Abstract
The effect of NaI on hydrogenation of diphenylacetylene catalyzed by the water-soluble [{RuCl(mtppms-Na)2}2(µ-Cl)2] (1) (mtppms-Na = meta-monosulfonated triphenylphosphine sodium salt) is reported. Hydrogenations were performed under mild conditions (P(H2) = 1 bar, T = 50–80 ℃) in aqueous–organic biphasic reaction mixtures wherein the catalyst was dissolved in aqueous phase of various pHs. In acidic solutions, addition of NaI to 1 + mtppms-Na increased the selective conversion of diphenylacetylene to stilbenes from 10% to 90% but did not effect the high Z-selectivity (up to 98%). In contrast, in basic solutions the major product was diphenylethane (up to 70%), and the yield of E-stilbene exceeded that of the Z-isomer. 1H and 31P NMR measurements revealed that depending on the absence or presence of NaI, the catalytically active Ru(II)-hydride species in acidic solutions was [RuHCl(mtppms-Na)3], 2, or [RuHI(mtppms-Na)3], 5, respectively, while in basic solutions, both 2 and 5 were hydrogenated further to yield the same hydride species, cis,fac-[RuH2(H2O)(mtppms-Na)3]. [RuHI(mtppms-Na)3] proved superior to [RuHCl(mtppms-Na)3] as a catalyst for the selective hydrogenation of cinnamaldehyde to dihydrocinamaldehyde. This finding was explained by a facile formation of a (putative) dihydrogen complex [Ru(H2)I2(H2O)(mtppms-Na)2] intermediate, resulting in fast heterolytic activation of H2.
1. Introduction
Selective catalytic hydrogenation of alkynes to alkenes attracts a great deal of interest, since alkenes are potential starting materials for production of chemicals containing almost any kind of substituents [1,2,3,4,5,6,7,8,9,10]. In addition, semihydrogenation of alkynes in olefins removes potentially harmful impurities but avoids the waste of alkenes via hydrogenation of C=C double bonds.
Most often, hydrogenation or transfer hydrogenation of internal alkynes yields Z-alkenes [4,6,11,12], while examples of E-selective hydrogenations are also known [13,14,15,16,17]. It is of interest that, in several cases, the E/Z selectivity can be reversed by small changes to the applied catalyst or reaction conditions [17,18,19,20,21,22,23]. For example, reduction of diphenylacetylene in dioxane with hydrogen transfer from neat formic acid (FA) with a catalyst formed in situ from [Pd2(dba)3] (dba = dibenzylideneacetone) and tricyclohexylphosphine (PCy3) yielded E-stilbene; however, Z-stilbene was obtained with 1,4-bis(diphenylphosphinobutane) (DPPB) instead of PCy3. In this system, water plays an important role, since with DPPB, but with a 25% aqueous formic acid instead of neat FA, E-stilbene was produced selectively [18].
Studies on alkyne hydrogenation in aqueous systems are scarce [7,24,25]. Previously, we have observed that in aqueous–organic biphasic systems, diphenylacetylene could be efficiently hydrogenated with [{RuCl(mtppms-Na)2}2(µ-Cl)2] (1) as the catalyst precursor (mtppms-Na = meta-monosulfonated triphenylphosphine sodium salt) in the presence of added mtppms-Na. The E/Z selectivity depended on the pH of the aqueous phase: under acidic conditions, Z-stilbene was produced with almost completely selective alkyne semihydrogenation, while with a basic aqueous phase, the major product was the fully saturated (overhydrogenated) product, diphenylethane, together with E-stilbene and small amounts of the Z-isomer (Scheme 1) [7].
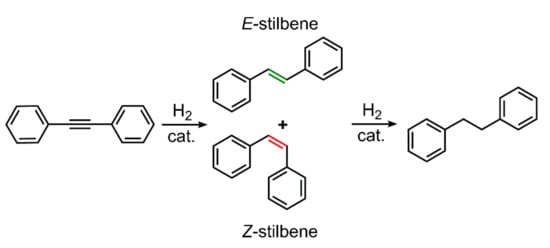
Scheme 1.
Hydrogenation of diphenylacetylene.
These investigations also revealed that the effective catalytic species formed under hydrogenation conditions were significantly more active for hydrogenation of the alkyne substrate than for reduction of the alkene product of semihydrogenation.
The Ru(II)-(mtppms-Na) or Ru(II)-(mtppts-Na3) (mtppts-Na3 = tris(meta-sulfonated)triphenylphosphine, Na-salt), as well as the related Os(II)-complexes with the same water-soluble phosphines, were studied in detail in selective hydrogenation of α,β-unsaturated aldehydes, such as cinnamaldehyde (Scheme 2). Similar to the case of alkyne hydrogenation, in aqueous–organic biphasic mixtures under acidic conditions, these catalysts facilitated only a slow (but exclusive) formation of the C=C hydrogenated product (dihydrocinnamaldehyde). Conversely, under basic conditions, only unsaturated alcohol (cinnamalcohol) was detected as a single product [26,27,28,29]. For the [{RuCl(mtppms-Na)2}2(µ-Cl)2] + mtppms-Na catalyst system it was later established, that this pH-dependent selectivity was due to formation of different hydride species under reaction conditions, i.e., [RuHCl(mtppms-Na)3], 2 at pH < 5 [30,31] and cis,fac-[RuH2(H2O)(mtppms-Na)3], 3 at pH > 7 [32].
