
The Fabrication of Cu2O-u/g-C3N4 Heterojunction and Its Application in CO2 Photoreduction

Abstract

1. Introduction
2. Results and Discussion
2.1. Characterization results of Cu2O-u/g-C3N4
2.2. Effects of Cu2O-u/g-C3N4 Heterojunction Composition on Catalytic Activity in CPR
2.3. Effects of Reaction Conditions on Product Selectivity and Reaction Rate in CPR
2.3.1. Effects of the Hole Scavenger
2.3.2. Effects of Reaction Medium
2.3.3. Effects of Reaction Time
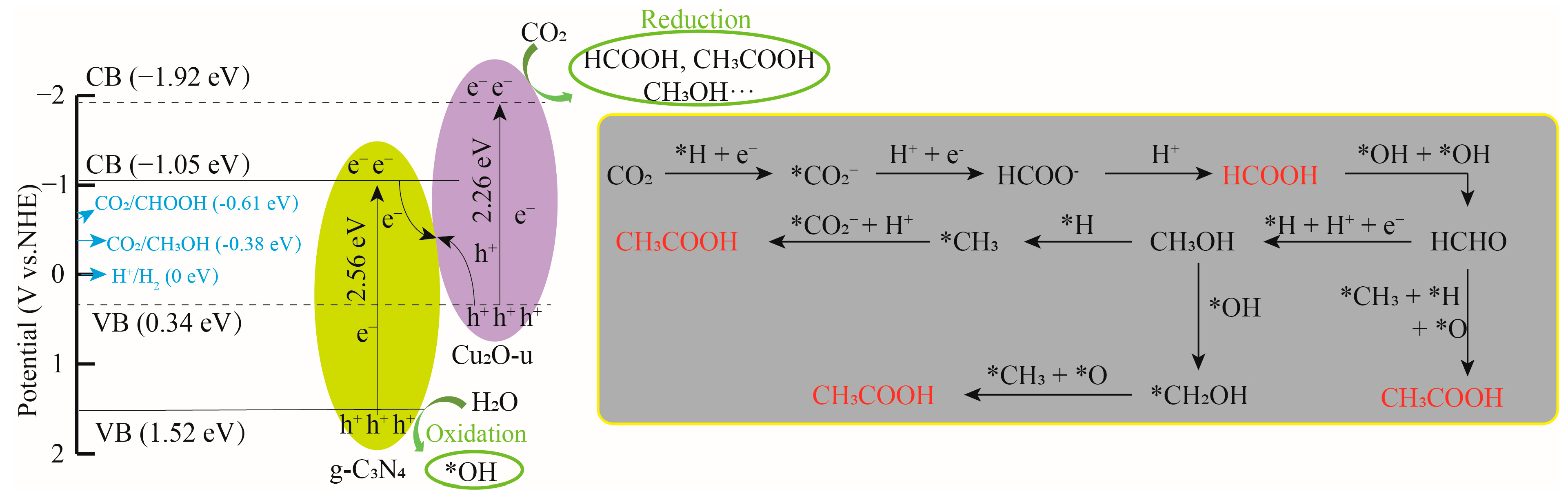
2.3.4. CPR Product Pathways of Cu2O-u/g-C3N4
2.4. Synthesis of Formic Acid and Acetic Acid by Cu2O-u/g-C3N4
2.4.1. Effects of Isopropanol and Cu2O-u/g-C3N4 Concentration
2.4.2. Effects of the Reaction System
2.5. Synthesis of Methanol by Cu2O-u/g-C3N4
2.5.1. Effects of TEOA Concentration
2.5.2. Effects of Reaction Time
2.5.3. Repeatability of Cu2O-u/g-C3N4 for Methanol Synthesis via CPR
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Cu2O-u/g-C3N4
3.3. Characterization of Cu2O-u/g-C3N4
3.4. Testing of CO2 Photoreduction
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cao, Y.; Guo, R.; Ma, M.; Huang, Z.; Zhou, Y. Effects of Electron Density Variation of Active Sites in CO2 Activation and Photoreduction: A Review. Xian Polytech Univ, Coll Environm & Chem Engn, Xian 710000, Peoples R China. Xinjiang Norm. Univ. Coll Chem. Chem. Engn. 2024, 40, 2303029. [Google Scholar] [CrossRef]
- Tedeeva, M.A.; Kustov, A.L.; Batkin, A.M.; Garifullina, C.; Zalyatdinov, A.A.; Yang, D.; Dai, Y.; Yang, Y.; Kustov, L.M. Catalytic systems for hydrogenation of CO2 to methanol. Mol. Catal. 2024, 566, 114403. [Google Scholar] [CrossRef]
- Reguero, M.; Claver, C.; Carrilho, R.M.B.; Masdeu-Bultó, A.M. Immobilized Molecular Catalysts for CO2 Photoreduction. Adv. Sustain. Syst. 2022, 6, 2100493. [Google Scholar] [CrossRef]
- Maximov, A.L.; Beletskaya, I.P. Carbon dioxide and “methanol” economy: Advances in the catalytic synthesis of methanol from CO2. Russ. Chem. Rev. 2024, 93, RCR5101. [Google Scholar] [CrossRef]
- Zulqarnain; Yusoff, M.H.M.; Keong, L.K.; Yasin, N.H.; Rafeen, M.S.; Hassan, A.; Srinivasan, G.; Yusup, S.; Shariff, A.M.; Jaafar, A.B. Recent development of integrating CO2 hydrogenation into methanol with ocean thermal energy conversion (OTEC) as potential source of green energy. Green Chem. Lett. Rev. 2023, 1, 16. [Google Scholar] [CrossRef]
- Rossini, F.D.; Jessup, R.S. Heat and free energy of formation of carbon dioxide. and of the transition between graphite and diamond. J. Res. Natl. Bur. Stand. 1938, 21, 491. [Google Scholar] [CrossRef]
