the outermost surface. Otherwise, there would have been an unreasonable number of S atoms per unit surface area. The small surface area of GC400 did not necessarily imply limited access to active sites in the esterification. Figure S2 shows the behaviour of GC400 and AC400 in n-butanol. It is surprising to find the volume of GC400 increased by approximately 1.5 times while there was barely a change in AC400 volume. This phenomenon indicated that GC400 possessed swelling capacity to incorporate reactants into the –SO3H sites located in the bulk phase. The swelling capacity is originated from the small and flexible aromatic carbon sheets in amorphous structure [31]. In contrast, the flexibility of carbon sheets in AC400 decreased due to the higher graphitization, leading to the poor swelling capacity in reaction solution. The total acid densities of two carbonaceous materials were determined by two different methods (NH3 adsorption and titration). The results were shown in Table S1. As we can see, the total acid densities of AC400 obtained by two methods were basically the same (0.44 vs. 0.46 mmol·g−1). However, the NH3 adsorption of GC400 resins resulted in a prediction of less than 10.9% of the real acid site density (1.1 vs. 1.22 mmol·g−1). This gap can be explained by the acid site accessibility in swollen GC400 being of more relevance than accessibility in the dry sample. On the other hand, the total acid densities of two carbonaceous materials were both higher than their –SO3H densities. The reason can be attributed to a certain amount of –COOH and phenolic –OH groups presenting in the two catalysts, which agrees with the following FT-IR results. It also can be seen from Table S1 that AC400 possessed a remarkably lower amount of –COOH and –OH groups than GC400, and the deficiency of these oxygenated groups on the carbon surface of AC400 is supposed to decrease its interaction ability with polar compounds in reaction mixture.
The IR spectra of two sulfonated carbon catalysts are depicted in Figure 2. As for GC400 catalyst, the bands at 1034 cm−1 and 1172 cm−1 were ascribed to the S=O symmetric and asymmetric stretching mode, providing the presence of –SO3H groups. The vibration bands at 1400 cm−1 and 1602 cm−1 were attributed to the aromatic C=C stretching mode, suggesting the existence of polycyclic aromatic rings, which were formed from the incomplete carbonization of the glucose substance [36]. Meanwhile, the band at 1706 cm−1 was related to the C=O stretching of the –COOH group. The bands in the range of 3000–3400 cm−1 were related to the O–H stretching of the –COOH and phenolic –OH groups. Therefore, the Brønsted acid sites (–SO3H and –COOH) and almost neutral phenolic –OH groups were confirmed to be attached onto the aromatic carbon structure of GC400. This is distinct from conventional solid acids with single functional groups. On the other hand, the weak bands at 1172 cm−1, 1706 cm−1 and 3365 cm−1 on AC400 revealed its deficiency of –SO3H, –COOH and –OH groups.
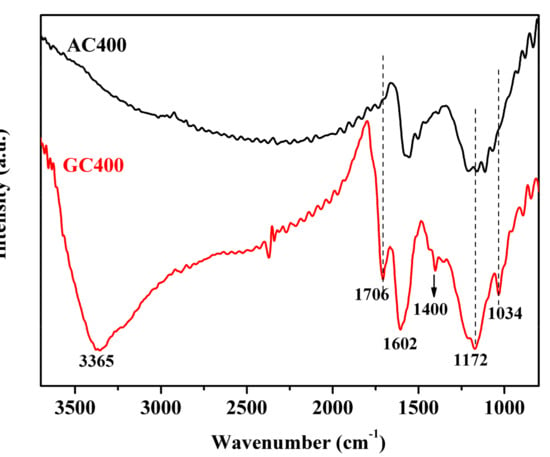
Figure 2.
FT-IR results of two sulfonated carbon materials.
The distribution of acid strength on carbon materials is shown in Figure 3. Two broad desorption peaks ranging from 400 to 700 K and 800 to 1000 K were observed on GC400. The former peak indicated the presence of weak acid sites due to interaction of NH3 with incompletely formed carbon sheets and –COOH groups [37]. The latter peak maximized at 950 K was ascribed to the presence of strong acid sites due to desorption of –SO3H groups [37]. The NH3-TPD result of unsulfonated GC400 further proved the above discussion (see Figure S3). However, only one desorption peak ranging from 400 to 570 K was detected on AC400, the missing desorption peaks at elevated temperature revealed its weak acid strength owing to the deficiency of –SO3H groups. The result was in accordance with the FT-IR and elemental analysis.
