Abstract
Transition metal catalysts play a vital role in a wide range of industrial organic processes. The large-scale production of chemicals relying on catalyzed organic reactions represents a sustainable approach to supply society with end products for many daily life applications. Homogeneous (mainly for academic uses) and heterogeneous (crucial in industrial processes) metal-based catalysts have been developed for a plethora of organic reactions. The search for more sustainable strategies has led to the development of a countless number of metal-supported catalysts, nanosystems, and electrochemical and photochemical catalysts. In this work, although a vast number of transition metals can be used in this context, special attention is devoted to Ir- and Pd-based catalysts in the industrial manufacture of pharmaceutical drugs. Pd is by far the most widely used and versatile catalyst not only in academia but also in industry. Moreover, Ir-based complexes have emerged as attractive catalysts, particularly in asymmetric hydrogenation reactions. Ir- and Pd-based asymmetric reductions, aminations, cross-coupling reactions, and C–H activation are covered herein in the production of biologically active compounds or precursors; adaptation to bulk conditions is particularly highlighted.
1. Introduction
Transition metal is the common term to refer to the d-block elements of the periodic table [1]. The main feature of these elements is their partially filled d subshell in the metallic state. This characteristic confers transition metals with the ability to adopt diverse and interchangeable oxidation states, which allows them to exchange electrons in a chemical reaction undergoing oxidation or reduction processes [2]. In addition, transition metals form complexes with numerous reaction reagents [3,4,5]. Transition metals are available either as homogeneous [6] or heterogeneous catalysts, including reactions based on electrochemical processes [7]. The former are largely employed in academic research, while the latter represent the preferred choice in industry, as they can be easily separated from the products using only standard filtration operations.
Inspired by nanosized metal catalysts (e.g., nanoclusters and nanoparticles) [8] or supported metals [9], the immobilization of homogeneous metal-based complexes emerged as a topic of interest. Many such systems were initially reported to exhibit a series of disadvantages that, in principle, hindered their industrial implementation: stability, costs, metal contamination of the final product, or low reaction rates and selectivities [10]. Nevertheless, in subsequent years, new immobilized metal catalysts with improved properties to overcome such problems were marketed [11]. In the search for processes endowed with atomic economy and sustainability, photoredox catalysts with metal complexes have also been developed [12].
Taken together, these properties make transition metals key elements for the catalysis of organic reactions not only in academia but also in industry [13,14]. Thus, several methods, ligands, and conditions for transition metal catalysis of organic reactions have been explored, reviewed, and increasingly reported in the literature in the past few decades. Numerous key industrial processes are based on transition-metal-catalyzed reactions, such as the Wacker process, metathesis reactions, carbonylations, or hydrogenations, among others [15].
A recent editorial pointed out the citation bias between industrial and academic research papers [16]. Additionally, literature reviews covering organic synthesis at the industrial level are scarce. Several factors can explain this difference despite the increasingly important role in the chemical synthesis of transition metal catalysts in the production of fine chemicals. One explanation might be the little knowledge of industrial chemistry taught in chemistry degrees in academia, which focuses mainly on the transformation of raw materials and the production of bulk chemicals. Thus, these transition-metal-catalyzed processes remain outside the scope of this literature review. In contrast, we focus on transition-metal-catalyzed organic reactions that are used in the large-scale production of investigational new drugs. In particular, palladium, which is by far the transition metal most widely used in catalysis in both academia and industry [17], and iridium are the bases of the review. The latter has emerged as a very versatile catalyst for C–H borylation of heteroarenes (starting materials for cross-coupling reactions), and particularly for asymmetric hydrogenation reactions of olefins and ketimines [18]. In the present review, key processes with Ir and Pd complexes are described in the industrial preparation of pharmaceutical drugs (asymmetric reductions, aminations, cross-coupling reactions, and C–H activation). The intention was not to perform a thorough literature review on the selected topic but to draw the attention of academic chemists to the relevance of scaling up transition-metal-catalyzed organic reactions in the pharmaceutical industry. The time frame selected was the last five years.
2. Asymmetric Reductions
2.1. Iridium
2.1.1. JAK Inhibitor Tofacitinib
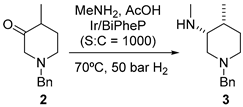
Tofacitinib (1) is a Janus-activated kinase (JAK) inhibitor approved for the treatment of rheumatoid arthritis (2012) and active psoriatic arthritis (2017) [19]. The challenge in the synthesis of 1 relies on obtaining the chiral piperidine fragment (Figure 1). The key step is the asymmetric reductive amination of 2 to obtain intermediate 3. The procedure described by Pfizer involves the reductive amination of 2 with methyl amine and sodium borohydride/acetic acid as the reducing reagent. Compound 3 is obtained in 92% yield as a mixture of the corresponding four diastereomeric forms. Thus, it must be submitted to diastereomeric crystallization (32% HCl in toluene/EtOH) followed by chiral resolution with di-p-toluoyl-l-tartaric acid (l-DTTA) or (+)-phencyphos to yield the almost enantiomerically pure salt (99% ee) with a poor overall yield (25%) [20].
