
Iridium- and Palladium-Based Catalysts in the Pharmaceutical Industry



Abstract
:1. Introduction
2. Asymmetric Reductions
2.1. Iridium
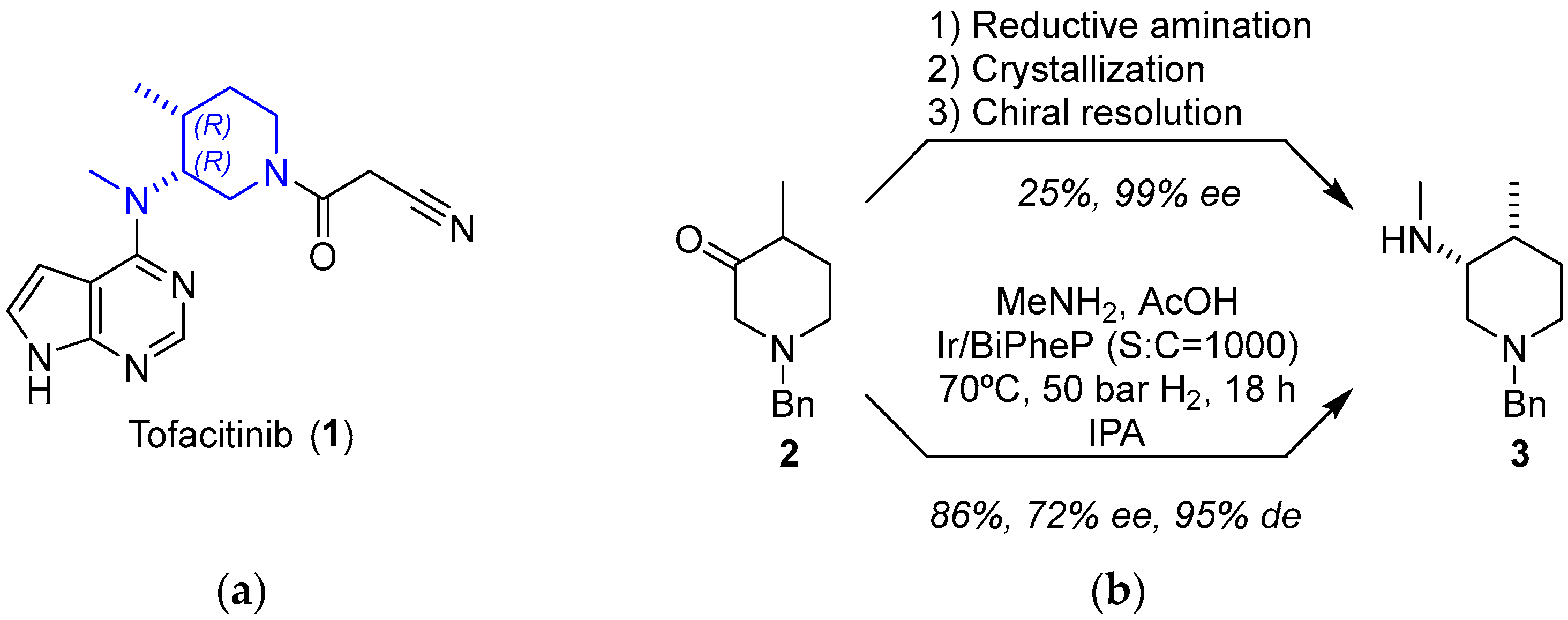
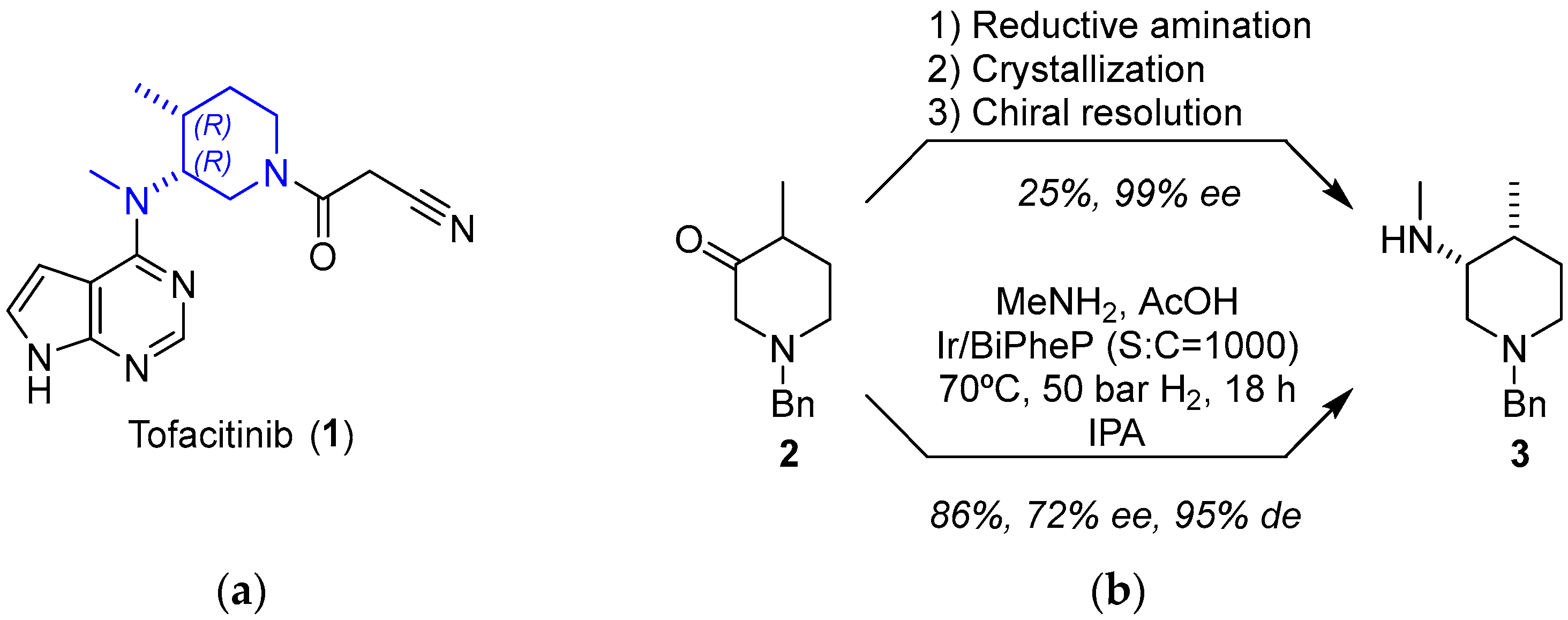
2.1.1. JAK Inhibitor Tofacitinib
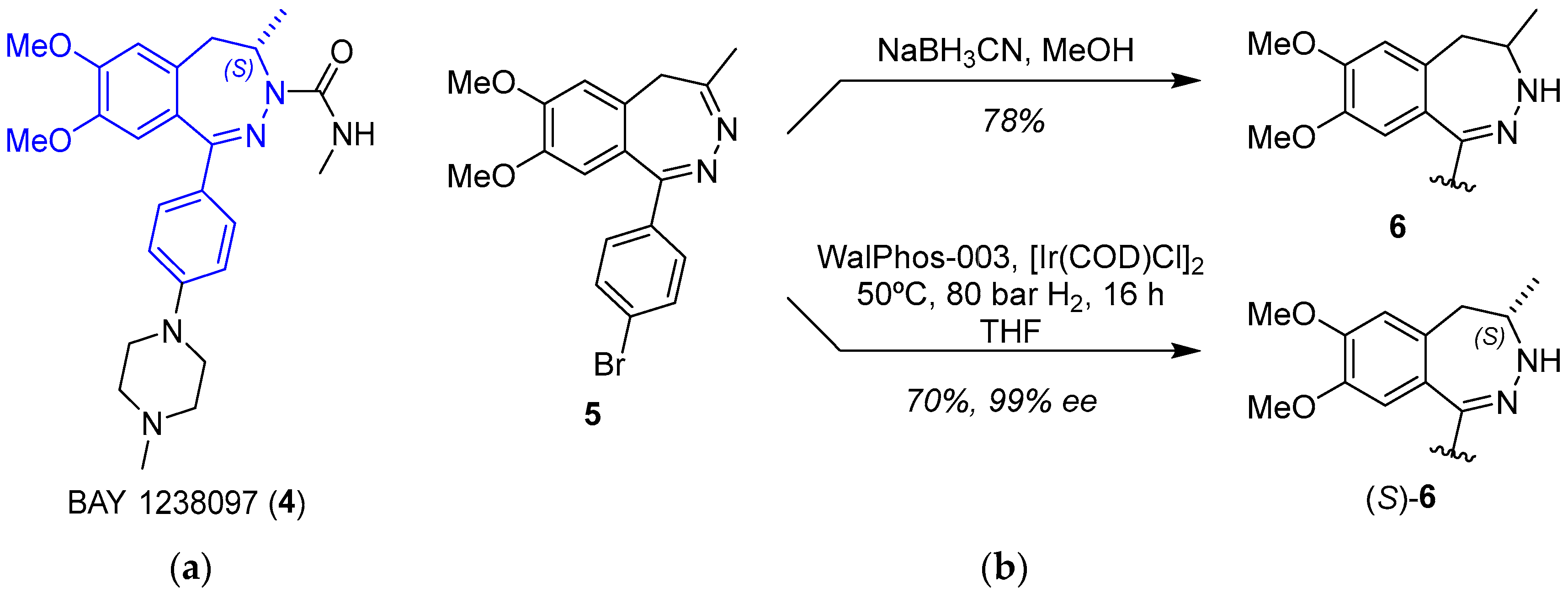
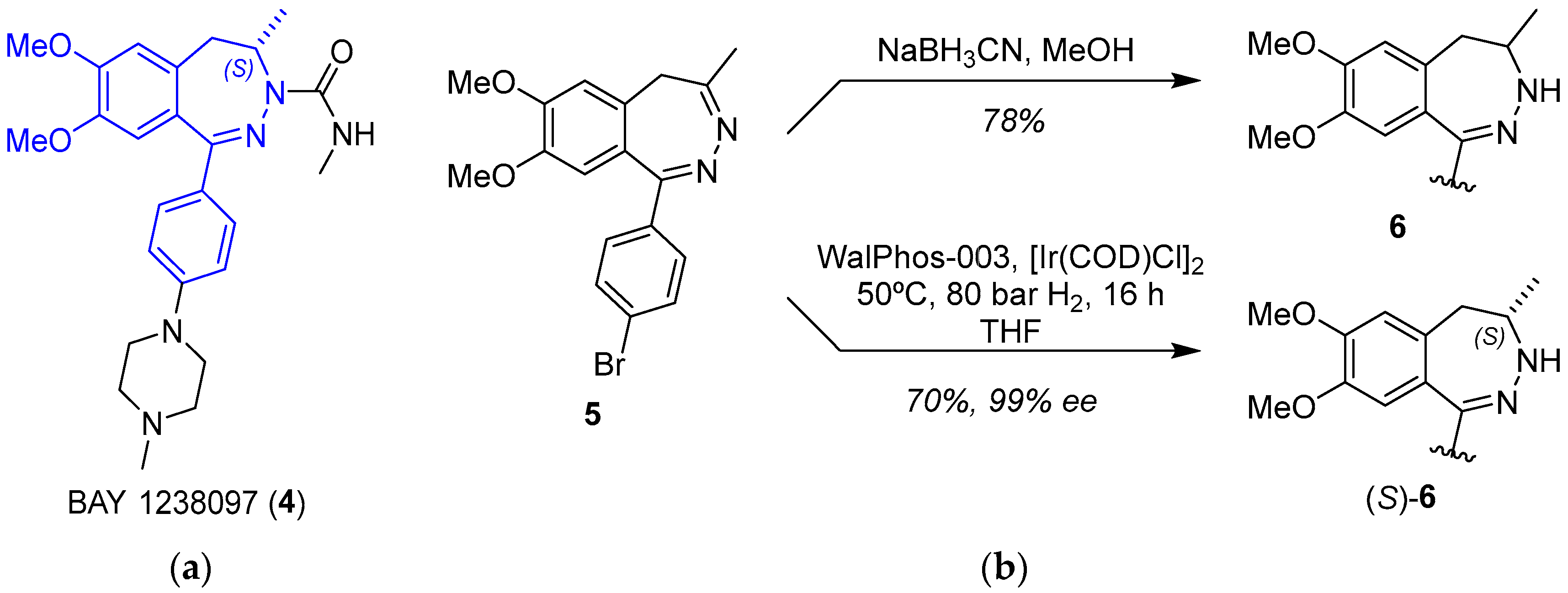
2.1.2. BET Inhibitor BAY 1238097
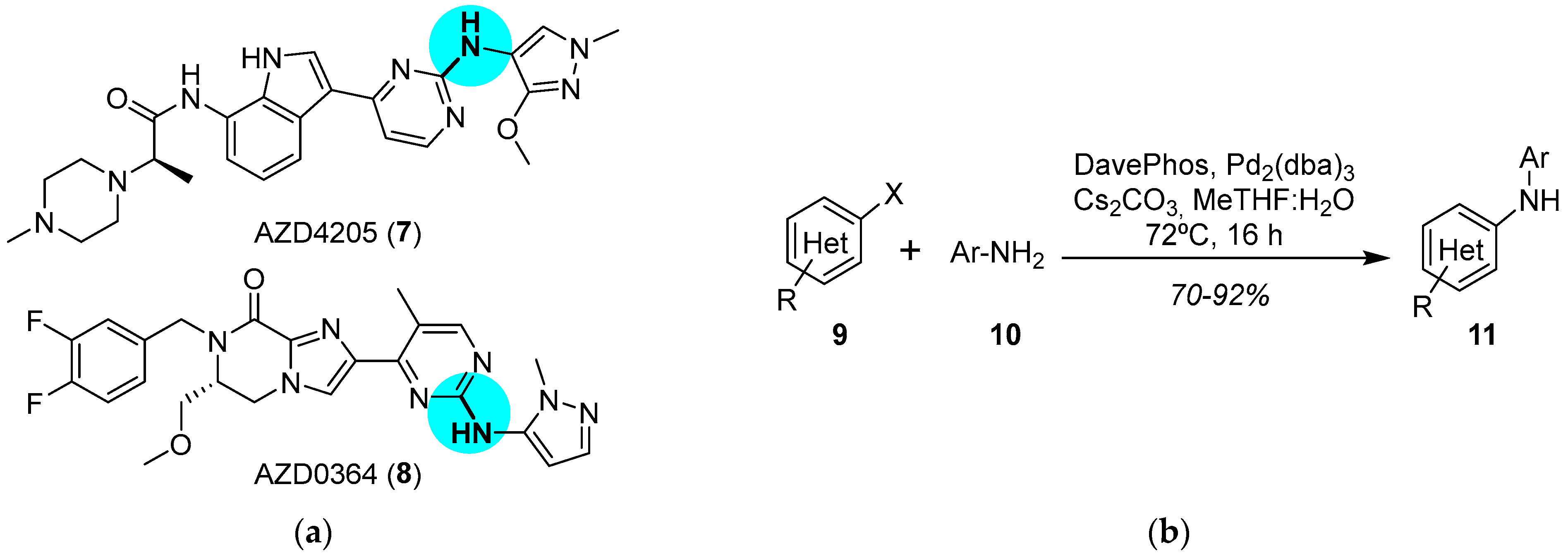
3. Aminations
3.1. Palladium
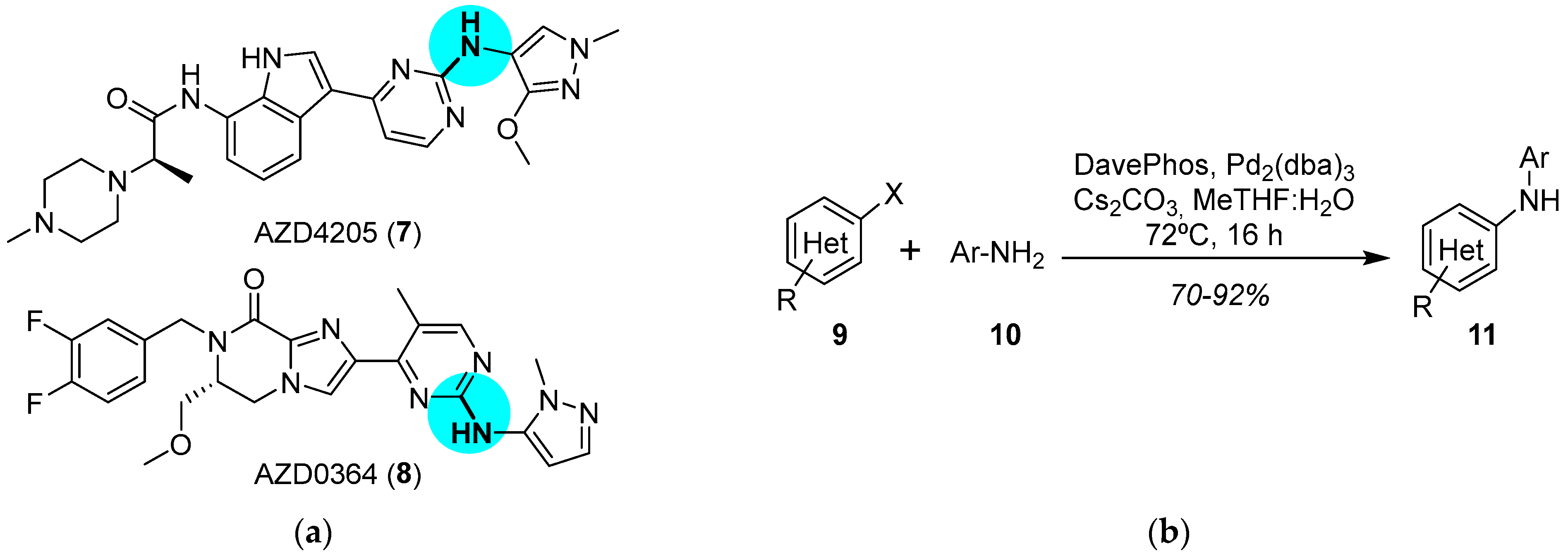
JAK and ERK Inhibitors
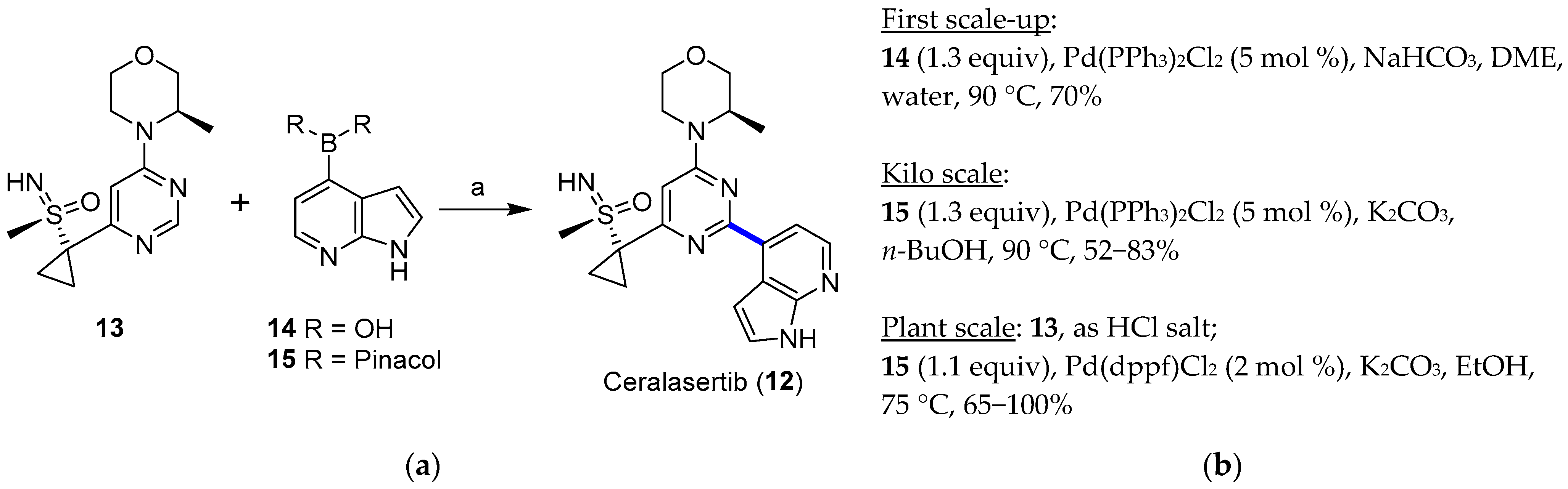
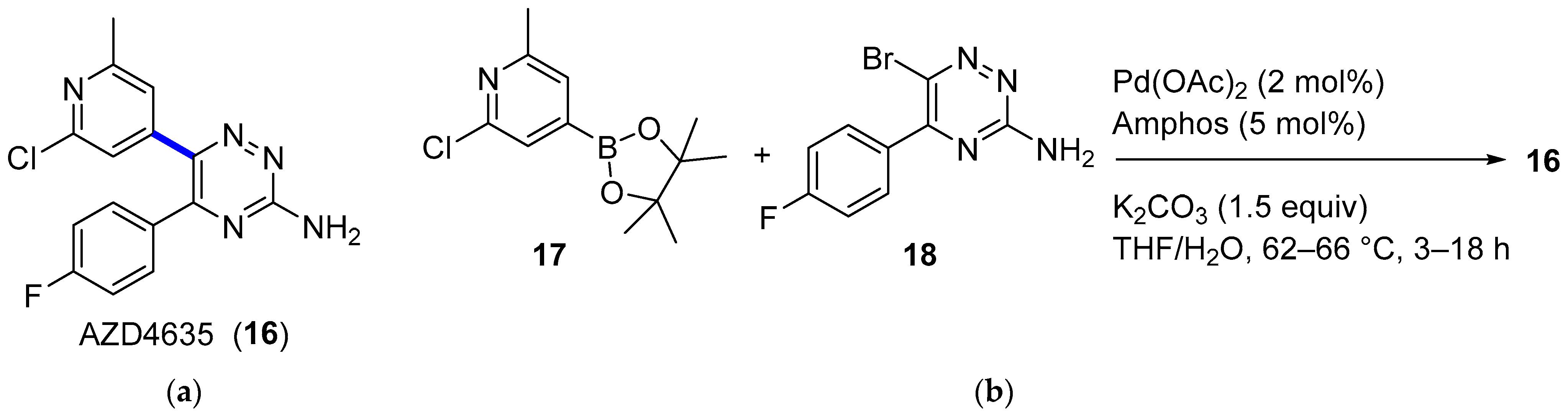
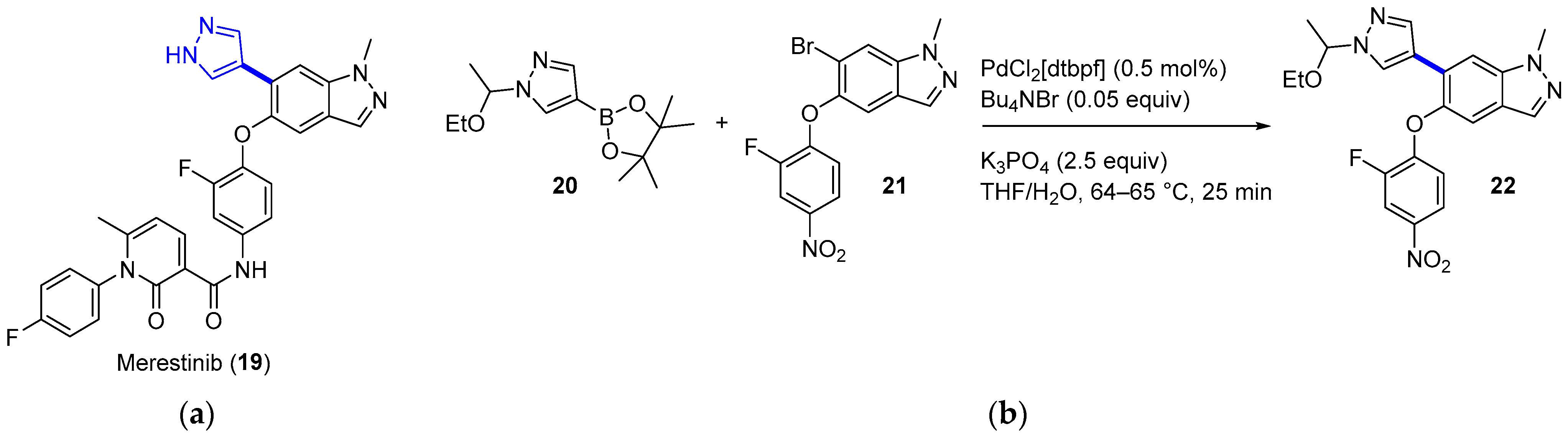
4. Cross-Coupling
4.1. Palladium
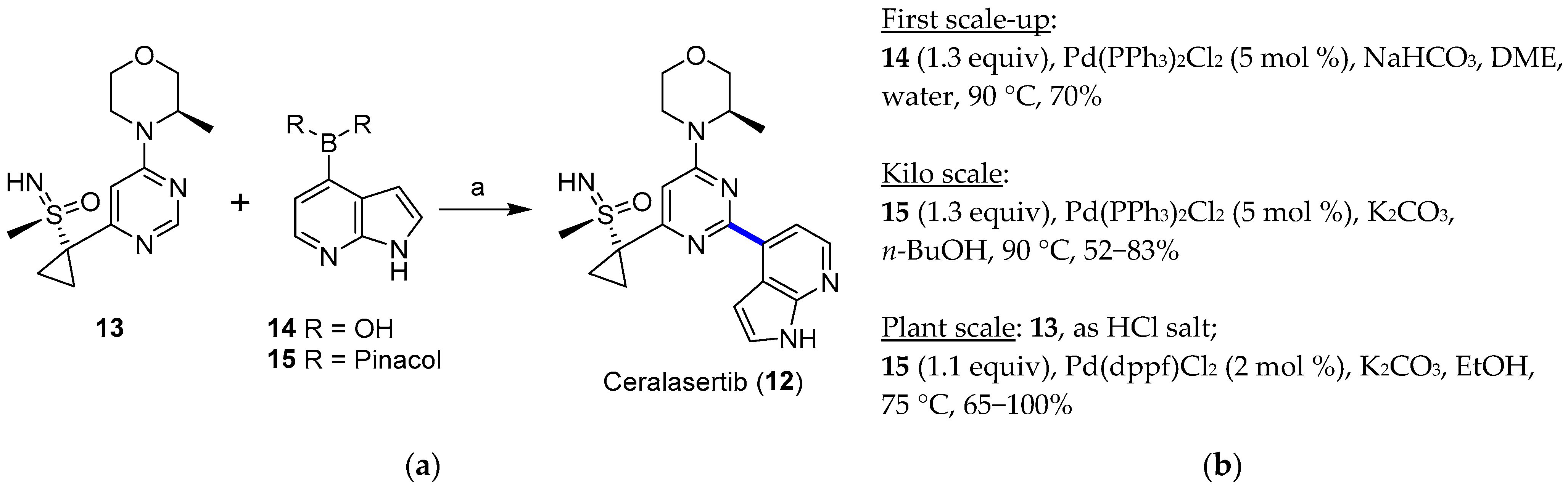
4.1.1. ATR Inhibitor Ceralasertib
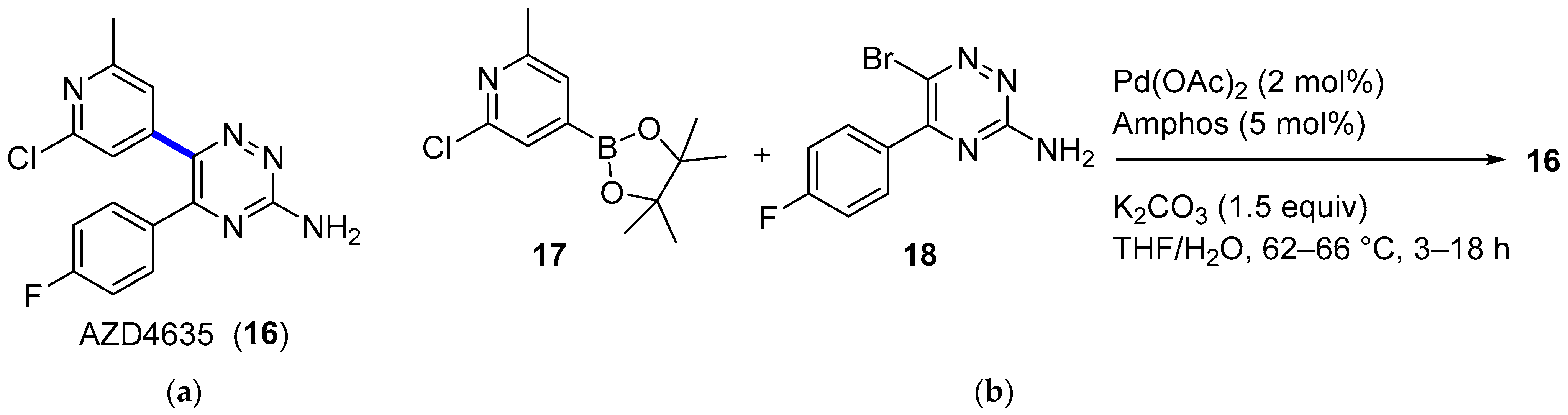
4.1.2. A2AR Antagonist AZD4635
4.1.3. MET Inhibitor Merestinib
5. C–H Activation
5.1. Palladium–Copper
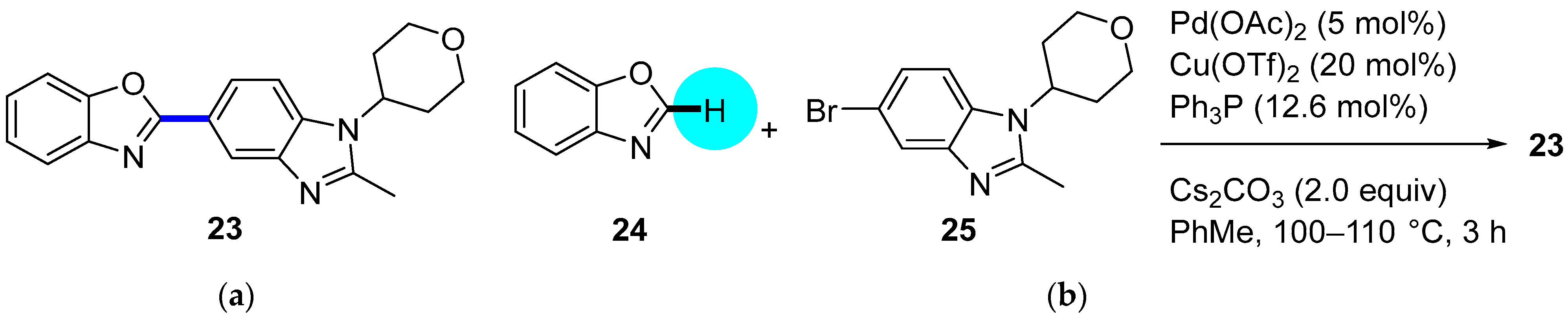
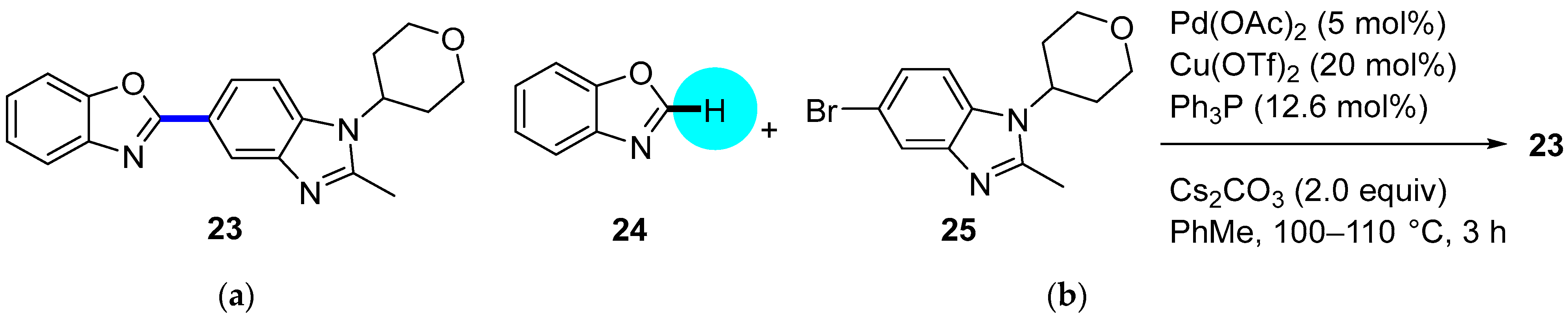
PDE4 Inhibitor
