
Promotional Effect of Pt-Doping on the Catalytic Performance of Pt−CeO2 Catalyst for CO Oxidation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
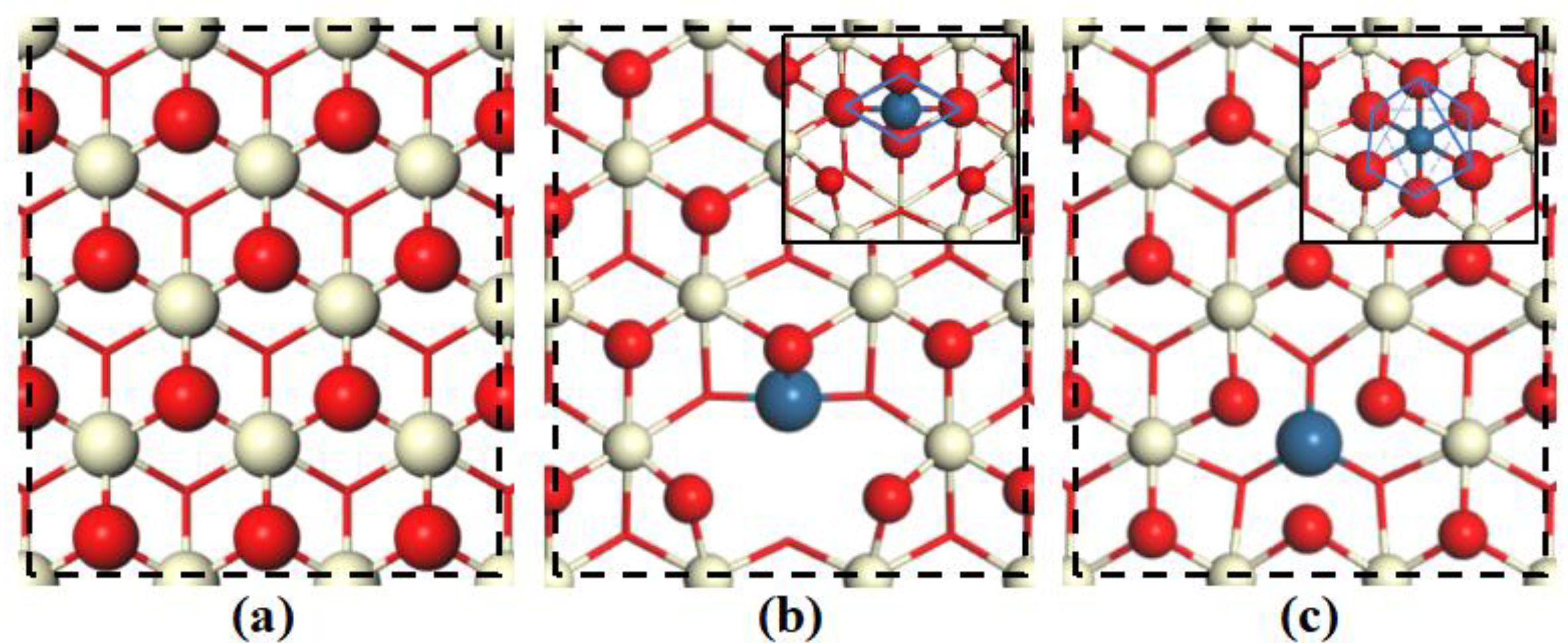
2.1. Structures of Pristine and Pt-doped CeO2(111) Surface
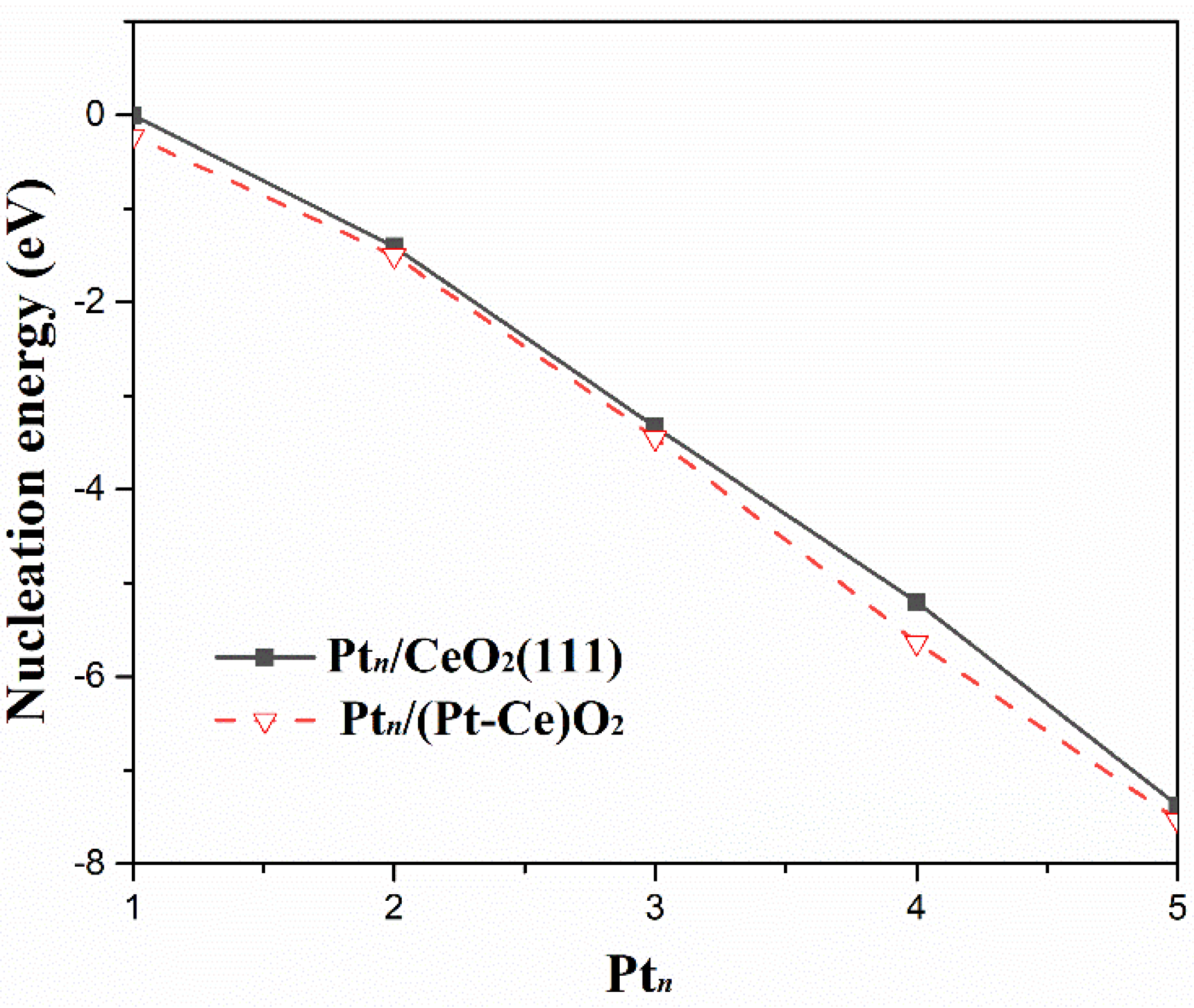
2.2. Stability of Ptn Clusters
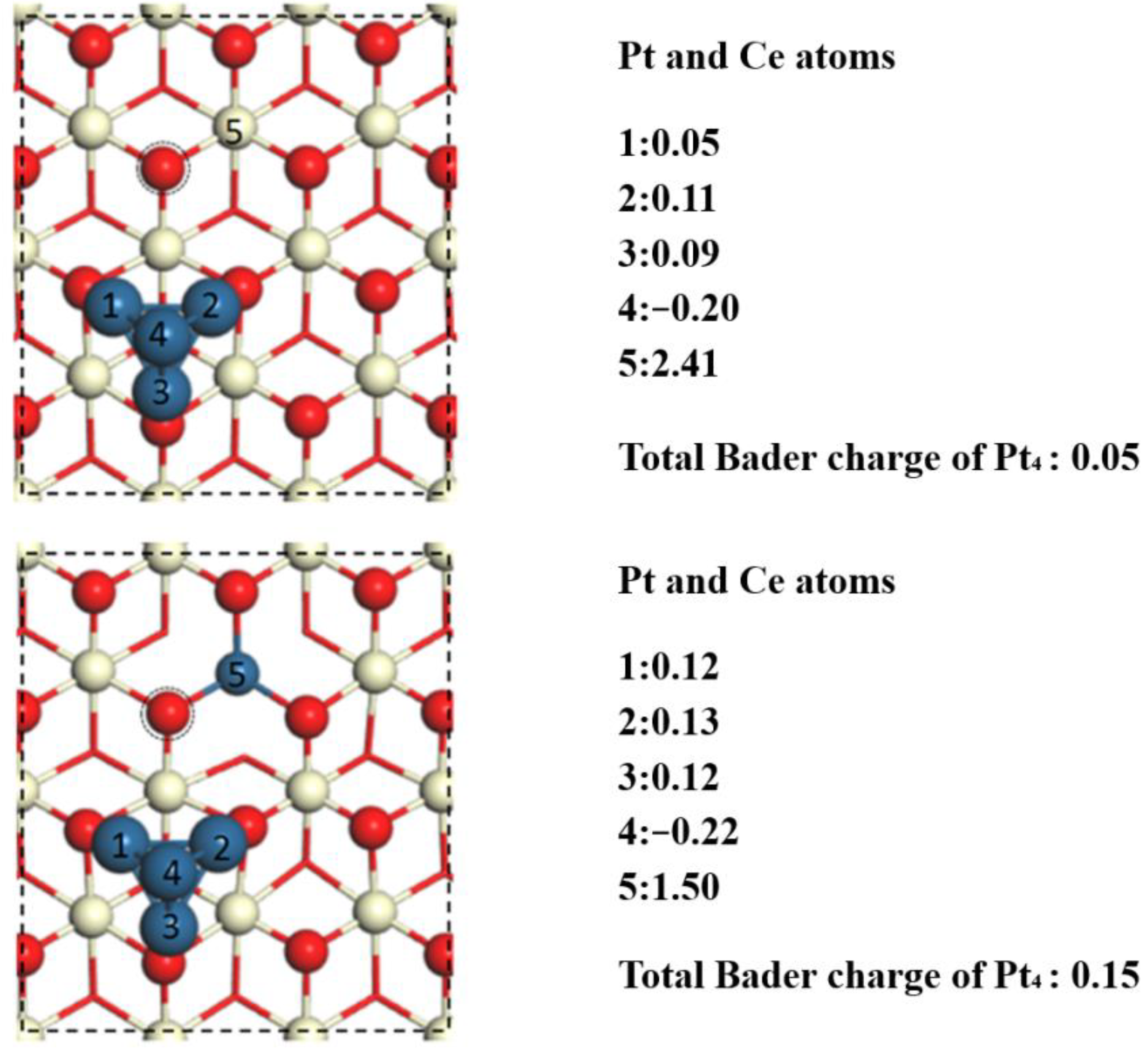
2.3. Oxygen Vacancy Formation
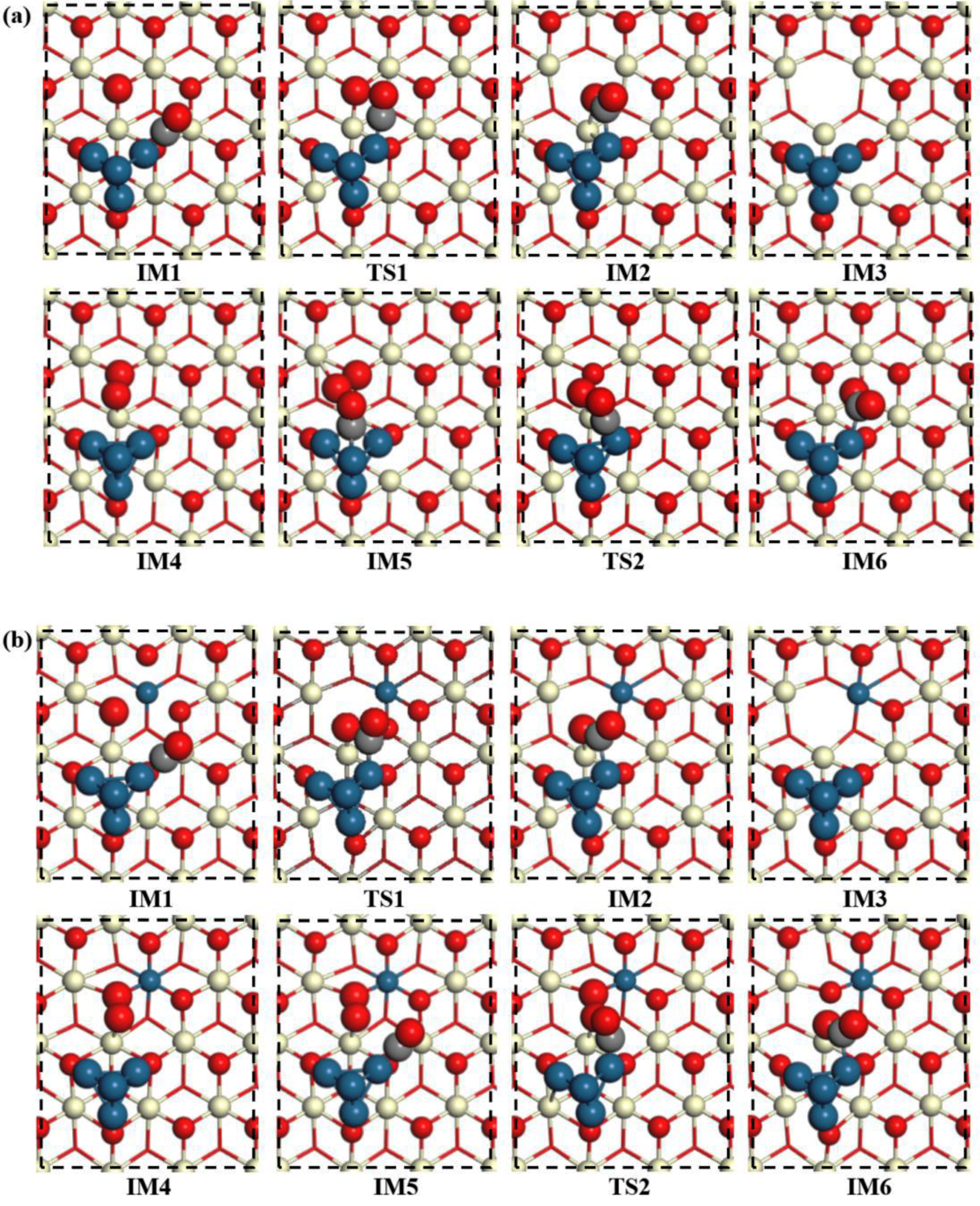
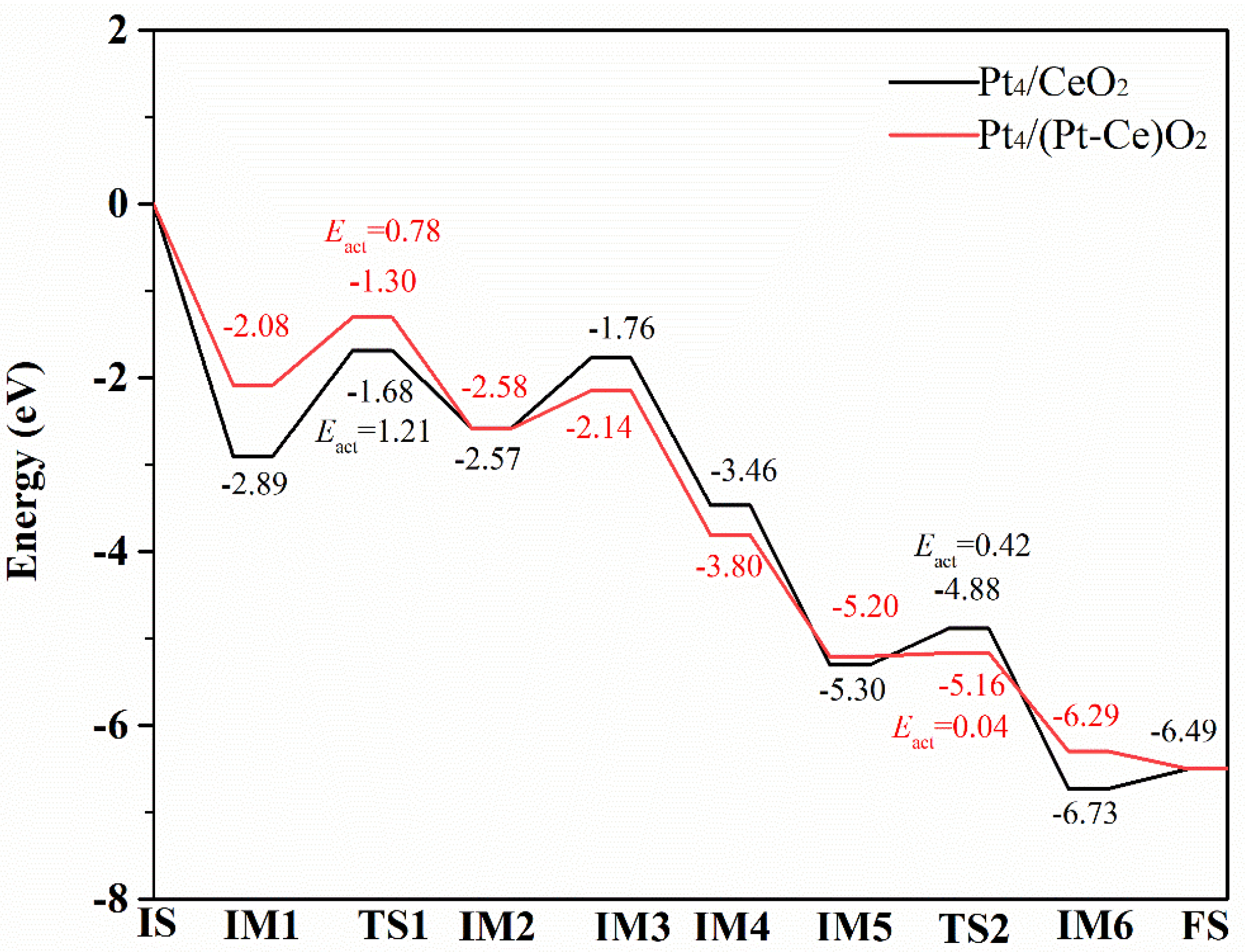
2.4. CO Oxidation on the Pt4/(Pt−Ce)O2 and Pt4/CeO2 Surfaces
3. Computation Details
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maciel, C.G.; de Freitas Silva, T.; Assaf, E.M.; Assaf, J.M. Hydrogen production and purification from the water-gas shift reaction on CuO/CeO2-TiO2 catalysts. Appl. Energy 2013, 112, 52–59. [Google Scholar] [CrossRef]
- Zhu, J.; Su, Y.; Chai, J.; Muravev, V.; Kosinov, N.; Hensen, E.J.M. Mechanism and nature of active sites for methanol synthesis from CO/CO2 on Cu/CeO2. ACS Catal. 2020, 10, 11532–11544. