
The Role of Catalytic Ozonation Processes on the Elimination of DBPs and Their Precursors in Drinking Water Treatment




Abstract
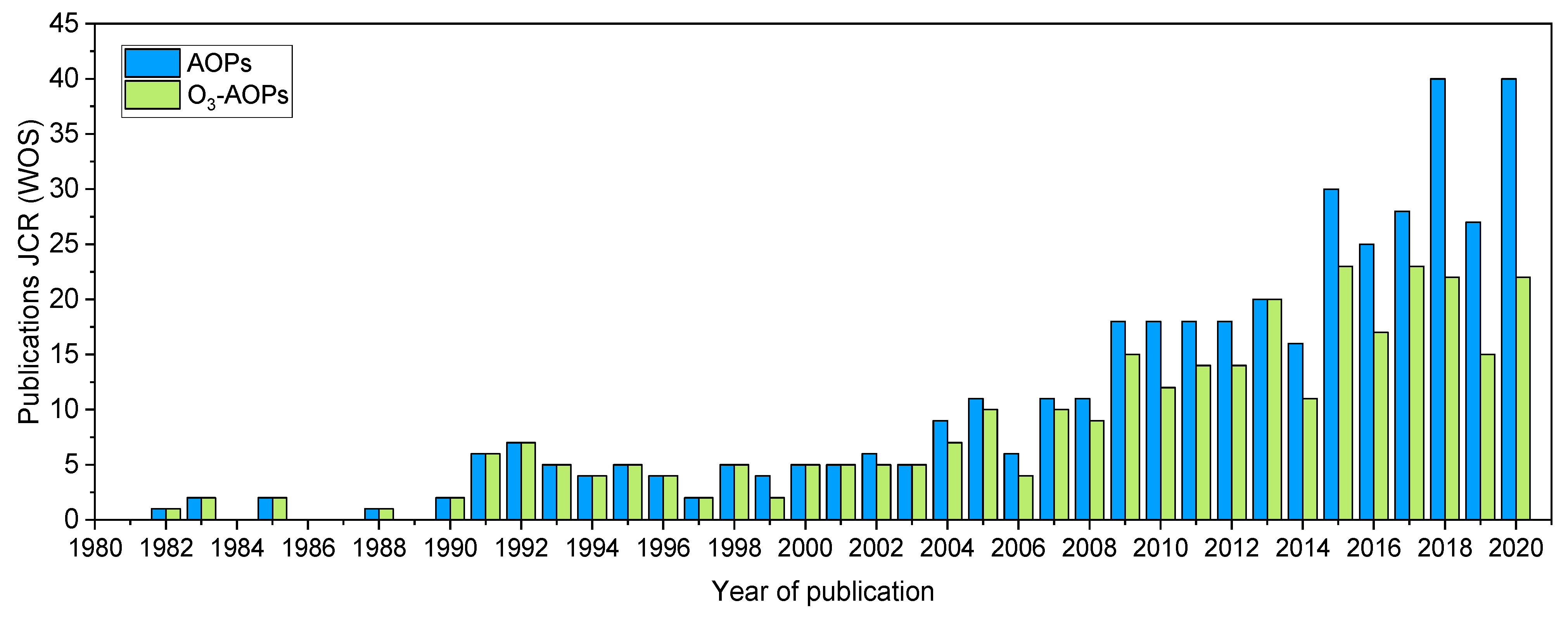
1. Introduction
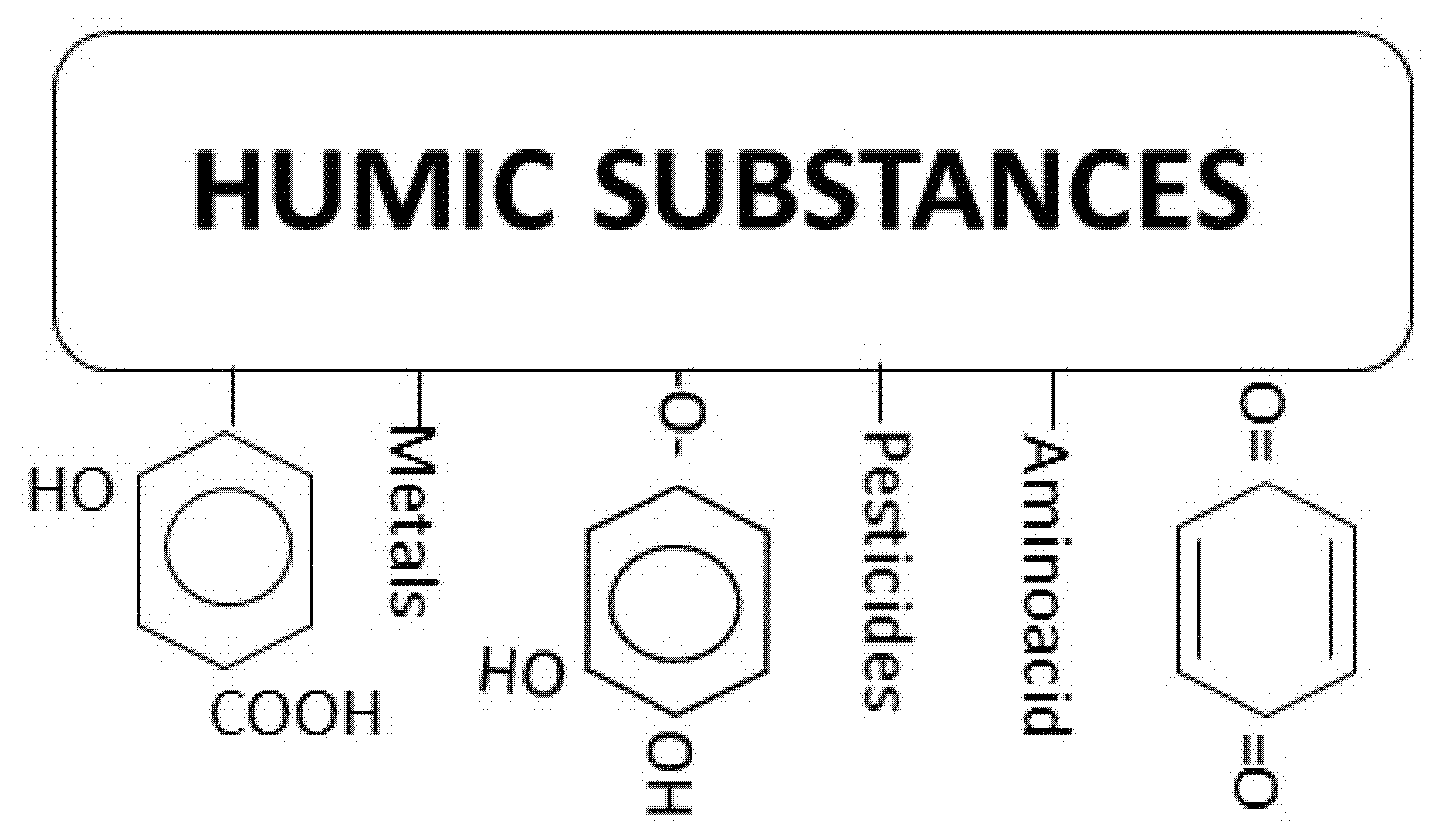
2. Nature of Chlorinated DBPs Precursors
3. Alternative Disinfectants to Chlorine
3.1. Ozone
3.2. Chlorine Dioxide
3.3. Chloramines
3.4. UV Disinfection
3.5. Other Disinfection Processes
4. Nature of DBPs from Classical Oxidant-Disinfectants Agents Used in DWTPs
5. Issues Related to DBPs Toxicity
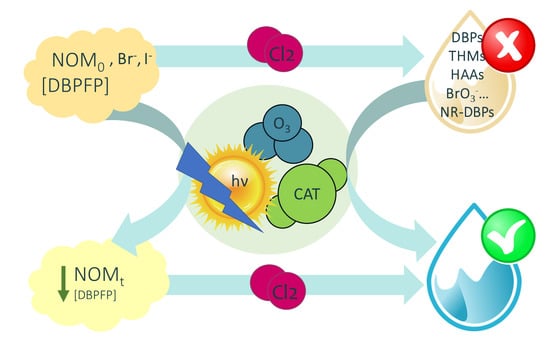
6. The Role of AOPs in the Removal of Precursors and DBPs
6.1. Elimination of Precursors or DBPs Formation Potential
6.1.1. Classic Ozonation Processes
6.1.2. Fenton Related Processes
6.1.3. Photocatalytic Oxidation
6.1.4. Sulfate-Radical Processes
6.1.5. Chlorine/UV Process
6.1.6. Electrochemical AOPs
6.1.7. Some Considerations on AOPs for DBPFP Removal
6.2. DBPs Removal
6.2.1. Ozone Based Processes
6.2.2. Ozone Free Processes
7. Elimination of DBP Precursors by Catalytic Ozonation Processes
7.1. Catalytic Ozonation
- Ozone adsorbed on the surface of the catalysts is decomposed into reactive species.
- Organic molecules are adsorbed on the surface of the catalysts with subsequent ozone attacks.