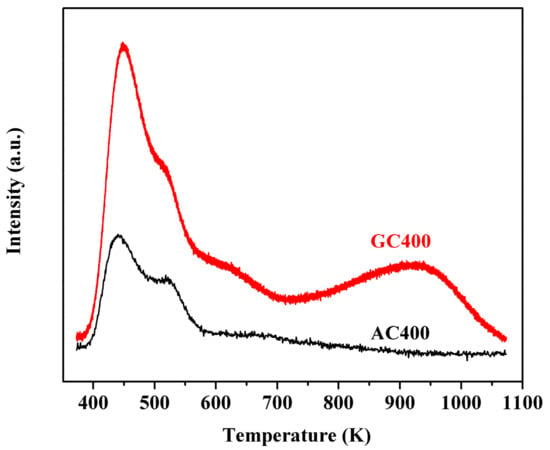
Figure 3.
NH3-TPD results of two sulfonated carbon materials.
2.2. Esterification Activity over Two Sulfonated Carbon Materials
Two sulfonated carbons were comparatively evaluated in the esterification of LA with n-butanol (Scheme 1) at 373 K for 4 h, n-butanol to LA molar ratio of 5:1, and catalyst loading of 10 wt % with respect to LA. In all cases, the selectivity of BL remained 100%. By-products like ethers coming from the intermolecular dehydration of alcohols were not observed under the moderate reaction conditions (see Figure S4). A blank experiment with 5.2% conversion of LA indicated that LA could catalyze the reaction as a weak acid catalyst [13]. Figure 4 shows that both sulfonated carbons exhibited a certain extent of catalytic activity, while a significantly higher reaction rate and LA conversion were obtained on GC400. The conversion could be consolidated through prolonging the reaction time within 4 h. After that, the promotion effect was weakened due to the equilibrium limitation. Finally, 90.5% conversion of LA on GC400 and 15.6% conversion of LA on AC400 were obtained. Therefore, the reaction time of 4 h was observed to be an optimum. The catalytic performances of two carbonaceous materials were quantified as turnover frequency (TOF) based on the amount of –SO3H sites and LA conversion at the initial stage. The calculated TOF value was 23.3 h−1 on AC400 and 95.8 h−1 on GC400.
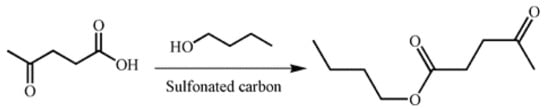
Scheme 1.
Sulfonated carbon catalyzed esterification of LA.
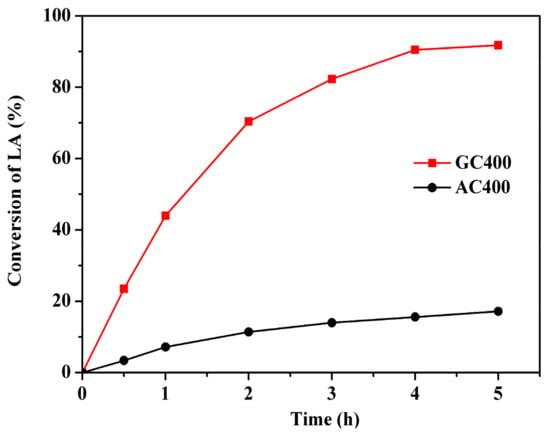
Figure 4.
Conversions of LA over two sulfonated carbon materials as the function of reaction time. Reaction conditions: 1.16 g LA, 3.7 g n-butanol and 0.116 g catalyst, 373 K, 4 h.
In order to explore the reasons for the distinct activities of two sulfonated carbons, a comparative test was carried out under the catalysis of H2SO4, H3PO4 and CH3COOH, which represent strong acid, medium acid and weak acid, respectively. The sequence for the conversion of LA was H2SO4 > H3PO4 > CH3COOH (see Figure S5), which was consistent with the acid strength of these acids. The result demonstrated that the esterification of LA and n-butanol was sensitive to the acid strength of catalyst. Strong acid was more active than weak acid for this reaction. Moreover, another test was conducted under the catalysis of different concentrations of H2SO4. The conversion of LA improved significantly with the increasing H2SO4 concentration from 1 to 20 wt % (see Figure S6). It further proved the positive effect of –SO3H density on esterification. Therefore, combining with the previous characterization results, the excellent catalytic performance of GC400 can be rationalized by its stronger acidity and higher –SO3H density. In addition, the swelling capacity of GC400 probably also contributed to the high catalytic performance because it provided good access of the reactants in solution to the –SO3H sites in the carbon material. Finally, the catalytic activities of two catalysts were studied with identical amounts of –SO3H groups in the reaction system (see Figure S7). Furthermore, 20% conversion of LA on GC400 and 6.2% conversion of LA on AC400 were obtained after 1.5 h. The activity difference further demonstrated the importance of strong acid strength and good accessibility of acid sites (due to the swelling capacity and abundant oxygenated functional groups) for the excellent activity of GC400.