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Alkhoori, A.A.; Efstathiou, A.M.; Jaoude, M.A.; Damaskinos, C.M.; Baker, M.A.; Almutawa, A.; Anjum, D.H.; Vasiliades, M.A.; Belabbes, A.; et al. Design aspects of doped CeO2 for low-temperature catalytic CO oxidation: Transient kinetics and DFT approach. ACS Appl. Mater. Interfaces 2021, 13, 22391–22415. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Grinter, D.C.; Liu, Z.; Palomino, R.M.; Senanayake, S.D. Ceria-based model catalysts: Fundamental studies on the importance of the metal-ceria interface in CO oxidation, the water-gas shift, CO2 hydrogenation, and methane and alcohol reforming. Chem. Soc. Rev. 2017, 46, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Aneggi, E.; De Leitenburg, C.; Trovarelli, A. On the role of lattice/surface oxygen in ceria-zirconia catalysts for diesel soot combustion. Catal. Today 2012, 181, 108–115. [Google Scholar] [CrossRef]
- Li, N.; Chen, Q.-Y.; Luo, L.-F.; Huang, W.-X.; Luo, M.-F.; Hu, G.S.; Lu, J.-Q. Kinetic study and the effect of particle size on low temperature CO oxidation over Pt/TiO2 catalysts. Appl. Catal. B Environ. 2013, 142–143, 523–532. [Google Scholar] [CrossRef]
- Slavinskaya, E.M.; Gulyaev, R.V.; Zadesenets, A.V.; Stonkus, O.A.; Zaikovskii, V.I.; Shubin, Y.V.; Korenev, S.V.; Boronin, A.I. Low-temperature CO oxidation by Pd/CeO2 catalysts synthesized using the coprecipitation method. Appl. Catal. B Environ. 2015, 166–167, 91–103. [Google Scholar] [CrossRef]