- Sudhakaran, A.; Singh, C.; Aaradhya, H.M.; Biradar, A.; Samal, A.K.; Chaudhari, N.K.; Jadhav, A.H. Advancements and Perspective of Environmentally Sustainable Technologies for Electrochemical Selective Conversion of CO2 to Methanol. Catal. Rev. 2024, 1–113. [Google Scholar] [CrossRef]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabéhère-Mallart, E.; Robert, M. Molecular catalysis of CO2 reduction: Recent advances and perspectives in electrochemical and light-driven processes with selected Fe. Ni and Co aza macrocyclic and polypyridine complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef]
- Adekoya, D.; Tahir, M.; Amin, N.A.S. Recent trends in photocatalytic materials for reduction of carbon dioxide to methanol. Renew. Sustain. Energy Rev. 2019, 116, 109389. [Google Scholar] [CrossRef]
- Fu, C.; Wan, Z.; Yang, X.; Zhang, J.; Zhang, Z. Artificial CO2 photoreduction: A review of photocatalyst design and product selectivity regulation. J. Mater. Chem. A 2024, 12, 28618–28657. [Google Scholar] [CrossRef]
- Cheng, S.-P.; Wei, L.-W.; Wang, H.-P. Photocatalytic Reduction of CO2 to Methanol by Cu2O/TiO2 Heterojunctions. Sustainability 2022, 14, 374. [Google Scholar] [CrossRef]
- Movahed, S.K.; Najinasab, A.; Nikbakht, R.; Dabiri, M. Visible light assisted photocatalytic reduction of CO2 to methanol using Fe3O4@N-C/Cu2O nanostructure photocatalyst. J. Photochem. Photobiol. A Chem. 2020, 401, 112763. [Google Scholar] [CrossRef]
- Li, J.; Chen, G.; Zhu, Y.; Liang, Z.; Pei, A.; Wu, C.-L.; Wang, H.; Lee, H.R.; Liu, K.; Chu, S.; et al. Efficient electrocatalytic CO2 reduction on a three-phase interface. Nat. Catal. 2018, 1, 592–600. [Google Scholar] [CrossRef]
- Jovanov, Z.P.; Hansen, H.A.; Varela, A.S.; Malacrida, P.; Peterson, A.A.; Nørskov, J.K.; Stephens, I.E.L.; Chorkendorff, I. Opportunities and challenges in the electrocatalysis of CO2 and CO reduction using bifunctional surfaces: A theoretical and experimental study of Au-Cd alloys. J. Catal. 2016, 343, 215–231. [Google Scholar] [CrossRef]
- Sahara, G.; Kumagai, H.; Maeda, K.; Kaeffer, N.; Artero, V.; Higashi, M.; Abe, R.; Ishitani, O. Photoelectrochemical Reduction of CO2 Coupled to Water Oxidation Using a Photocathode with a Ru(II)–Re(I) Complex Photocatalyst and a CoOx/TaON Photoanode. J. Am. Chem. Soc. 2016, 138, 14152–14158. [Google Scholar] [CrossRef]
- Kuk, S.K.; Singh, R.K.; Nam, D.H.; Singh, R.; Lee, J.-K.; Park, C.B. Photoelectrochemical Reduction of Carbon Dioxide to Methanol through a Highly Efficient Enzyme Cascade. Angew. Chem. Int. Ed. 2017, 56, 3827–3832. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, Y.; Zong, Y.; Li, J.; Yang, N.; Zhang, M.; Guo, Z.; Song, H. Construction of Functionally Compartmental Inorganic Photocatalyst–Enzyme System via Imitating Chloroplast for Efficient Photoreduction of CO2 to Formic Acid. ACS Appl. Mater. Interfaces 2020, 12, 34795–34805. [Google Scholar] [CrossRef]