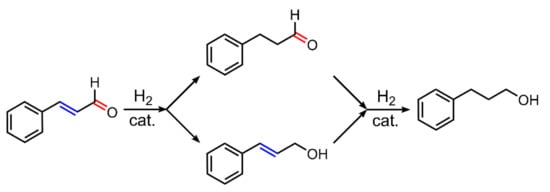
Scheme 2.
Hydrogenation of cinnamaldehyde.
Basset et al. have found that in aqueous solutions and in the presence of free mtppts-Na3, [{RuCl(mtppts-Na3)2}2(µ-Cl)2], 4 actively catalyzed the hydrogenation of propanal to propanol at 100 °C and 50 bar H2 pressure but was inactive at 35 ℃ (other conditions unchanged). However, addition of NaI to the same reaction mixture already initiated very fast hydrogenation at this low temperature (initial TOF > 2000 h–1 at 35 °C; TOF = (mol reacted substrate) × (mol catalyst)–1 × time–1). Although it was shown by 1H and 31P NMR measurements that under reaction conditions, the catalyst precursor was transformed to [RuHI(mtppts-Na3)3], interestingly, in the absence of NaI, the isolated iodo-complex did not catalyze the hydrogenation of propanal at 35 °C [33,34].
On the basis of the above findings, we conceived that addition of iodide may change both the rate and selectivity of hydrogenation of internal alkynes catalyzed by [{RuCl(mtppms-Na)2}2(µ-Cl)2], 1 in aqueous systems. Therefore, we carried out hydrogenation of diphenylacetylene with 1 as the catalyst precursor in the presence of NaI and determined the rate (conversion) and chemo- and stereoselectivity of the reaction as a function of the pH of the aqueous phase. 1H and 31P NMR measurements were also carried out to establish the molecular state of the effective catalyst.
2. Results and Discussion
In our previous work, hydrogenation of diphenylacetylene as a function of pH was studied with the use of [{Ru(mtppms-Na)2}2(µ-Cl)2], 1 and mtppms-Na dissolved in aqueous phosphate buffer (Na2HPO4/NaH2PO4/H3PO4) of appropriate pH [24]. In the present communication, we disclose the results of diphenylacetylene hydrogenation with the same catalyst system supplemented with NaI ([NaI]/[Ru] = 10). Figure 1 shows the observed conversions both in the absence and presence of NaI.
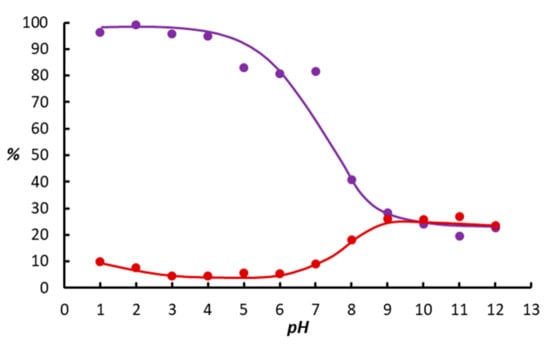
Figure 1.
Conversion of the hydrogenation of diphenylacetylene as a function of the pH in the absence (●) and presence of NaI (●). Conditions: [{Ru(mtppms-Na)2}2(µ-Cl)2] = 6.6 mg (6.79 × 10–3 mmol Ru), mtppms = 8.1 mg (2.03 × 10–2 mmol), alkyne = 0.5 mmol, P(H2) = 1 bar, V(toluene) = 1 mL, V (0.2 M aqueous Na-phosphate buffer) = 2 mL, T = 50 °C, t = 3 h.
It is seen from Figure 1, that on addition of NaI, the reaction proceeded with a high rate, and conversions above 90% were observed in strongly acidic solutions in contrast with the conversions below 10% in the absence of NaI under otherwise identical conditions. However, Figure 1 also reveals, that in basic solutions (9 ≥ pH ≥ 12), conversions around 25% were achieved independent of the absence or presence of NaI in the reaction mixture.
The selectivity of diphenylacetylene hydrogenation with the 1 + mtppms-Na catalyst precursor is shown on Figure 2 both in the absence (A) and in the presence (B) of NaI.
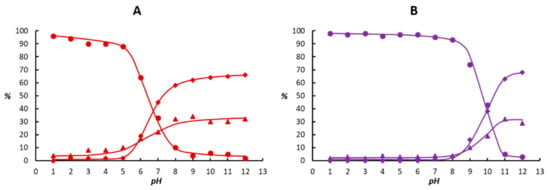
Figure 2.
Product distribution in the hydrogenation of diphenylacetylene as a function of the pH in the absence (A) and in the presence (B) of NaI. Conditions: (A) [{Ru(mtppms-Na)2}2(µ-Cl)2] = 6.6 mg (6.79 × 10–3 mmol Ru), mtppms = 8.1 mg (2.03 × 10–2 mmol), alkyne = 89.1 mg (0.5 mmol), P(H2) = 1 bar, V(chlorobenzene) = 1 mL, V(0.2 M aqueous Na-phosphate buffer) = 2 mL, T = 50 °C, t = 3 h. (B) Same as A, but with addition of 6.79 × 10–2 mmol NaI, and 1 mL of toluene instead of 1 mL chlorobenzene. ●● Z-stilbene; ▲▲ E-stilbene; ♦♦ 1,2-diphenylethane.