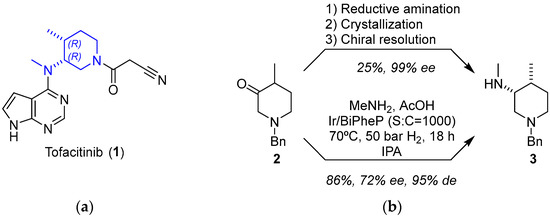
Figure 1.
(a) Structure of tofacitinib (1) highlighting the piperidine fragment; (b) key step of the synthetic procedure showing Pfizer’s procedure (top) and the improved approach using iridium as catalyst (bottom).
An elegant solution is the one-pot asymmetric reductive amination (ARA) of 2 under dynamic kinetic resolution (DKR) [21]. In that study, Verzijl and coworkers screened the reaction conditions, including the ligand (12 different chiral phosphine ligands), solvent (CH2Cl2, EtOAc, MeOH, and isopropyl alcohol), the use or not of I2 as an additive (commonly used in combination with Ir catalysts), and the ratio of substrate to catalyst using high-throughput experimentation. Using these combinations, the authors tested 96 different catalyst mixtures in a liquid-handling robot in order to optimize the process. They discovered that imine synthesis could take place in AcOH without the need to remove the water generated during the reaction; such mildly acidic conditions were proved to be useful not only to promote the formation of the corresponding transient imine but also to accomplish the epimerization of the imine, as well as 2, a prerequisite for achieving the DKR process. The use of strongly basic conditions (t-BuOK) unexpectedly led to low yields of 3, together with low ee values, presumably due to decomposition of the catalyst into achiral species. Remarkably, the mildly acidic environment was fully compatible with the iridium-catalyzed asymmetric hydrogenation step. As a result, 3 was obtained in 86% yield and 72% ee under optimized conditions using a combination of ARA and DKR processes with the bidentate phosphine ligand BiPheP, featuring axial chirality. Although the enantiomeric purity of 3 was lower than in the existing procedure, the yield significantly surpassed the theoretical maximum of 50% without epimerization.
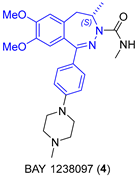
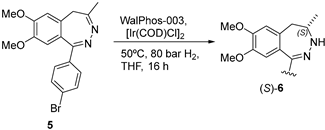
2.1.2. BET Inhibitor BAY 1238097
BAY 1238097 (4) belongs to the family of inhibitors of bromodomain and extraterminal (BET) proteins, which are potential druggable anticancer compounds [22]. The synthetic procedure developed at Bayer included the reduction of 5 as the key step, which was carried out with sodium cyanoborohydride, leading to racemic 6 (Figure 2) [23]. However, a cost-efficient process for the asymmetric hydrogenation of prochiral intermediate 5 on a kilogram scale was greatly needed. Verzijl and coworkers initially attempted to accomplish a metal-catalyzed (Noyori Ru catalysts) proton transfer; however, no complete conversion was observed, and accordingly, this approach was revealed to be useless to develop a scalable and low-cost procedure. Based on their expertise in scalable enantioselective C=N double-bond hydrogenation reactions, they then shifted to hydrogenations, and in particular, Ir catalysts were analyzed. High-throughput experimentation and parallel screening of reaction conditions and ligands led to an unoptimized solution to the problem. A plethora of experimental conditions were tested in the search for the best conversions and ee values after four different screenings; they included a total of 60 chiral ligands, solvent (DCM, THF, TFE, AcOH, i-PrOH, EtOAc, and toluene), temperature (rt and 60 °C), and the presence or absence of I2 as additive. In the initial screening, the most promising ligands fell into three categories: WalPhos (bisphosphine-derived ferrocene), UBAPhox (oxazoline containing a phosphinite motif), and POX (phosphine-based ferrocene with an oxazoline moiety), with the latter having generally poorer enantioselectivities. Although some of the WalPhos and UBAPhox ligands led to the highest enantioselectivities, no complete conversion was achieved; increasing the temperature to 60 °C led to complete conversion without detrimental effects on the enantioselectivity. Taking this into consideration, ligand WalPhos-003 was selected for the scale-up process, using [Ir(COD)Cl]2 as the metal source; the catalyst did not have to be preformed, but chelation occurred under a H2 atmosphere, and the use of corrosive I2 could be avoided in this case. A catalytic load of 1.5% was used in order to avoid the displacement of the chiral ligand by 6 observed at a high substrate–catalyst ratio. Moreover, all of the tested reactions proceeded with complete regioselectivity, as no hydrogenation of C=N on the 1,2-positions was observed. Thus, the asymmetric hydrogenation of 5 using Ir/WalPhos-003 as a catalyst allowed the preparation of the chiral intermediate (S)-6 in a batchwise fashion (27 kg, 69.4% average yield) and in excellent enantiomeric purity (99% ee) [24].