- Chakrabortty, S.; Nayak, J.; Ruj, B.; Pal, P.; Kumar, R.; Banerjee, S.; Sardar, M.; Chakraborty, P. Photocatalytic conversion of CO2 to methanol using membrane-integrated Green approach: A review on capture, conversion and purification. J. Environ. Chem. Eng. 2020, 8, 103935. [Google Scholar] [CrossRef]
- Uddin, M.R.; Khan, M.R.; Rahman, M.W.; Yousuf, A.; Cheng, C.K. Photocatalytic reduction of CO2 into methanol over CuFe2O4/TiO2 under visible light irradiation. Reaction Kinetics. Mech. Catal. 2015, 116, 589–604. [Google Scholar] [CrossRef]
- Mori, K.; Matsuo, J.; Kondo, Y.; Hata, H.; Yamashita, H. Photoreduction of Carbon Dioxide to Formic Acid with Fe-Based MOFs: The Promotional Effects of Heteroatom Doping and Alloy Nanoparticle Confinement. ACS Appl. Energy Mater. 2021, 4, 11634–11642. [Google Scholar] [CrossRef]
- Prasetya, N.; Ladewig, B.P. New Azo-DMOF-1 MOF as a Photoresponsive Low-Energy CO2 Adsorbent and Its Exceptional CO2/N2 Separation Performance in Mixed Matrix Membranes. ACS Appl. Mater. Interfaces 2018, 10, 34291–34301. [Google Scholar] [CrossRef]
- Ahmed, S.; Khan, M.K.; Kim, J. Revolutionary advancements in carbon dioxide valorization via metal-organic framework-based strategies. Carbon Capture Sci. Technol. 2025, 15, 100405. [Google Scholar] [CrossRef]
- Rao, H.; Schmidt, L.C.; Bonin, J.; Robert, M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 2017, 548, 74–77. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons; Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Yisilamu, G.; Maimaiti, H.; Awati, A.; Zhang, D.; Sun, F.; Xu, B. Preparation of Cuprous Oxide Nanoparticles Coated with Aminated Cellulose for the Photocatalytic Reduction of Carbon Dioxide to Methanol. Energy Technol. 2018, 6, 1168–1177. [Google Scholar] [CrossRef]
- Maciá-Agulló, J.A.; Corma, A.; Garcia, H. Photobiocatalysis: The Power of Combining Photocatalysis and Enzymes. Chem.—A Eur. J. 2015, 21, 10940–10959. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, S.; Kang, H.; He, R.; Ning, Y.; Yu, Y.; Wang, M.; Chen, B. Construction of multi-enzyme cascade biomimetic carbon sequestration system based on photocatalytic coenzyme NADH regeneration. Renew. Energy 2020, 156, 107–116. [Google Scholar] [CrossRef]
- Amao, Y.; Takahara, S.; Sakai, Y. Visible-light induced hydrogen and formic acid production from biomass and carbon dioxide with enzymatic and artificial photosynthesis system. Int. J. Hydrog. Energy 2014, 39, 20771–20776. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Li, P.; Liu, L.; An, W.; Wang, H.; Cui, W. Efficient photothermal catalytic CO2 reduction to CH3CH2OH over Cu2O/g-C3N4 assisted by ionic liquids. Appl. Surf. Sci. 2021, 565, 150448. [Google Scholar] [CrossRef]
- Adekoya, D.O.; Tahir, M.; Amin, N.A.S. g-C3N4/(Cu/TiO2) nanocomposite for enhanced photoreduction of CO2 to CH3OH and HCOOH under UV/visible light. J. CO2 Util. 2017, 18, 261–274. [Google Scholar] [CrossRef]