Figure 2 demonstrates that the pH of the aqueous phase (containing the dissolved catalyst) influenced the product selectivity both in the absence and presence of NaI in a similar, although not identical manner. In acidic solutions, the reaction was selective to formation of stilbenes accompanied by negligible formation of diphenylethane. In addition, in the lower pH region (approximately pH 1–5 without NaI and pH 1–8 with NaI) the hydrogenation was also stereoselective to formation of Z-stilbene, with GC yields around 95% (A) or 98% (B). However, at pH 5 (A) and 8 (B), formation of E-stilbene and diphenylethane set on with a concomitant decrease in the yield of the Z-alkene. Finally, at pH 12, the overhydrogenated product, diphenylethane, became the major species in the reaction mixture (close to 70% in both cases, A and B). Of the two isomeric stilbenes, E-stilbene was preferred at pH 12 (approximately 30%), while the yield of Z-stilbene did not exceed a few %. What is characteristic, though, is the pH value at which diphenylethane became the major product: approximately pH 7 in the absence and pH 10 in the presence of NaI. It can be concluded from these results that addition of NaI was advantageous not only to provide reasonably faster reactions under acidic conditions, but also to extend the pH region of chemo- and stereoselectivity by about 3 pH units (from 5 to 8). However, in basic solutions, no significant effect of NaI on the reaction rate and selectivity could be observed.
In an attempt to determine the molecular composition of the immediate catalytic species, the reaction of 1, mtppms and NaI under an H2 atmosphere was investigated by spectroscopic methods. In acidic solutions and at room temperature, 1 + mtppms + H2 (atmospheric pressure) yielded a cherry red solution which turned purple on addition of NaI ([NaI]/[Ru] = 10). Conversely, under basic conditions, yellow solutions were obtained independent of the absence or presence of NaI. The processes could be quantitatively described on the basis of 1H and 31P NMR spectroscopies and with the use of pH-potentiometry. It was reported earlier that in acidic solutions, the major species was [RuHCl(mtppms-Na)3], 2 (together with < 10% [{RuH(mtppms-Na)2}2(µ-Cl)2], 6) [30,31], while in basic solutions cis,fac-[RuH2(H2O)(mtppms-Na)3], 3 could be identified as single species [32]. In contrast, when the acidic solutions of 1 + mtppms-Na also contained NaI, instead of the signals of 2 (δ = –17.94 ppm, q) and 6 (δ = –8.71 ppm, t), the 1H NMR spectrum contained only a resonance due to [RuHI(mtppms-Na)3], 5 at δ = –15.0 ppm (q) [32], which is very close to that of [RuHI(mtppts-Na3)3], δ = –15.4 ppm (q), known from the literature [34]. In more basic solutions (pH > 6), the resonance of cis,fac-[H2Ru(H2O)(mtppms-Na)3], 3 also appeared, and became dominant at high pH. These changes are depicted on Figure 3. The effect of I– is shown not only by the lack of the Ru(II)-monohydride dimer (the iodo analogue of 6), but also by the shift of the Ru(II)-dihydride/Ru(II)-monohydride equilibrium to more basic conditions.
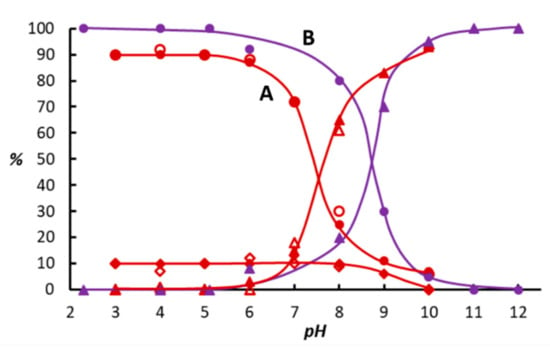
Figure 3.
Distribution of the various Ru(II)-hydride species formed in aqueous solutions of [{RuCl(mtppms-Na)2}2(µ-Cl)2], 1 + mtppms-Na under H2, as a function of pH in the absence (red, A) and in the presence (purple, B) of NaI, determined by 1H NMR (open symbols) and 31P NMR spectroscopy (filled symbols). [RuHCl(mtppms-Na)3], 2 (●,◯); [{RuH(mtppms-Na)2}2(µ-Cl)2], 6 (♦,⋄); [RuHI(mtppms-Na)3], 5 (●); cis,fac- [RuH2(H2O)(mtppms-Na)3], 3 (▲,△,▲). Conditions: A) [Ru] = 2.4 × 10−2 M, [mtppms-Na]/[Ru] = 3, T = 50 °C, P(H2) = 1 bar,; B) same as A but with [NaI] = 0.2 M. The pH of solutions for spectral measurements were adjusted by appropriate amounts of HCl and KOH.