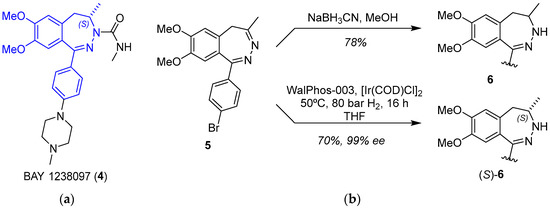
Figure 2.
(a) Structure of BAY 1238097 (4) highlighting the benzodiazepine fragment; (b) key step of the synthetic procedure showing Bayer’s procedure (top) and the improved approach using iridium as catalyst (bottom).
3. Aminations
3.1. Palladium
JAK and ERK Inhibitors
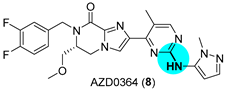
JAK has been identified as an upstream activator of the extracellular signal-regulated kinase (ERK) pathway [25]. As promising anticancer drugs, JAK1 inhibitor AZD4205 (7) [26] and ERK1/2 inhibitor AZD0364 (8) [27] recently entered Phase I/II and Phase I clinical trials, respectively [28]. Both compounds have an aryl-NH-aryl’ moiety in common (Figure 3a), which can be prepared through C–N bonding. The palladium-catalyzed Buchwald–Hartwig amination remains the gold standard for the construction of C–N bonds [29], which are frequently found in drugs. The main issues to be solved are the appropriate choice of solvent (in order to avoid the precipitation of reagents, the formation of emulsions, or the need to use an aqueous work-up) and base (critical for allowing compatibility of other functionalities added to the substrates). In order to scale up the reaction for its application in the production of drugs, Pithani and coworkers established mildly basic aqueous biphasic reaction conditions (Figure 3b) [30]. The use of other solvents in the optimization step, such as ethereal solvents or DMF, led to precipitation of some of the components or to the formation of emulsions, respectively, in the work-up. In their approach, the reaction runs in 2-methyltetrahydrofuran (MeTHF) and uses water as a cosolvent. The addition of water produces a positive impact on the outcome of the reaction since it solubilizes the inorganic salt. Furthermore, water affords a homogeneous biphasic reaction mixture, which prevents scalability issues. The best conditions were determined to be the use of Pd2(dba)3 and the biaryl ligand DavePhos in the biphasic medium; attempts to replace MeTHF with other solvents, such as dioxane or MeCN, failed. The palladium-catalyzed Buchwald–Hartwig amination allowed for the multikilogram (hectogram to >50 kg) synthesis of diverse JAK1 (e.g., AZD4205, 7) and ERK1/2 (e.g., AZD0364, 8) inhibitors.
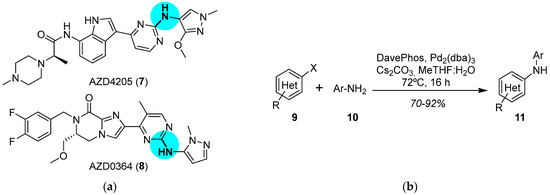
Figure 3.
(a) Structure of JAK1 inhibitor AZD4205 (7) and ERK1/2 inhibitor AZD0364 (8) highlighting the C–N bond formed; (b) palladium-catalyzed Buchwald–Hartwig amination under basic aqueous biphasic reaction conditions.