- Both ozone and organic molecules are adsorbed on the catalyst surface and surface reactions take place.
7.1.1. Catalysts
7.1.2. Catalytic Activity, Stability, and Reusability
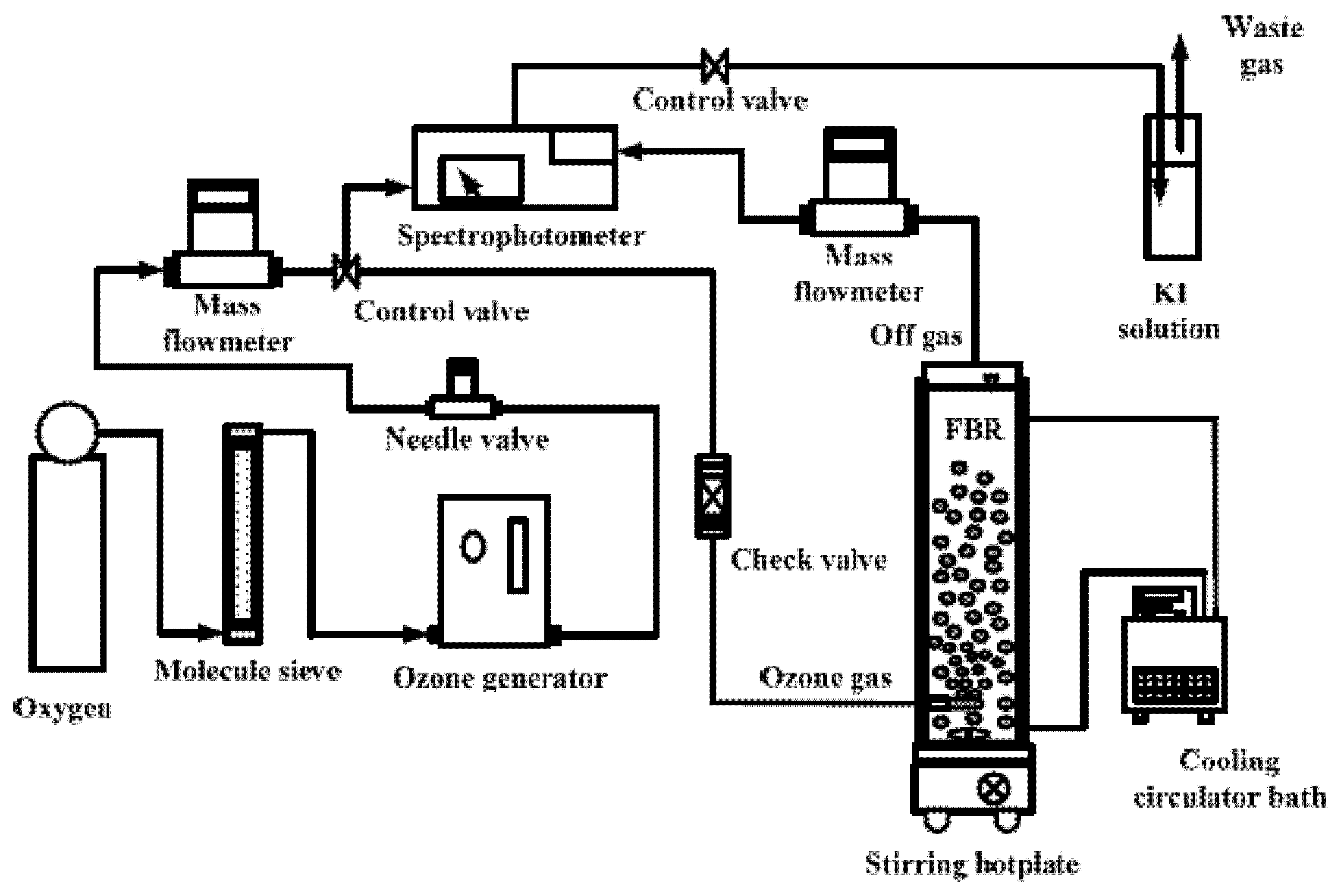
7.1.3. Reactors and Variables Studied
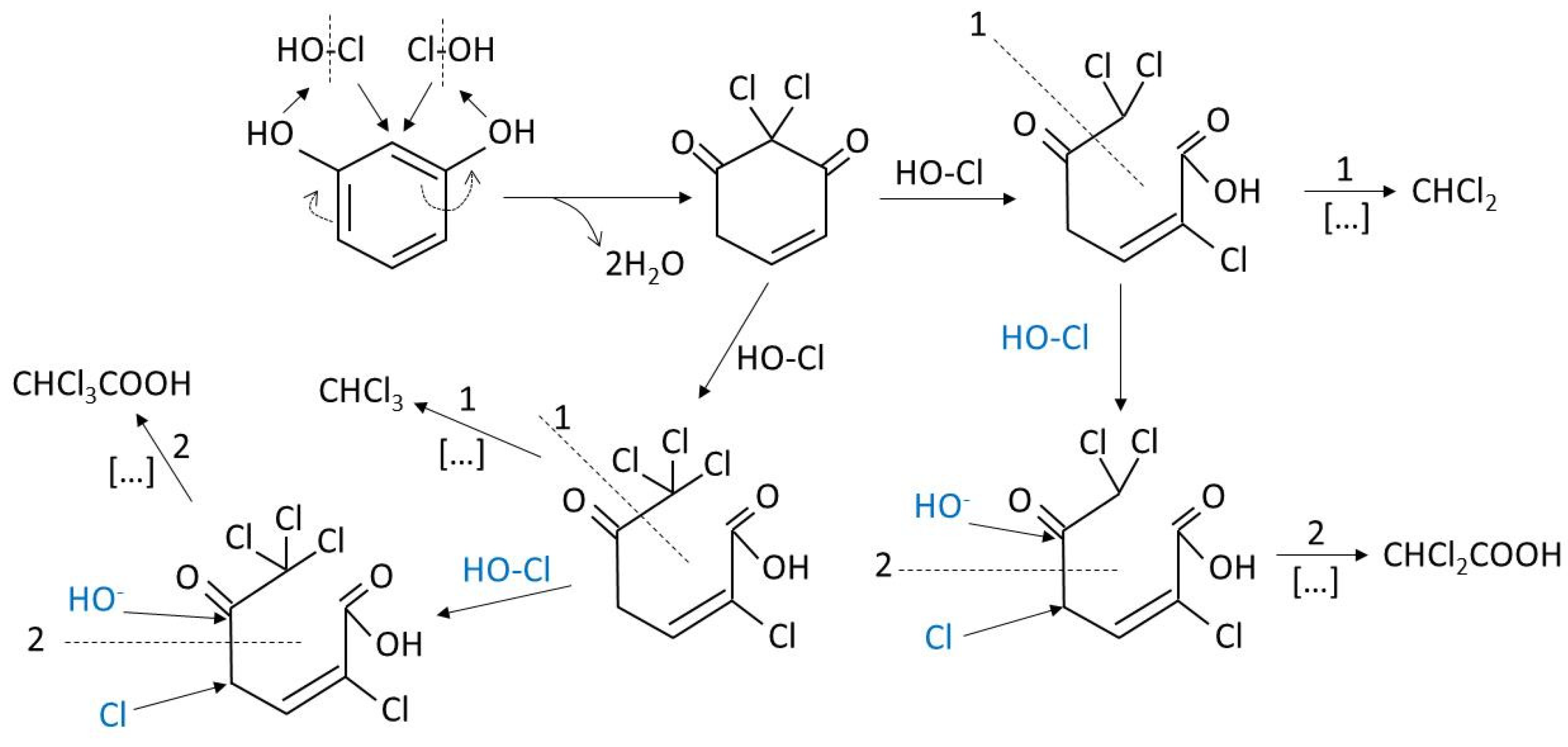
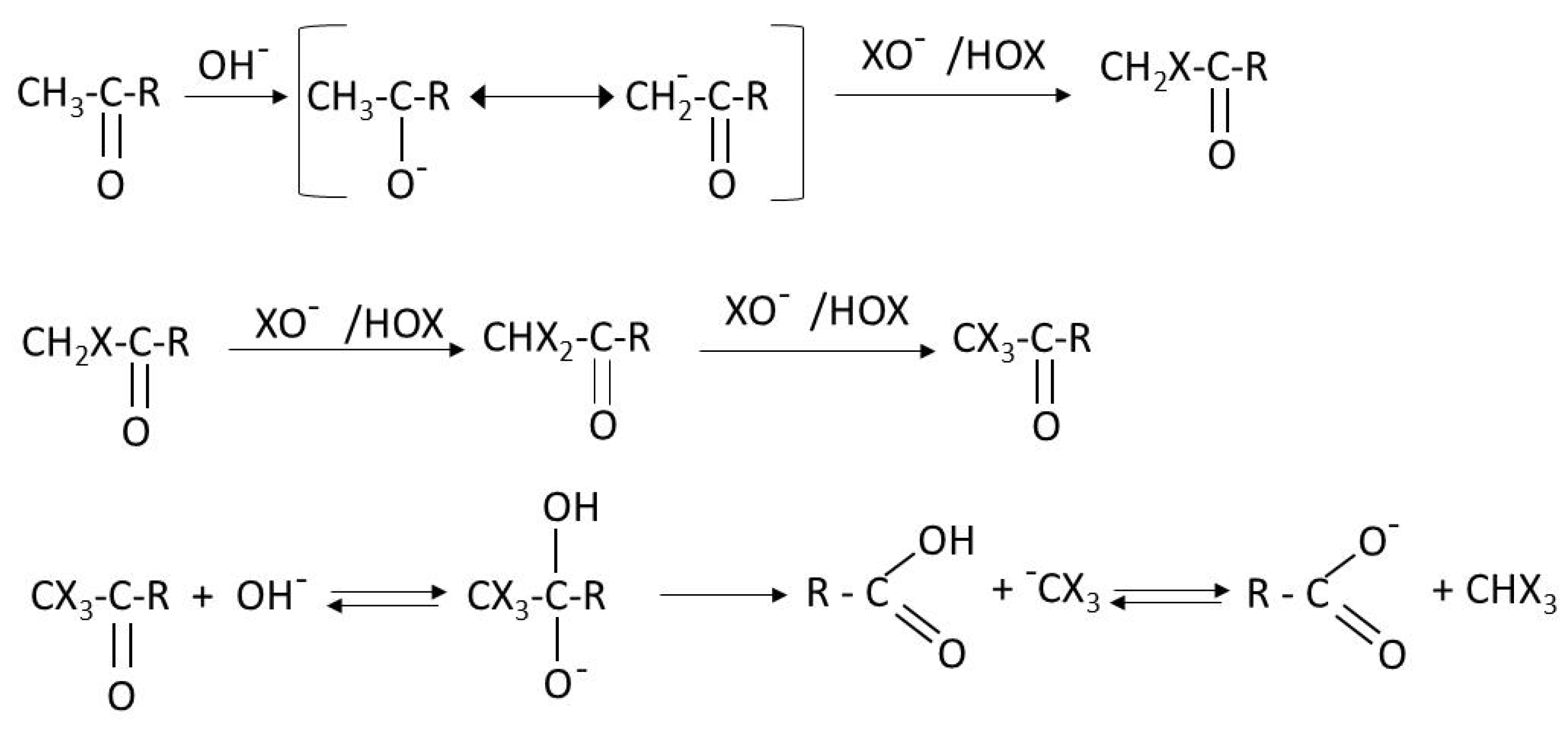
7.1.4. Mechanisms and Kinetics for the Removal of DBPs Precursors
7.2. Photocatalytic Ozonation
8. Elimination of DBPs by Catalytic/Photocatalytic Ozonation Processes
8.1. Catalytic Ozonation
8.1.1. Catalysts
8.1.2. Catalytic Activity, Stability, and Reusability
8.1.3. Reactors and Variables Studied
8.1.4. Kinetics and Mechanisms
8.2. Photocatalytic Ozonation
8.2.1. Catalysts and Radiation Use
8.2.2. Catalytic Activity, Stability, and Reusability
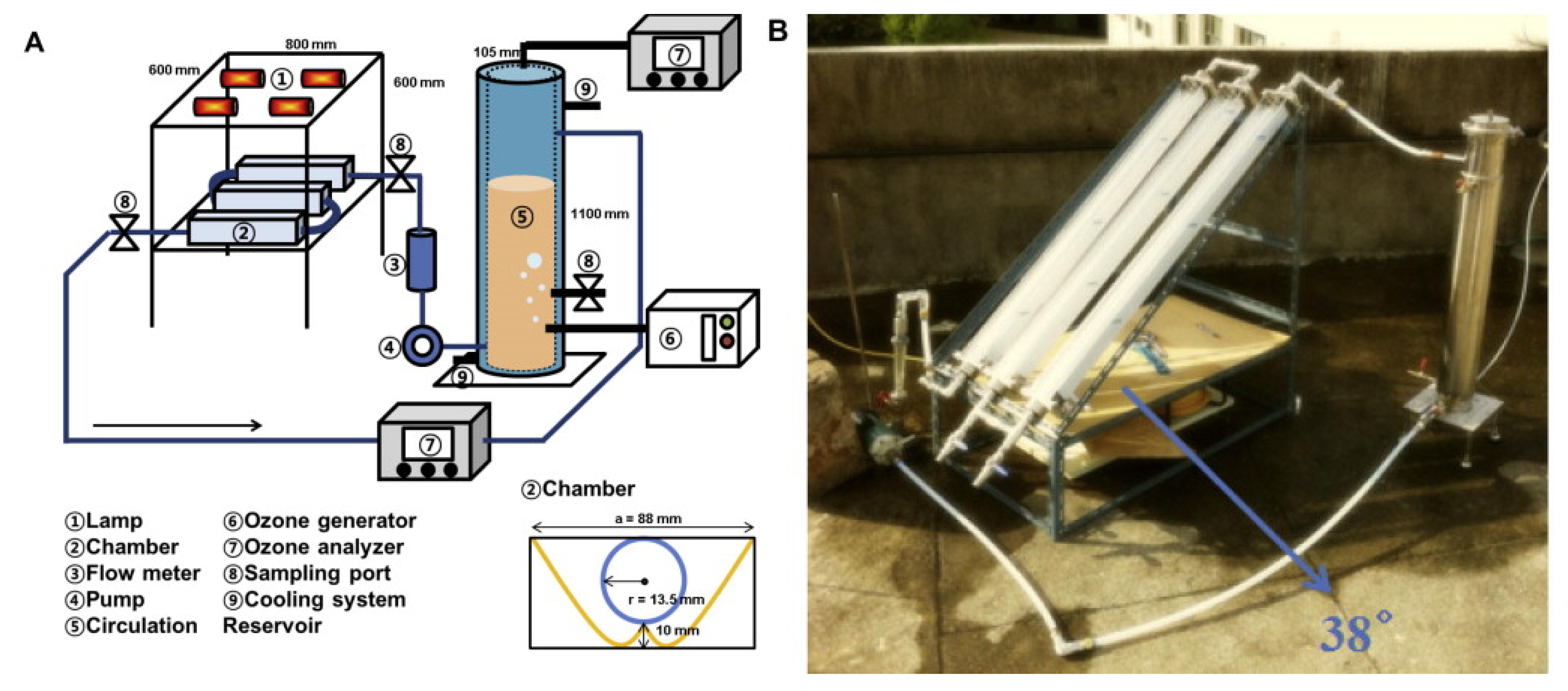
8.2.3. Reactors, Radiation Source, and Variables Studied
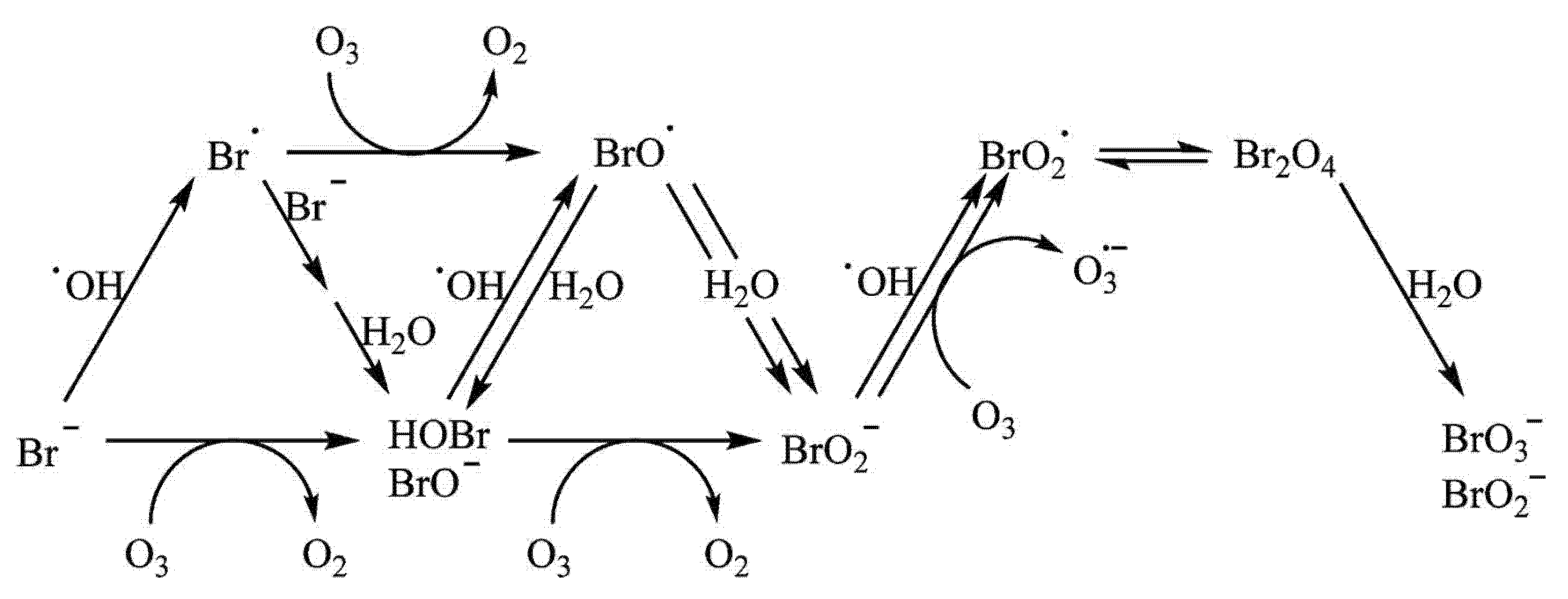
9. The Case of Bromate in Ozonation Processes
10. Concluding Remarks and Future Challenges
Author Contributions
Funding
Conflicts of Interest
Nomenclature
| AC | Activated carbon |
| AOPs | Advanced oxidation processes |
| AORPs | Advanced oxidation/reduction processes |
| AOX | Adsorbable organic halides |
| BAC | Biological activated carbon |
| BC | Before Christ |