Comparison of the pH-dependent concentration distribution of the Ru(II)-hydride species (Figure 3) and the reaction rates and selectivities of diphenylacetylene hydrogenation (Figure 1 and Figure 2) allows the following conclusions. First, [RuHI(mtppms-Na)3], 5, dominant in acidic solutions, seems to be a more potent catalyst for the hydrogenation of the alkyne triple bond than its chloro analogue, [RuHCl(mtppms-Na)3], 2, since in this pH region, conversions of diphenylacetylene to stilbenes in the presence of NaI far exceed those determined in its absence. Second, in basic solutions, both [RuHCl(mtppms-Na)3], 2 and [RuHI(mtppms-Na)3], 5 are hydrogenated further to the same halide-free species, cis,fac-[RuH2(H2O)(mtppms-Na)3], 3. Consequently, the conversion of diphenylacetylene and the product distribution become independent of the absence or presence of NaI and converge to the same values. Nevertheless, in comparison with 2, the transformation of 5 to 3 takes place in slightly more basic solutions, therefore the region of stereoselective formation of Z-stilbene is also shifted to a higher pH. The latter phenomenon may be the consequence of the stepwise formation of Ru(II)-dihydride 3 in two consecutive heterolytic H2 activation steps (Equations (1)–(3), X− = Cl− or I−) rather than by oxidative addition of H2 onto a Ru(II)-precursor species, as demonstrated earlier also by pH potentiometric studies [30,31].
½ [{RuCl(mtppms-Na)2}2(µ-Cl)2] + mtppms-Na + H2 = [RuHCl(mtppms-Na)3] + H+ + Cl–
½ [{RuCl(mtppms-Na)2}2(µ-Cl)2] + mtppms-Na + I– + H2 = [RuHI(mtppms-Na)3] + H+ + 2Cl–
[RuHX(mtppms-Na)3] + H2 + H2O = cis,fac-[RuH2(H2O)(mtppms-Na)3] + H+ + X– (X = Cl or I)
Formation of the Ru(II)-dihydride catalyst 3, which shows a preference for the formation of E-stilbene and overhydrogenation to diphenylethane, requires release of one H+ and one X– (Cl– or I–). Obviously, this reaction (Equation (3)) is facilitated in basic solutions. Conversely, the hydration enthalpy of iodide (–298 kJ/mol) is less than that of chloride (–369 kJ/mol) [35]; therefore, solvation of the released iodide contributes less to the overall reaction enthalpy and makes reaction (3) less facile compared to hydrogenation of the chloro-complex 2.
For comparison, we have briefly studied the hydrogenation of cinnamaldehyde catalyzed by 1. Similar observations were made to those in the case of diphenylacetylene hydrogenation. In basic solutions, cinnamaldehyde was reduced to cinnamalcohol independent of the absence or presence of NaI. Earlier, we established that this reaction was also catalyzed by the halide-free cis,fac-[H2Ru(H2O)(mtppms-Na)3], 3 [30,31], which was shown in the present study (Figure 3) to be the major species in solutions containing NaI. However, in acidic solutions, the rate of hydrogenation of the C=C bond in cinnamaldehyde increased substantially (Figure 4) in the presence of NaI (25% vs. 2% conversion to 3-phenylpropanal at pH 3).
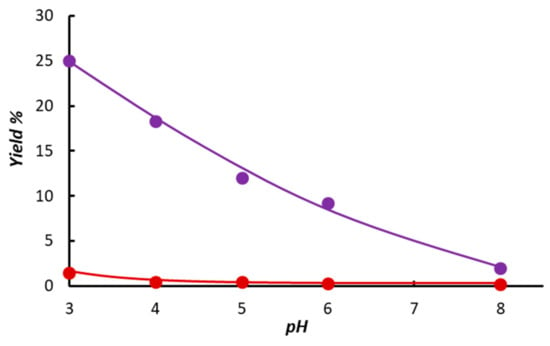
Figure 4.
Yield of 3-phenylpropanal in hydrogenation of cinnamaldehyde catalyzed by [{RuCl(mtppms-Na)2}2(µ-Cl)2], 1 + mtppms in the absence (●) and presence of NaI (●). Conditions: [Ru] = 3.3 × 10−3 M, [mtppms-Na]/[Ru] = 10, [alkyne]/[Ru] = 40, T = 80 °C, P(H2) = 1 bar, t = 1 h, V(0.2 M aqueous Na-phosphate buffer) = 3 mL; [NaI] = 0 M (●) or 33 × 10–3 M (●).
For an explanation of the accelerating effect of NaI on hydrogenation of the C=C bond in cinnamaldehyde one should consider the general mechanism suggested for alkene hydrogenations catalyzed by 1 + mtppms-Na in acidic aqueous solutions [36].