4. Cross-Coupling
4.1. Palladium
4.1.1. ATR Inhibitor Ceralasertib
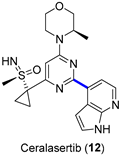
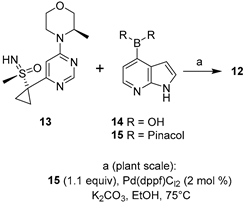
Ataxia-telangiectasia and the RAD3-related (ATR) kinase inhibitor ceralasertib (12, AZD6738) entered Phase II clinical trials in 2019 (NCT03682289) [28] to study how the drug works alone or in combination with olaparib in treating diverse solid tumors [31]. The chemistry to manufacture ceralasertib (12) represented a challenge in the scale-up from the gram to multikilogram scale [32]. Scientists had to address several synthetic issues while meeting the demands of materials for clinical trials; the main drawbacks in the initial synthetic pathway were the lack of stereoselectivity at the sulfoxide stage, the low reproducibility of the formation of the sulfoximine, and the cyclopropanation step, together with the requirement for several chromatographic separations, which hindered the scale-up for the industrial demand. Compared to the initial medicinal chemistry route (overall yield = 6.5%), the improvement in three key steps, namely, sulfoximine formation, cyclopropanation, and the final Suzuki–Miyaura coupling, allowed the preparation of several kilograms of ceralasertib (12) in an overall yield of 18%. Figure 4 shows how the Suzuki–Miyaura coupling was applied at the diverse phases of the scaling-up process. In order to limit the formation of a homodimer from boronated derivative 25, oxygen had to be purged from the reaction vessel (degasification with N2 for 2 h). When using Pd(dppf)Cl2 as the best catalyst and keeping its load at 2 mol %, the temperature could be reduced to 75 °C, and the detected amount of the homocoupled product from 15 was just 0.04%. Target derivative 12 could be isolated after a series of filtrations, extractions, and crystallization from butan-1-ol/n-heptane mixtures. At the plant scale, such optimized conditions allowed the reaction of 61.6 and 53.5 kg of 13 and 15, respectively, to furnish 12 (45 kg, 65% yield).
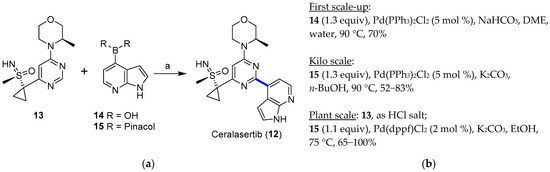
Figure 4.
(a) Palladium-catalyzed Suzuki–Miyaura coupling to prepare ceralasertib; (b) scaling-up conditions and outcome.
4.1.2. A2AR Antagonist AZD4635
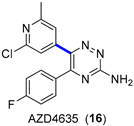
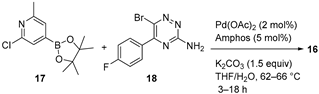
Adenosine signaling through the adenosine 2A receptor (A2AR) on immune cells elicits a range of immunosuppressive effects that promote tumor growth and limit the efficacy of immune checkpoint inhibitors. AZD4635 (16) (Figure 5a) is a potent and selective A2AR inhibitor that has shown indirect antitumor effects on immune cells [33], and it is currently being evaluated in diverse Phase I (NCT02740985, NCT03381274) and Phase II (NCT04495179, NCT04089553) clinical trials [28]. Therefore, a multikilogram process was developed, with the key step being the Suzuki–Miyaura coupling of pinacol boronate ester 17 (4.11 kg) and aryl bromide 18 (4.37 kg) (Figure 5b) [34]. Although the production of 16 was accomplished in high yields, the main drawback of the process was related to the unexpected corrosion observed inside the reaction vessel of a thermopocket constructed from tantalum. This metal is commonly used to repair damaged glass-lined steel vessels, which are widely prevalent in manufacturing units; nevertheless, the use of methanolic solutions of bromine (5%) with less than 5% water dissolves the protective oxide coating, thus leading to the observed corrosion. The resulting intermediate 18 contained 558 ppm of tantalum, which is unacceptable according to the specifications required for active pharmaceutical ingredients (APIs). Fortunately, the insoluble tantalum material could be removed with the implementation of an in-line filtration prior to isolation, leading to a concentration lower than 1 ppm. Besides accidental tantalum, API 16 contained high levels of palladium (121–129 ppm) from the Suzuki–Miyaura coupling and iridium (66–69 ppm) from the synthesis of 17. Both palladium and iridium metals were partially removed through crystallization of 16 without the need for scavenging, leading to final concentrations of 16 and 29 ppm, respectively. This work highlights the relevance of controlling traces of (heavy) metals in APIs.
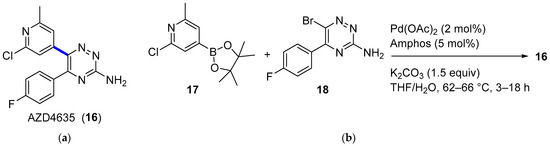
Figure 5.
(a) Structure of AZD4635 (16) highlighting the key C–C bond formation; (b) key Suzuki–Miyaura coupling.