| BDCA | Bromodichloroacetic acid |
| BET | Brunauer, Emmet, and Teller (referred to isotherm and surface area) |
| Br-DBPs | Brominated disinfection by-products |
| Ci | Concentration of i |
| CAAs | Chloroacetic acids |
| Cat | Catalyst |
| CDBA | Chlorodibromoacetic acid |
| CH | Chloral hydrate |
| CPC | Compound parabolic collector |
| CT | Parameter used in disinfection with C concentration of chlorine, T contact time |
| D | Diameter (particle size or reactor diameter) |
| DBPFP | Disinfection by-products formation potential |
| DBPs | Disinfection by-products |
| DCA | Dichloroacetic acid |
| DCAN | Dichloroacetonitrile |
| DFT | Density functional theory |
| DLS | Dynamic light scattering |
| DMP | Dimethyl phthalate |
| DOC | Dissolved organic carbon |
| DWT | Drinking water treatment |
| DWTP | Drinking water treatment plant |
| EAOPs | Electrochemical advanced oxidation processes |
| ECA | European Chemical Agency |
| EDS | Dispersive X-Ray spectroscopy |
| EPA | Environmental Protection Agency |
| E-peroxone | Electro-peroxone |
| EPR | Electron paramagnetic resonance |
| ER | Eley-Rideal mechanism |
| FMOG | Flower-like nanocomposite |
| FTIR | Fourier transformed infrared spectroscopy |
| GAC | Granular activated carbon |
| HA | Humic acid |
