

Generating Novel and Soluble Class II Fructose-1,6-Bisphosphate Aldolase with ProteinGAN


Abstract
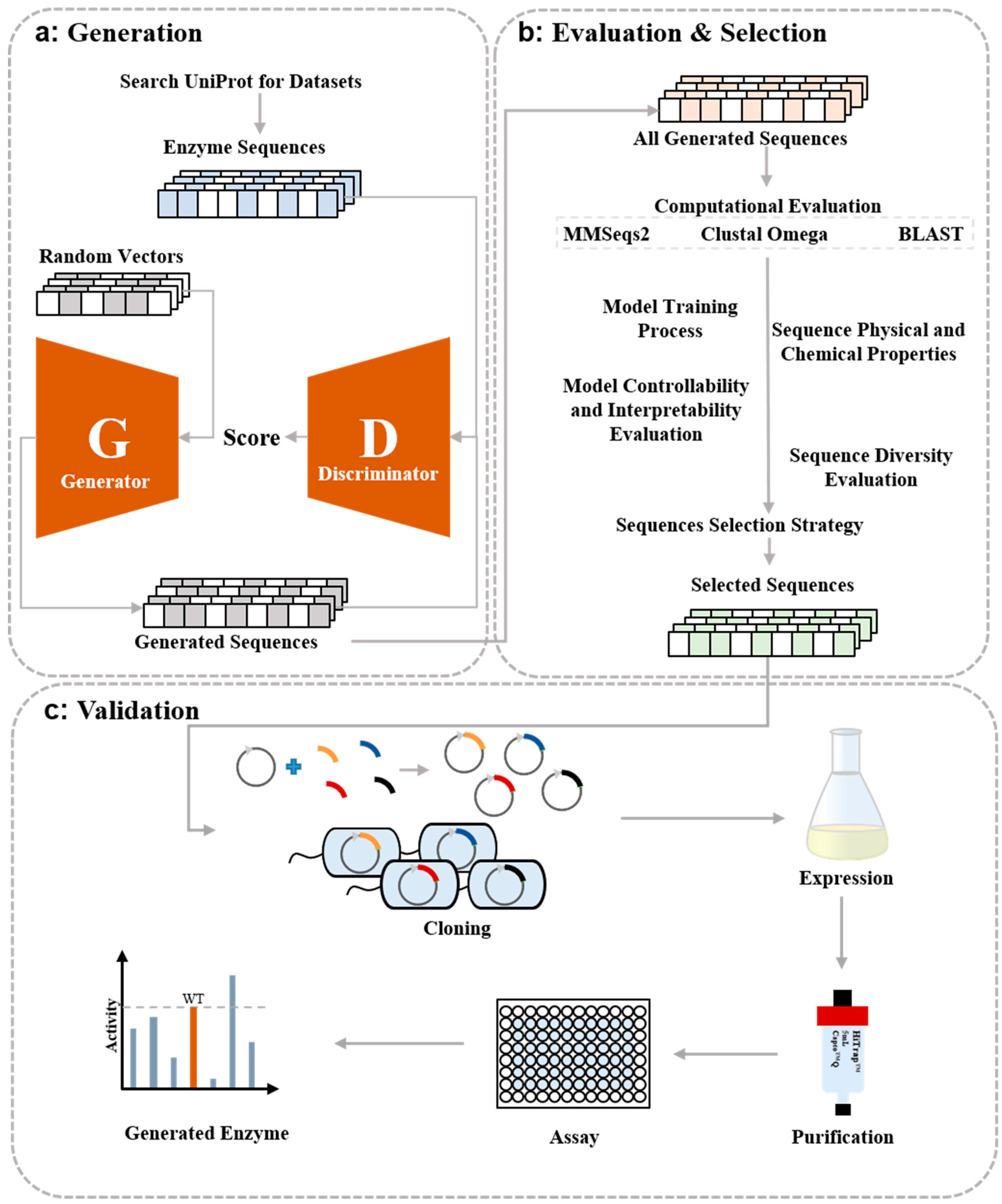
:1. Introduction
2. Results
2.1. Replication of ProteinGAN and Validation Using MDH
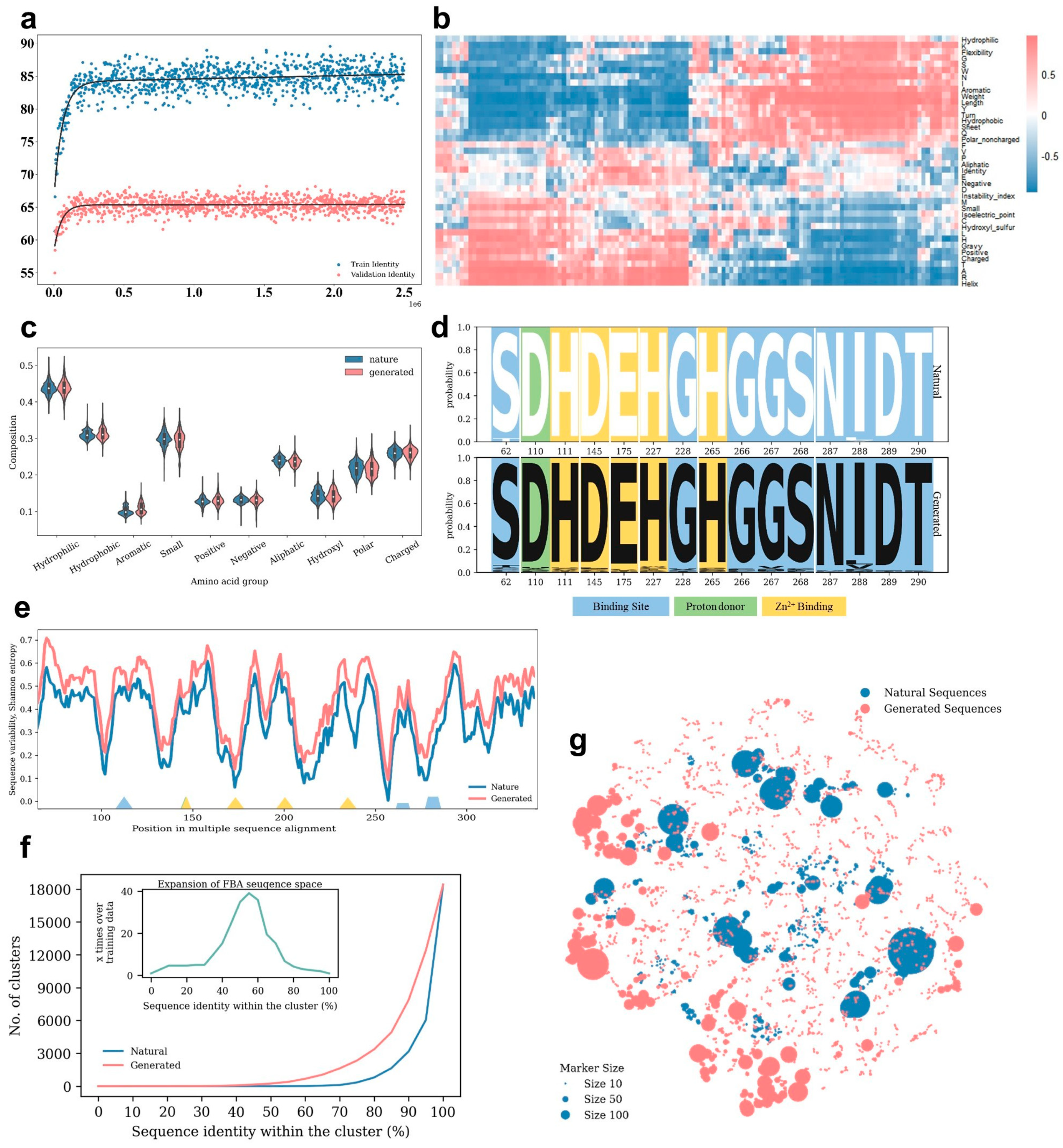
2.2. Computational Evaluation of Generated FBA Sequences
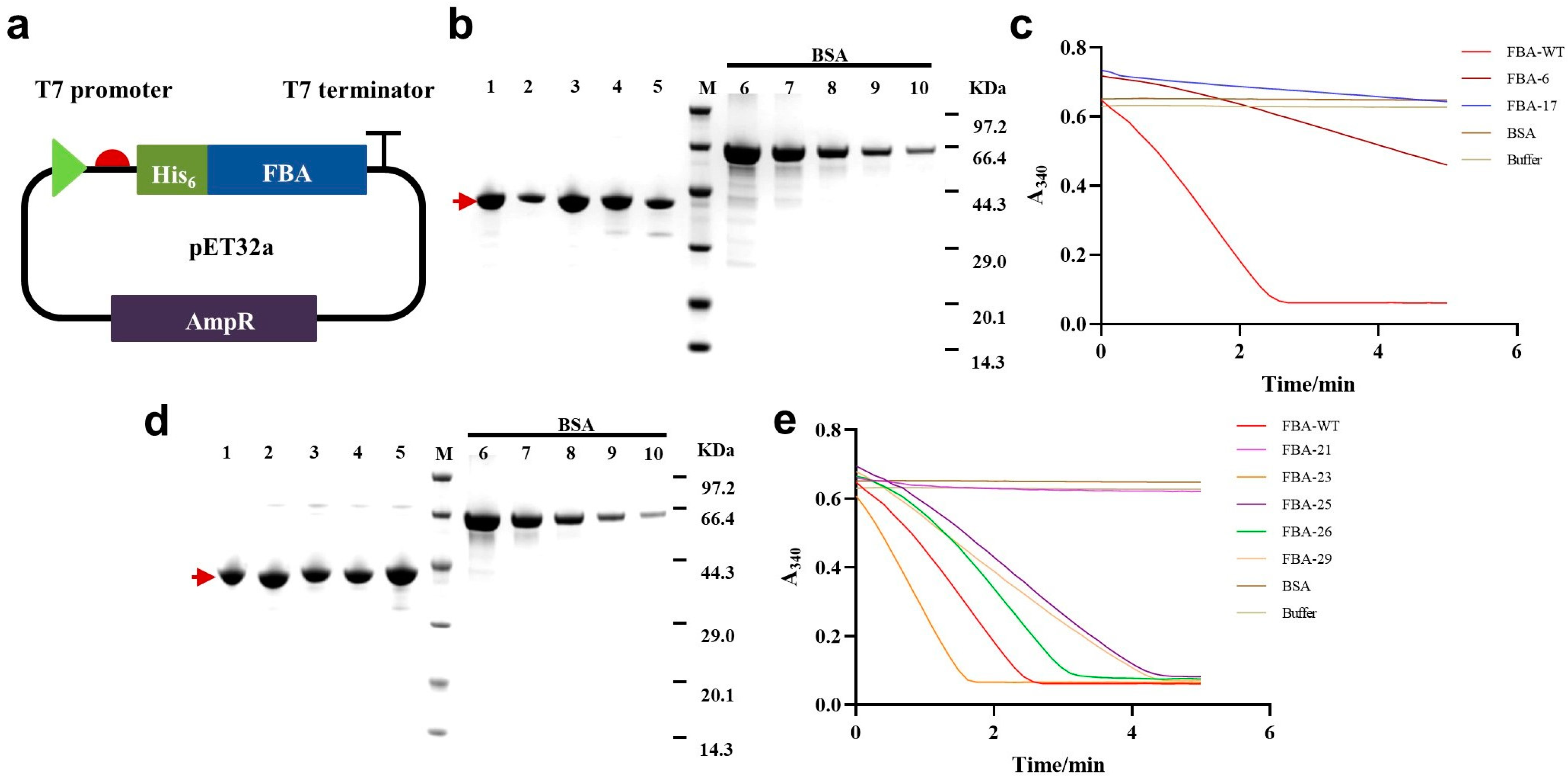
2.3. Experimental Validation of Generated FBA Sequences
2.4. Experimental Validation of Re-Selected FBA Sequences
2.5. Sequence Diversity Analysis of FBA−23
3. Discussion
4. Materials and Methods
4.1. Model Training
4.1.1. FBA Datasets
4.1.2. Architecture of the Model
4.1.3. Training Process
4.1.4. Interpolation
4.2. Computational Evaluation
4.2.1. Amino Acids and a Group of Amino Acids Composition
4.2.2. Shannon Entropy
4.2.3. Sequence Logo
4.2.4. Sequence Diversity
4.3. Experimental Validation of Generated Enzymes
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schink, S.J.; Christodoulou, D.; Mukherjee, A.; Athaide, E.; Brunner, V.; Fuhrer, T.; Bradshaw, G.A.; Sauer, U.; Basan, M. Glycolysis/gluconeogenesis specialization in microbes is driven by biochemical constraints of flux sensing. Mol. Syst. Biol. 2022, 18, e10704. [Google Scholar] [CrossRef] [PubMed]
- Genee, H.J.; Bali, A.P.; Petersen, S.D.; Siedler, S.; Bonde, M.T.; Gronenberg, L.S.; Kristensen, M.; Harrison, S.J.; Sommer, M.O.A. Functional mining of transporters using synthetic selections. Nat. Chem. Biol. 2016, 12, 1015–1022. [Google Scholar] [CrossRef]
- Uchiyama, T.; Miyazaki, K. Substrate-induced gene expression screening: A method for high-throughput screening of metagenome libraries. In Metagenomics: Methods and Protocols; Streit, W.R., Daniel, R., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 153–168. [Google Scholar] [CrossRef]