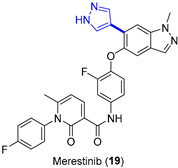
4.1.3. MET Inhibitor Merestinib
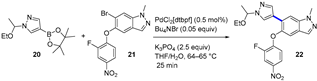
Merestinib (LY2801653, 19, Figure 6a), initially developed to target the MET receptor tyrosine kinase, is an oral tyrosine and serine/threonine kinase multitarget inhibitor with antiproliferative and antiangiogenic activity [35]. With a yearly demand of circa 2000 kg for Phase II clinical trials, the production of merestinib at the demonstration stage was switched from conventional continuous manufacturing to a hybrid method that included four small-volume continuous processes [36]. The reasons behind this change were related to the risks due to known and potential genotoxic impurities when producing kilograms of merestinib (19), including some intermediates and the starting nitrobenzene derivative 21. In addition to running reactions in continuous flow, the synthetic sequence was reordered to avoid the use of ZnBr2, which could produce undesired dehalogenation as a side effect. In the new route, the Suzuki–Miyaura cross-coupling reaction between pyrazole 20 and bromide 21 (Figure 6b) was placed first in the sequence (intermittent-flow stirred tank), thus avoiding the use of ZnBr2 for inhibiting debromination during the hydrogenation step. Impurities were kept to acceptable levels by carrying out a continuous extractive metal treatment, as well as a crystallization process. The flow performance of the Suzuki coupling reactions was proved to furnish a similar yield and product purity compared to a batch process; interestingly, reduced Pd impurities were detected in the final product (<20 ppm vs. 30 ppm). From the demonstration campaign, 13.7 kg/day was expected for compound 22. Subsequently, the nitro-group hydrogenolysis of 22 (trickle-bed reactor platform), amide bond formation, and deprotection (heated flow reactor) took place. High conversion of the nitro reduction step was mandatory to limit the amount of reactive intermediates. Minimization of impurities was also implemented in the amide bond formation. For that reason, a mixed anhydride was used instead of classical coupling reagents. This allowed the complete solubilization of the reagents, as well as an increase in the reaction kinetics.
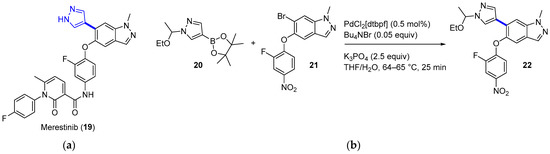
Figure 6.
(a) Structure of merestinib (19) highlighting the key C–C bond formation; (b) Suzuki–Miyaura coupling.
5. C–H Activation
5.1. Palladium–Copper
PDE4 Inhibitor
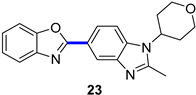
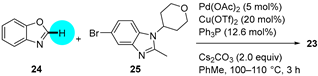
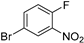
Although phosphodiesterase 4 (PDE4) inhibitors appear to be potential agents for asthma and chronic obstructive pulmonary disease (COPD) [37], they have also begun to be investigated for CNS diseases, such as memory impairments or multiple sclerosis, among others [38]. This is the case for compound 23 (Figure 7a), whose fumarate salt was considered as a potential candidate for the treatment of Alzheimer’s disease [39]. This PDE4 inhibitor represents a noteworthy example of the differences in drug development between academia and industry. Clinical trials demand kilogram amounts of APIs, and the time to provide this material is generally short. In contrast to academic researchers, medicinal chemists at companies first develop fast but inefficient synthetic routes in order to comply with timing. However, an improvement in the synthetic pathway of the API is constantly sought. In the particular case of PDE4 inhibitor 23, the so-called second-generation synthesis involved the direct coupling of benzoxazole 24 with the heteroaryl bromide 25 (obtained from 4-bromo-1-fluoro-2-nitrobenzene in five steps) using a combination of Pd(OAc)2/Cu(OTf)2/Ph3P to effect C–H/C–Br coupling (Figure 7b); this kind of approach avoids the need to prepare stoichiometric amounts of aryl-metal reagents. The optimization of the reaction conditions involved the addition of the appropriate Cu salt and the effects of the ligand and the solvent. The coupling was significantly accelerated by the addition of CuOTf, increasing the yield of 23 to 87%, while reducing half the amount of Pd(OAc)2 to 5 mol%. Thus, the higher Lewis acidities of Cu salts increase the reactivity, probably due to the activation of 24. Among the panel of ligands tested (P(o-Tol)3, PtBu3, cataCXium A (a di-adamantane-based phosphine), XPhos type, dppf, or PEPPI-IPr), Ph3P was the preferred one; in the absence of ligand, no reaction took place, and Ph3P allowed short reaction times and resulted in the lowest amounts of residual benzimidazole 25. A 2.5 Ph3P/Pd ratio was found to be optimal for suppressing unknown impurities in the outcome of the reaction. Moreover, the solvent could be changed from N-methyl-2-pyrrolidone (NMP) to toluene. Notably, the coupling reaction allowed a decrease in the number of synthetic steps for accessing 23 and, more importantly, circumvention of the use of 2-aminophenol, which is known to be mutagenic.