The higher catalytic hydrogenation activity of [RuHI(mtppms-Na)3] in comparison to [RuHCl(mtppms-Na)3] may be explained by the suggested reaction mechanism (Figure 5). As mentioned earlier, 1H and 31P NMR measurements showed that under acidic conditions, the dominant Ru(II)-hydride species is [RuHX(mtppms-Na)3], X=Cl– (2), or I– (5) in the presence of NaI (Figure 3). It is also known that in several cases (e.g., in hydrogenation of crotonic acid) excess mtppms-Na inhibits the reaction [36]; therefore, it is reasonable to assume that Ru(II) enters the catalytic cycle as a bisphosphine species [RuHX(H2O)P2] (P = mtppms-Na). Replacement of the solvent by the alkene substrate (Step 1) yields [RuHX(alkene)P2], and in this reaction we do not expect a significant difference between X– = Cl– or I–. In Step 2, hydride migration and solvent coordination supplies [RuX(H2O)(alkyl)P2]. Protonation of this Ru(II)-alkyl intermediate and halide coordination results in formation of the alkane product (Step 3) with concomitant formation of [RuX2(H2O)P2]. Since under acidic conditions, the proton concentration is higher than that of the Ru(II)-catalyst, again, no significant rate difference is expected between the protonation of [RuX(H2O)(alkyl)P2] with X– = Cl– or I–. Reaction of [RuX2(H2O)P2] with H2 (Step 4) may yield the dihydrogen complex [Ru(H2)X2(H2O)P2] (or the pentacoordinate [Ru(H2)X2P2]), which in Step 5 releases a proton and a halide to yield [RuHX(H2O)P2] and initiates a new catalytic cycle. Inclusion of a dihydrogen complex into the reaction mechanism may seem somewhat speculative, but not unreasonable. We did not directly observe [Ru(H2)X2(H2O)P2] in hydrogenation of C=C unsaturated substrates (alkenoic acids [36], or unsaturated aldehydes); however, this does not exclude their kinetic role. In fact, in aqueous solutions, coordination of dihydrogen (as η2-H2) was found more favourable thermodynamically than that of H2O [37]. Furthermore, formation of dihydrogen complexes (non-classical hydrides) in Ru(II)-mtppms-Na systems was unambiguously detected in aqueous solutions at pH ≥ 7.6 and P(H2) = 5 bar pressure [32]. We believe that it is the formation of [Ru(H2)I2(H2O)P2] (Step 4) which leads to higher catalytic activities in the presence of NaI. The driving force of the formation of dihydrogen complexes mostly arises from back donation from the metal to the H2 ligand, and the back donation of Ru(II) in [Ru(H2)I2(H2O)P2] is expected to be stronger than in [Ru(H2)Cl2(H2O)P2] due to the more electron-rich iodide ligands.
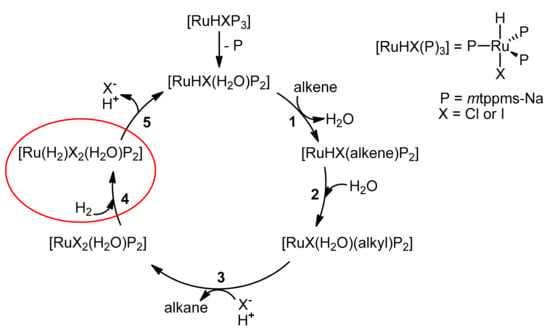
Figure 5.
Suggested general mechanism of alkene hydrogenation catalyzed by [RuHX(mtppms-Na)3], in acidic aqueous solutions (X = Cl or I).
The suggested catalytic cycle also allows an explanation of the chemoselectivity of diphenylacetylene hydrogenation to stilbenes, with the assumption that in Step 1, the alkyne coordinates preferentially over the already formed alkene(s). This assumption has been verified experimentally through semihydrogenation of alkynes [1,2].
3. Materials and Methods
All reagents of commercial quality were used without further purification. Diphenylacetylene was purchased from Sigma-Aldrich (St. Louis, MO, USA); chlorobenzene and toluene from VWR International (West Chester, PA, USA). mtppms-Na [38] and [{RuCl(mtppms-Na)2}2(µ-Cl)2], 1 [38] were prepared by known procedures. All manipulations were performed under inert atmosphere using standard Schlenk techniques. Reaction mixtures were analyzed by gas chromatography (HP5890, Agilent Technologies (Santa Clara, CA, USA); Chrompack WCOT Fused Silica 30 m × 32 mm CP WAX52CB column; FID; carrier gas: argon). The products were quantified by their peak areas. They were identified by their retention times compared to those of authentic samples and by 1H NMR spectroscopy (Bruker Avance 360 instrument, CDCl3 solutions). Ru(II)-hydrides were identified in solution by their 1H and 31P NMR spectra (Bruker AV360 instrument, Bruker (Billerica, MA, USA) recorded in D2O solutions. Chemical shifts (δ, ppm) were referenced to 3-(trimethylsilyl)-1-propanesulfonic acid Na-salt, DSS (1H NMR) and to 85% H3PO4 (31P NMR).
Catalytic hydrogenation of diphenylacetylene: In a typical reaction, 2 mL of 0.2 M aqueous buffer (NaH2PO4/Na2HPO4) containing 10.4 mg (0.069 mmol) NaI was purged with H2 for 15 min at room temperature. 6.6 mg (0.0069 mmol) [{RuCl(mtppms-Na)2}2(µ-Cl)2] and 8.1 mg (0.020 mmol) mtppms-Na were dissolved in this solution and the reaction mixture was heated to 50 °C. Depending on the actual pH, the characteristic purple color of [RuHI(mtppms-Na)3] or yellow color of cis,fac-[RuH2(H2O)(mtppms-Na)3] developed in about 3 min. At this point, 0.5 mmol of diphenylacetylene dissolved in 1 mL of toluene was added and the mixture was stirred further vigorously for 3 h. Samples withdrawn from the organic phase of the reaction mixture were passed through a small silica plug and were analyzed by gas chromatography using the equipment described above with T(injector and detector) = 250 °C, T(oven) = 200 °C.