- Cao, S.; Li, Y.; Zhu, B.; Jaroniec, M.; Yu, J. Facet effect of Pd cocatalyst on photocatalytic CO2 reduction over g-C3N4. J. Catal. 2017, 349, 208–217. [Google Scholar] [CrossRef]
- Bika, P.; Papailias, I.; Giannakopoulou, T.; Tampaxis, C.; Steriotis, T.A.; Trapalis, C.; Dallas, P. Prominent COFg-C3N4, and Their Heterojunction Materials for Selective Photocatalytic CO2 Reduction. Catalysts 2023, 13, 1331. [Google Scholar] [CrossRef]
- Chen, P.; Dong, X.A.; Huang, M.; Li, K.; Xiao, L.; Sheng, J.; Chen, S.; Zhou, Y.; Dong, F. Rapid Self-Decomposition of g-C3N4 During Gas–Solid Photocatalytic CO2 Reduction and Its Effects on Performance Assessment. ACS Catal. 2022, 12, 4560–4570. [Google Scholar] [CrossRef]
- Patnaik, S.; Sahoo, D.P.; Parida, K. Recent advances in anion doped g-C3N4 photocatalysts: A review. Carbon 2021, 172, 682–711. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, H.; Wu, Z.; Wang, L. g-C3N4 based composite photocatalysts for photocatalytic CO2 reduction. Catal. Today 2018, 300, 160–172. [Google Scholar] [CrossRef]
- Wang, K.; Li, Q.; Liu, B.; Cheng, B.; Ho, W.; Yu, J. Sulfur-doped g-C3N4 with enhanced photocatalytic CO2-reduction performance. Appl. Catal. B Environ. 2015, 1761–1777, 44–52. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, H.; Wei, J.; Zhang, H.-X.; Wu, X.; Li, Y.; Li, C.; Zhang, J.; Ye, J. Integrating the g-C3N4 Nanosheet with B–H Bonding Decorated Metal–Organic Framework for CO2 Activation and Photoreduction. ACS Nano 2018, 12, 5333–5340. [Google Scholar] [CrossRef]
- Chen, J.; Shen, S.; Guo, P.; Wang, M.; Wu, P.; Wang, X.; Guo, L. In-situ reduction synthesis of nano-sized Cu2O particles modifying g-C3N4 for enhanced photocatalytic hydrogen production. Appl. Catal. B Environ. 2014, 152–153, 335–341. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, T.; Zeng, H. Design of Cu-Cu2O/g-C3N4 nanocomponent photocatalysts for hydrogen evolution under visible light irradiation using water-soluble Erythrosin B dye sensitization. Appl. Surf. Sci. 2017, 391, 404–414. [Google Scholar] [CrossRef]
- Wen, J.; Xie, J.; Chen, X.; Li, X. A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 2017, 391, 72–123. [Google Scholar] [CrossRef]
- Peng, B.; Zhang, S.; Yang, S.; Wang, H.; Yu, H.; Zhang, S.; Peng, F. Synthesis and characterization of g-C3N4/Cu2O composite catalyst with enhanced photocatalytic activity under visible light irradiation. Mater. Res. Bull. 2014, 56, 19–24. [Google Scholar] [CrossRef]
- Li, D.; Zuo, S.; Xu, H.; Zan, J.; Sun, L.; Han, D.; Liao, W.; Zhang, B.; Xia, D. Synthesis of a g-C3N4-Cu2O heterojunction with enhanced visible light photocatalytic activity by PEG. J. Colloid Interface Sci. 2018, 531, 28–36. [Google Scholar] [CrossRef]