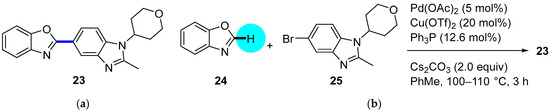
Figure 7.
(a) Structure of PDE4 inhibitor 23 highlighting the key C–C bond formation; (b) Pd–Cu-catalyzed coupling step.
As a summary, Table 1 depicts all reactions covered herein, the target pharmaceutical drug, and the reported scale and yields.

Table 1.
Summary of the optimization of Ir- and Pd-catalyzed bulk reactions useful in pharmaceutical industry.
6. Conclusions
Concerned by the great importance of metal catalysis in industry, in this review, we focus on the application of Ir and Pd catalysts for the large-scale production of drug substances. The choice of the metals to cover this review is due, on the one hand, to the extraordinary versatility of Pd complexes for catalyzing an ample number of organic reactions and, on the other hand, to the emerging importance of Ir, particularly for catalyzing asymmetric hydrogenation reactions. The benefits of the catalyzed pathways are highlighted. Thus, relevant pharmaceutical drugs are discussed in which one of the key steps is a metal complex-catalyzed reaction: asymmetric reductions, Buchwald–Hartwig amination, Suzuki–Miyaura, and C–H/C–Br coupling reactions. In all of the analyzed cases, high yields and (if so) ee values were observed, and the title reactions could be executed up to a multikilogram scale, suitable for the industrial manufacturing process. Special emphasis has been made on the scale-up optimization and the elimination of contaminating metal traces so as to fulfill the legal requirements of purity. The pharmaceutical industry, while based on academic findings and discoveries on synthetic processes, has evolved to comply with regulations such as current Good Manufacturing Practices (cGMP) in order to deliver APIs that meet the required specifications. Thus, industrial chemists do not only quest for high-yield reactions, but they also have to deal with a number of regulatory issues, such as total impurities in drug substances, crystal form, residual metals, and residual solvents, to mention a few. In this scenario, new concepts and innovations should provide solutions to regulatory constraints. In the past two decades, the pharmaceutical industry and the U.S. Food and Drug Administration (FDA) have recognized continuous manufacturing as an economically and environmentally transformative means of drug production. While continuous manufacturing develops as a plausible alternative to batch processes, another point of intervention related to sustainability should be considered for the future of industrial catalysis, i.e., the replacement of transition metals by greener systems [40]. We hope that this work will encourage academic scientists to explore the hitherto less developed organocatalytic processes in industry, with the aim of overcoming the limitations of metal-catalyzed reactions in terms of cost and residue generation.
Author Contributions
Writing—review and editing, Ó.L. and J.M.P.; funding acquisition, Ó.L. and J.M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by ULL and Consejería de Economía, Conocimiento y Empleo, grant number CEI Canarias-ULL, SD-19/02. Ó.L. also thanks the Spanish Government for grant PID2020-116460RB-I00 funded by MCIN/AEI/10.13039/501100011033. The APC was waived by the editors.
Acknowledgments
We acknowledge the kind invitation of Gianluigi Broggini and Michail Christodoulou to contribute to the Special Issue “Towards the Transition Metal Catalysis in Organic Synthesis”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 2nd ed.; Pearson Prentice-Hall: Essex, UK, 2005; pp. 20–21. [Google Scholar]
- Santoro, S.; Kalek, M.; Huang, G.; Himo, F. Elucidation of Mechanisms and Selectivities of Metal-Catalyzed Reactions using Quantum Chemical Methodology. Acc. Chem. Res. 2016, 49, 1006–1018. [Google Scholar] [CrossRef]
- Nandy, A.; Duan, C.; Taylor, M.G.; Liu, F.; Steeves, A.H.J.; Kulik, H.J. Computational Discovery of Transition-metal Complexes: From High-throughput Screening to Machine Learning. Chem. Rev. 2021, 121, 9927–10000. [Google Scholar] [CrossRef]
- Desnoyera, A.N.; Love, J.A. Recent advances in well-defined, late transition metal complexes that make and/or break C–N, C–O and C–S bonds. Chem. Soc. Rev. 2017, 46, 197–238. [Google Scholar] [CrossRef]
- Campos, J.; López-Serrano, J.; Peloso, R.; Carmona, E. Methyl Complexes of the Transition Metals. Chem. Eur. J. 2016, 22, 6432–6457. [Google Scholar] [CrossRef] [Green Version]
- Mondal, R.; Guin, A.K.; Chakrabortya, G.; Paul, N.D. Metal–ligand cooperative approaches in homogeneous catalysis using transition metal complex catalysts of redox noninnocent ligands. Org. Biomol. Chem. 2022, 20, 296–328. [Google Scholar] [CrossRef]