| HAAs | Haloacetic acids |
| HAcAm | Haloacetamides |
| HANs | Haloacetonitriles |
| HIA | Hydrophilic acid |
| HIB | Hydrophilic base |
| HKs | Haloketones |
| HOA | Hydrophobic acid |
| HON | Hydrophobic neutral |
| HS | Humic substances |
| ICP | Inductively coupled plasma |
| ICZ | Iron coated zeolite |
| I-DBPs | Iodinated disinfection by-products |
| JCR | Journal Citation Report |
| k | Kinetic constant |
| L | Length |
| LED | Light emitting diodes |
| LHHW | Langmuir–Hinshelwood–Hougen–Watson mechanism |
| MBA | Bromoacetic acid |
| MCA | Monochloroacetic acid |
| MCL | Maximum concentration level |
| MOFs | Metal organic frameworks |
| NB | Nitrobencene |
| N-DBPs | Nitrogen containing disinfection by-products |
| NDBA | N-nitrosodibuthylamine |
| NDMA | N-nitrosodimethylamine |
| NOM | Natural organic matter |
| NR-DBPs | Non-regulated disinfection by-products |
| NTU | Nephelometric turbidity unit |
| OA | Oxalic acid |
| PACL | Poly-aluminium chloride |
| PAG | Pilkington ActiveTM glass |
| pCBA | p-Chlorobenzoic acid |
| PMO | Pure manganese oxide |
| PMS | Peroxymonosulfate |
| PS | Persulfate |
| PZC | Potential of zero charge |
| Q | Volumetric flow |
| Rct | Ratio of the hydroxyl radicals to the ozone exposure during ozone processes |