2.3. Effect of Reaction Temperature over Two Sulfonated Carbon Materials
Figure 5 shows the conversion of LA varied with reaction temperature over two sulfonated carbons at n-butanol to LA molar ratio of 5:1 and catalyst loading of 10 wt %. Both catalysts exhibited a growing trend in catalytic activity with the increasing reaction temperature. In general, elevated temperature is conducive to the forward reaction, as esterification reaction is a reversible endothermic process. The optimal reaction temperature was found to be 373 K; at this time, the conversion of LA over GC400 was observed to increase up to 90.5% from 31.6% at reaction temperature of 333 K. With a further increment to 393 K, there was no appreciable enhancement in LA conversion. AC400 still demonstrated a poor activity even at elevated temperature of 393 K, and the effect of mass transfer limitation has been excluded (see Figure S8). Therefore, GC400 was selected for the further optimization.
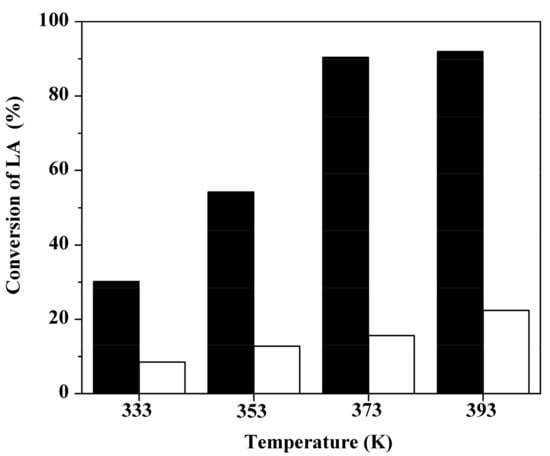
Figure 5.
Conversions of LA over GC400 (black) and AC400 (white) at different reaction temperature. Reaction conditions: 1.16 g LA, 3.7 g n-butanol and 0.116 g catalyst, 4 h.
2.4. Effect of Reactant Molar Ratio and Catalyst Loading over GC400
The n-butanol to LA molar ratio was varied between 1:1 to 7:1 over GC400 at reaction temperature 373 K, reaction time 4 h and catalyst loading 10 wt %. As shown in Figure 6a, LA conversion increased gradually from 45.2 to 90.5% with an increase in n-butanol to LA ratio from 1:1 to 5:1. A further increase in molar ratio did not significantly increase the conversion of LA, and even had a negative effect. Increasing the alcohol amount is an advantage to accelerate the forward reaction, as esterification is reversible. However, excessive dilution in the reaction system may reduce the frequency of collision [38] and flood the active sites of catalyst [39], thereby restricting a further increase in conversion. A similar phenomenon was observed by Maheria et al. [22] in the esterification catalyzed by zeolites. In the present case, the n-butanol to LA molar ratio of 5 was observed to be an optimum.
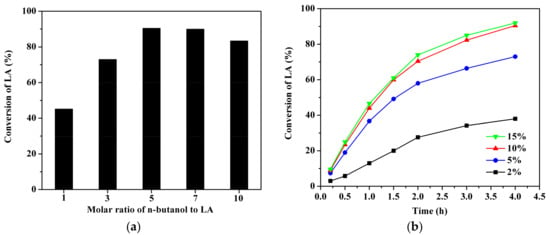
Figure 6.
Conversions of LA over GC400 as the function of molar ratio of n-butanol to LA (a) or as the function of catalyst loading (b). Reaction conditions: (a) 1.16 g LA, 0.116 g catalyst, 373 K, 4 h; (b) 1.16 g LA, 3.7 g n-butanol, 373 K, 4 h.
The effect of catalyst loading on the conversion of LA was also studied at a temperature of 373 K, reaction time of 4 h and an n-butanol to LA molar ratio of 5:1. The dosage of GC400 catalyst was varied in the range of 2–15 wt % relative to the weight of LA. Figure 6b shows that a higher reaction rate and conversion of LA were observed with an increase in catalyst loading from 2 to 10 wt %, as the number of available sites for the reaction increased. Further heightening the catalyst amount to 15 wt % made no distinct increment in esterification efficiency. This marginal increase in LA conversion may be due to catalyst agglomeration or decrease in contact opportunities between the reactants.