- Franco, F.; Rettenmaier, C.; Jeon, H.S.; Roldán Cuenya, B. Transition metal-based catalysts for the electrochemical CO2 reduction: From atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884–6946. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Mandoli, A. Supported Metal Catalysts and Their Applications in Fine Chemicals. Catalysts 2021, 11, 791. [Google Scholar] [CrossRef]
- Hübner, S.; de Vries, J.G.; Farina, V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts? Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pagliaro, M.; Luque, R. Heterogeneous catalysis under flow for the 21st century fine chemical industry. Green Energy Environ. 2021, 6, 161–166. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Melchiorre, P. Chemistry glows green with photoredox catalysis. Nat. Commun. 2020, 11, 803. [Google Scholar] [CrossRef]
- Keim, W. Concepts for the Use of Transition Metals in Industrial Fine Chemical Synthesis. In Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed.; Beller, M., Bolm, C., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 15–25. [Google Scholar]
- Andreatta, J.R.; Dilip, M. Transition Metal Catalysis for Organic Synthesis. In Catalysis for Sustainability: Goals, Challenges, and Impacts; Umile, T.P., Ed.; CRC Press: Boca Raton, FL, USA, 2015; pp. 23–45. [Google Scholar]
- Malinowski, J.; Zych, D.; Jacewicz, D.; Gawdzik, B.; Drzeżdżon, J. Application of Coordination Compounds with Transition Metal Ions in the Chemical Industry—A Review. Int. J. Mol. Sci. 2020, 21, 5443. [Google Scholar] [CrossRef]
- Rossen, K.; Miller, K.A. Citation Bias in Organic Chemistry Research: Are Industry-Affiliated Papers Cited Less Often? Org. Process Res. Dev. 2021, 25, 167–168. [Google Scholar] [CrossRef]
- Bhilare, S.; Shet, H.; Sanghvi, Y.S.; Kapdi, A.R. Discovery, Synthesis, and Scale-up of Efficient Palladium Catalysts Useful for the Modification of Nucleosides and Heteroarenes. Molecules 2020, 25, 1645. [Google Scholar] [CrossRef] [Green Version]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, I.J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry Research Areas from a Pharmaceutical Manufacturers’ Perspective Revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.C.R.; Lourenço, A.; Ferreira, L.M.; Branco, P.S. Tofacitinib Synthesis—An Asymmetric Challenge. Eur. J. Org. Chem. 2019, 2019, 615–624. [Google Scholar] [CrossRef]
- Ripin, D.H.B.; Abele, S.; Cai, W.; Blumenkopf, T.; Casavant, J.M.; Doty, J.L.; Flanagan, M.; Koecher, C.; Laue, K.W.; McCarthy, K.; et al. Development of a Scalable Route for the Production Of cis-N-Benzyl-3-Methylamino-4-Methylpiperidine. Org. Process Res. Dev. 2003, 7, 115–120. [Google Scholar] [CrossRef]
- Verzijl, G.K.M.; Schuster, C.; Dax, T.; de Vries, A.H.M.; Lefort, L. Asymmetric Synthesis of a Key Intermediate for Tofacitinib via a Dynamic Kinetic Resolution-Reductive Amination Protocol. Org. Process Res. Dev. 2018, 22, 1817–1822. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.-C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-Human Phase I Study of the Bromodomain and Extraterminal Motif Inhibitor BAY 1238097: Emerging Pharmacokinetic/Pharmacodynamic Relationship and Early Termination Due to Unexpected Toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef]
- Siegel, S.; Bäeurle, S.; Cleve, A.; Haendler, B.; Fernández-Montalván, A.E.; Mönning, U.; Krause, S.; Lejeune, P.; Busemann, M.; Kuhnke, J. Bicyclo 2,3-Benzodiazepines and Spirocyclically Substituted 2,3-Benzodiazepines. PCT Int. Appl. WO 2014128067 A1, 28 August 2014. [Google Scholar]
- Verzijl, G.K.M.; Hassfeld, J.; de Vries, A.H.M.; Lefort, L. Enantioselective Synthesis of a 2,3-Benzodiazepine Intermediate of BET Inhibitor BAY 1238097 via Catalytic Asymmetric Hydrogenation. Org. Process Res. Dev. 2020, 24, 255–260. [Google Scholar] [CrossRef]
- Dhillon, A.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Su, Q.; Banks, E.; Bebernitz, G.; Bell, K.; Borenstein, C.F.; Chen, H.; Chuaqui, C.E.; Deng, N.; Ferguson, A.D.; Kawatkar, S.; et al. Discovery of (2R)-N-[3-[2-[(3-Methoxy-1-Methyl-Pyrazol-4-yl)Amino]Pyrimidin-4-yl]-1H-Indol-7-yl]-2-(4-Methylpiperazin-1-yl)Propenamide (AZD4205) as a Potent and Selective Janus Kinase 1 Inhibitor. J. Med. Chem. 2020, 63, 4517–4527. [Google Scholar] [CrossRef]
- Ward, R.A.; Anderton, M.J.; Bethel, P.; Breed, J.; Cook, C.; Davies, E.J.; Dobson, A.; Dong, Z.; Fairley, G.; Farrington, P.; et al. Discovery of a Potent and Selective Oral Inhibitor of ERK1/2 (AZD0364) That Is Efficacious in Both Monotherapy and Combination Therapy in Models of Non-small Cell Lung Cancer (NSCLC). J. Med. Chem. 2019, 62, 11004–11018. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 10 January 2022).