- Woolfson, D.N. A brief history of de novo protein design: Minimal, rational, and computational. J. Mol. Biol. 2021, 433, 167160. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Amrein, B.A.; Steffen-Munsberg, F.; Szeler, I.; Purg, M.; Kulkarni, Y.; Kamerlin, S.C. CADEE: Computer-aided directed evolution of enzymes. IUCrJ 2017, 4, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Siedhoff, N.E.; Schwaneberg, U.; Davari, M.D. Machine learning-assisted enzyme engineering. Methods Enzymol. 2020, 643, 281–315. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Strokach, A.; Becerra, D.; Corbi-Verge, C.; Perez-Riba, A.; Kim, P.M. Fast and flexible protein design using deep graph neural networks. Cell Syst. 2020, 11, 402–411. [Google Scholar] [CrossRef]
- Hawkins-Hooker, A.; Depardieu, F.; Baur, S.; Couairon, G.; Chen, A.; Bikard, D. Generating functional protein variants with variational autoencoders. PLoS Comput. Biol. 2021, 17, e1008736. [Google Scholar] [CrossRef]
- Repecka, D.; Jauniskis, V.; Karpus, L.; Rembeza, E.; Rokaitis, I.; Zrimec, J.; Poviloniene, S.; Laurynenas, A.; Viknander, S.; Abuajwa, W.; et al. Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 2021, 3, 324–333. [Google Scholar] [CrossRef]
- Lv, G.Y.; Guo, X.G.; Xie, L.P.; Xie, C.G.; Zhang, X.H.; Yang, Y.; Xiao, L.; Tang, Y.Y.; Pan, X.L.; Guo, A.G.; et al. Molecular characterization, gene evolution, and expression analysis of the fructose-1, 6-bisphosphate aldolase (FBA) gene family in wheat (Triticum aestivum L.). Front. Plant Sci. 2017, 8, 1030. [Google Scholar] [CrossRef]
- Iwaki, T.; Wadano, A.; Yokota, A.; Himeno, M. Aldolase—An important enzyme in controlling the ribulose 1,5-bisphosphate regeneration rate in photosynthesis. Plant Cell Physiol. 1991, 32, 1083–1091. [Google Scholar] [CrossRef]