2.5. Comparison with Other Typical Solid Acids
Various typical solid acid catalysts (HZSM-5, Hβ zeolite, Amberlyst-15 and Nafion-212 resin) were chosen to evaluate their catalytic performance under the same reaction conditions. The acid strength, acid densities and activities of various samples were summarized in Table 2. The acid strength was represented by the Hammett acidity function (–H0) values of these catalysts. The TOF values were calculated according to the initial LA conversion rate and the –SO3H amount. As we can see, the TOF decreased in the order of GC400 > Nafion-212 > Amberlyst-15 > Hβ > HZSM-5. The result agrees with the recent study by Fernandes et al. [13], who found that the activity of zeolites in esterification of LA were generally lower than that of sulfated oxides. The poor activity of zeolites is related to the formation of the transition state inside the channels rather than to their acidity. This may explain the activity gap between Hβ and HZSM-5 due to their different pore channels.
Table 2.
Hammett acidy function (–H0) values, acid density and catalytic activity of the solid acids.
Amberlyst-15 is a polymer-based solid acid with moderate acidity and high –SO3H density. 76% conversion of LA could be obtained on Amberlyst-15, which was lower than that on GC400 with lower –SO3H density but stronger acidity. The result furthermore indicates that the esterification of LA and n-butanol was sensitive to the acid strength of catalyst. Nafion-212 resin is a kind of perfluorosulfonic acid-based catalyst, which has a strong Hammett acidity similar to sulfuric acid. It is worth noting that, although Nafion-212 had stronger acidity (–H0 = 11–13 vs. 8–11) and higher –SO3H density (0.9 mmol g−1 vs. 0.66 mmol·g−1) than GC400, the TOF value obtained on Nafion-212 (59.0 h−1) was significantly lower than that obtained on GC400 (95.8 h−1). Therefore, the activity difference could not be simply explained by the acid strength or density. One possible origin of the high efficiency is the specific phenolic –OH and –COOH groups on GC400. As Nafion-212 and GC400 are both non-porous materials with swelling capacity, increasing the accessibility of acid sites in the bulk structure has a strong effect on catalyst activity. Even if phenolic –OH and –COOH groups on GC400 contribute little due to their insufficient acidity, they are supposed to increase interaction ability with hydrophilic reactants in solution and provide good access to the –SO3H sites. In this case, higher esterification reaction rate was observed on GC400 at the early stage of esterification (see Figure S9). Further detailed study will be necessary to determine how these functional groups contribute to the reaction.
2.6. Stability Test
The reusability of GC400 catalyst was investigated under the optimal reaction conditions. The catalyst was recovered, washed and dried before another reaction was conducted. As shown in Figure 7, GC400 was observed to retain a remarkably high proportion (92%) of its original catalytic activity after five cycles. A hot filtration test was conducted to prove the stability of –SO3H groups. By using the reaction mixture filtered at reaction time of 2 h, we run another batch of reaction. No change in LA conversion was noticed after reacting for another 2 h. The result indicated that GC400 catalyst was behaving in a truly heterogeneous manner and no catalytic activity could be attributed to the leached species in the solution. The slight decline in activity after three cycles was due to the catalyst mass being lost in the filtration and washing steps. As GC400 is composed of polycyclic aromatic carbon sheets, the inherent hydrophobicity of carbon can prevent the hydration of –OH groups. Meanwhile, the electron-withdrawing –COOH groups in GC400 are supposed to increase the electron density between carbon and sulfur atoms, resulting in the tight attachment of –SO3H groups to carbon framework. This might partially explain for the observation that the glucose-derived catalyst was especially robust.
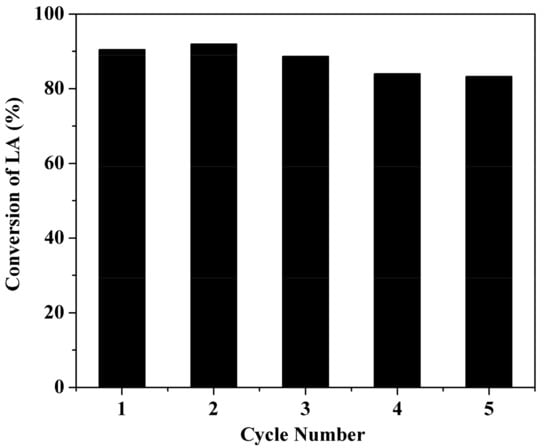
Figure 7.