| R-DBPs | Regulated disinfection by-products |
| RGO | Reduced graphene oxide |
| RO | Reverse osmosis |
| ROS | Reactive oxidizing species |
| SCE | Saturated calomel electrode |
| SEM | Scanning electron microscopy |
| SHE | Standard hydrogen electrode |
| TBA | Tribromoacetic acid |
| TCA | Trichloroacetic acid |
| TCNM | Trichloronitromethane |
| THAAs | Total haloacetic acids |
| THAAFP | Total haloacetic acids formation potential |
| THMs | Trihalomethanes |
| THMFP | Trihalomethane formation potential |
| TNT | Titanate nanotubes |
| TOC | Total organic carbon |
| TOX | Total organic halogen |
| TOXFP | Total organic halogen formation potential |
| TTHMFP | Total trihalomethane formation potential |
| WHO | World Health Organization |
| USEPA | United States Environmental Protection Agency |
| USA | United States of America |
| UV | Ultraviolet radiation |
| UVA | Ultraviolet A radiation |
| UVC | Ultraviolet C radiation |
| UV-Vis-DRS | Ultraviolet-visible diffuse reflectance spectroscopy |
| UV254 | Referred to absorbance at 254 nm |
| V | Volume |
| WOS | Web of Science |
| X | Halogen |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
| XRF | X-ray fluorescence |
References
- Danil De Namor, A.F. Water purification: From ancient civilization to the XXI Century. Water Sci. Technol. Water Supply 2007, 7, 33–39. [Google Scholar] [CrossRef]