- Rutter, W.J.; Hunsley, J.R.; Groves, W.E.; Calder, J.; Rajkumar, T.; Woodfin, B. Fructose diphosphate aldolase. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1966; Volume 9, pp. 479–498. [Google Scholar]
- Mendonca, M.; Moreira, G.M.; Conceicao, F.R.; Hust, M.; Mendonca, K.S.; Moreira, A.N.; Franca, R.C.; da Silva, W.P.; Bhunia, A.K.; Aleixo, J.A. Fructose 1,6-bisphosphate aldolase, a novel immunogenic surface protein on Listeria species. PLoS ONE 2016, 11, e0160544. [Google Scholar] [CrossRef]
- Ocampos, D.; Bacila, M. Purification and properties of fructose-1, 6-diphosphate aldolase from beef heart muscle. An. Acad. Bras. Cienc. 1975, 47, 551–556. [Google Scholar] [PubMed]
- Daher, R.; Coincon, M.; Fonvielle, M.; Gest, P.M.; Guerin, M.E.; Jackson, M.; Sygusch, J.; Therisod, M. Rational design, synthesis, and evaluation of new selective inhibitors of microbial class II (zinc dependent) fructose bis-phosphate aldolases. J. Med. Chem. 2010, 53, 7836–7842. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef]
- Plater, A.R.; Berry, A.; Zgiby, S.M.; Thomson, G.J. Conserved residues in the mechanism of the E. coli class II FBP-aldolase. J. Mol. Biol. 1999, 285, 843–855. [Google Scholar] [CrossRef]
- Wehmeier, U.F. Molecular cloning, nucleotide sequence and structural analysis of the Streptomyces galbus DSM40480 fda gene: The S. galbus fructose-1,6-bisphosphate aldolase is a member of the class II aldolase. FEMS Microbiol. Lett. 2001, 197, 53–58. [Google Scholar] [CrossRef]
- Rukseree, K.; Thammarongtham, C.; Palittapongarnpim, P. One-step purification and characterization of a fully active histidine-tagged class II fructose-1,6-bisphosphate aldolase from Mycobacterium tuberculosis. Enzym. Microb. Technol. 2008, 43, 500–506. [Google Scholar] [CrossRef]