Catalytic hydrogenations of cinnamaldehyde were carried out with the same procedure under conditions of Figure 4, with 3 mL of Na-phosphate buffer as the aqueous phase. Samples were analyzed by gas chromatography using the equipment described above with T(injector and detector) = 250 °C, T(oven) = 130 °C isotherm (15 min), ramp to 200 °C (35 °C/min), hold at 200 °C (5 min).
4. Conclusions
In this work, we established that in acidic aqueous solutions, addition of NaI strongly increased the rate of hydrogenation of diphenylacetylene to Z-stilbene (and that of cinnamaldehyde to dihydrocinnamaldehyde) catalyzed by the water-soluble Ru(II)-complex with sulfonated phosphine ligands, [{RuCl(mtppms-Na)2}2(µ-Cl)2], 1. Conversely, in basic solutions, neither the reaction rate nor the product selectivity changed in the presence of NaI. 1H and 31P NMR measurements revealed that in acidic solutions, the rate changes were brought about by the change in the actual catalytic species, from [RuHCl(mtppms-Na)3], 2 to [RuHI(mtppms-Na)3], 5, in the absence and presence of NaI, respectively. In contrast, in basic solutions, the catalytically active Ru(II)-hydride was cis,fac-[RuH2(H2O)(mtppms-Na)3], 3, both in the absence and presence of NaI. It is suggested that the higher catalytic activity of the iodo-Ru(II)-hydride, 5, compared to its chloro-analogue, 2, may be due to easier formation of a putative dihydrogen complex, [Ru(H2)I2(H2O)P2] resulting in faster heterolytic activation of dihydrogen. This iodide effect extends the pH region of high Z-selectivity by about 3 pH units and provides an example of the importance of metal-halide moieties [39] in transition-metal organometallic catalysis.
Author Contributions
Conceptualization, Á.K. and F.J.; methodology, H.H.H. and G.P.; investigation, H.H.H. and G.P.; writing—original draft preparation, Á.K. and F.J.; writing—review and editing, all authors; visualization, H.H.H. and G.P.; funding acquisition, Á.K., F.J. and G.P. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported by the EU and co-financed by the European Regional Development Fund under the projects GINOP-2.3.3–15-2016–00004 and GINOP 2.3.2–15-2016–00008. Project TKP2020-NKA-04 has been implemented with support provided by the National Research, Development and Innovation Fund of Hungary, financed under the 2020-4.1.1-TKP2020 funding scheme. The financial support of the Hungarian National Research, Development and Innovation Office (FK-128333) is greatly acknowledged.
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors are grateful to Julianna Molnár for her participation in some parts of the experimental work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kluwer, A.M.; Elsevier, C.J. Homogeneous Hydrogenation of Alkynes and Dienes. In Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Volume 1, pp. 375–411. [Google Scholar] [CrossRef]
- Chaloner, P.A.; Esteruelas, M.A.; Joó, F.; Oro, L.A. Homogeneous Hydrogenation; Kluwer: Dordrecht, The Netherlands, 1994; pp. 5–85. [Google Scholar] [CrossRef]
- Wienhöfer, G.; Westerhaus, F.Á.; Jagadeesh, R.V.; Junge, K.; Junge, H.; Beller, M. Selective iron-catalyzed transfer hydrogenation of terminal alkynes. Chem. Commun. 2012, 48, 4827–4829. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Reddy, A.S.; Sandeep, K.; Kalyani, A. Advances of chemoselective and/or stereoselective semihydrogenation of alkynes. Tetrahedron Lett. 2018, 50, 419–429. [Google Scholar] [CrossRef]
- Decker, D.; Drexler, H.-J.; Heller, D.; Beveries, T. Homogeneous catalytic transfer semihydrogenation of alkynes—An overview of hydrogen sources, catalysts and reaction mechanisms. Catal. Sci. Technol. 2020, 10, 6449–6463. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, Y.; Leng, X.; Huang, Z. An Amine-Assisted Ionic Monohydride Mechanism Enables Selective Alkyne cis-Semihydrogenation with Ethanol: From Elementary Steps to Catalysis. J. Am. Chem. Soc. 2021, 143, 4824–4836. [Google Scholar] [CrossRef] [PubMed]
- Joó, F.; Kathó, Á. Two-Phase Aqueous Hydrogenations. In Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Volume 3, pp. 1327–1359. [Google Scholar] [CrossRef]