- Boronin, A.I.; Slavinskaya, E.M.; Figueroba, A.; Stadnichenko, A.I.; Kardash, T.Y.; Stonkus, O.A.; Fedorova, E.A.; Muravev, V.V.; Svetlichnyi, V.A.; Bruix, A.; et al. CO oxidation activity of Pt/CeO2 catalysts below 0 °C: Platinum loading effects. Appl. Catal. B Environ. 2021, 286, 119931. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, Z.; Henkelman, G.; Song, W. Computational design of a CeO2-supported pd-based bimetallic nanorod for CO oxidation. J. Phys. Chem. C 2016, 120, 5557–5564. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Li, T.; Zhao, Z.; Gong, X.Q.; Chen, Y.; Duan, A.; Jiang, G.; Wei, Y. Interfacial effects of CeO2-supported pd nanorod in catalytic CO oxidation: A theoretical study. J. Phys. Chem. C 2015, 119, 12923–12934. [Google Scholar] [CrossRef]
- Sun, C.; Li, H.; Chen, L. Nanostructured ceria-based materials: Synthesis, properties, and applications. Energy Environ. Sci. 2012, 5, 8475–8505. [Google Scholar] [CrossRef]
- Qin, Y.Y.; Su, Y.Q. A DFT study on heterogeneous Pt/CeO2 (110) single atom catalysts for CO oxidation. ChemCatChem 2021, 13, 3857–3863. [Google Scholar] [CrossRef]