- Baldwin, S.A.; Perham, R.N.; Stribling, D. Purification and characterization of the class II d-fructose 1,6-bisphosphate aldolase from Escherichia coli (crookes’ strain). Biochem. J. 1978, 169, 633–641. [Google Scholar] [CrossRef]
- Berry, A.; Marshall, K.E. Identification of zinc-binding ligands in the class II fructose-l, 6-bisphosphate aldolase of Escherichia coli. FEBS 1993, 318, 11–16. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Peng, C.; Shi, Y.; Li, H.; Xu, Z.; Zhu, W. D3DistalMutation: A database to explore the effect of distal mutations on enzyme activity. J. Chem. Inf. Model. 2021, 61, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Miyato, T.; Koyama, M. cGANs with projection discriminator. arXiv 2018, arXiv:1802.05637. [Google Scholar] [CrossRef]
- Anand, N.; Eguchi, R.; Mathews, I.I.; Perez, C.P.; Derry, A.; Altman, R.B.; Huang, P.S. Protein sequence design with a learned potential. Nat. Commun. 2022, 13, 746. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st Annual Conference on Neural Information Processing Systems (NIPS), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Zhang, H.; Goodfellow, I.; Metaxas, D.; Odena, A. Self-attention generative adversarial networks. In Proceedings of the International Conference on Machine Learning, Long Beach, CA, USA, 9–15 June 2019; pp. 7354–7363. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Miyato, T.; Kataoka, T.; Koyama, M.; Yoshida, Y. Spectral normalization for generative adversarial networks. arXiv 2018, arXiv:1802.05957. [Google Scholar]
- Mescheder, L.; Geiger, A.; Nowozin, S. Which training methods for GANs do actually converge? In Proceedings of the International Conference on Machine Learning, Stockholm, Sweden, 10–15 July 2018; pp. 3481–3490. [Google Scholar]