Stability test of GC400. Reaction conditions: 1.16 g LA, 3.7 g n-butanol, 0.116 g catalyst, 373 K, 4 h.
2.7. Merits of the Present Method
According to previous literature, very few reports are available on the esterification of LA with n-butanol. Dharne et al. [21] has claimed that 97% conversion of LA could be obtained using H3PW12O40/K10 catalyst in the esterification of LA and n-butanol, but the activity suffered a drastic decrease after the third run due to the leaching of heteropolyacid from supports. Maheria et al. [22] has reported at a higher temperature of 393 K and a higher n-butanol ratio of 7:1, 82.2% conversion of LA could be obtained on H-BEA zeolite. It is also reported by Cirujano et al. [24] that LA was almost quantitatively converted to esters on Zr-containing metal–organic frameworks formed by either terephthalate or 2-aminoterephthalate ligands. In addition, Al-modified MCM-41 exhibited 90% yield of BL after a long reaction time of 8 h in the esterification of LA with n-butanol, and the calcination step was inevitable to keep its activity during each cycle [25]. As for immobilized lipase catalysts, despite them being active, solvent washing after each run led to difficulties in operation [29]. As we can see, comparing with those reported solid acids, sulfonated glucose-derived carbon possesses great advantages with respect to operation cost, catalytic activity and reusability.
3. Materials and Methods
3.1. Catalyst Preparation
Sulfonated glucose-derived carbon (denoted as GC400) was prepared according to Hara’s method [23] with minor modifications. All the chemicals were purchased from Sinopharm Chemcial Reagent Company (Beijing, China). 1 g glucose powder was heated for 12 h at 673 K under N2 flow to produce a brown-black solid. The solid was grounded to powder and heated in 10 cm3 of concentrated H2SO4 at 423 K for one day. The black precipitate was washed thoroughly with hot distilled water until no sulfate ions were detected in the filtrate. The prepared catalyst was dried at 373 K in a vacuum oven. The particle size of GC400 was around 5~20 μm (see Figure S10). Sulfonated active carbon (denoted as AC400) was prepared by sulfonating grinded active carbon (coconut shell carbon, NORIT Company, Boston, MA, USA) in concentrated H2SO4 at 423 K for one day. HZSM-5 (SiO2/Al2O3 = 25) and Hβ (SiO2/Al2O3 = 25) zeolites were provided by Nankai University. Nafion-212 resin was purchased from Dupont. Amberlyst-15 resin was purchased from Sigma-Aldrich (St. Louis, MO, USA).
3.2. Catalyst Characterization
The specific surface areas (SBET) of the catalysts were measured by N2 adsorption, which was carried out at 77 K using an ASAP 2010 apparatus (Micromeritics, Norcross, GA, USA). Before each measurement, the sample was evacuated at 573 K for 3 h. The structure of the catalyst was characterized by with a PANalytical X’Pert-Pro X-ray diffractometer (Bruker, Karlsruhe, Germany), using Cu Kα monochromatized radiation (λ = 0.1541 nm) at a scan speed of 5° min−1. Elemental composition of the prepared catalysts was determined by elemental analysis (EA) using an Elementar vario EL b apparatus (Langenselbold, Germany). HRTEM images were collected on an electron microscope (JEM-2100F, JEOL, Tokyo, Japan), using a 200 kV accelerating voltage. FT-IR spectra were recorded by a VERTEX 70 (Bruker, Karlsruhe, Germany) with the KBr pellet ranging from 500 to 4000 cm−1. The acid strength of carbon materials was characterized by NH3-TPD, which was carried out with a Micromeritics AutoChem II 2920 Automated Catalyst Characterization System (Micromeritics, Norcross, GA, USA) coupling with a mass spectrometry (MS) detector (Pfeiffer Vacuum, Nashua, NH, USA). The detailed procedure has been described in our previous works [44]. Before each test, 0.1 g sample was placed in a quartz reactor, pretreated in He flow at 393 K for 1 h and cooled down in Helium flow to 373 K. After the saturated adsorption NH3, the sample was heated at 373 K in He for 30 min to remove the physically adsorbed NH3. Desorption of NH3 was conducted in He flow from 373 K to 1073 K at a heating rate of 10 K min−1. The desorbed NH3 molecules were detected by a MS OminiStar (Pfeiffer Vacuum, Nashua, NH, USA) equipped with the software quadstar 32-bit (Pfeiffer Vacuum, Nashua, NH, USA).
