
Study of Intermolecular Interaction between Small Molecules and Carbon Nanobelt: Electrostatic, Exchange, Dispersive and Inductive Forces



Abstract
:1. Introduction
2. Results and Discussion
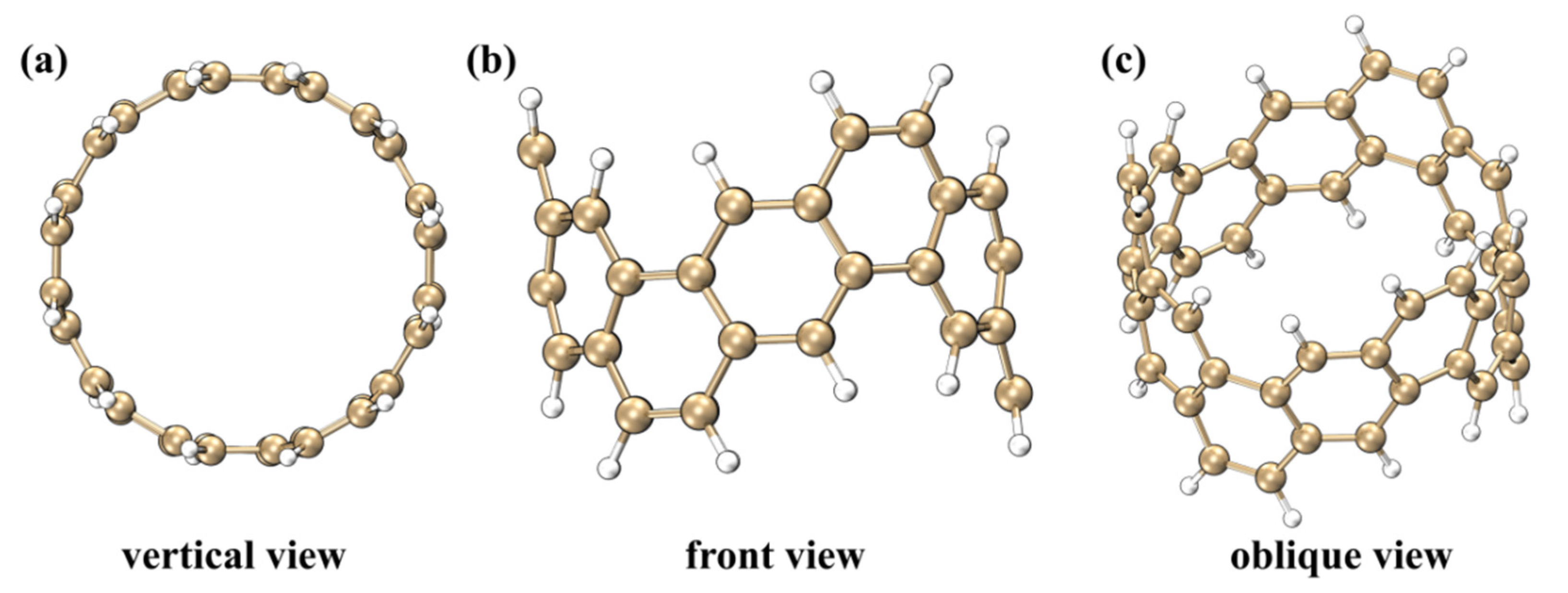
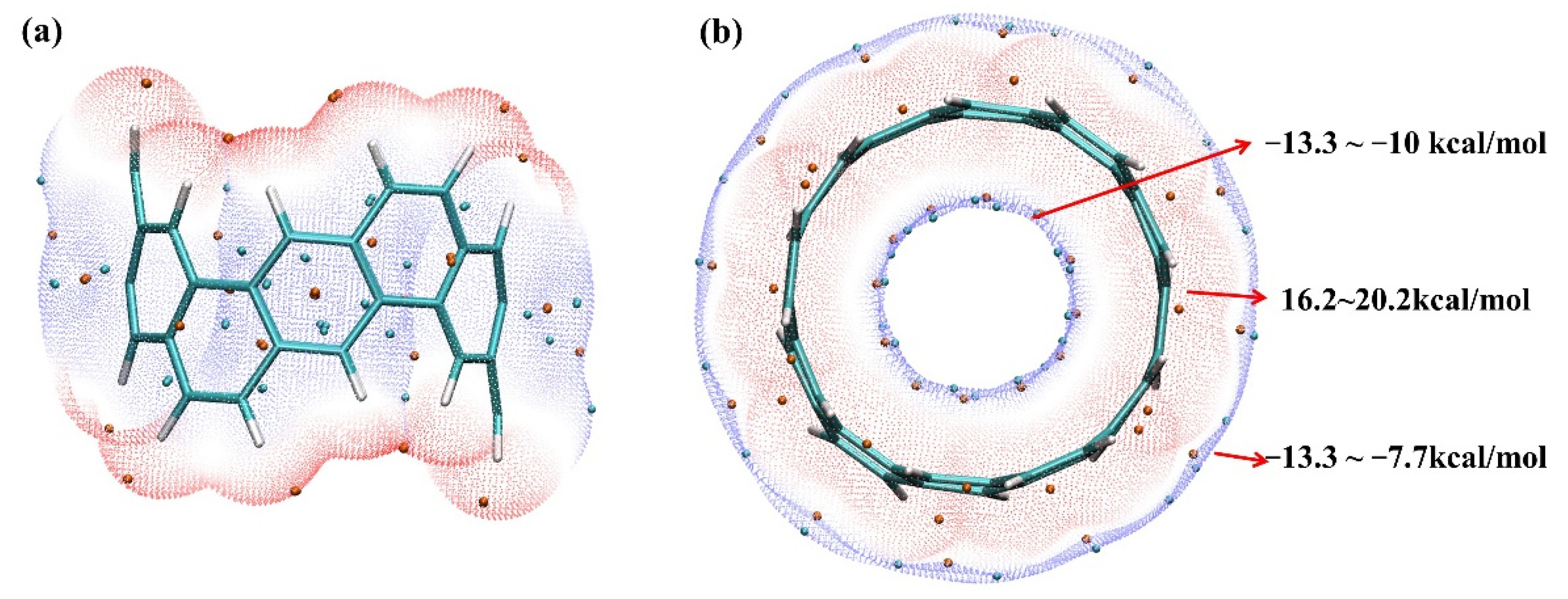
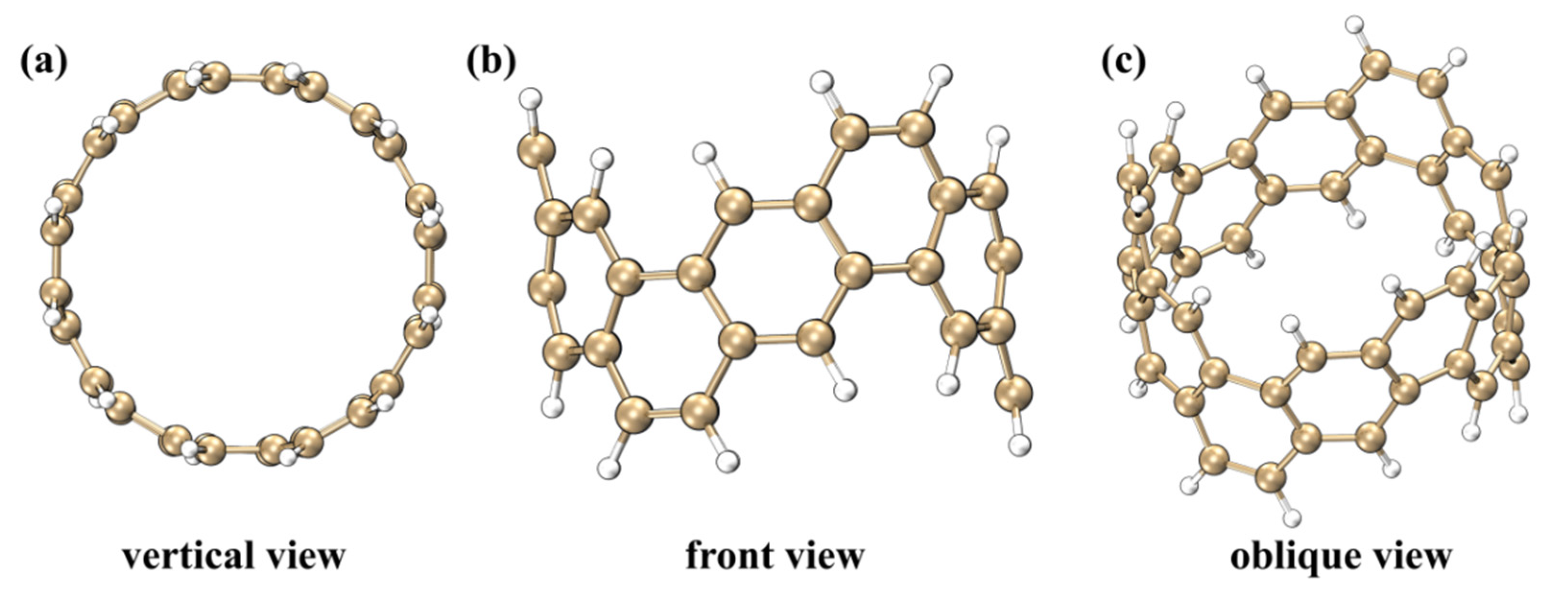
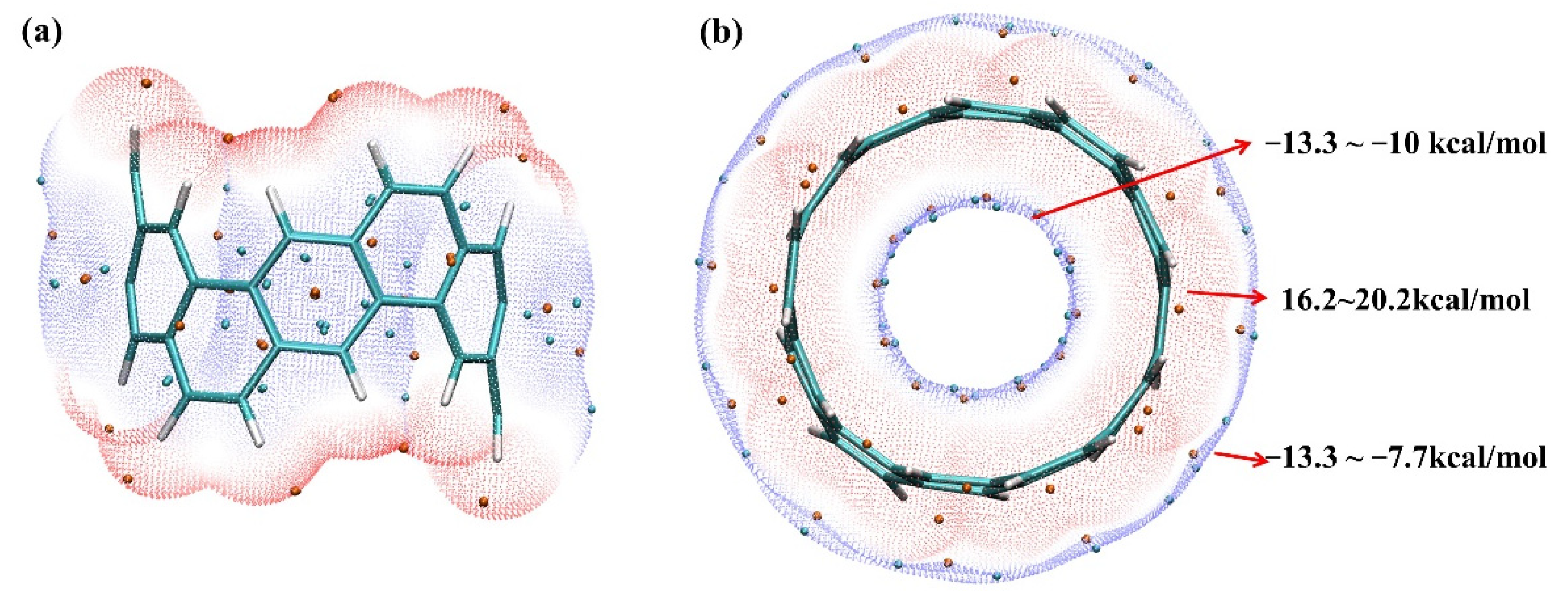
2.1. Electrostatic Potential and Van Der Waals Potential
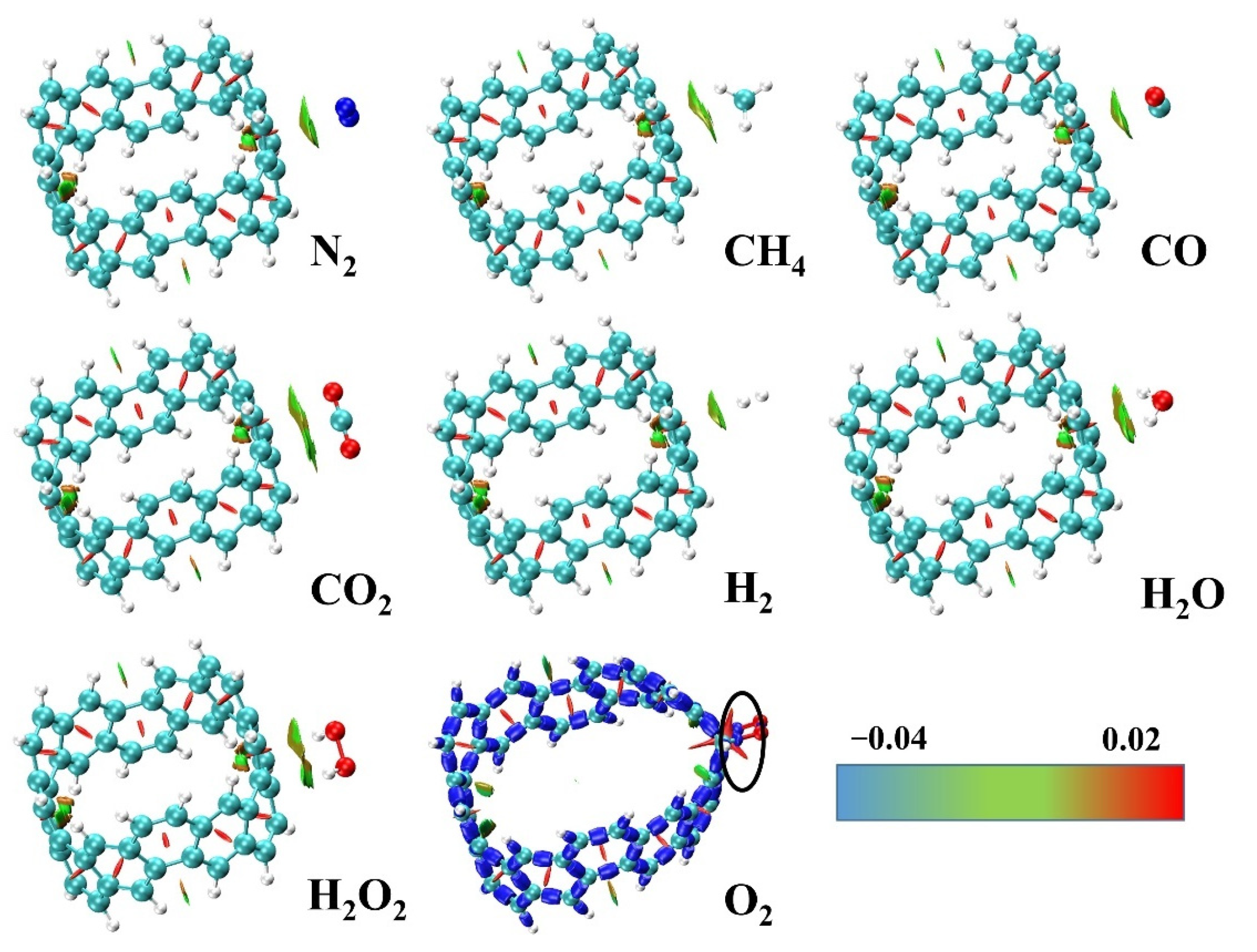
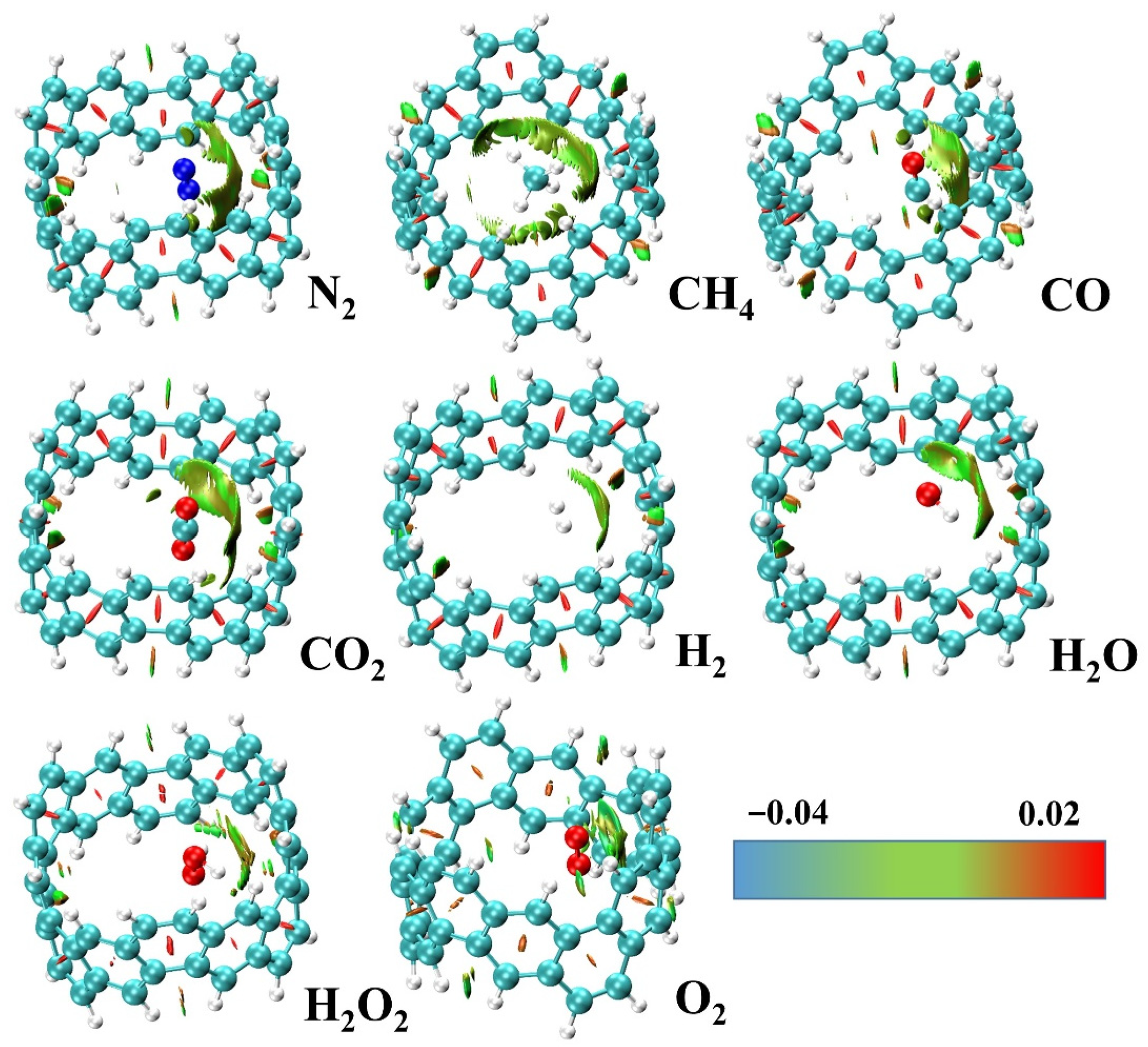
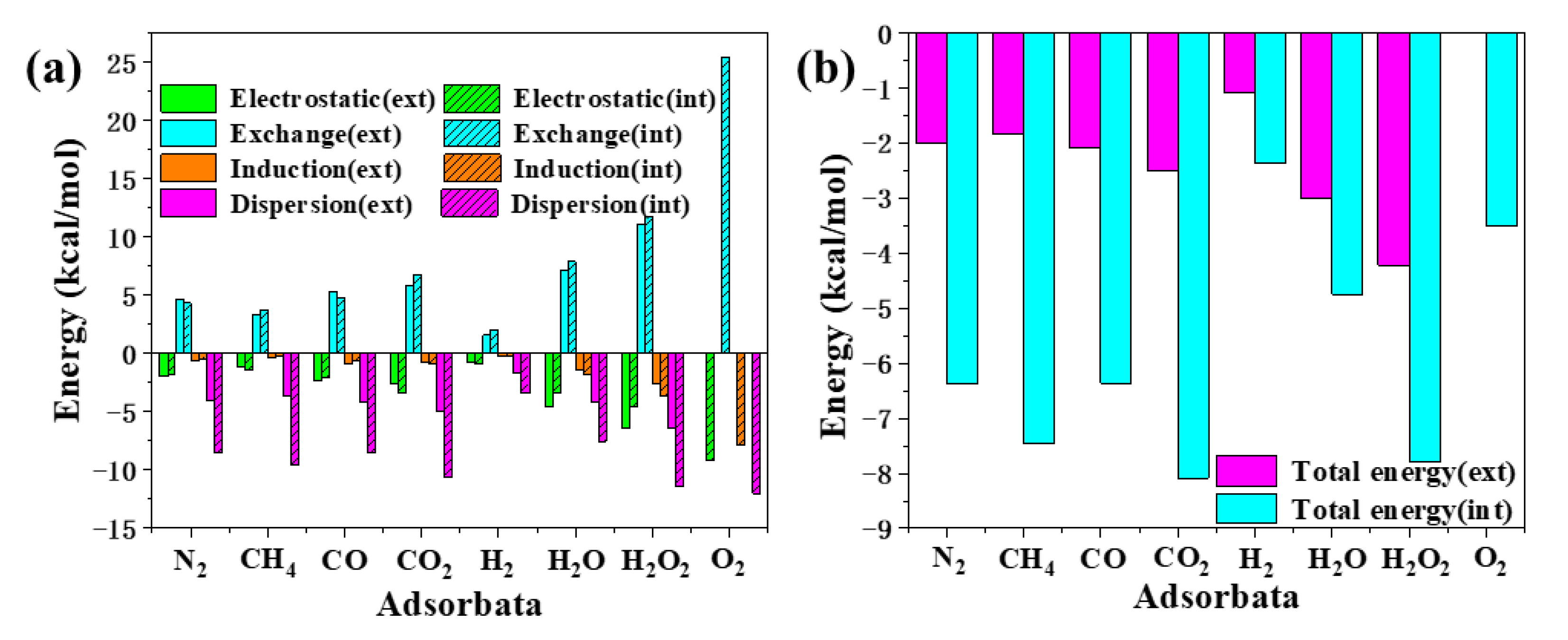
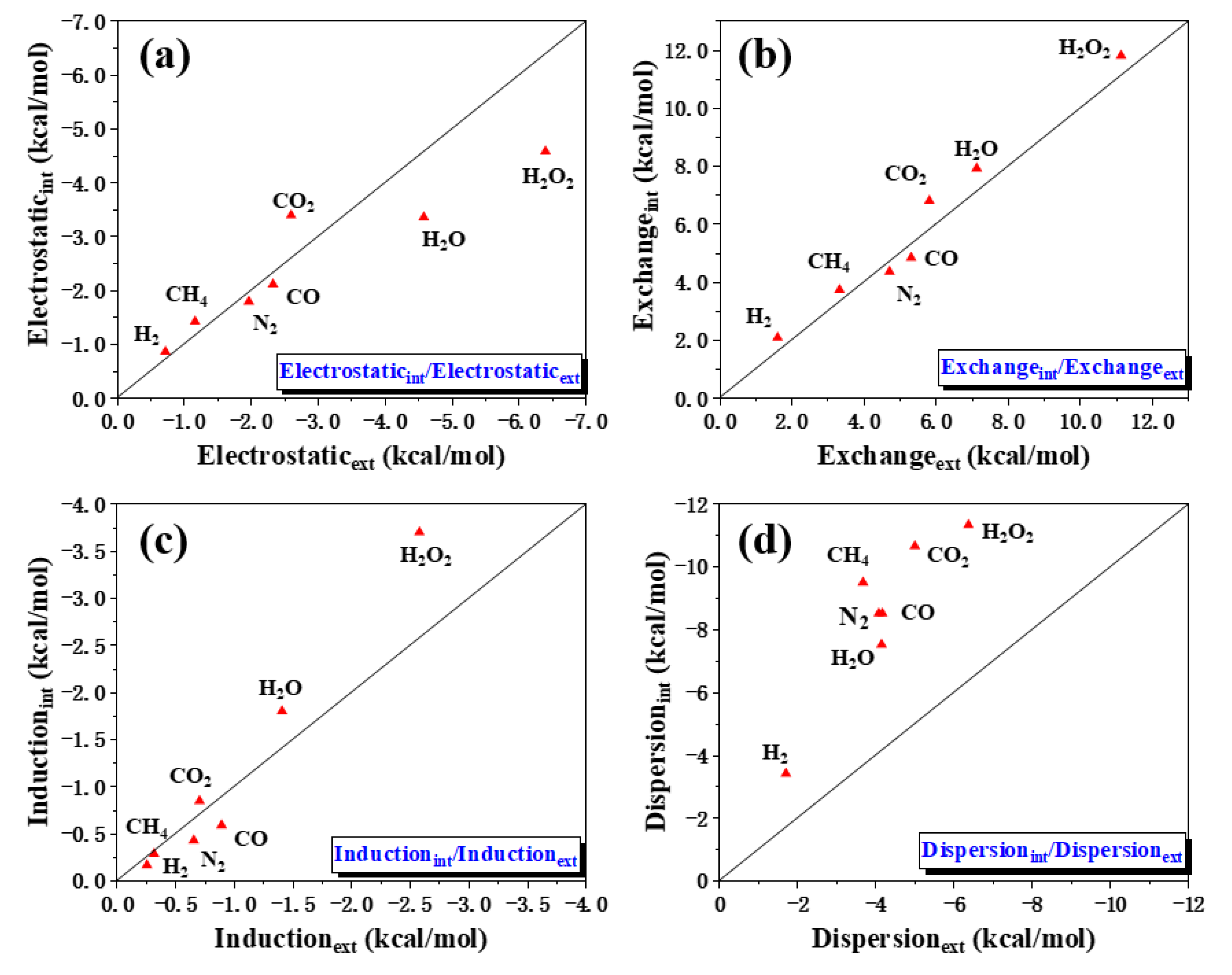
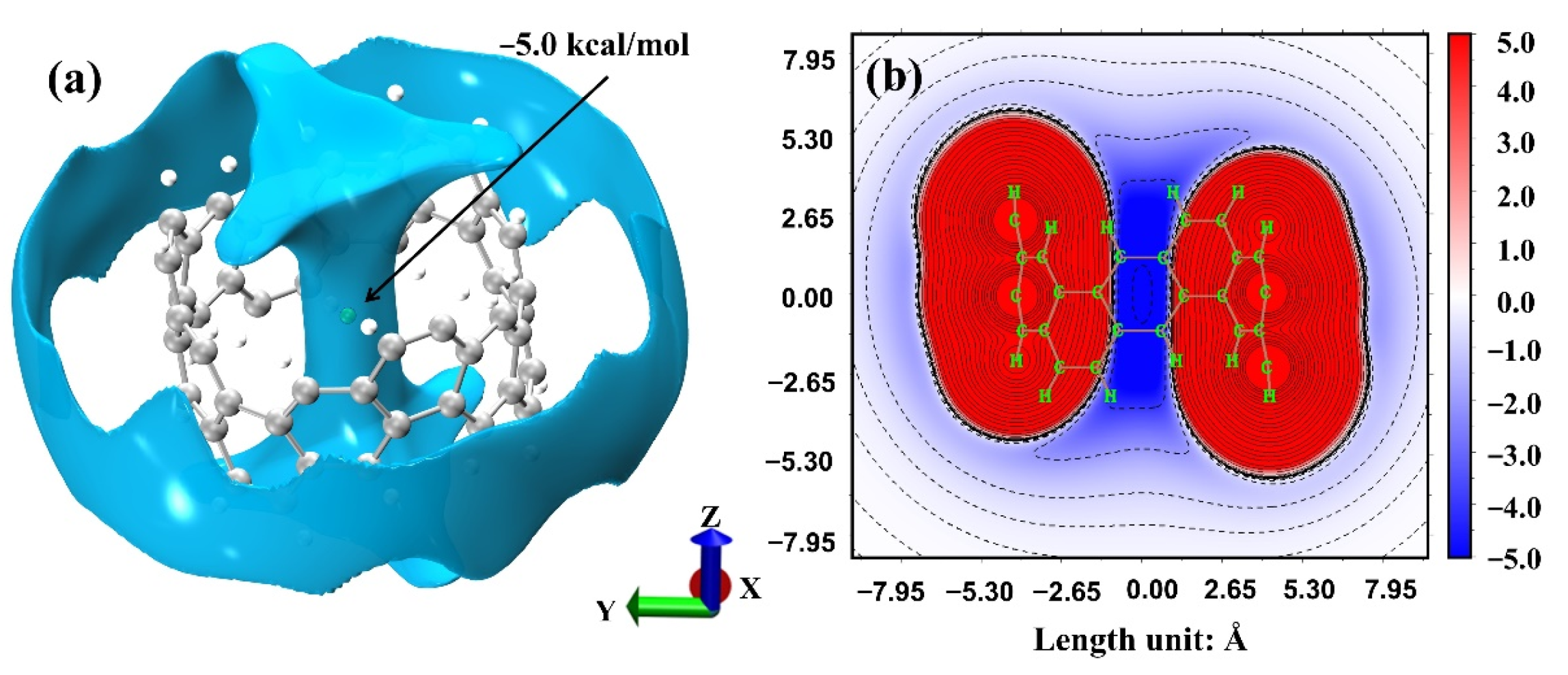
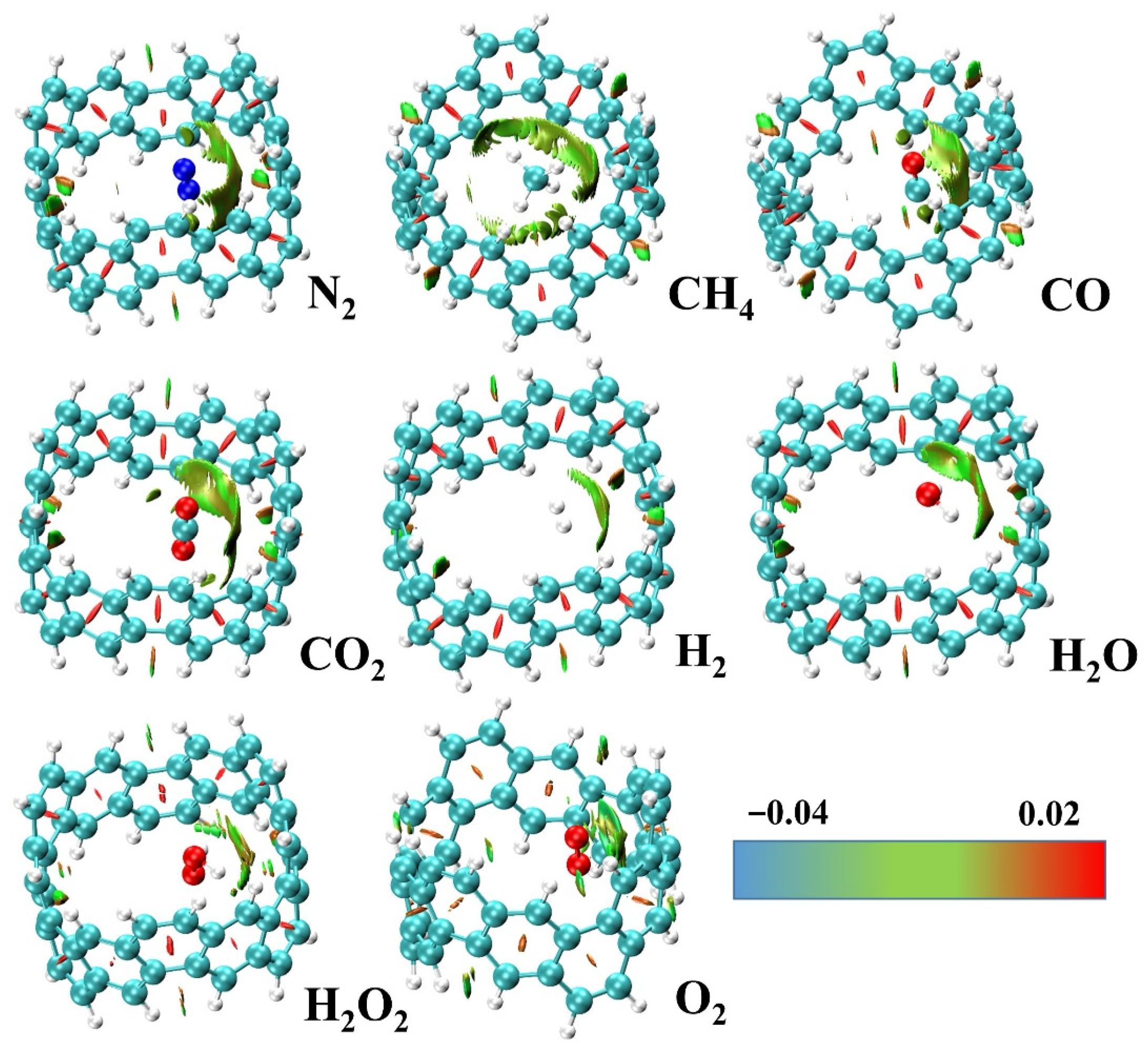
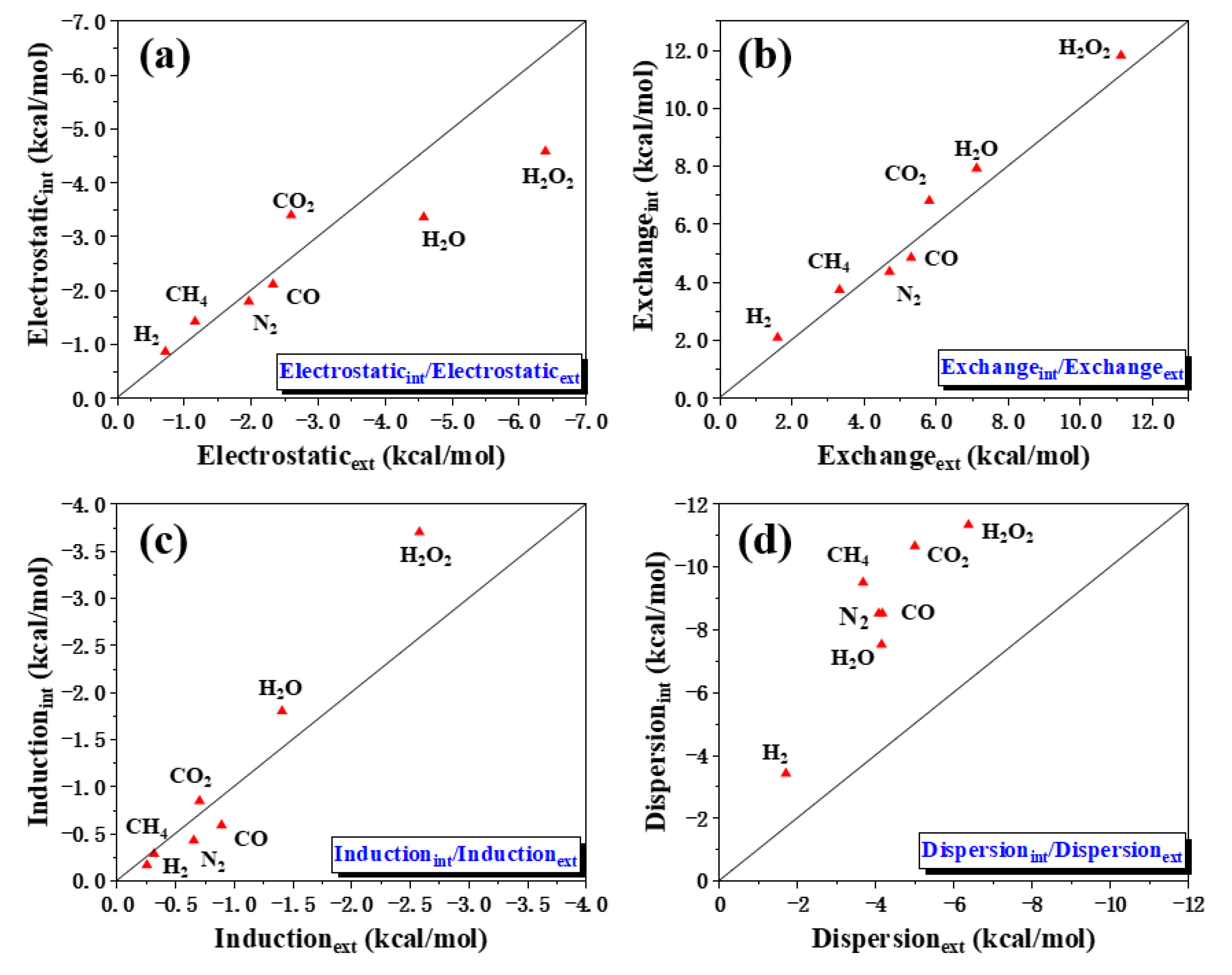
2.2. Adsorption and Energy Decomposition between [6, 6] CNB and Small Molecules
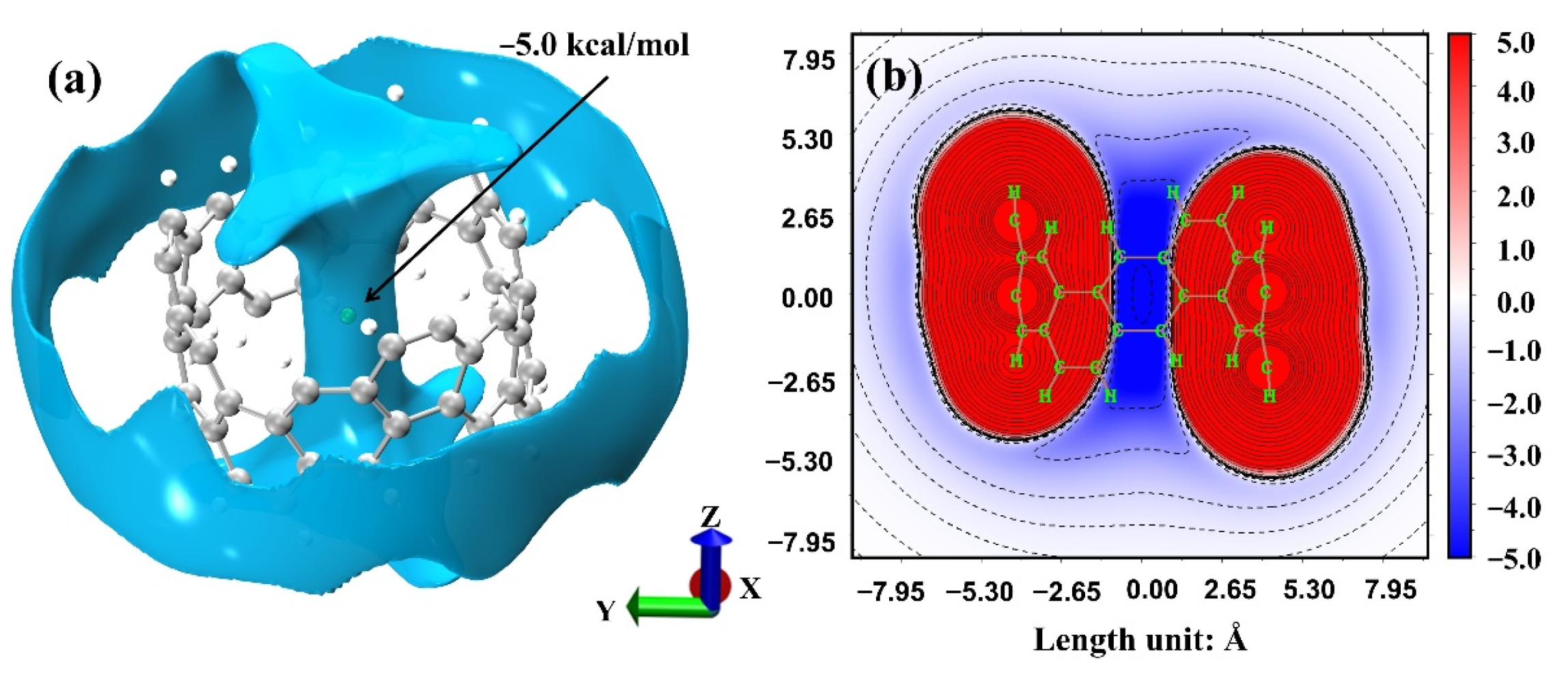
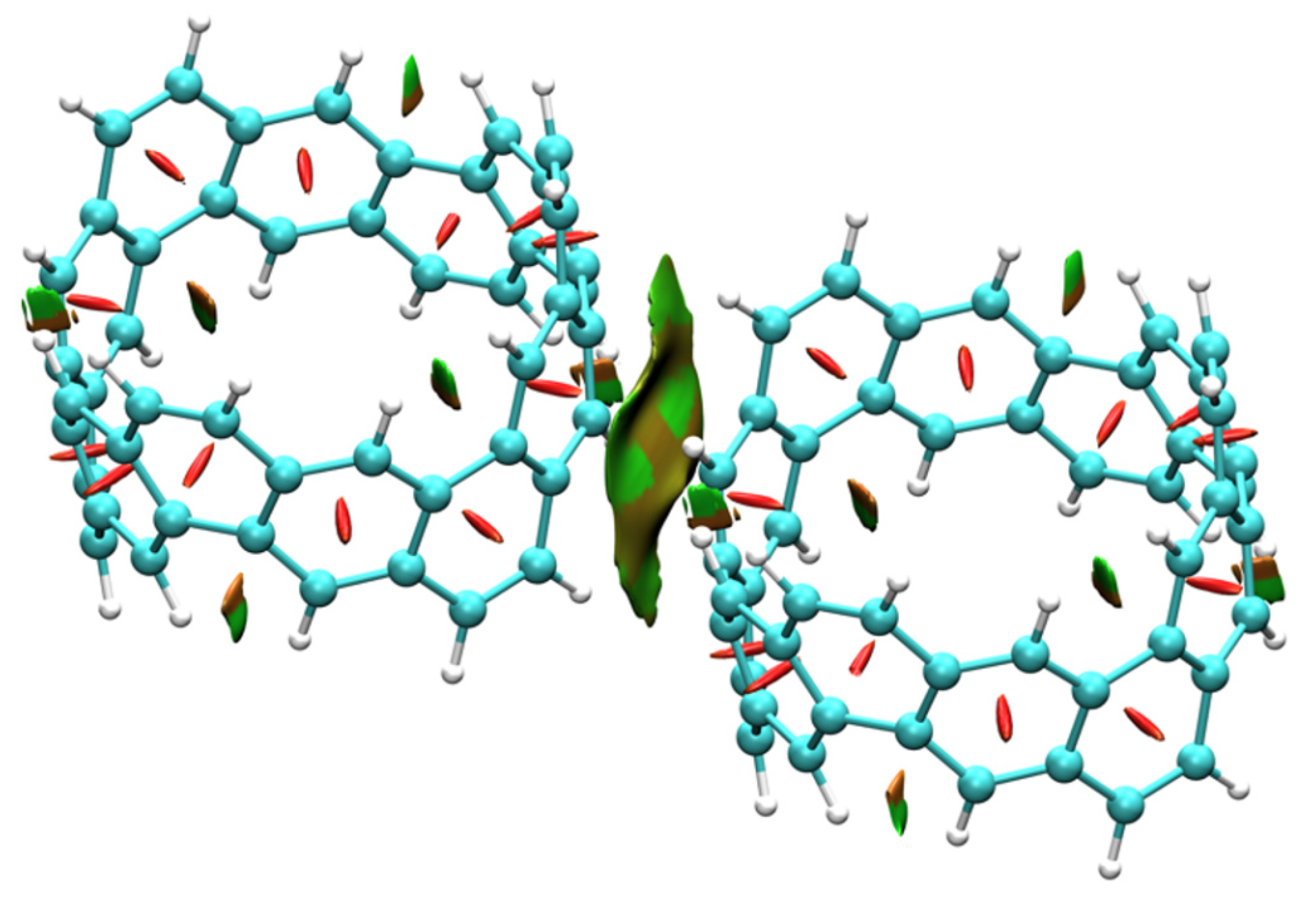
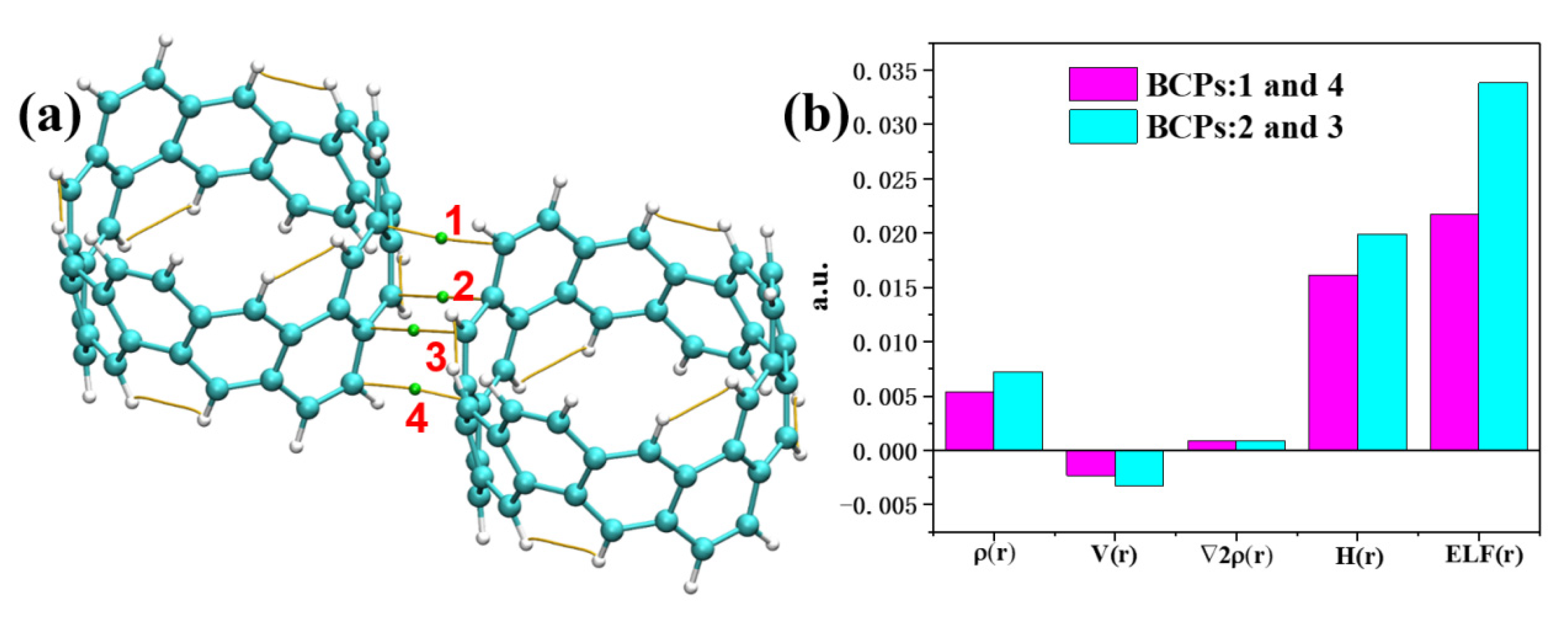
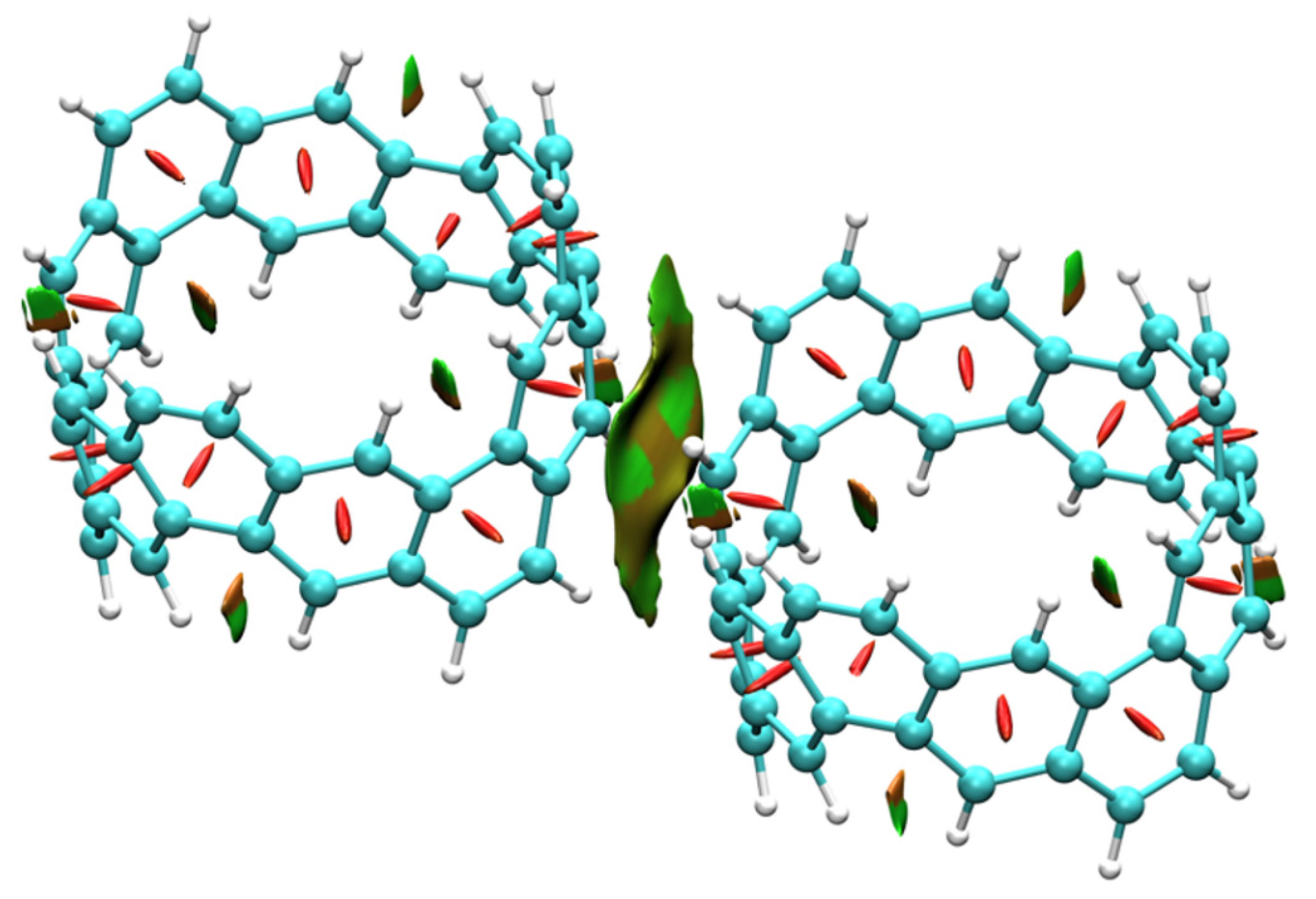
2.3. Intermolecular Interactions of [6, 6] CNB Dimer
3. Calculation Method
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dahl, J.A.; Maddux, B.L.S.; Hutchison, J.E. Toward greener nanosynthesis. Chem. Rev. 2007, 107, 2228–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Wang, Y.; Liu, Y.; Chen, L.; Feng, B.; Jin, X.; Zhou, Y.; Huang, T.; Xiao, M.; Huang, F.; et al. Conferring poly (ionic liquid) s with high surface areas for enhanced catalytic activity. ACS Sustain. Chem. Eng. 2021, 9, 2115–2128. [Google Scholar] [CrossRef]