- Garbe, M.; Budweg, S.; Papa, V.; Wei, Z.; Hornke, H.; Bachmann, S.; Scalone, M.; Spannenberg, A.; Jiao, H.; Junge, K. Chemoselective Semihydrogenation of Alkynes Catalyzed by Manganese(I)-PNP Pincer Complexes. Catal. Sci. Technol. 2020, 10, 3994–4001. [Google Scholar] [CrossRef]
- Both, N.F.; Spannenberg, A.; Junge, K.; Beller, M. Low-Valent Molybdenum PNP Pincer Complexes as Catalysts for the Semihydrogenation of Alkynes. Organometallics 2022. [Google Scholar] [CrossRef]
- Thiel, N.O.; Teichert, J.F. Stereoselective alkyne semihydrogenations with an air-stable copper(I) catalyst. Org. Biomol. Chem. 2016, 14, 10660–10666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.M.; Gouda, C.; Gonnade, R.G.; Punji, B. Room temperature Z-selective hydrogenation of alkynes by hemilabile and non-innocent (NNN)Co(ii) catalysts. Catal. Sci. Technol. 2022, 12, 1843–1849. [Google Scholar] [CrossRef]
- Zubar, V.; Sklyaruk, J.; Brzozowska, A.; Rueping, M. Chemoselective Hydrogenation of Alkynes to (Z)-Alkenes Using an Air-Stable Base Metal Catalyst. Org. Lett. 2020, 22, 5423–5428. [Google Scholar] [CrossRef]
- Ekebergh, A.; Begon, R.; Kann, N. Ruthenium-Catalyzed E-Selective Alkyne Semihydrogenation with Alcohols as Hydrogen Donors. J. Org. Chem. 2020, 85, 2966–2975. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Dutta, I.; Saha, S.; Das, S.; Pati, S.K.; Choudhury, J.; Bera, J.K. An Annelated Mesoionic Carbene (MIC) Based Ru(II) Catalyst for Chemo- and Stereoselective Semihydrogenation of Internal and Terminal Alkynes. Organometallics 2020, 39, 3212–3223. [Google Scholar] [CrossRef]
- Fetzer, M.N.A.; Tavakoli, G.; Klein, A.; Prechtl, M.H.G. Ruthenium-Catalyzed E-Selective Partial Hydrogenation of Alkynes under Transfer-Hydrogenation Conditions using Paraformaldehyde as Hydrogen Source. ChemCatChem 2021, 13, 1317–1325. [Google Scholar] [CrossRef]
- Hale, D.J.; Ferguson, M.J.; Turculet, L. (PSiP)Ni-Catalyzed (E)-Selective Semihydrogenation of Alkynes with Molecular Hydrogen. ACS Catal. 2022, 12, 146–155. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Weber, S.; Csendes, Z.; Ammaturo, A.; Fleissner, S.; Hoffmann, H.; Veiros, L.F.; Kirchner, K. E-Selective Manganese-Catalyzed Semi hydrogenation of Alkynes with H2 Directly Employed or In Situ-Generated. ACS Catal. 2022, 12, 2253–2260. [Google Scholar] [CrossRef]
- Shen, R.; Chen, T.; Zhao, Y.; Qiu, R.; Zhou, Y.; Yin, S.; Wang, X.; Goto, M.; Han, L.B. Facile Regio- and Stereoselective Hydrometalation of Alkynes with a Combination of Carboxylic Acids and Group 10 Transition Metal Complexes: Selective Hydrogenation of Alkynes with Formic Acid. J. Am. Chem. Soc. 2011, 133, 17037–17044. [Google Scholar] [CrossRef]
- Fu, S.; Chen, Y.; Liu, X.; Shao, Z.; Luo, S.P.; Liu, Q. Ligand-Controlled Cobalt-Catalyzed Transfer Hydrogenation of Alkynes: Stereodivergent Synthesis of Z- and E-Alkenes. J. Am. Chem. Soc. 2016, 138, 8588–8594. [Google Scholar] [CrossRef]
- Luo, F.; Pan, C.; Wang, W.; Ye, Z.; Cheng, J. Palladium-catalyzed reduction of alkynes employing HSiEt3: Stereoselective synthesis of trans- and cis-alkenes. Tetrahedron 2010, 66, 1399–1403. [Google Scholar] [CrossRef]
- Richmond, E.; Moran, J. Ligand Control of E/Z Selectivity in Nickel-Catalyzed Transfer Hydrogenative Alkyne Semireduction. J. Org. Chem. 2015, 80, 6922–6929. [Google Scholar] [CrossRef]
- Chen, K.; Zhu, H.; Li, Y.; Peng, Q.; Guo, Y.; Wang, X. Dinuclear Cobalt Complex-Catalyzed Stereodivergent Semireduction of Alkynes: Switchable Selectivities Controlled by H2O. ACS Catal. 2021, 11, 13696–13705. [Google Scholar] [CrossRef]
- Tokmic, K.; Fout, A.R. Alkyne Semihydrogenation with a Well Defined Nonclassical Co-H2 Catalyst: A H2 Spin on Isomerization and E-selectivity. J. Am. Chem. Soc. 2016, 138, 13700–13705. [Google Scholar] [CrossRef]
- Horváth, H.H.; Joó, F. Stereoselective homogeneous catalytic hydrogenation of disubstituted alkynes in aqueous-organic biphasic media. React. Kinet. Catal. Lett. 2005, 85, 355–360. [Google Scholar] [CrossRef]