- Farmer, J.A.; Campbell, C.T. Ceria maintains smaller metal catalyst particles by strong metal-support bonding. Science 2010, 329, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Gulec, A.; Johnson, A.M.; Schweitzer, N.M.; Stucky, G.D.; Marks, L.D.; Stair, P.C. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science 2015, 350, 189–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gänzler, A.M.; Casapu, M.; Vernoux, P.; Loridant, S.; Aires, F.J.C.S.; Epicier, T.; Betz, B.; Hoyer, R.; Grunwaldt, J.D. Tuning the structure of platinum particles on ceria in situ for enhancing the catalytic performance of exhaust gas catalysts. Angew. Chem. Int. Ed. 2017, 56, 13078–13082. [Google Scholar] [CrossRef]
- Lu, Y.; Thompson, C.; Kunwar, D.; Datye, A.K.; Karim, A.M. Origin of the high CO oxidation activity on CeO2 supported Pt nanoparticles: Weaker binding of CO or facile oxygen transfer from the support? ChemCatChem 2020, 12, 1726–1733. [Google Scholar] [CrossRef]
- Ghosh, P.; Camellone, M.F.; Fabris, S. Fluxionality of Au clusters at ceria surfaces during CO oxidation: Relationships among reactivity, size, cohesion, and surface defects from DFT simulations. J. Phys. Chem. Lett. 2013, 4, 2256–2263. [Google Scholar] [CrossRef]