- Scida, K.; Stege, P.W.; Haby, G.; Messina, G.A.; García, C.D. Recent applications of carbon-based nanomaterials in analytical chemistry: Critical review. Anal. Chim. Acta 2011, 691, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Wang, L.; Mu, X.; Wang, J. The magical photoelectric and optoelectronic properties of graphene nanoribbons and their applications. J. Mater. Chem. C 2021, 9, 13600–13616. [Google Scholar] [CrossRef]
- Wang, J.; Mu, X.; Sun, M.; Mu, T. Optoelectronic properties and applications of graphene-based hybrid nanomaterials and van der waals heterostructures. Appl. Mater. Today 2019, 16, 1–20. [Google Scholar] [CrossRef]
- Wang, J.; Song, J.; Mu, X.; Sun, M. Optoelectronic and photoelectric properties and applications of graphene-based nanostructures. Mater. Today Phys. 2020, 13, 100196. [Google Scholar] [CrossRef]
- Wang, J.; Mu, X.; Wang, L.; Sun, M. Properties and applications of new superlattice: Twisted bilayer graphene. Mater. Today Phys. 2019, 9, 100099. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Z.; Feng, N.; Wang, J. Electric field induced twisted bilayer graphene infrared plasmon spectrum. Nanomaterials 2021, 11, 2433. [Google Scholar] [CrossRef]
- Wang, J.; Bo, W.; Ding, Y.; Wang, X.; Mu, X. Optical, optoelectronic, and photoelectric properties in moire superlattices of twist bilayer graphene. Mater. Today Phys. 2020, 14, 100238. [Google Scholar] [CrossRef]