- Merlet, N. Contribution á l’Etude du Mécanisme de Formation des Trihalométhanes et des Composés Organohalogénés non Volatils Lors de la Chloration de Molecules Modèles. Ph.D. Thesis, Université de Poitiers, Poitiers, France, 1986. [Google Scholar]
- White, G.C. Current chlorination and dechlorination practices in the treatment of potable water, wastewater and cooling water. In Water Chlorination: Environmental Impacts and Health Effects; Ann Arbor Science Publishers: Ann Arbor, MI, USA, 1978; Volume 1, pp. 1–18. [Google Scholar]
- Symons, J.M.; Bellar, T.A.; Carswell, J.K. National organics reconnaissance survey for halogenated organics. J. Am. Water Work. Assoc. 1975, 67, 634–647. [Google Scholar] [CrossRef]
- Rook, J.J. Formation of Haloforms during Chlorination of Natural Waters. Water Treat. Exam. 1974, 23, 234–243. [Google Scholar]
- Bellar, T.A.; Lichtenberg, J.J.; Kroner, R.C. Occurrence of Organohalides in Chlorinated Drinking Waters. J. Am. Water Work. Assoc. 1974, 66, 703–706. [Google Scholar] [CrossRef]
- US Environmental Protection Agency. Lower Mississippi River Facility. New Orleans Area Water Supply Study; US Environmental Protection Agency: Whasington, DC, USA, 1974.