- Le, T.X.; Merola, J.S. Synthesis and Reaction Chemistry of Water-Soluble mer-(Me3P)3Ir(H)(H)Cl: Activation by Water of Alkyne Insertion into an Ir-H Bond. Organometallics 1993, 12, 3798–3799. [Google Scholar] [CrossRef]
- Bényei, A.; Joó, F. Organometallic catalysis in aqueous solutions: The biphasic transfer hydrogenation of aldehydes catalyzed by water-soluble phosphine complexes of ruthenium, rhodium and iridium. J. Mol. Catal. 1990, 58, 151–163. [Google Scholar] [CrossRef]
- Grosselin, J.M.; Mercier, C.; Allmang, G.; Grass, F. Selective Hydrogenations of α,β-Unsaturated Aldehydes in Aqueous-Organic Two-Phase Solvent System Using Ruthenium or Rhodium Complexes of Sulfonated Phosphines. Organometallics 1991, 10, 2126–2133. [Google Scholar] [CrossRef]
- Sánchez-Delgado, R.A.; Medina, M.; López-Linares, F.; Fuentes, A. The chemistry and catalytic properties of ruthenium and osmium compounds. Part 7. Regioselective hydrogenation of cinnamaldehyde (3-phenyl-2-propenal) catalyzed by Ru and Os triphenylphosphine complexes in homogeneous solution and by meta-sulfonatophenyl-diphenyldiphosphine (TPPMS) and tris-meta-sulfonato-phenylphosphine (TPPTS) derivatives in an aqueous biphasic system. J. Mol. Catal. A Chem. 1997, 116, 167–177. [Google Scholar] [CrossRef]
- Lopez-Linares, F.; Gonzalez, M.G.; Paez, D.E. The regioselective biphasic hydrogenation of trans-cinnaldehyde by meta sulfonatophenyl-diphenylphosphine (TPPMS) Ru(II) and Os(II) species. The influence of ionic strength, ligand tensoactivity and metal nature in the selective production of the unsaturated alcohol. J. Mol. Catal. A Chem. 1999, 145, 61–68. [Google Scholar] [CrossRef]
- Joó, F.; Kovács, J.; Bényei, A.C.; Kathó, Á. Solution pH: A Selectivity Switch in Aqueous Organometallic Catalysis—Hydrogenation of Unsaturated Aldehydes Catalyzed by Sulfonatophenylphosphane—Ru Complexes. Angew. Chem. Int. Ed. 1998, 37, 969–970. [Google Scholar] [CrossRef]
- Joó, F.; Kovács, J.; Bényei, A.C.; Kathó, Á. The effects of pH on the molecular distribution of water soluble ruthenium(II) hydrides and its consequences on the selectivity of the catalytic hydrogenation of unsaturated aldehydes. Catal. Today 1998, 42, 441–448. [Google Scholar] [CrossRef]
- Papp, G.; Horváth, H.; Laurenczy, G.; Szatmári, I.; Kathó, Á.; Joó, F. Classical and non-classical phosphine-Ru(II)-hydrides in aqueous solutions: Many, various, and useful. Dalton Trans. 2013, 42, 521–529. [Google Scholar] [CrossRef]
- Fache, E.; Santini, C.; Senocq, F.; Basset, J.M. Homogeneous catalysis in water Part III. The catalytic hydrogenation of propionaldehyde with (RuCl2L2)2, RuHClL3, RuH(OAc)L3, RuH2L4, RuHlL3, RuCl2(CO)2L2 and [Ru(OAc)(CO)2L]2, (L = P(C6H4-mSO3Na)3. 3H2O): A kinetic investigation of the salt effect in water. J. Mol. Catal. 1992, 72, 337–350. [Google Scholar] [CrossRef]
- Fache, E.; Santini, C.; Senocq, F.; Basset, J.M. Homogeneous catalysis in water Part II. Synthesis and characterization of ruthenium water-soluble complexes. J. Mol. Catal. 1992, 72, 331–336. [Google Scholar] [CrossRef]
- Burgess, J. Ions in Solution; Woodhead Publishing: Sawston, UK, 1999; pp. 45–61. [Google Scholar] [CrossRef]
- Tóth, Z.; Joó, F.; Beck, M.T. Homogeneous Hydrogenations in Aqueous Solutions Catalyzed by Ruthenium-Phosphine Complexes. Inorg. Chim. Acta 1980, 42, 153–161. [Google Scholar] [CrossRef]
- Kubas, G.J.; Burns, C.J.; Khaisha, G.R.K.; Van Der Sluys, L.S.; Kiss, G.; Hoff, C.D. Dihydrogen: A Better Ligand Than Water? IR and X-Ray Evidence for Aquo Coordination in W(CO)3(PR3)2(H2O), Thermodynamics of H2O versus η2-H2 Binding, and H2O/D2 Isotopic Exchange. Implications on the Biological Activation of Hydrogen. Organometallics 1992, 11, 3390–3404. [Google Scholar] [CrossRef]
- Joó, F.; Kovács, J.; Kathó, Á.; Bényei, A.C.; Decuir, T.; Darensbourg, D.J. (Meta-Sulfonatophenyl)diphenylphosphine sodium salt and its complexes with rhodium(I), ruthenium(II) and iridium(I). Inorg. Synth. 1998, 32, 1–8. [Google Scholar] [CrossRef]
- Fagnou, K.; Lautens, M. Halide Effects in Transition Metal Catalysis. Angew. Chem. Int. Ed. 2002, 41, 26–47. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).