- Kim, H.Y.; Henkelman, G. CO oxidation at the interface between doped CeO2 and supported Au nanoclusters. J. Phys. Chem. Lett. 2012, 3, 2194–2199. [Google Scholar] [CrossRef]
- Nie, L.; Mei, D.; Xiong, H.; Peng, B.; Ren, Z.; Hernandez, X.I.P.; DeLaRiva, A.; Wang, M.; Engelhard, M.H.; Kovarik, L.; et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 2017, 358, 1419–1423. [Google Scholar] [CrossRef] [Green Version]
- Krcha, M.D.; Mayernick, A.D.; Janik, M.J. Periodic trends of oxygen vacancy formation and C-H bond activation over transition metal-doped CeO2 (111) surfaces. J. Catal. 2012, 293, 103–115. [Google Scholar] [CrossRef]
- Dutta, G.; Gupta, A.; Waghmare, U.V.; Hegde, M.S. CO adsorption on ionic Pt, Pd and Cu sites in Ce1-xMxO2-δ (M=Pt2+, Pd2+, Cu2+). J. Chem. Sci. 2011, 123, 509–516. [Google Scholar] [CrossRef]
- Hu, Z.; Li, B.; Sun, X.Y.; Metiu, H. Chemistry of Doped oxides: The activation of surface oxygen and the chemical compensation effect. J. Phys. Chem. C 2011, 115, 3065–3074. [Google Scholar] [CrossRef]