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 2nd ed.; Pearson Prentice-Hall: Essex, UK, 2005; pp. 20–21. [Google Scholar]
- Santoro, S.; Kalek, M.; Huang, G.; Himo, F. Elucidation of Mechanisms and Selectivities of Metal-Catalyzed Reactions using Quantum Chemical Methodology. Acc. Chem. Res. 2016, 49, 1006–1018. [Google Scholar] [CrossRef]
- Nandy, A.; Duan, C.; Taylor, M.G.; Liu, F.; Steeves, A.H.J.; Kulik, H.J. Computational Discovery of Transition-metal Complexes: From High-throughput Screening to Machine Learning. Chem. Rev. 2021, 121, 9927–10000. [Google Scholar] [CrossRef]
- Desnoyera, A.N.; Love, J.A. Recent advances in well-defined, late transition metal complexes that make and/or break C–N, C–O and C–S bonds. Chem. Soc. Rev. 2017, 46, 197–238. [Google Scholar] [CrossRef]
- Campos, J.; López-Serrano, J.; Peloso, R.; Carmona, E. Methyl Complexes of the Transition Metals. Chem. Eur. J. 2016, 22, 6432–6457. [Google Scholar] [CrossRef] [Green Version]
- Mondal, R.; Guin, A.K.; Chakrabortya, G.; Paul, N.D. Metal–ligand cooperative approaches in homogeneous catalysis using transition metal complex catalysts of redox noninnocent ligands. Org. Biomol. Chem. 2022, 20, 296–328. [Google Scholar] [CrossRef]
- Franco, F.; Rettenmaier, C.; Jeon, H.S.; Roldán Cuenya, B. Transition metal-based catalysts for the electrochemical CO2 reduction: From atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884–6946. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Mandoli, A. Supported Metal Catalysts and Their Applications in Fine Chemicals. Catalysts 2021, 11, 791. [Google Scholar] [CrossRef]
- Hübner, S.; de Vries, J.G.; Farina, V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts? Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pagliaro, M.; Luque, R. Heterogeneous catalysis under flow for the 21st century fine chemical industry. Green Energy Environ. 2021, 6, 161–166. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Melchiorre, P. Chemistry glows green with photoredox catalysis. Nat. Commun. 2020, 11, 803. [Google Scholar] [CrossRef]