- Chang, S.L. The safety of water disinfection. Annu. Rev. Public Health 1982, 3, 393–418. [Google Scholar] [CrossRef]
- Glaze, W.H.; Henderson, J.E., IV. Formation of organochlorine compounds from the chlorination of a municipal secondary effluent. J. Water Pollut. Control Fed. 1975, 47, 2511–2515. [Google Scholar]
- Schnitzer, M.; Khan, S.U. Humic Subtances in the Environment; Marcel Dekker Inc.: New York, NY, USA, 1972. [Google Scholar]
- Steelink, C. Humates and other natural organic substances in the aquatic environment. J. Chem. Educ. 1977, 54, 599–603. [Google Scholar] [CrossRef]
- Croué, J.P. Contribution á l’étude de l’Oxydation par le Chlore et L’ozone D’acides Fulviques Naturels Extraits D’eaux de Surface. Ph.D. Thesis, Université de Poitiers, Poitiers, France, 1987. [Google Scholar]
- Corin, N.; Backhand, P.; Kulovaara, M. Degradation products formed during UV-irradiation of humic waters. Chemosphere 1996, 33, 245–255. [Google Scholar] [CrossRef]
- Beckett, R. The Surface Chemistry of Humic Substances in Aquatic Systems. In Surface and Colloid Chemistry in Natural Waters and Water Treatment; Beckett, R., Ed.; Plenum: New York, NY, USA, 1990; pp. 3–16. [Google Scholar]
- Sirivedhin, T.; Gray, K.A., II. Comparison of the disinfection by-product formation potentials between a wastewater effluent and surface waters. Water Res. 2005, 39, 1025–1036. [Google Scholar] [CrossRef]
- Doré, M.; Goichon, J. Etude d’une methode d’evaluation globale des precurseurs de la reaction haloforme. Water Res. 1980, 14, 657–663. [Google Scholar] [CrossRef]
- Gilca, A.F.; Teodosiu, C.; Fiore, S.; Musteret, C.P. Emerging disinfection byproducts: A review on their occurrence and control in drinking water treatment processes. Chemosphere 2020, 259, 127476. [Google Scholar] [CrossRef]
- Sun, S.; Jiang, T.; Lin, Y.; Song, J.; Zheng, Y.; An, D. Characteristics of organic pollutants in source water and purification evaluations in drinking water treatment plants. Sci. Total Environ. 2020, 733, 139277. [Google Scholar] [CrossRef]
- Gonsioroski, A.; Mourikes, V.E.; Flaws, J.A. Endocrine disruptors in water and their effects on the reproductive system. Int. J. Mol. Sci. 2020, 21, 1929. [Google Scholar] [CrossRef] [PubMed]
- Tak, S.; Vellanki, B.P. Natural organic matter as precursor to disinfection byproducts and its removal using conventional and advanced processes: State of the art review. J. Water Health 2018, 16, 681–703. [Google Scholar] [CrossRef]