- Hu, Z.; Metiu, H. Effect of Dopants on the energy of oxygen-vacancy formation at the surface of ceria: Local or global? J. Phys. Chem. C 2011, 115, 17898–17909. [Google Scholar] [CrossRef]
- Su, Y.Q.; Zhang, L.; Muravev, V.; Hensen, E.J.M. Lattice oxygen activation in transition metal doped ceria. Chin. J. Catal. 2020, 41, 977–984. [Google Scholar] [CrossRef]
- Su, Y.Q.; Filot, I.A.W.; Liu, J.X.; Hensen, E.J.M. Stable Pd-doped ceria structures for CH4 activation and CO oxidation. ACS Catal. 2018, 8, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Liu, N.; Chen, B.; Li, J.; Mei, D. Nucleation of Cun (n = 1–5) clusters and equilibrium morphology of Cu particles supported on CeO2 surface: A density functional theory study. J. Phys. Chem. C 2018, 122, 27402–27411. [Google Scholar] [CrossRef]
- Liu, J.X.; Su, Y.; Filot, I.A.W.; Hensen, E.J.M. A linear scaling relation for CO oxidation on CeO2-supported Pd. J. Am. Chem. Soc. 2018, 140, 4580–4587. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wan, Q.; Xiong, H.; Zhou, S.; Chen, X.; Hernandez, X.I.P.; Wang, Y.; Lin, S.; Datye, A.K.; Guo, H. Correlating DFT calculations with CO oxidation reactivity on Ga-doped Pt/CeO2 single-atom catalysts. J. Phys. Chem. C 2018, 122, 22460–22468. [Google Scholar] [CrossRef]