- Keim, W. Concepts for the Use of Transition Metals in Industrial Fine Chemical Synthesis. In Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed.; Beller, M., Bolm, C., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 15–25. [Google Scholar]
- Andreatta, J.R.; Dilip, M. Transition Metal Catalysis for Organic Synthesis. In Catalysis for Sustainability: Goals, Challenges, and Impacts; Umile, T.P., Ed.; CRC Press: Boca Raton, FL, USA, 2015; pp. 23–45. [Google Scholar]
- Malinowski, J.; Zych, D.; Jacewicz, D.; Gawdzik, B.; Drzeżdżon, J. Application of Coordination Compounds with Transition Metal Ions in the Chemical Industry—A Review. Int. J. Mol. Sci. 2020, 21, 5443. [Google Scholar] [CrossRef]
- Rossen, K.; Miller, K.A. Citation Bias in Organic Chemistry Research: Are Industry-Affiliated Papers Cited Less Often? Org. Process Res. Dev. 2021, 25, 167–168. [Google Scholar] [CrossRef]
- Bhilare, S.; Shet, H.; Sanghvi, Y.S.; Kapdi, A.R. Discovery, Synthesis, and Scale-up of Efficient Palladium Catalysts Useful for the Modification of Nucleosides and Heteroarenes. Molecules 2020, 25, 1645. [Google Scholar] [CrossRef] [Green Version]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, I.J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry Research Areas from a Pharmaceutical Manufacturers’ Perspective Revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, L.C.R.; Lourenço, A.; Ferreira, L.M.; Branco, P.S. Tofacitinib Synthesis—An Asymmetric Challenge. Eur. J. Org. Chem. 2019, 2019, 615–624. [Google Scholar] [CrossRef]
- Ripin, D.H.B.; Abele, S.; Cai, W.; Blumenkopf, T.; Casavant, J.M.; Doty, J.L.; Flanagan, M.; Koecher, C.; Laue, K.W.; McCarthy, K.; et al. Development of a Scalable Route for the Production Of cis-N-Benzyl-3-Methylamino-4-Methylpiperidine. Org. Process Res. Dev. 2003, 7, 115–120. [Google Scholar] [CrossRef]