3.3. Esterification of LA and N-Butanol
Esterification reaction was carried out in a Parr batch reactor (Parr, Moline, IL, USA). Typically, 1.16 g (0.01 mol) LA, 3.7 g (0.05 mol) n-butanol and 0.116 g catalyst were put into the reactor. The mixture was stirred at 373 K for 4 h with stirrer speed of 200 rpm. The liquid product was filtered and analyzed by an Agilent 1100 HPLC (Santa Clara, CA, USA) equipped with a ZORBAX SB-C18 (4.6 × 150 mm, 5 mm) column (Santa Clara, CA, USA) and refractive index detector (RID). The solution of methanol and water (7:3 v/v) was used as the eluent. The LA conversion was calculated by its consumption. The reusability of glucose-derived carbon was investigated under the optimal reaction conditions. After each cycle of use, the catalyst was filtered, thoroughly washed with acetone, and vacuum dried at 373 K before another batch reaction. All the chemicals were purchased from Sinopharm Chemcial Reagent Company (Beijing, China).
4. Conclusions
N-butyl levulinate fuel additive was successfully synthesized through the esterification of LA and n-butanol over a sulfonated carbon (GC400) bearing –SO3H, –COOH and phenolic –OH groups. Compared with the commercial active carbon, the amorphous carbon prepared from incomplete carbonization of glucose possessed smaller carbon sheets that were easier to be sulfonated, resulting in higher –SO3H density of GC400. The unique swelling capacity of GC400 could provide good access of reactants to the –SO3H sites located in the carbon matrix, despite the small surface area. Under the optimized conditions (373 K, 4 h, n-butanol to LA molar ratio of 5:1, catalyst loading of 10%), GC400 displayed a much higher catalytic efficiency than sulfonated active carbon, HZSM-5 and Nafion-212 for esterification. The strong acid strength and high –SO3H density, probably in cooperation with weak acid sites (–COOH and phenolic –OH) attached on the carbon surface, are the main factors for its excellence.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/8/1/14/s1, Figure S1: HRTEM images of GC400 (a) and AC400 (b), Figure S2: The behavior of GC400 (a) and AC400 (b) in contact with n-butanol, Figure S3: NH3-TPD results of GC400 and unsulfonated GC400, Figure S4: HPLC chromatograms of the liquid products from the esterification of LA and n-butanol over GC400 (a) and AC400 (b), Figure S5: Conversions of LA catalyzed by different liquid acids, Figure S6: Conversions of LA catalyzed by different concentrations of H2SO4 solution, Figure S7: Conversions of LA over GC400 and AC400 when identical amounts of –SO3H sites in reaction mixture were utilized, Figure S8: Effect of stirrer speed (a) and particle size (b) on esterification of LA over AC400. Figure S9: Conversions of LA over GC400 and Nafion-212 as the function of reaction time, Figure S10: SEM images of GC400 sample, Table S1: Acid amount of different functional groups on two samples.
Acknowledgments
This work was supported by the Natural Science Foundation Research Project of Shaanxi Province (No. 2017JQ2015), and the Scientific Research Foundation of Shaanxi University of Science and Technology (No. 2015BJ-27).
Author Contributions
Jinfan Yang conceived and designed the experiments; Guangyi Li and Lulu Zhang performed the experiments; Jinfan Yang analyzed the data; Sufeng Zhang contributed reagents and analysis tools; and Jinfan Yang wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12, 1493–1513. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Nikitin, E.B. High-yield conversion of plant biomass into the key value-added feedstocks 5-(hydroxymethyl)furfural, levulinic acid, and levulinic esters via 5-(chloromethyl)furfural. Green Chem. 2010, 12, 370–373. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of energy’s “top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Antonetti, C.; Licursi, D.; Fulignati, S.; Valentini, G.; Raspolli Galletti, A. New frontiers in the catalytic synthesis of levulinic acid: From sugars to raw and waste biomass as starting feedstock. Catalysts 2016, 6, 196. [Google Scholar] [CrossRef]