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Pithani, S.; Malmgren, M.; Aurell, C.-J.; Nikitidis, G.; Friis, S.D. Biphasic Aqueous Reaction Conditions for Process-Friendly Palladium-Catalyzed C–N Cross-Coupling of Aryl Amines. Org. Process Res. Dev. 2019, 23, 1752–1757. [Google Scholar] [CrossRef]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing Effective Combination Therapy for Pancreatic Cancer: An Overview. Pharmacol. Res. 2020, 155, 104740. [Google Scholar] [CrossRef]
- Goundry, W.R.F.; Dai, K.; Gonzalez, M.; Legg, D.; O’Kearney-McMullan, A.; Morrison, J.; Stark, A.; Siedlecki, P.; Tomlin, P.; Yang, J. Development and Scale-up of a Route to ATR Inhibitor AZD6738. Org. Process Res. Dev. 2019, 23, 1333–1342. [Google Scholar] [CrossRef]
- Kotulová, J.; Hajdúch, M.; Džubák, P. Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose? Int. J. Mol. Sci. 2021, 22, 12569. [Google Scholar] [CrossRef]
- Douglas, J.J.; Adams, B.W.V.; Benson, H.; Broberg, K.; Gillespie, P.M.; Hoult, O.; Ibraheem, A.K.; Janbon, S.; Janin, G.; Parsons, C.D.; et al. Multikilogram-Scale Preparation of AZD4635 via C–H Borylation and Bromination: The Corrosion of Tantalum by a Bromine/Methanol Mixture. Org. Process Res. Dev. 2019, 23, 62–68. [Google Scholar] [CrossRef]
- He, A.R.; Cohen, R.B.; Denlinger, C.S.; Sama, A.; Birnbaum, A.; Hwang, J.; Sato, T.; Lewis, N.; Mynderse, M.; Niland, M.; et al. First-in-Human Phase I Study of Merestinib, an Oral Multikinase Inhibitor, in Patients with Advanced Cancer. Oncologist 2019, 24, e930–e942. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.P.; Reizman, B.J.; Hess, M.; Groh, J.M.; Laurila, M.E.; Cope, R.F.; Campbell, B.M.; Forst, M.B.; Burt, J.L.; Maloney, T.D.; et al. Small-Volume Continuous Manufacturing of Merestinib. Part 1. Process Development and Demonstration. Org. Process Res. Dev. 2019, 23, 858–869. [Google Scholar] [CrossRef]
- Martin, C.; Burgel, P.-R.; Roche, N. Inhaled Dual Phosphodiesterase 3/4 Inhibitors for the Treatment of Patients with COPD: A Short Review. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 2363–2373. [Google Scholar] [CrossRef]
- Blokland, A.; Heckman, P.; Vanmierlo, T.; Schreiber, R.; Paes, D.; Prickaerts, J. Phosphodiesterase Type 4 Inhibition in CNS Diseases. Trends Pharmacol. Sci. 2019, 40, 971–985. [Google Scholar] [CrossRef]
- Kuroda, K.; Tsuyumine, S.; Kodama, T. Direct Synthesis of a PDE4 Inhibitor by Using Pd–Cu-Catalyzed C–H/C–Br Coupling of Benzoxazole with a Heteroaryl Bromide. Org. Process Res. Dev. 2016, 20, 1053–1058. [Google Scholar] [CrossRef]
- Caruso, M.; Petroselli, M.; Cametti, M. Design and Synthesis of Multipurpose Derivatives for N-Hydroxyimide and NHPI-based Catalysis Applications. ChemistrySelect 2021, 6, 12975. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).