- Jang, E.; Gu, S.; Poole, B. Categorical reparameterization with gumbel-softmax. arXiv 2016, arXiv:1611.01144. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Mult. Seq. Alignment Methods 2014, 1079, 105–116. [Google Scholar] [CrossRef]
- Xia, J.; Xin, W.; Wang, F.; Xie, W.; Liu, Y.; Xu, J. Cloning and characterization of fructose-1,6-bisphosphate aldolase from Euphausia superba. Int. J. Mol. Sci. 2022, 23, 10478. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| ID | Total Protein a (mg) | Yield b (mg/L) | Purity c (%) |
|---|---|---|---|
| FBA−WT | 2.76 | 27.63 | 91 |
| FBA−1 | 11.29 | 112.91 | 95 |
| FBA−6 | 7.32 | 73.16 | 93 |
| FBA−17 | 1.89 | 18.87 | 86 |
| FBA−21 | 3.68 | 36.81 | 89 |
| FBA−23 | 1.23 | 12.35 | 87 |
| FBA−25 | 2.33 | 23.35 | 85 |
| FBA−26 | 3.10 | 31.02 | 86 |
| FBA−29 | 0.48 | 4.78 | 88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, F.; Ren, M.; Li, X.; Lin, Z.; Yang, X. Generating Novel and Soluble Class II Fructose-1,6-Bisphosphate Aldolase with ProteinGAN. Catalysts 2023, 13, 1457. https://doi.org/10.3390/catal13121457
Tang F, Ren M, Li X, Lin Z, Yang X. Generating Novel and Soluble Class II Fructose-1,6-Bisphosphate Aldolase with ProteinGAN. Catalysts. 2023; 13(12):1457. https://doi.org/10.3390/catal13121457
Chicago/Turabian StyleTang, Fangfang, Mengyuan Ren, Xiaofan Li, Zhanglin Lin, and Xiaofeng Yang. 2023. "Generating Novel and Soluble Class II Fructose-1,6-Bisphosphate Aldolase with ProteinGAN" Catalysts 13, no. 12: 1457. https://doi.org/10.3390/catal13121457
APA StyleTang, F., Ren, M., Li, X., Lin, Z., & Yang, X. (2023). Generating Novel and Soluble Class II Fructose-1,6-Bisphosphate Aldolase with ProteinGAN. Catalysts, 13(12), 1457. https://doi.org/10.3390/catal13121457