- Hayes, D.J. An examination of biorefining processes, catalysts and challenges. Catal. Lett. 2009, 145, 138–151. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Sonar, S.K.; Niphadkar, P.S.; Joshi, P.N.; Deshpande, S.S.; Patil, V.S.; Bokade, V.V. Catalytic upgrading of renewable levulinic acid to ethyl levulinate biodiesel using dodecatungstophosphoric acid supported on desilicated H-ZSM-5 as catalyst. Appl. Catal. A Gen. 2013, 460–461, 90–98. [Google Scholar] [CrossRef]
- Yan, K.; Wu, G.; Wen, J.; Chen, A. One-step synthesis of mesoporous H4SiW12O40-SiO2 catalysts for the production of methyl and ethyl levulinate biodiesel. Catal. Commun. 2013, 34, 58–63. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Niphadkar, P.S.; Deshpande, S.S.; Bokade, V.V. Esterification of renewable levulinic acid to ethyl levulinate biodiesel catalyzed by highly active and reusable desilicated H-ZSM-5. J. Chem. Technol. Biotechnol. 2014, 89, 1507–1515. [Google Scholar] [CrossRef]
- Patil, C.R.; Niphadkar, P.S.; Bokade, V.V.; Joshi, P.N. Esterification of levulinic acid to ethyl levulinate over bimodal micro–mesoporous H/BEA zeolite derivatives. Catal. Commun. 2014, 43, 188–191. [Google Scholar] [CrossRef]
- Fernandes, D.R.; Rocha, A.S.; Mai, E.F.; Mota, C.J.A.; Teixeira da Silva, V. Levulinic acid esterification with ethanol to ethyl levulinate production over solid acid catalysts. Appl. Catal. A Gen. 2012, 425–426, 199–204. [Google Scholar] [CrossRef]
- Osatiashtiani, A.; Durndell, L.J.; Manayil, J.C.; Lee, A.F.; Wilson, K. Influence of alkyl chain length on sulfated zirconia catalysed batch and continuous esterification of carboxylic acids by light alcohols. Green Chem. 2016, 18, 5529–5535. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Teixeira da Silva, V. Sulfonated carbon nanotubes as catalysts for the conversion of levulinic acid into ethyl levulinate. Catal. Today 2014, 234, 257–263. [Google Scholar] [CrossRef]
- Budarin, V.L.; Clark, J.H.; Luque, R.; Macquarrie, D.J. Versatile mesoporous carbonaceous materials for acid catalysis. Chem. Commun. 2007, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Ma, L.; Song, D.; Zhang, X.; Guo, Y. Design of a highly ordered mesoporous H3PW12O40/ZrO2-Si(Ph)Si hybrid catalyst for methyl levulinate synthesis. Green Chem. 2013, 15, 885–890. [Google Scholar] [CrossRef]
- Christensen, E.; Yanowitz, J.; Ratcliff, M.; McCormick, R.L. Renewable oxygenate blending effects on gasoline properties. Energy Fuels 2011, 25, 4723–4733. [Google Scholar] [CrossRef]
- Shrivastav, G.; Khan, T.S.; Agarwal, M.; Haider, M.A. Reformulation of gasoline to replace aromatics by biomass-derived alkyl levulinates. ACS Sustain. Chem. Eng. 2017, 5, 7118–7127. [Google Scholar] [CrossRef]
- Earl Christensen, A.W.; Paul, S.; Burton, S.; McCormick, A.R.L. Properties and performance of levulinate esters as diesel blend components. Energy Fuels 2011, 25, 5422–5428. [Google Scholar] [CrossRef]
- Dharne, S.; Bokade, V.V. Esterification of levulinic acid to n-butyl levulinate over heteropolyacid supported on acid-treated clay. J. Nat. Gas Chem. 2011, 20, 18–24. [Google Scholar] [CrossRef]
- Maheria, K.C.; Kozinski, J.; Dalai, A. Esterification of levulinic acid to n-butyl levulinate over various acidic zeolites. Catal. Lett. 2013, 143, 1220–1225. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Bokade, V.V. Esterification of renewable levulinic acid to n-butyl levulinate over modified h-zsm-5. Chem. Eng. Technol. 2015, 38, 246–252. [Google Scholar] [CrossRef]
- Cirujano, F.G.; Corma, A.; Llabrés i Xamena, F.X. Conversion of levulinic acid into chemicals: Synthesis of biomass derived levulinate esters over Zr-containing MOFs. Chem. Eng. Sci. 2015, 124, 52–60. [Google Scholar] [CrossRef]
- Najafi Chermahini, A.; Nazeri, M. Esterification of the levulinic acid with n-butyl and isobutyl alcohols over aluminum-containing MCM-41. Fuel Process. Technol. 2017, 167, 442–450. [Google Scholar] [CrossRef]
- Tejero, M.A.; Ramírez, E.; Fité, C.; Tejero, J.; Cunill, F. Esterification of levulinic acid with butanol over ion exchange resins. Appl. Catal. A Gen. 2016, 517, 56–66. [Google Scholar] [CrossRef]