- Chen, X.; Lu, C.; Wang, L.; Wang, J.; Spectroscopy, B. Angle-resolved one and two-photon absorption spectrum in twisted bilayer graphene quantum dots. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 271, 120894. [Google Scholar] [CrossRef]
- Kong, W.; Liu, F.; Liu, Y. Design of nitrogen-doped graphitized 2d hierarchical porous carbons as efficient solid base catalysts for transesterification to biodiesel. Green Chem. 2020, 22, 903–912. [Google Scholar] [CrossRef]
- Peyvandi, A.; Soroushian, P.; Abdol, N.; Balachandra, A.M. Surface-modified graphite nanomaterials for improved reinforcement efficiency in cementitious paste. Carbon 2013, 63, 175–186. [Google Scholar] [CrossRef]
- Mauter, M.S.; Elimelech, M. Environmental applications of carbon-based nanomaterials. Environ. Sci. Technol. 2008, 42, 5843–5859. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Huang, Y.; Wang, K.; Xu, T.; Li, S.; Zhao, M.; Wang, Y.; Chen, Q. Novel hexagonal Bi2O2CO3 porous nanoplate/nitrogen-doped graphene nanomaterials with enhanced electrochemical properties for oxygen reduction reaction in acidic media for fuel cells. Carbon 2019, 152, 459–473. [Google Scholar] [CrossRef]