- Zuo, S.; Xu, H.; Liao, W.; Sun, L.; Han, D.; Zan, J.; Zhang, B.; Li, D.; Xia, D. Acid-treated g-C3N4-Cu2O composite catalyst with enhanced photocatalytic activity under visible-light irradiation. Appl. Organomet. Chem. 2018, 32, e4448. [Google Scholar] [CrossRef]
- Paul, A.M.; Sajeev, A.; Nivetha, R.; Gothandapani, K.; Bhardwaj, P.; Raghavan, V.; Jacob, G.; Sellapan, R.; Jeong, S.K.; Grace, A.N. Cuprous oxide (Cu2O)/graphitic carbon nitride (g-C3N4) nanocomposites for electrocatalytic hydrogen evolution reaction. Diam. Relat. Mater. 2020, 107, 107899. [Google Scholar] [CrossRef]
- Thompson, W.A.; Fernandez, E.S.; Maroto-Valer, M.M. Review and Analysis of CO2 Photoreduction Kinetics. ACS Sustain. Chem. Eng. 2020, 8, 4677–4692. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.; Chen, Z.; Li, M.; Wang, L.; Wu, S.; Zhang, J. Photocatalytic conversion of carbon dioxide on triethanolamine: Unheeded catalytic performance of sacrificial agent. Appl. Catal. B Environ. 2023, 326, 122338. [Google Scholar] [CrossRef]
- Fang, S.; Rahaman, M.; Bharti, J.; Reisner, E.; Robert, M.; Ozin, G.A.; Hu, Y.H. Photocatalytic CO2 reduction. Nat. Rev. Methods Primers 2023, 3, 61. [Google Scholar] [CrossRef]
- Wang, S.; Lin, J.; Wang, X. Semiconductor–redox catalysis promoted by metal–organic frameworks for CO2 reduction. Phys. Chem. Chem. Phys. 2014, 16, 14656–14660. [Google Scholar] [CrossRef]
- Sato, S.; Morikawa, T.; Saeki, S.; Kajino, T.; Motohiro, T. Visible-Light-Induced Selective CO2 Reduction Utilizing a Ruthenium Complex Electrocatalyst Linked to a p-Type Nitrogen-Doped Ta2O5 Semiconductor. Angew. Chem. Int. Ed. 2010, 49, 5101–5105. [Google Scholar] [CrossRef]
- Bahadori, E.; Tripodi, A.; Villa, A.; Pirola, C.; Prati, L.; Ramis, G.; Rossetti, I. High Pressure Photoreduction of CO2: Effect of Catalyst Formulation. Hole Scavenger Addition and Operating Conditions. Catalysts 2018, 8, 430. [Google Scholar] [CrossRef]
- Das, R.; Chakraborty, S.; Peter, S.C. Systematic Assessment of Solvent Selection in Photocatalytic CO2 Reduction. ACS Energy Lett. 2021, 6, 3270–3274. [Google Scholar] [CrossRef]
- Gross, P.; Höppe, H.A. Biuret—A Crucial Reaction Intermediate for Understanding Urea Pyrolysis To Form Carbon Nitrides: Crystal-Structure Elucidation and In Situ Diffractometric. Vibrational and Thermal Characterisation. Chem.—A Eur. J. 2020, 26, 14366–14376. [Google Scholar] [CrossRef]
- Yang, W.; Jia, L.; Wu, P.; Zhai, H.; He, J.; Liu, C.; Jiang, W. Effect of thermal program on structure–activity relationship of g-C3N4 prepared by urea pyrolysis and its application for controllable production of g-C3N4. J. Solid State Chem. 2021, 304, 122545. [Google Scholar] [CrossRef]
- Kroke, E.; Schwarz, M.; Horath-Bordon, E.; Kroll, P.; Noll, B.; Norman, A.D. Tri-s-triazine derivatives. Part I. From trichloro-tri-s-triazine to graphitic C3N4 structures. New J. Chem. 2002, 26, 508–512. [Google Scholar] [CrossRef]
