
Rational Engineering of 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase for a Biomimetic Nicotinamide Mononucleotide Cofactor

 and
and 
Abstract
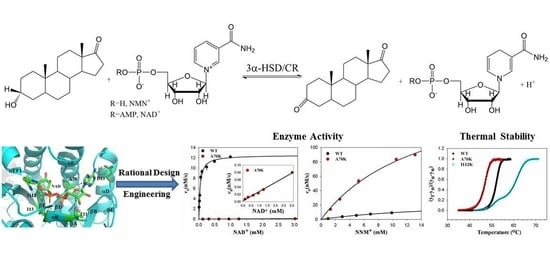
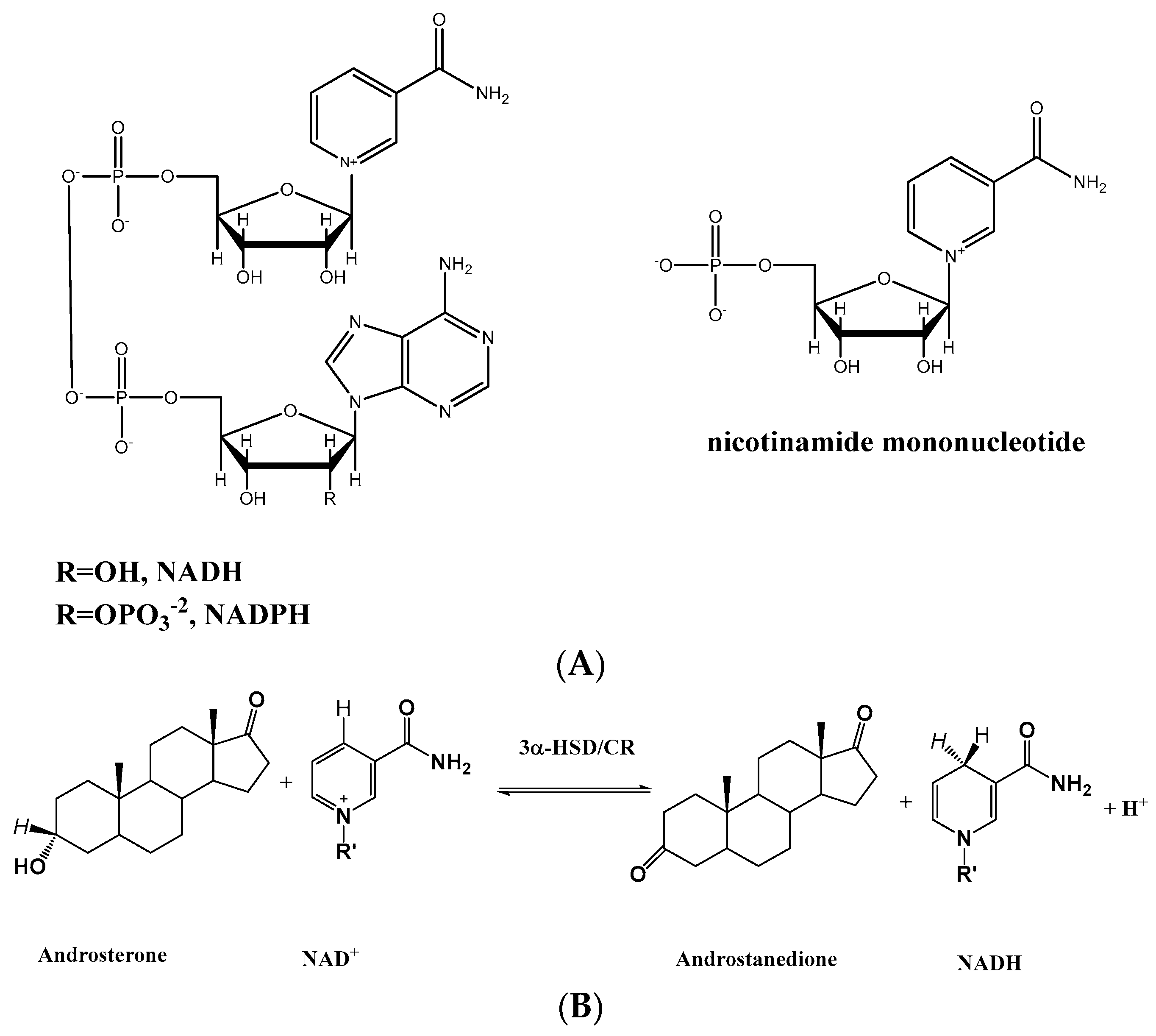
1. Introduction
2. Results
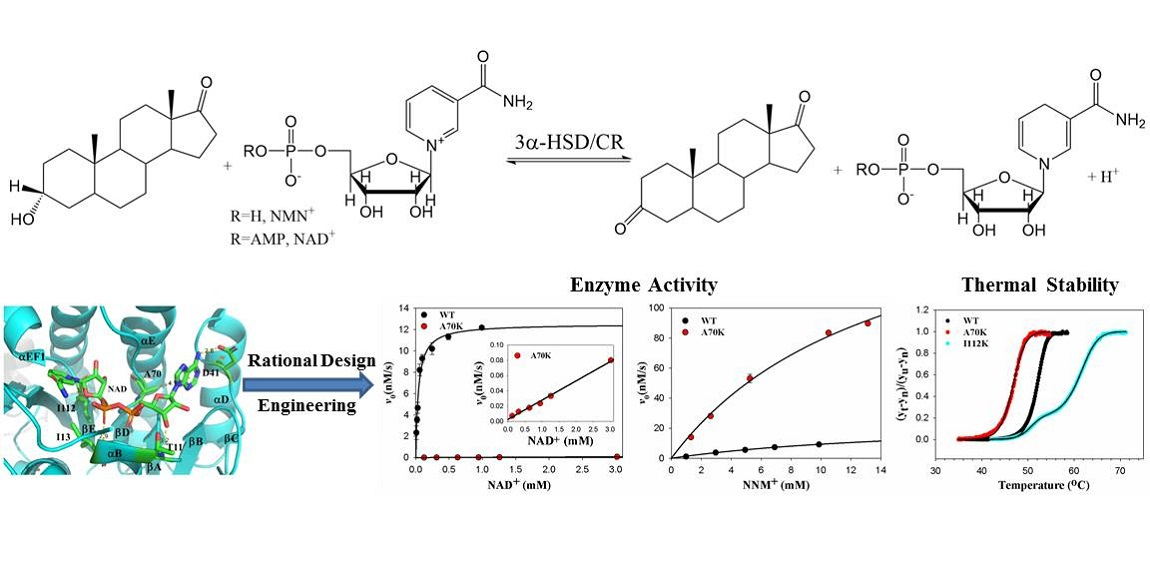
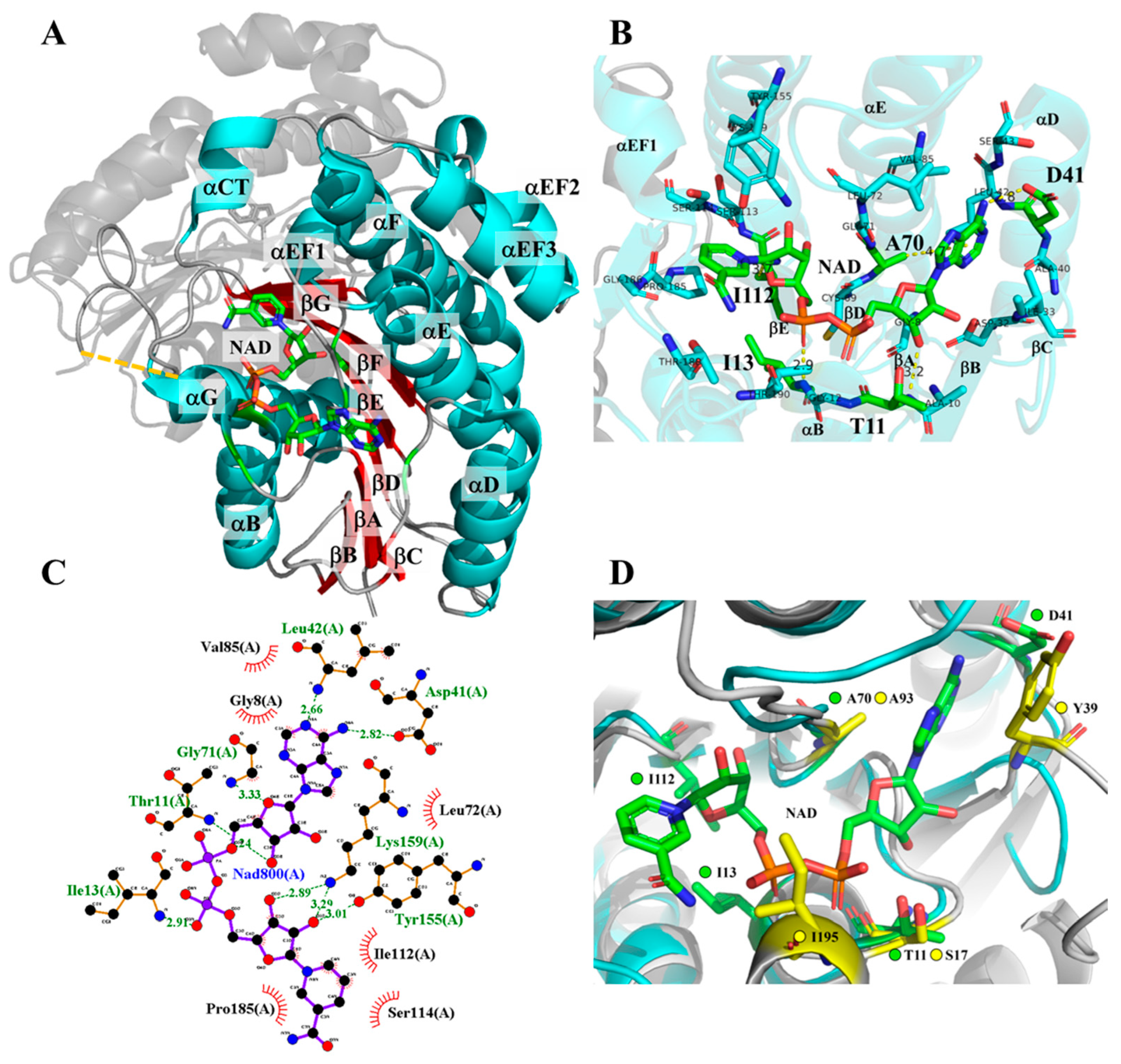
2.1. Residue Selection of 3α-HSD/CR Engineering by Rational Design
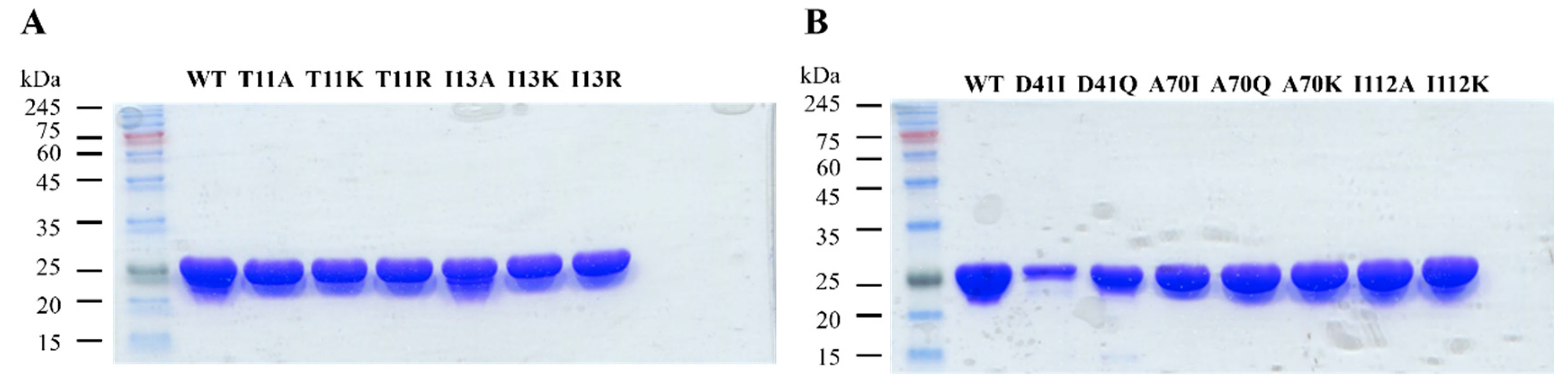
2.2. Protein Preparation and Characterization
2.2.1. Site-Directed Mutagenesis
2.2.2. Overexpression and Purification of Wild-Type and Mutant 3α-HSD/CRs
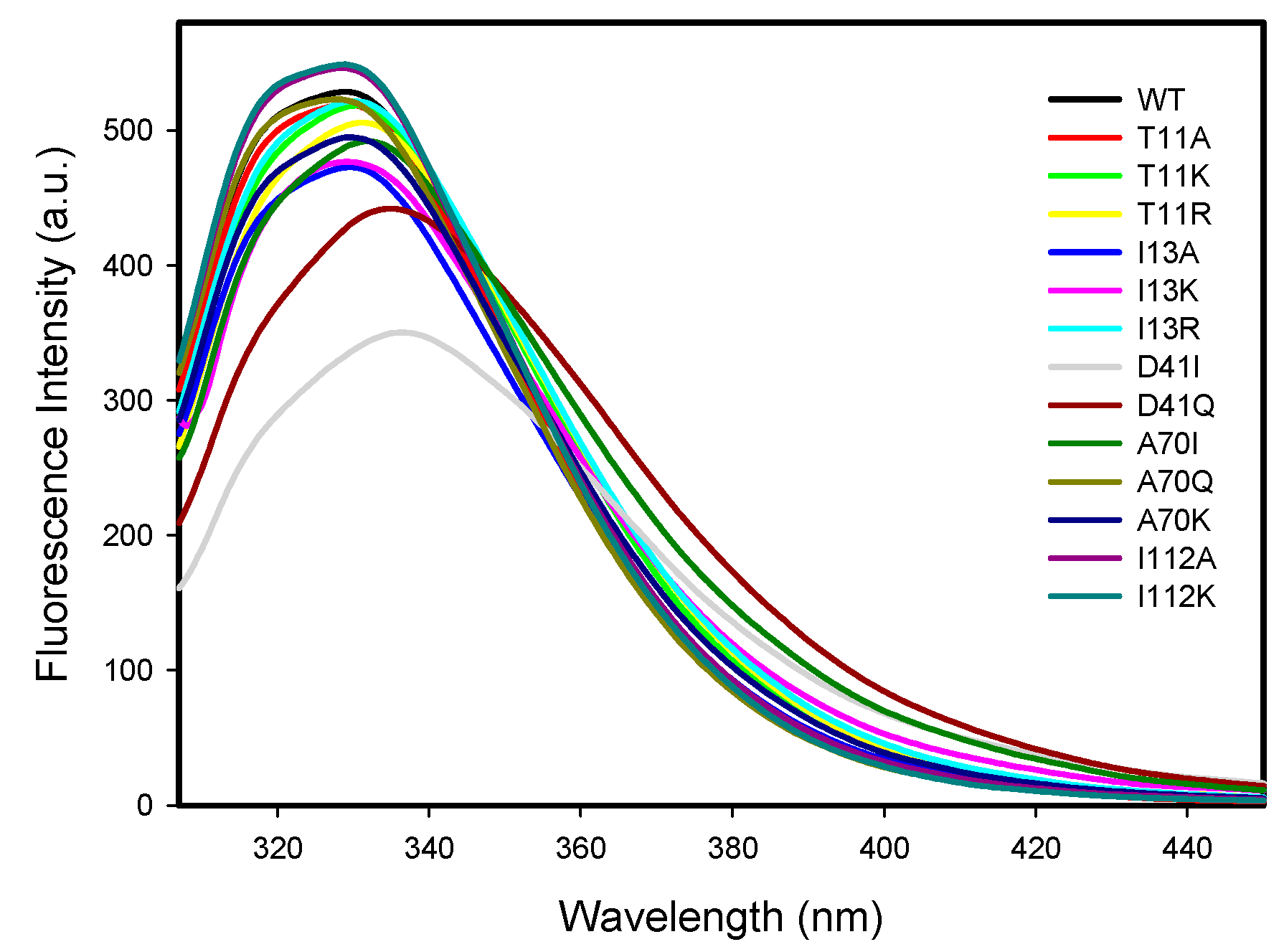
2.2.3. Protein Intrinsic Fluorescence
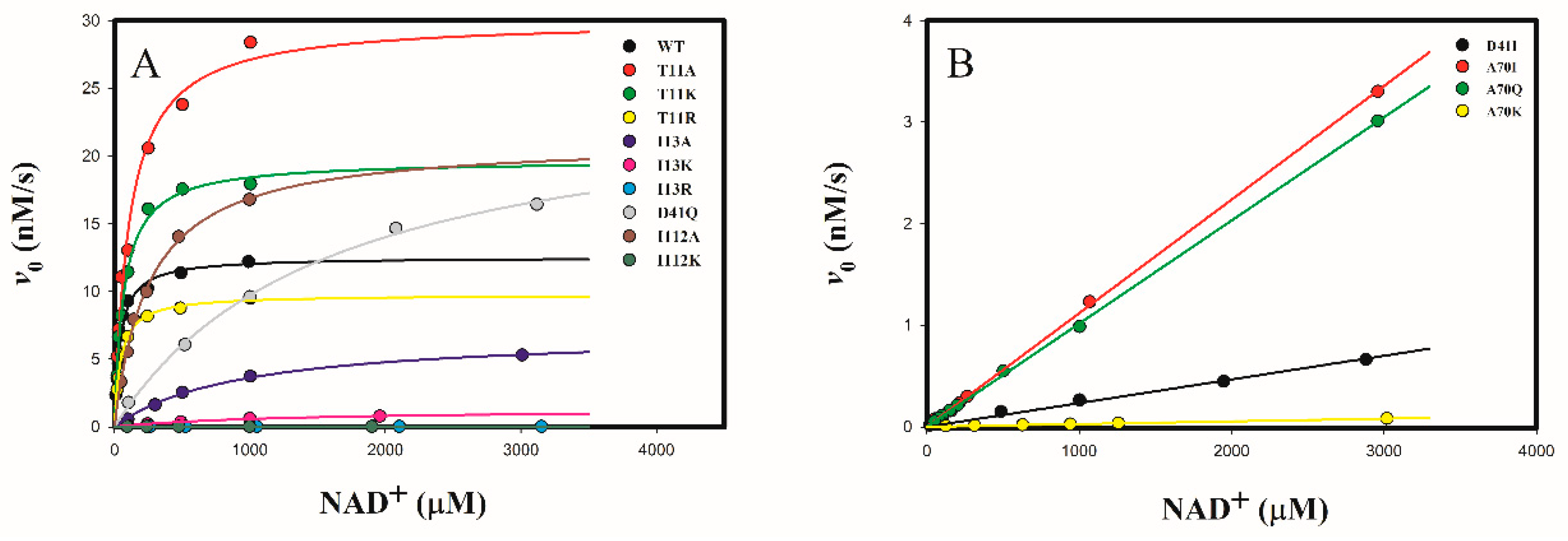
2.3. Steady-State Kinetics of Wild-Type and Mutant 3α-HSD/CRs
2.3.1. Determine the Cofactor Specificity of Wild-Type and Mutant 3α-HSD/CRs for NAD+
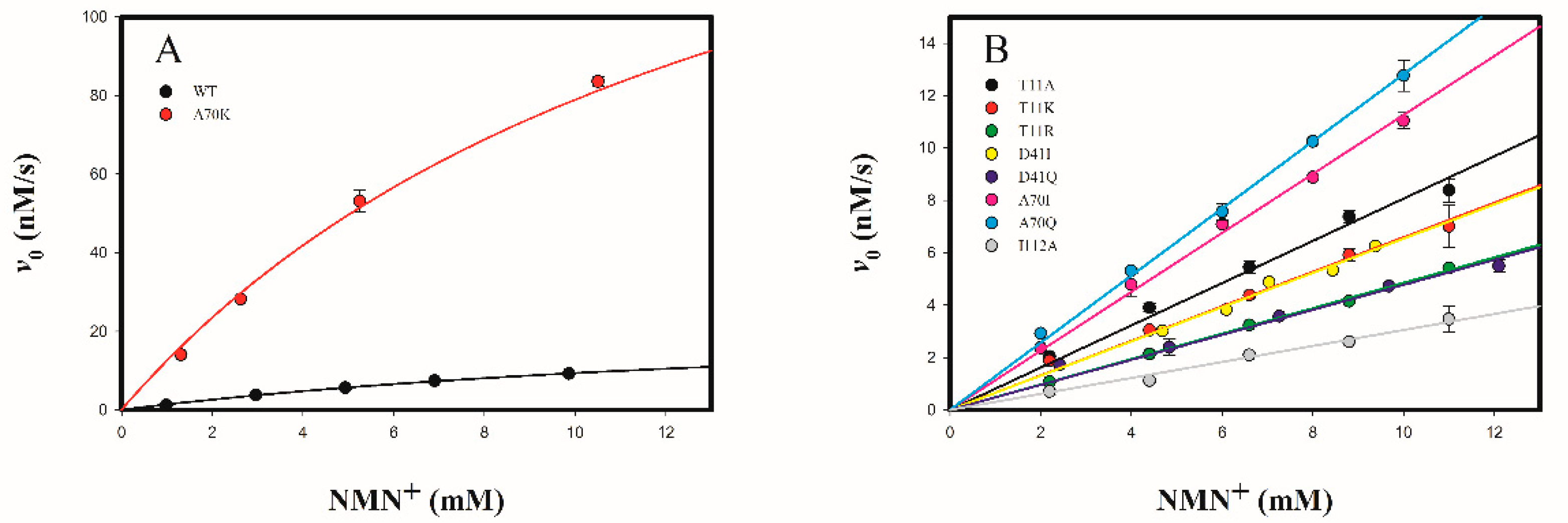
2.3.2. Determine the Cofactor Specificity of Wild-Type and Mutant 3α-HSD/CRs for NMN+
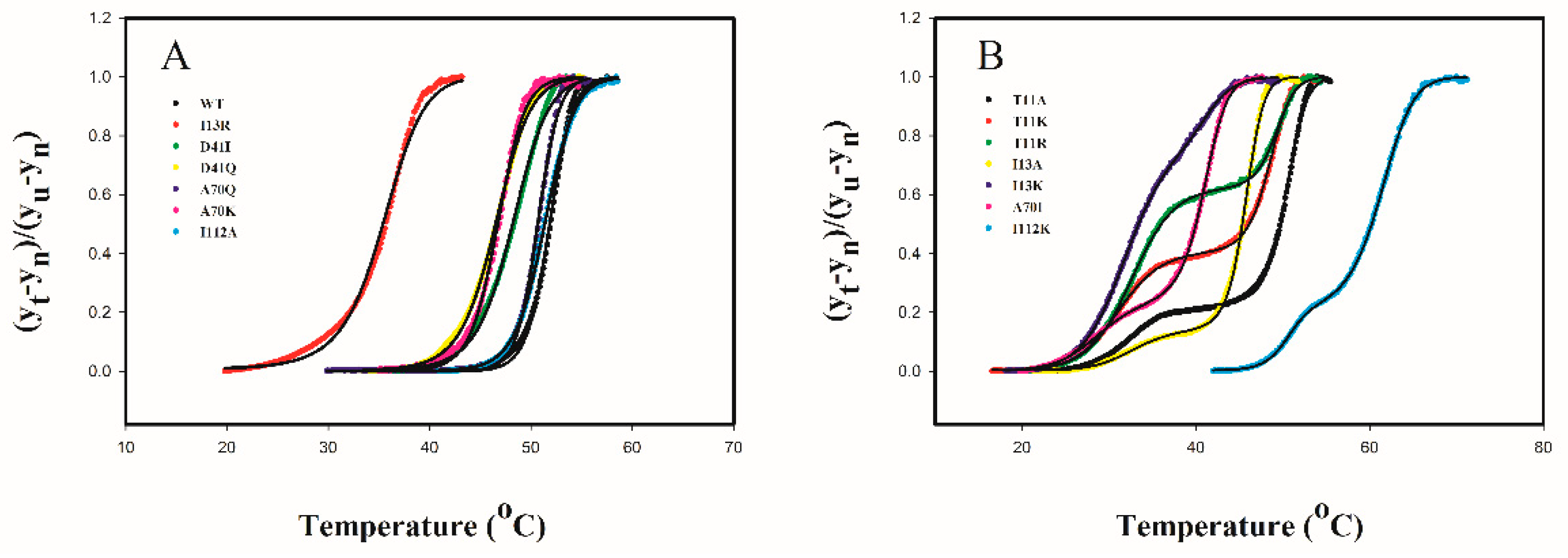
2.4. Protein Thermal Stability
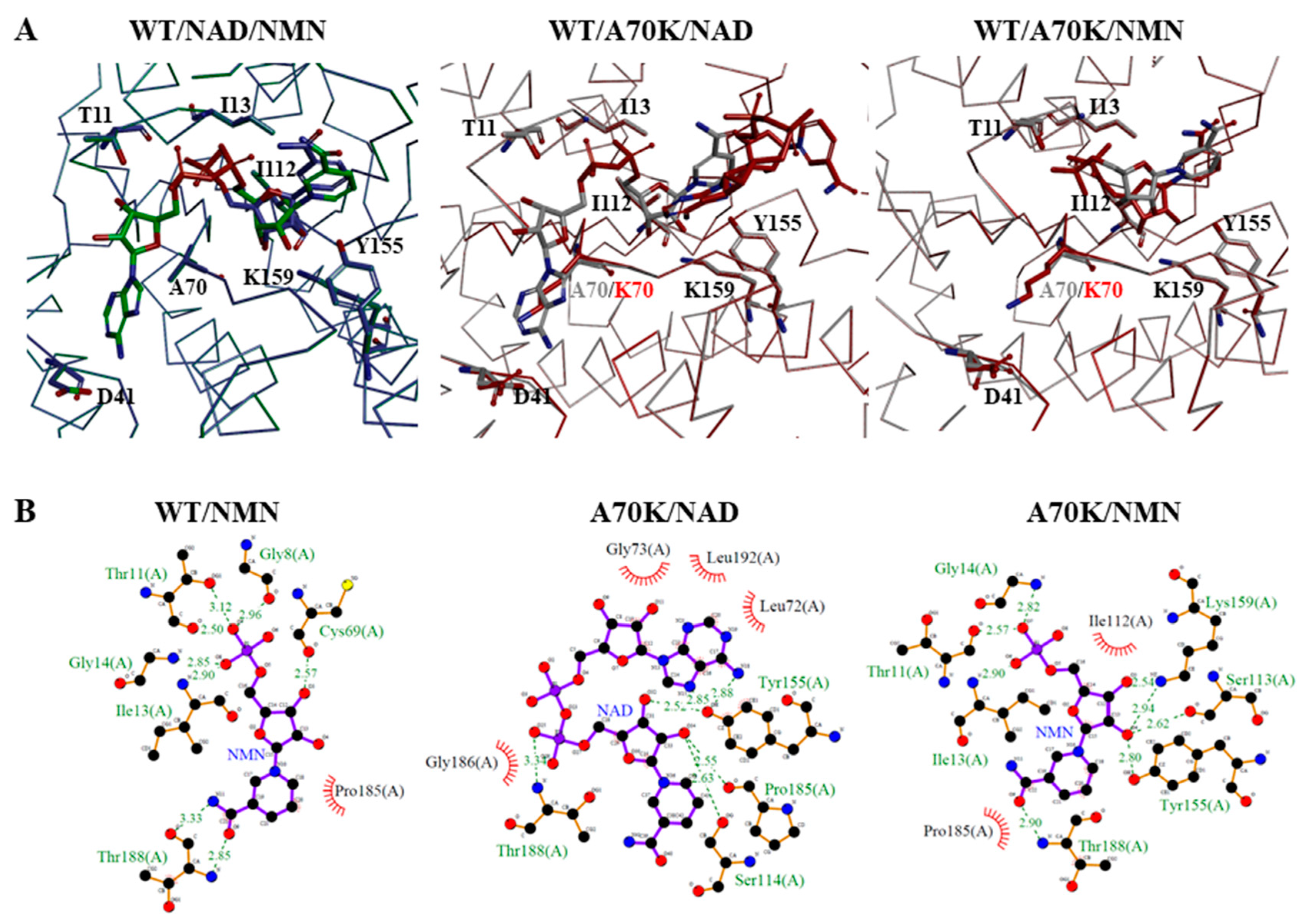
2.5. Molecular Docking Analysis of NAD+ and NMN+ for Wild-Type and Mutant 3α-HSD/CRs
3. Discussion
3.1. Roles of T11 and I13 in 3α-HSD/CR Catalysis and Cofactor Specificity
3.2. Roles of D41 in 3α-HSD/CR Catalysis and Cofactor Specificity
3.3. Roles of A70 in 3α-HSD/CR Catalysis and Cofactor Specificity
3.4. Roles of I112 in 3α-HSD/CR Catalysis and Cofactor Specificity
4. Materials and Methods
4.1. Site-Directed Mutagenesis, and the Overexpression and Purification of 3α-HSD/CR Variants