- Li, D.; Hu, X.; Zhang, S. Biodegradation of graphene-based nanomaterials in blood plasma affects their biocompatibility, drug delivery, targeted organs and antitumor ability. Biomaterials 2019, 202, 12–25. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, W.; Alvarez, P.J.J. Manganese peroxidase degrades pristine but not surface-oxidized (carboxylated) single-walled carbon nanotubes. Environ. Sci. Technol. 2014, 48, 7918–7923. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Rideal, E.K.; Wright, W.M. Clxxxiv.—Low temperature oxidation at charcoal surfaces. Part I. The behaviour of charcoal in the absence of promoters. J. Chem. Soc. Trans. 1925, 127, 1347–1357. [Google Scholar] [CrossRef]
- Fan, F.R.; Wang, R.; Zhang, H.; Wu, W. Emerging beyond-graphene elemental 2d materials for energy and catalysis applications. Chem. Soc. Rev. 2021, 50, 10983–11031. [Google Scholar] [CrossRef]
- Povie, G.; Segawa, Y.; Nishihara, T.; Miyauchi, Y.; Itami, K. Synthesis of a carbon nanobelt. Science 2017, 356, 172–175. [Google Scholar] [CrossRef]
- Abd El Sater, M.; Jaber, N.; Schulz, E. Chiral salen complexes for asymmetric heterogeneous catalysis: Recent examples for recycling and cooperativity. ChemCatChem 2019, 11, 3662–3687. [Google Scholar] [CrossRef] [Green Version]
- Machado, I.V.; Dos Santos, J.R.N.; Januario, M.A.P.; Corrêa, A.G. Greener organic synthetic methods: Sonochemistry and heterogeneous catalysis promoted multicomponent reactions. Ultrason. Sonochemistry 2021, 78, 105704. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Amin, N.S. Advances in visible light responsive titanium oxide-based photocatalysts for co2 conversion to hydrocarbon fuels. Energy Convers. Manag. 2013, 76, 194–214. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and perspectives of co 2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Peng, B.; Peng, T. Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Wu, D.; Deng, K.; Hu, B.; Lu, Q.; Liu, G.; Hong, X. Plasmon-assisted photothermal catalysis of low-pressure CO2 hydrogenation to methanol over Pd/ZnO catalyst. ChemCatChem 2019, 11, 1598–1601. [Google Scholar] [CrossRef]
- Hou, W.; Hung, W.H.; Pavaskar, P.; Goeppert, A.; Aykol, M.; Cronin, S.B. Photocatalytic conversion of co2 to hydrocarbon fuels via plasmon-enhanced absorption and metallic interband transitions. ACS Catal. 2011, 1, 929–936. [Google Scholar] [CrossRef]
- Koshy, D.M.; Nathan, S.S.; Asundi, A.S.; Abdellah, A.M.; Dull, S.M.; Cullen, D.A.; Higgins, D.; Bao, Z.; Bent, S.F.; Jaramillo, T.F. Bridging thermal catalysis and electrocatalysis: Catalyzing co2 conversion with carbon-based materials. Angew. Chem. Int. Ed. 2021, 60, 17472–17480. [Google Scholar] [CrossRef]
- Xiao, G.; Wang, L.; Mu, X.; Zou, X.; Wu, Y.; Cao, J. Lead and iodide fixation by thiol copper (II) porphyrin for stable and environmental-friendly perovskite solar cells. CCS Chem. 2021, 3, 25–36. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, L.; Mu, X.; Chen, B.; Xiong, Q.; Wang, W.D.; Ding, J.; Gao, P.; Wu, Y.; Cao, J. Grain boundary engineering with self-assembled porphyrin supramolecules for highly efficient large-area perovskite photovoltaics. J. Am. Chem. Soc. 2021, 143, 18989–18996. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, L.; Mu, X.; Chen, C.C.; Wu, Y.; Cao, J.; Tang, Y. Intramolecular electric field construction in metal phthalocyanine as dopant-free hole transporting material for stable perovskite solar cells with> 21% efficiency. Angew. Chem. Int. Ed. 2021, 60, 6294–6299. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Hou, J.; Zhang, S.; Liang, Y.; Yang, G.; Yang, Y.; Yu, L.; Yue, W.; Gang, L. Polymer solar cells with enhanced open-circuit voltage and efficiency. Nat. Photonics 2009, 3, 649–653. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, Q.; Li, G.; Luo, S.; Song, T.-B.; Duan, H.-S.; Hong, Z.; You, J.; Liu, Y.; Yang, Y. Interface engineering of highly efficient perovskite solar cells. Science 2014, 345, 542–546. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chen, C.C.; Dou, L.; Murase, S.; Duan, H.S.; Hawks, S.A.; Xu, T.; Son, H.J.; Yu, L.; Li, G. Metal oxide nanoparticles as an electron-transport layer in high-performance and stable inverted polymer solar cells. Adv. Mater. 2012, 24, 5267–5272. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Van der waals potential: An important complement to molecular electrostatic potential in studying intermolecular interactions. J. Mol. Modeling 2020, 26, 1–9. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo [18] carbon: Focusing on molecular adsorption and stacking. Carbon 2021, 171, 514–523. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Henon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction region indicator: A simple real space function clearly revealing both chemical bonds and weak interactions. Chem. -Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction methods for noncovalent interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Szalewicz, K. Symmetry-adapted perturbation theory of intermolecular forces. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 254–272. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16, Revision c. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. Iv. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with london dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.; Simmonett, A.C.; DePrince III, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M. Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Burns, L.A.; Marshall, M.S.; Sherrill, C.D. Appointing silver and bronze standards for noncovalent interactions: A comparison of spin-component-scaled (SCS), explicitly correlated (F12), and specialized wavefunction approaches. J. Chem. Phys. 2014, 141, 234111. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| attrdis/attrtot | N2 | CH4 | CO | CO2 | H2 | H2O | H2O2 | O2 |
|---|---|---|---|---|---|---|---|---|
| ext | 0.61 | 0.71 | 0.56 | 0.60 | 0.64 | 0.41 | 0.42 | -- |
| int | 0.79 | 0.85 | 0.76 | 0.72 | 0.77 | 0.59 | 0.58 | 0.41 |
| Component | Value (kcal/mol) |
|---|---|
| Electrostatics | −6.28 (23%) |
| Exchange | 17.95 |
| Induction | −1.98 (7%) |
| Dispersion | −19.30 (70%) |
| Total | −9.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Chen, P.; Li, C.; Wang, J. Study of Intermolecular Interaction between Small Molecules and Carbon Nanobelt: Electrostatic, Exchange, Dispersive and Inductive Forces. Catalysts 2022, 12, 561. https://doi.org/10.3390/catal12050561
Lu C, Chen P, Li C, Wang J. Study of Intermolecular Interaction between Small Molecules and Carbon Nanobelt: Electrostatic, Exchange, Dispersive and Inductive Forces. Catalysts. 2022; 12(5):561. https://doi.org/10.3390/catal12050561
Chicago/Turabian StyleLu, Chen, Panpan Chen, Cunlei Li, and Jingang Wang. 2022. "Study of Intermolecular Interaction between Small Molecules and Carbon Nanobelt: Electrostatic, Exchange, Dispersive and Inductive Forces" Catalysts 12, no. 5: 561. https://doi.org/10.3390/catal12050561
APA StyleLu, C., Chen, P., Li, C., & Wang, J. (2022). Study of Intermolecular Interaction between Small Molecules and Carbon Nanobelt: Electrostatic, Exchange, Dispersive and Inductive Forces. Catalysts, 12(5), 561. https://doi.org/10.3390/catal12050561