- Verzijl, G.K.M.; Schuster, C.; Dax, T.; de Vries, A.H.M.; Lefort, L. Asymmetric Synthesis of a Key Intermediate for Tofacitinib via a Dynamic Kinetic Resolution-Reductive Amination Protocol. Org. Process Res. Dev. 2018, 22, 1817–1822. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.-C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-Human Phase I Study of the Bromodomain and Extraterminal Motif Inhibitor BAY 1238097: Emerging Pharmacokinetic/Pharmacodynamic Relationship and Early Termination Due to Unexpected Toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef]
- Siegel, S.; Bäeurle, S.; Cleve, A.; Haendler, B.; Fernández-Montalván, A.E.; Mönning, U.; Krause, S.; Lejeune, P.; Busemann, M.; Kuhnke, J. Bicyclo 2,3-Benzodiazepines and Spirocyclically Substituted 2,3-Benzodiazepines. PCT Int. Appl. WO 2014128067 A1, 28 August 2014. [Google Scholar]
- Verzijl, G.K.M.; Hassfeld, J.; de Vries, A.H.M.; Lefort, L. Enantioselective Synthesis of a 2,3-Benzodiazepine Intermediate of BET Inhibitor BAY 1238097 via Catalytic Asymmetric Hydrogenation. Org. Process Res. Dev. 2020, 24, 255–260. [Google Scholar] [CrossRef]
- Dhillon, A.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Su, Q.; Banks, E.; Bebernitz, G.; Bell, K.; Borenstein, C.F.; Chen, H.; Chuaqui, C.E.; Deng, N.; Ferguson, A.D.; Kawatkar, S.; et al. Discovery of (2R)-N-[3-[2-[(3-Methoxy-1-Methyl-Pyrazol-4-yl)Amino]Pyrimidin-4-yl]-1H-Indol-7-yl]-2-(4-Methylpiperazin-1-yl)Propenamide (AZD4205) as a Potent and Selective Janus Kinase 1 Inhibitor. J. Med. Chem. 2020, 63, 4517–4527. [Google Scholar] [CrossRef]
- Ward, R.A.; Anderton, M.J.; Bethel, P.; Breed, J.; Cook, C.; Davies, E.J.; Dobson, A.; Dong, Z.; Fairley, G.; Farrington, P.; et al. Discovery of a Potent and Selective Oral Inhibitor of ERK1/2 (AZD0364) That Is Efficacious in Both Monotherapy and Combination Therapy in Models of Non-small Cell Lung Cancer (NSCLC). J. Med. Chem. 2019, 62, 11004–11018. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 10 January 2022).