- Shi, W.; Guo, X.; Cui, C.; Jiang, K.; Li, Z.; Qu, L.; Wang, J.-C. Controllable synthesis of Cu2O decorated WO3 nanosheets with dominant (0 0 1) facets for photocatalytic CO2 reduction under visible-light irradiation. Appl. Catal. B Environ. 2019, 243, 236–242. [Google Scholar] [CrossRef]
- Wang, D.; Dong, N.; Niu, Y.; Hui, S. A Review of Urea Pyrolysis to Produce NH3 Used for NOx Removal. J. Chem. 2019, 2019, 6853638. [Google Scholar] [CrossRef]
- Rangel, L.S.; de la Rosa, J.R.; Ortiz, C.L.; Castaldi, M.J. Pyrolysis of urea and guanidinium salts to be used as ammonia precursors for selective catalytic reduction of NOx. J. Anal. Appl. Pyrolysis 2015, 113, 564–574. [Google Scholar] [CrossRef]
- Li, P.; Liu, L.; An, W.; Wang, H.; Guo, H.; Liang, Y.; Cui, W. Ultrathin porous g-C3N4 nanosheets modified with AuCu alloy nanoparticles and C-C coupling photothermal catalytic reduction of CO2 to ethanol. Appl. Catal. B Environ. 2020, 266, 118618. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M. FTIR study of methanol decomposition on gold catalyst for fuel cells. J. Power Sources 2003, 118, 304–310. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, X.; Wang, H.; Zhang, J.; Pan, B.; Xie, Y. Enhanced Photoresponsive Ultrathin Graphitic-Phase C3N4 Nanosheets for Bioimaging. J. Am. Chem. Soc. 2013, 135, 18–21. [Google Scholar] [CrossRef]
- Tammer, M.G. Sokrates: Infrared and Raman characteristic group frequencies: Tables and charts. Colloid Polym. Sci. 2004, 283, 235. [Google Scholar] [CrossRef]
- Guo, D.; Wang, L.; Du, Y.; Ma, Z.; Shen, L. Preparation of octahedral Cu2O nanoparticles by a green route. Mater. Lett. 2015, 160, 541–543. [Google Scholar] [CrossRef]
- Raziq, F.; Sun, L.; Wang, Y.; Zhang, X.; Humayun, M.; Ali, S.; Bai, L.; Qu, Y.; Yu, H.; Jing, L. Synthesis of Large Surface-Area g-C3N4 Comodified with MnOx and Au-TiO2 as Efficient Visible-Light Photocatalysts for Fuel Production. Adv. Energy Mater. 2018, 8, 1701580. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, K.; Qiu, X.; Yu, J.; Liu, M. Product selectivity of photocatalytic CO2 reduction reactions. Mater. Today 2020, 32, 222–243. [Google Scholar] [CrossRef]
- Di, J.; Hao, G.; Liu, G.; Zhou, J.; Jiang, W.; Liu, Z. Defective materials for CO2 photoreduction: From C1 to C2+ products. Coord. Chem. Rev. 2023, 482, 215057. [Google Scholar] [CrossRef]
- Bosch, H.; Versteeg, G.F.; Van Swaaij, W.P.M. Gas—Liquid mass transfer with parallel reversible reactions—I. Absorption of CO2 into solutions of sterically hindered amines. Chem. Eng. Sci. 1989, 44, 2723–2734. [Google Scholar] [CrossRef]
- Alhebshi, A.; Aldeen, E.S.; Mim, R.S.; Tahir, B.; Tahir, M. Recent advances in constructing heterojunctions of binary semiconductor photocatalysts for visible light responsive CO2 reduction to energy efficient fuels: A review. Int. J. Energy Res. 2022, 46, 5523–5584. [Google Scholar] [CrossRef]