4.2. Overexpression and Purification of Wild-Type and Mutant 3α-HSD/CRs
4.3. Steady-State Kinetics
4.4. Protein Intrinsic Tryptophan Fluorescence
4.5. Thermal Denaturation Assay by Differential Scanning Fluorimetry
4.6. Molecular Docking Analysis
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sharma, A.; Gupta, G.; Ahmad, T.; Mansoor, S.; Kaur, B. Enzyme engineering: Current trends and future perspectives. Food Rev. Int. 2021, 37, 121–154. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Pereira, P.C. Biocatalysis engineering: The big picture. Chem. Soc. Rev. 2017, 46, 2678–2691. [Google Scholar] [CrossRef]
- Vidal, L.S.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. BBA-Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Mordhorst, S.; Andexer, J.N. Round, round we go—Strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef] [PubMed]
- Graef, M.R.D.; Alexeeva, S.; Snoep, J.L.; Mattos, M.J.T.d. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 1999, 181, 2351–2357. [Google Scholar] [CrossRef]
- Wang, M.; Chen, B.; Fang, Y.; Tan, T. Cofactor engineering for more efficient production of chemicals and biofuels. Biotechnol. Adv. 2017, 35, 1032–1039. [Google Scholar] [CrossRef]
- Cahn, J.K.B.; Werlang, C.A.; Baumschlager, A.; Brinkrnann-Chen, S.; Mayo, S.L.; Arnold, F.H. A general tool for engineering the NAD/NADP cofactor preference of oxidoreductases. Acs Synth. Biol. 2017, 6, 326–333. [Google Scholar] [CrossRef]
- You, C.; Huang, R.; Wei, X.L.; Zhu, Z.G.; Zhang, Y.-H.P. Protein engineering of oxidoreductases utilizing nicotinamide-based coenzymes, with applications in synthetic biology. Synth. Syst. Biotechnol. 2017, 2, 208–218. [Google Scholar] [CrossRef]