- Ren, Z.; Peng, F.; Chen, B.; Mei, D.; Li, J. A combined experimental and computational study of water-gas shift reaction over rod-shaped Ce0.75M0.25O2 (M = Ti, Zr, and Mn) supported Cu catalysts. Int. J. Hydrog. Energy 2017, 42, 30086–30097. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, A.; Ren, Z.; Qu, Y.; Zhang, Y.; Li, J. Promotional Effect of Pt-Doping on the Catalytic Performance of Pt−CeO2 Catalyst for CO Oxidation. Catalysts 2022, 12, 529. https://doi.org/10.3390/catal12050529
Jiang A, Ren Z, Qu Y, Zhang Y, Li J. Promotional Effect of Pt-Doping on the Catalytic Performance of Pt−CeO2 Catalyst for CO Oxidation. Catalysts. 2022; 12(5):529. https://doi.org/10.3390/catal12050529
Chicago/Turabian StyleJiang, Angran, Zhibo Ren, Yaqi Qu, Yanjun Zhang, and Jianwei Li. 2022. "Promotional Effect of Pt-Doping on the Catalytic Performance of Pt−CeO2 Catalyst for CO Oxidation" Catalysts 12, no. 5: 529. https://doi.org/10.3390/catal12050529
APA StyleJiang, A., Ren, Z., Qu, Y., Zhang, Y., & Li, J. (2022). Promotional Effect of Pt-Doping on the Catalytic Performance of Pt−CeO2 Catalyst for CO Oxidation. Catalysts, 12(5), 529. https://doi.org/10.3390/catal12050529