- Kianička, J.; Čík, G.; Šeršeň, F.; Špánik, I.; Sokolík, R.; Filo, J. Photo-Reduction of CO2 by VIS Light on Polythiophene-ZSM-5 Zeolite Hybrid Photo-Catalyst. Molecules 2019, 24, 992. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, C.; Guan, A.; Kan, M.; Zheng, G.; CO, P. Photocatalytic CO2 conversion: From C1 products to multi-carbon oxygenates. Nanoscale 2022, 14, 10268–10285. [Google Scholar] [CrossRef]
- Gong, S.; Niu, Y.; Liu, X.; Xu, C.; Chen, C.; Meyer, T.J.; Chen, Z. Selective CO2 Photoreduction to Acetate at Asymmetric Ternary Bridging Sites. ACS Nano 2023, 17, 4922–4932. [Google Scholar] [CrossRef]
- Kovačič, Ž.; Likozar, B.; Huš, M. Photocatalytic CO2 Reduction: A Review of Ab Initio Mechanism. Kinetics, and Multiscale Modeling Simulations. ACS Catal. 2020, 10, 14984–15007. [Google Scholar] [CrossRef]
- Gamayurova, V.S.; Zinov’eva, M.E.; Shnaider, K.L.; Davletshina, G.A. Lipases in Esterification Reactions: A Review. Catal. Ind. 2021, 13, 58–72. [Google Scholar] [CrossRef]
- Mendieta-Reyes, N.E.; Cheuquepán, W.; Rodes, A.; Gómez, R. Spectroelectrochemical Study of CO2 Reduction on TiO2 Electrodes in Acetonitrile. ACS Catal. 2020, 10, 103–113. [Google Scholar] [CrossRef]
- Xie, B.; Wong, R.J.; Tan, T.H.; Higham, M.; Gibson, E.K.; Decarolis, D.; Callison, J.; Aguey-Zinsou, K.-F.; Bowker, M.; Catlow, C.R.A.; et al. Synergistic ultraviolet and visible light photo-activation enables intensified low-temperature methanol synthesis over copper/zinc oxide/alumina. Nat. Commun. 2020, 11, 1615. [Google Scholar] [CrossRef]
- Ostad, M.I.; Shahrak, M.N.; Galli, F. Photocatalytic carbon dioxide reduction to methanol catalyzed by ZnO. Pt, Au, and Cu nanoparticles decorated zeolitic imidazolate framework-8. J. CO2 Util. 2021, 43, 101373. [Google Scholar] [CrossRef]
- Lashgari, M.; Soodi, S. CO2 conversion into methanol under ambient conditions using efficient nanocomposite photocatalyst/solar-energy materials in aqueous medium. RSC Adv. 2020, 10, 15072–15078. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, R.; Hu, W.; Lin, L.; Liu, J.; Wang, Q.; Wang, D.; Wu, Z.; Zhang, J. High-performance photothermal conversion of sludge derived biochar and its potential for peroxydisulfate-based advanced oxidation processes. Sep. Purif. Technol. 2022, 303, 122214. [Google Scholar] [CrossRef]
- Yuan, J.; Gu, C.; Ding, W.; Hao, C. Correction to Photo-electrochemical Reduction of Carbon Dioxide into Methanol at CuFeO2 Nanoparticle-Decorated CuInS2 Thin-Film Photocathodes. Energy Fuels 2021, 35, 10944. [Google Scholar] [CrossRef]
- Bafaqeer, A.; Tahir, M.; Amin, N.A.S. Well-designed ZnV2O6/g-C3N4 2D/2D nanosheets heterojunction with faster charges separation via pCN as mediator towards enhanced photocatalytic reduction of CO2 to fuels. Appl. Catal. B Environ. 2019, 242, 312–326. [Google Scholar] [CrossRef]