- Campbell, E.; Meredith, M.; Minteer, S.D.; Banta, S. Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem. Commun. 2012, 48, 1898–1900. [Google Scholar] [CrossRef]
- King, E.; Maxel, S.; Li, H. Engineering natural and noncanonical nicotinamide cofactor-dependent enzymes: Design principles and technology development. Curr. Opin. Biotechnol. 2020, 66, 217–226. [Google Scholar] [CrossRef]
- Black, W.B.; Zhang, L.; Mak, W.S.; Maxel, S.; Cui, Y.; King, E.; Fong, B.; Martinez, A.S.; Siegel, J.B.; Li, H. Engineering a nicotinamide mononucleotide redox cofactor system for biocatalysis. Nat. Chem. Biol. 2020, 16, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Meyer, M.M.; Meinhold, P.; Otey, C.R.; MacMillan, D.; Arnold, F.H. Evolving strategies for enzyme engineering. Curr. Opin. Struct. Biol. 2005, 15, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Zeymer, C.; Hilvert, D. Directed evolution of protein catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. [Google Scholar] [CrossRef] [PubMed]
- Kuchner, O.; Arnold, F.H. Directed evolution of enzyme catalysts. Trends Biotechnol. 1997, 15, 523–530. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Tuning the activity of an enzyme for unusual environments: Sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc. Natl. Acad. Sci. USA 1993, 90, 5618–5622. [Google Scholar] [CrossRef]
- Goldsmith, M.; Tawfik, D.S. Enzyme engineering: Reaching the maximal catalytic efficiency peak. Curr. Opin. Struct. Biol. 2017, 47, 140–150. [Google Scholar] [CrossRef]