- Trombettoni, V.; Bianchi, L.; Zupanic, A.; Porciello, A.; Cuomo, M.; Piermatti, O.; Marrocchi, A.; Vaccaro, L. Efficient catalytic upgrading of levulinic acid into alkyl levulinates by resin-supported acids and flow reactors. Catalysts 2017, 7, 235. [Google Scholar] [CrossRef]
- Nakhate, A.V.; Yadav, G.D. Synthesis and characterization of sulfonated carbon-based graphene oxide monolith by solvothermal carbonization for esterification and unsymmetrical ether formation. ACS Sustain. Chem. Eng. 2016, 4, 1963–1973. [Google Scholar] [CrossRef]
- Badgujar, K.C.; Bhanage, B.M. Thermo-chemical energy assessment for production of energy-rich fuel additive compounds by using levulinic acid and immobilized lipase. Fuel Process. Technol. 2015, 138, 139–146. [Google Scholar] [CrossRef]
- Toda, M.; Takagaki, A.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Green chemistry: Biodiesel made with sugar catalyst. Nature 2005, 438, 178. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Takagaki, A.; Toda, M.; Kondo, J.N.; Domen, K.; Tatsumi, T.; Hara, M.; Hayashi, S. Acid-catalyzed reactions on flexible polycyclic aromatic carbon in amorphous carbon. Chem. Mater. 2006, 18, 3039–3045. [Google Scholar] [CrossRef]
- Lou, W.; Zong, M.; Duan, Z. Efficient production of biodiesel from high free fatty acid-containing waste oils using various carbohydrate-derived solid acid catalysts. Bioresour. Technol. 2008, 99, 8752–8758. [Google Scholar] [CrossRef] [PubMed]
- Lokman, I.M.; Rashid, U.; Taufiqyap, Y.H.; Yunus, R. Methyl ester production from palm fatty acid distillate using sulfonated glucose-derived acid catalyst. Renew. Energy 2015, 81, 347–354. [Google Scholar] [CrossRef]
- Cao, X.; Sun, S.; Sun, R. Application of biochar-based catalysts in biomass upgrading: A review. RSC Adv. 2017, 7, 48793–48805. [Google Scholar] [CrossRef]
- Dandekar, A.; Baker, R.T.K.; Vannice, M.A. Characterization of activated carbon, graphitized carbon fibers and synthetic diamond powder using TPD and drifts. Carbon 1998, 36, 1821–1831. [Google Scholar] [CrossRef]
- Chen, G.; Fang, B. Preparation of solid acid catalyst from glucose–starch mixture for biodiesel production. Bioresour. Technol. 2011, 102, 2635–2640. [Google Scholar] [CrossRef] [PubMed]
- Dawodu, F.A.; Ayodele, O.; Xin, J.; Zhang, S.; Yan, D. Effective conversion of non-edible oil with high free fatty acid into biodiesel by sulphonated carbon catalyst. Appl. Energy 2014, 114, 819–826. [Google Scholar] [CrossRef]
- Shu, Q.; Gao, J.; Nawaz, Z.; Liao, Y.; Wang, D.; Wang, J. Synthesis of biodiesel from waste vegetable oil with large amounts of free fatty acids using a carbon-based solid acid catalyst. Appl. Energy 2010, 87, 2589–2596. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, S.; Li, J. Activity of the carbon-based heterogeneous acid catalyst derived from bamboo in esterification of oleic acid with ethanol. Energy Convers. Manag. 2016, 114, 188–196. [Google Scholar] [CrossRef]
- Okuhara, T. Water-tolerant solid acid catalysts. Chem. Rev. 2002, 102, 3641–3666. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, M.; Bringué, R.; Fité, C.; Ramírez, E.; Cunill, F. Liquid-Phase Oligomerization of 1-Hexene Catalyzed by Macroporous Ion-Exchange Resins. Top. Catal. 2011, 54, 998–1008. [Google Scholar] [CrossRef]
- Suganuma, S.; Nakajima, K.; Kitano, M.; Yamaguchi, D.; Kato, H.; Hayashi, S. Hydrolysis of cellulose by amorphous carbon bearing SO3H, COOH, and OH groups. J. Am. Chem. Soc. 2008, 130, 12787–12793. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, A.; Toda, M.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Esterification of higher fatty acids by a novel strong solid acid. Catal. Today 2006, 116, 157–161. [Google Scholar] [CrossRef]
- Yang, J.; Li, N.; Li, S.; Wang, W.; Li, L.; Wang, A.; Wang, X.; Cong, Y.; Zhang, T. Synthesis of diesel and jet fuel range alkanes with furfural and ketones from lignocellulose under solvent free conditions. Green Chem. 2014, 16, 4879–4884. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).