- Shao, X.; Yin, X.; Wang, J. Nanoheterostructures of potassium tantalate and nickel oxide for photocatalytic reduction of carbon dioxide to methanol in isopropanol. J. Colloid Interface Sci. 2018, 512, 466–473. [Google Scholar] [CrossRef]
- Li, J.; Luo, D.; Yang, C.; He, S.; Chen, S.; Lin, J.; Zhu, L.; Li, X. Copper(II) imidazolate frameworks as highly efficient photocatalysts for reduction of CO2 into methanol under visible light irradiation. J. Solid State Chem. 2013, 203, 154–159. [Google Scholar] [CrossRef]
- Zhang, S.; Yin, X.; Zheng, Y. Enhanced photocatalytic reduction of CO2 to methanol by ZnO nanoparticles deposited on ZnSe nanosheet. Chem. Phys. Lett. 2018, 693, 170–175. [Google Scholar] [CrossRef]
- Sewify, G.H.; El-Hout, S.I. Ag2O-supported FePO4 heterojunctions: Facile fabrication and fast visible-light carbon dioxide photoreduction into methanol with superb recyclability. Mater. Sci. Semicond. Process. 2025, 187, 109160. [Google Scholar] [CrossRef]
- Li, M.; Liu, D.; Chen, X.; Yin, Z.; Shen, H.; Aiello, A.; McKenzie, K.R., Jr.; Jiang, N.; Li, X.; Wagner, M.J.; et al. Radical-Driven Decomposition of Graphitic Carbon Nitride Nanosheets: Light Exposure Matters. Environ. Sci. Technol. 2021, 55, 12414–12423. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalysts | Yield of Methanol (μmol·h−1·gcat−1) | References |
|---|---|---|
| Cu-ZnO-Al2O3 | 7512 | [75] |
| Pt/ZIF-8, ZnO/ZIF-8, Cu/ZIF-8, Au/ZIF-8 | 6843.0 ± 342.1 | [76] |
| CNT/NiO/Fe2O3 | 4380 | [77] |
| Pd/ZnO | 4000 | [78] |
| Cu2O-u/g-C3N4 | 3061.64 | This work |
| CuFeO2/CuInS2 | 2618 | [79] |
| ZnV2O6/protonated g-C3N4 | 1871 | [80] |
| KTaO3-NiO | 1815 | [81] |
| Cu (II) ZIF | 1712.7 | [82] |
| ZnO/ZnSe | 1581.2 | [83] |
| Ag2O- FePO4 | 179.1 | [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Zhang, Y.; Xiao, F.; Liu, Z.; Li, Y.; Shi, G.; Zhang, H. The Fabrication of Cu2O-u/g-C3N4 Heterojunction and Its Application in CO2 Photoreduction. Catalysts 2025, 15, 715. https://doi.org/10.3390/catal15080715
Lu J, Zhang Y, Xiao F, Liu Z, Li Y, Shi G, Zhang H. The Fabrication of Cu2O-u/g-C3N4 Heterojunction and Its Application in CO2 Photoreduction. Catalysts. 2025; 15(8):715. https://doi.org/10.3390/catal15080715
Chicago/Turabian StyleLu, Jiawei, Yupeng Zhang, Fengxu Xiao, Zhikai Liu, Youran Li, Guiyang Shi, and Hao Zhang. 2025. "The Fabrication of Cu2O-u/g-C3N4 Heterojunction and Its Application in CO2 Photoreduction" Catalysts 15, no. 8: 715. https://doi.org/10.3390/catal15080715
APA StyleLu, J., Zhang, Y., Xiao, F., Liu, Z., Li, Y., Shi, G., & Zhang, H. (2025). The Fabrication of Cu2O-u/g-C3N4 Heterojunction and Its Application in CO2 Photoreduction. Catalysts, 15(8), 715. https://doi.org/10.3390/catal15080715