- Tokuriki, N.; Jackson, C.J.; Afriat-Jurnou, L.; Wyganowski, K.T.; Tang, R.M.; Tawfik, D.S. Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme. Nat. Commun. 2012, 3, 1257. [Google Scholar] [CrossRef]
- Persson, B.; Kallberg, Y.; Bray, J.E.; Bruford, E.; Dellaporta, S.L.; Favia, A.D.; Duarte, R.G.; Jornvall, H.; Kavanagh, K.L.; Kedishvili, N.; et al. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem. Biol. Interact. 2009, 178, 94–98. [Google Scholar] [CrossRef]
- Kavanagh, K.L.; Jornvall, H.; Persson, B.; Oppermann, U. Medium- and short-chain dehydrogenase/reductase gene and protein families: The SDR superfamily: Functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef]
- Hwang, C.C.; Chang, Y.H.; Hsu, C.N.; Hsu, H.H.; Li, C.W.; Pon, H.I. Mechanistic roles of Ser-114, Tyr-155, and Lys-159 in 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. J. Biol. Chem. 2005, 280, 3522–3528. [Google Scholar] [CrossRef]
- Chang, Y.H.; Chuang, L.Y.; Hwang, C.C. Mechanism of proton transfer in the 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. J. Biol. Chem. 2007, 282, 34306–34314. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.C.; Chang, Y.H.; Lee, H.J.; Wang, T.P.; Su, Y.M.; Chen, H.W.; Liang, P.H. The catalytic roles of P185 and T188 and substrate-binding loop flexibility in 3 alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. PLoS ONE 2013, 8, e63594. [Google Scholar]