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Pithani, S.; Malmgren, M.; Aurell, C.-J.; Nikitidis, G.; Friis, S.D. Biphasic Aqueous Reaction Conditions for Process-Friendly Palladium-Catalyzed C–N Cross-Coupling of Aryl Amines. Org. Process Res. Dev. 2019, 23, 1752–1757. [Google Scholar] [CrossRef]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing Effective Combination Therapy for Pancreatic Cancer: An Overview. Pharmacol. Res. 2020, 155, 104740. [Google Scholar] [CrossRef]
- Goundry, W.R.F.; Dai, K.; Gonzalez, M.; Legg, D.; O’Kearney-McMullan, A.; Morrison, J.; Stark, A.; Siedlecki, P.; Tomlin, P.; Yang, J. Development and Scale-up of a Route to ATR Inhibitor AZD6738. Org. Process Res. Dev. 2019, 23, 1333–1342. [Google Scholar] [CrossRef]
- Kotulová, J.; Hajdúch, M.; Džubák, P. Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose? Int. J. Mol. Sci. 2021, 22, 12569. [Google Scholar] [CrossRef]
- Douglas, J.J.; Adams, B.W.V.; Benson, H.; Broberg, K.; Gillespie, P.M.; Hoult, O.; Ibraheem, A.K.; Janbon, S.; Janin, G.; Parsons, C.D.; et al. Multikilogram-Scale Preparation of AZD4635 via C–H Borylation and Bromination: The Corrosion of Tantalum by a Bromine/Methanol Mixture. Org. Process Res. Dev. 2019, 23, 62–68. [Google Scholar] [CrossRef]
- He, A.R.; Cohen, R.B.; Denlinger, C.S.; Sama, A.; Birnbaum, A.; Hwang, J.; Sato, T.; Lewis, N.; Mynderse, M.; Niland, M.; et al. First-in-Human Phase I Study of Merestinib, an Oral Multikinase Inhibitor, in Patients with Advanced Cancer. Oncologist 2019, 24, e930–e942. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.P.; Reizman, B.J.; Hess, M.; Groh, J.M.; Laurila, M.E.; Cope, R.F.; Campbell, B.M.; Forst, M.B.; Burt, J.L.; Maloney, T.D.; et al. Small-Volume Continuous Manufacturing of Merestinib. Part 1. Process Development and Demonstration. Org. Process Res. Dev. 2019, 23, 858–869. [Google Scholar] [CrossRef]
- Martin, C.; Burgel, P.-R.; Roche, N. Inhaled Dual Phosphodiesterase 3/4 Inhibitors for the Treatment of Patients with COPD: A Short Review. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 2363–2373. [Google Scholar] [CrossRef]
- Blokland, A.; Heckman, P.; Vanmierlo, T.; Schreiber, R.; Paes, D.; Prickaerts, J. Phosphodiesterase Type 4 Inhibition in CNS Diseases. Trends Pharmacol. Sci. 2019, 40, 971–985. [Google Scholar] [CrossRef]
- Kuroda, K.; Tsuyumine, S.; Kodama, T. Direct Synthesis of a PDE4 Inhibitor by Using Pd–Cu-Catalyzed C–H/C–Br Coupling of Benzoxazole with a Heteroaryl Bromide. Org. Process Res. Dev. 2016, 20, 1053–1058. [Google Scholar] [CrossRef]
- Caruso, M.; Petroselli, M.; Cametti, M. Design and Synthesis of Multipurpose Derivatives for N-Hydroxyimide and NHPI-based Catalysis Applications. ChemistrySelect 2021, 6, 12975. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Catalyzed Reaction | Yield (%) | ee/ de (%) | Scale | Ref |
|---|---|---|---|---|---|
 |  Asymmetric reduction | 86 | 72/ 95 | 0.2 mmol | [21] |
 |  Asymmetric reduction | 70 | 99 | 1.2 kg (2.66 mol) | [24] |
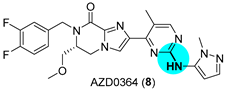 |  Buchwald–Hartwig amination | 70 | --- | 104.7 g (225.6 mmol) | [30] |
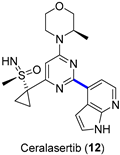 |  Suzuki–Miyaura coupling | 65 | --- | 61.6 kg of 13, 53.5 kg of 15, | [32] |
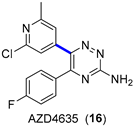 |  Suzuki–Miyaura coupling | 70 | --- | 4.11 kg of 17, 4.37 kg of 18 | [34] |
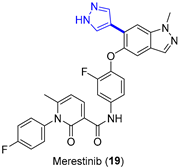 | 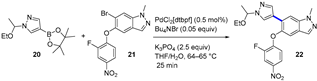 Suzuki–Miyaura coupling | --- | --- | 13.7 kg/day of 22 (Expected) | [36] |
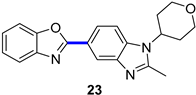 | 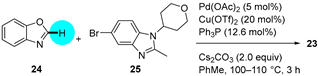 C–H/C–Br coupling | 87 | --- | 330 g of 23 from 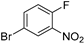 | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, Ó.; Padrón, J.M. Iridium- and Palladium-Based Catalysts in the Pharmaceutical Industry. Catalysts 2022, 12, 164. https://doi.org/10.3390/catal12020164
López Ó, Padrón JM. Iridium- and Palladium-Based Catalysts in the Pharmaceutical Industry. Catalysts. 2022; 12(2):164. https://doi.org/10.3390/catal12020164
Chicago/Turabian StyleLópez, Óscar, and José M. Padrón. 2022. "Iridium- and Palladium-Based Catalysts in the Pharmaceutical Industry" Catalysts 12, no. 2: 164. https://doi.org/10.3390/catal12020164
APA StyleLópez, Ó., & Padrón, J. M. (2022). Iridium- and Palladium-Based Catalysts in the Pharmaceutical Industry. Catalysts, 12(2), 164. https://doi.org/10.3390/catal12020164