- Hwang, C.C.; Chang, P.R.; Wang, T.P. Contribution of remote substrate binding energy to the enzymatic rate acceleration for 3 alpha-hydroxysteroid dehydrogenase/carbonyl reductase. Chem.-Biol. Interact. 2017, 276, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Maser, E.; Mobus, E.; Klebe, G.; Reuter, K.; Ficner, R. The crystal structure of 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni shows a novel oligomerization pattern within the short chain dehydrogenase/reductase family. J. Biol. Chem. 2000, 275, 41333–41339. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Huang, T.J.; Chuang, L.Y.; Hwang, C.C. Role of S114 in the NADH-induced conformational change and catalysis of 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. Biochim. Biophys. Acta-Proteins Proteom. 2009, 1794, 1459–1466. [Google Scholar] [CrossRef]
- Filling, C.; Berndt, K.D.; Benach, J.; Knapp, S.; Prozorovski, T.; Nordling, E.; Ladenstein, R.; Jornvall, H.; Oppermann, U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 2002, 277, 25677–25684. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Chang, Y.H.; Wang, Y.L.; Lin, J.Y.; Chuang, L.Y.; Hwang, C.C. Expression, purification, and characterization of a human recombinant 17 beta-hydroxysteroid dehydrogenase type 1 in Escherichia coli. Mol. Biotechnol. 2010, 44, 133–139. [Google Scholar] [CrossRef]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef]
- Matulis, D.; Kranz, J.K.; Salemme, F.R.; Todd, M.J. Thermodynamic stability of carbonic anhydrase: Measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005, 44, 5258–5266. [Google Scholar] [CrossRef] [PubMed]
- Wolfenden, R.; Snider, M.J. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001, 34, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Amyes, T.L.; Richard, J.P.; Tait, J.J. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 2005, 127, 15708–15709. [Google Scholar] [CrossRef] [PubMed]
- Amyes, T.L.; Richard, J.P. Specificity in transition state binding: The Pauling model revisited. Biochemistry 2013, 52, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. Binding energy, specificity, and enzymic catalysis: The circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. [Google Scholar]
- Yao, X.; Bleile, D.W.; Yuan, Y.; Chao, J.; Sarathy, K.P.; Sanders, D.A.; Pinto, B.M.; O’Neill, M.A. Substrate directs enzyme dynamics by bridging distal sites: UDP-galactopyranose mutase. Proteins 2009, 74, 972–979. [Google Scholar] [CrossRef]
- Chen, C.H.; Namanja, A.T.; Chen, Y. Conformational flexibility and changes underlying activation of the SUMO-specific protease SENP1 by remote substrate binding. Nat. Commun. 2014, 5, 4968. [Google Scholar] [CrossRef]
- Johnson, T.A.; McLeod, M.J.; Holyoak, T. Utilization of substrate intrinsic binding energy for conformational change and catalytic function in phosphoenolpyruvate carboxykinase. Biochemistry 2016, 55, 575–587. [Google Scholar] [CrossRef]
- Hwang, C.-C.; Chang, P.-R.; Hsieh, C.-L.; Chou, Y.-H.; Wang, T.-P. Thermodynamic analysis of remote substrate binding energy in 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase catalysis. Chem.-Biol. Interact. 2019, 302, 183–189. [Google Scholar] [CrossRef]
- Sicsic, S.; Durand, P.; Langrene, S.; Legoffic, F. A new approach for using cofactor dependent enzymes—Example of alcohol-dehydrogenase. FEBS Lett. 1984, 176, 321–324. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Residues | Interacted Portion of NAD+ | Designed Mutants | Expected Function of Mutant |
|---|---|---|---|
| D41, A70 | Adenine | D41I, D41Q A70I, A70Q, A70K | (I) T11A, I13A, and I112A: Relaxation of cofactor specificity by decreasing the size of the residue |
| T11 | The ribose of adenosine | T11A, T11K, T11R | (II) D41I, D41Q, A70I, and A70Q: Size reduction in the cofactor binding pocket by increasing the size of the residue |
| I13 | Pyrophosphate | I13A, I13K, I13R | |
| I112 | Nicotinamide | I112A, I112K | (III) T11R, T11K, I13R, I13K, A70K, and I112K: Install polar interactions for NMN by incorporating the charge or polar group |
| 3α-HSD/CR | NAD+ | NMN+ | ||||
|---|---|---|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (M−1s−1 × 106) | kcat/Km (M−1s−1) | CSR e | RCS f | |
| WT | 135 ± 5 | 41.4 ± 5.2 | 3.2 ± 0.3 | 1.46 ± 0.06 | 4.6 × 10−7 | 1 |
| T11A | 329 ± 19 [0.4] | 108 ± 16 [2.6] | 2.8 ± 0.4 [1.1] | 0.78 ± 0.02 [1.8] | 2.8 × 10−7 | 0.61 |
| T11K | 219 ± 6 [0.6] | 68.4 ± 6.3 [1.7] | 3.2 ± 0.2 [1] | 0.64 ± 0.03 [2.3] | 2.0 × 10−7 | 0.43 |
| T11R | 105 ± 2 [1.3] | 45.9 ± 3.6 [1.1] | 2.3 ± 0.2 [1.4] | 0.47 ± 0.01 [3.1] | 2.0 × 10−7 | 0.43 |
| I13A | 75 ± 3 [1.8] | 923 ± 85 [22.3] | 0.081 ± 0.005 [40] | N.D. | ||
| I13K | 14 ± 1 [9.6] | 1288 ± 217 [31.1] | 0.011 ± 0.001 [291] | N.D. | ||
| I13R | 0.11 ± 0.01 [1225] | 1027 ±136 [24.8] | (1.1 ± 0.1) × 10−5 [29,091] | N.D. | ||
| D41I | N.A. | N.A. | (6.4 ± 1.5) × 10−3 [500] | 0.64 ± 0.01 [2.3] | 1.0 × 10−4 | 217 |
| D41Q | 269 ± 12 [0.5] | 1580 ± 158 [38.2] | 0.17 ± 0.01 [19] | 0.46 ± 0.02 [3.2] | 2.7 × 10−6 | 5.9 |
| A70I | N.A. | N.A. | 0.012 ± 0.001 [267] | 1.09 ± 0.02 [1.4] | 9.1 × 10−5 | 198 |
| A70Q | N.A. | N.A. | 0.011 ± 0.001 [291] | 1.24 ± 0.01 [1.2] | 1.1 × 10−4 | 239 |
| A70K | N.A. | N.A. | (2.9 ± 0.1) × 10−4 [11,034] | 12.7 ± 0.7 [0.11] | 4.4 × 10−2 | 9.6 × 104 |
| I112A | 227 ± 5 [0.6] | 255 ± 15 [6.2] | 0.89 ± 0.03 [3.6] | 0.29 ± 0.01 [5.0] | 3.3 × 10−7 | 0.72 |
| I112K | 0.12 ± 0.01 [1123] | 653 ± 85 [15.8] | (1.8 ± 0.2) × 10−4 [17,778] | N.D. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-L.; Chou, Y.-H.; Hsieh, C.-L.; Chiou, S.-J.; Wang, T.-P.; Hwang, C.-C. Rational Engineering of 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase for a Biomimetic Nicotinamide Mononucleotide Cofactor. Catalysts 2022, 12, 1094. https://doi.org/10.3390/catal12101094
Chen Y-L, Chou Y-H, Hsieh C-L, Chiou S-J, Wang T-P, Hwang C-C. Rational Engineering of 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase for a Biomimetic Nicotinamide Mononucleotide Cofactor. Catalysts. 2022; 12(10):1094. https://doi.org/10.3390/catal12101094
Chicago/Turabian StyleChen, Yan-Liang, Yun-Hao Chou, Chia-Lin Hsieh, Shean-Jaw Chiou, Tzu-Pin Wang, and Chi-Ching Hwang. 2022. "Rational Engineering of 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase for a Biomimetic Nicotinamide Mononucleotide Cofactor" Catalysts 12, no. 10: 1094. https://doi.org/10.3390/catal12101094
APA StyleChen, Y.-L., Chou, Y.-H., Hsieh, C.-L., Chiou, S.-J., Wang, T.-P., & Hwang, C.-C. (2022). Rational Engineering of 3α-Hydroxysteroid Dehydrogenase/Carbonyl Reductase for a Biomimetic Nicotinamide Mononucleotide Cofactor. Catalysts, 12(10), 1094. https://doi.org/10.3390/catal12101094