- Miller, G.W.; Rice, R.G.; Robson, C.M.; Scullin, R.L.; Kuhn, W.; Wolf, H. An Assessment of Ozone and Chlorine Dioxide Technologies for Treatment of Municipal Water Supplies; Environmental Protection Agency, Office of Research and Development, Municipal Environmental Research Laboratory: Cincinnati, OH, USA, 1978; Executive Summary EPA Report; EPA-600/8-78-018 October 1978.
- Doré, M. Chimie des Oxydants et Traitement des Eaux. Tech. Doc. Paris 1989. [Google Scholar]
- Langlais, B.; Reckhow, D.A.; Brink, D.R. Ozone in Water Treatment: Application and Engineering; Langlais, B., Reckhow, D.A., Brink, D.R., Eds.; Lewis Publishers: Chelsea, MI, USA, 1991. [Google Scholar]
- Glaze, W.H.; Kang, J.W.; Chapin, D.H. The chemistry of water treatment processes involving ozone, hydrogen peroxide and ultraviolet radiation. Ozone Sci. Eng. 1987, 9, 335–352. [Google Scholar] [CrossRef]
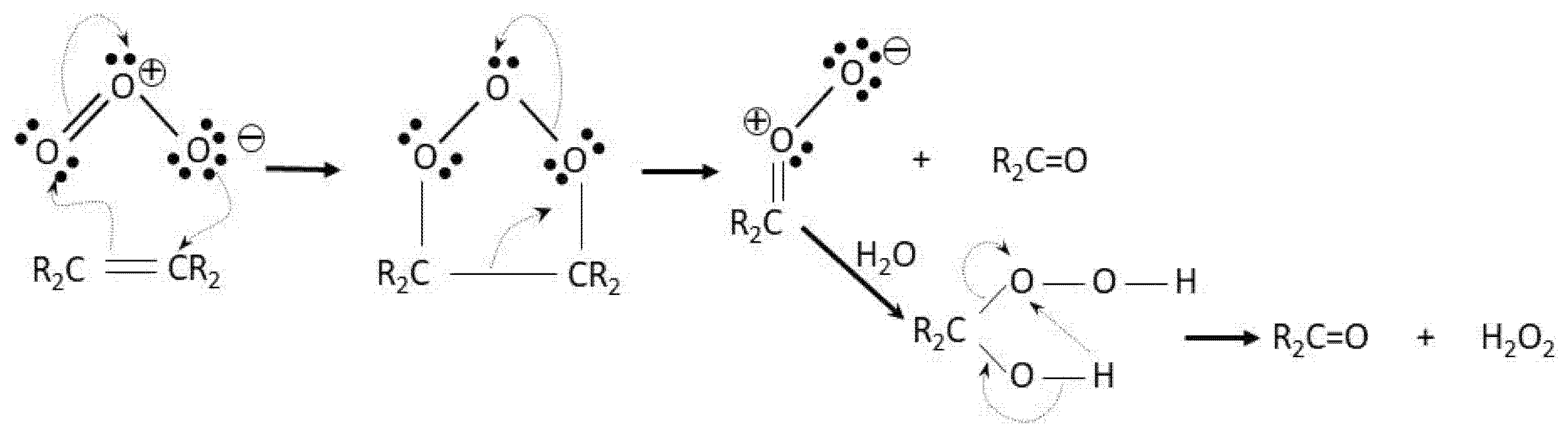
- Bailey, P.S. The Reactions Of Ozone With Organic Compounds. Chem. Rev. 1958, 58, 925–1010. [Google Scholar] [CrossRef]
- Staehelin, J.; Hoigne, J. Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions. Environ. Sci. Technol. 1985, 19, 1206–1213. [Google Scholar] [CrossRef]
- Beltrán, F.J. Ozone Reaction Kinetics for Water and Wastewater Systems; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Bader, H.; Hoigné, J. Rate Constants of Reactions of Ozone with Organic and Inorganic Compounds in Water -II. Dissociating Organic Compounds. Water Res. 1983, 17, 185–194. [Google Scholar]
- Hoigné, J.; Bader, H. Rate constants of reactions of ozone with organic and inorganic compounds in water-I. Non-dissociating organic compounds. Water Res. 1983, 17, 173–183. [Google Scholar] [CrossRef]
- Kuczkowski, R.L. Ozone and carbonyl oxides. In 1,3, Dipolar Cycloaddition Chemistry; John Wiley and Sons: New York, NY, USA, 1984; Volume 2A, pp. 197–277. [Google Scholar]
- Beltran, F.J.; Gonzalez, M.; GarcĺA-Araya, J.F.; Cabrera, J.L. The use of ozonation to reduce the potential for forming trihalomethane compounds in chlorinating resorcinol, phloroglucinol and 1,3 cyclohexanedione. Chem. Eng. Commun. 1990, 96, 321–339. [Google Scholar] [CrossRef]
- Cavanagh, J.E.; Weinberg, H.S.; Avram, G.; Sangalah, R.; Dean, M.; Glaze, W.H.; Collette, T.W.; Richardson, S.D.; Thruston, A.D. Ozonation Byproducts: Identification of Bromohydrins from the Ozonation of Natural Waters with Enhanced Bromide Levels. Environ. Sci. Technol. 1992, 26, 1658–1662. [Google Scholar] [CrossRef]
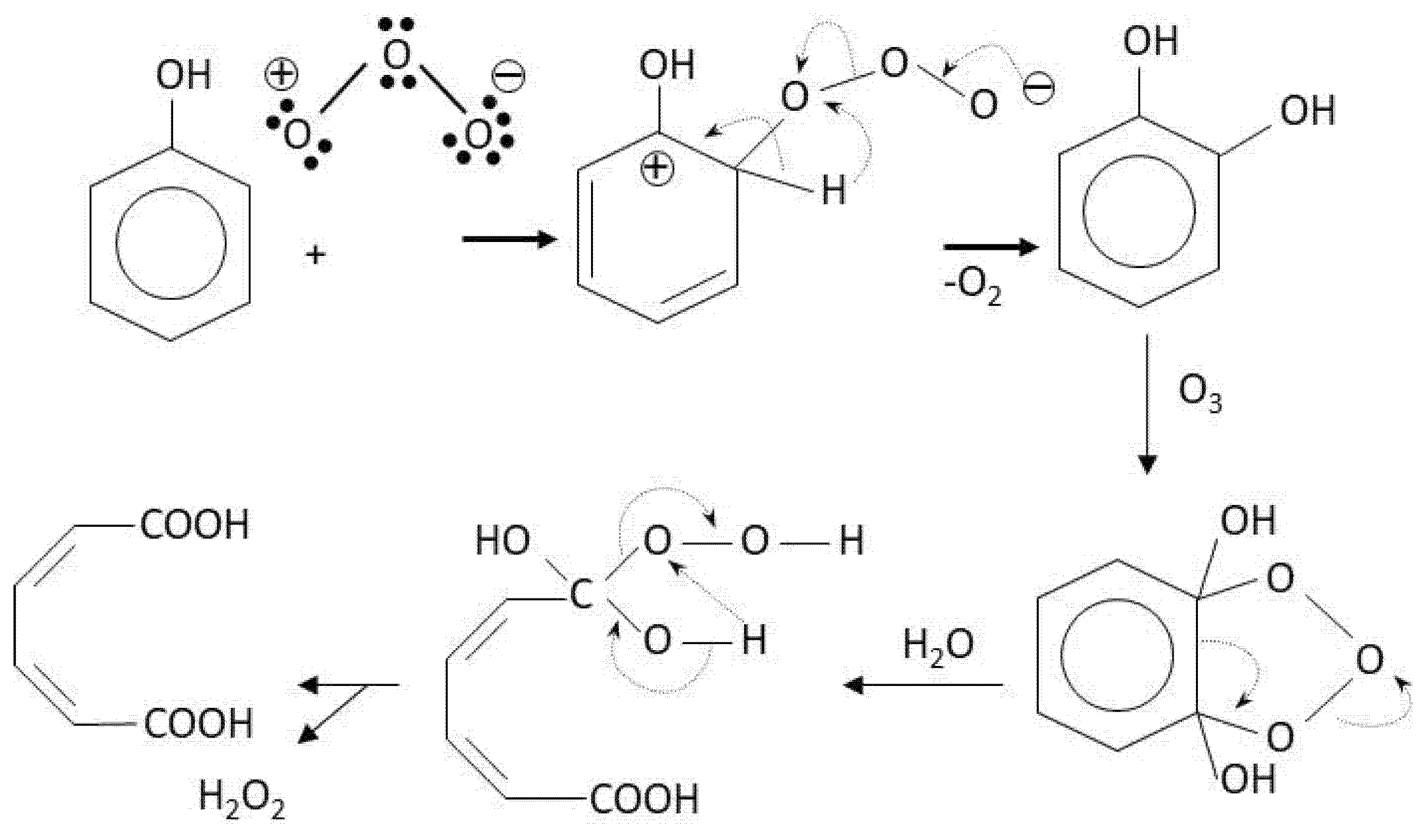
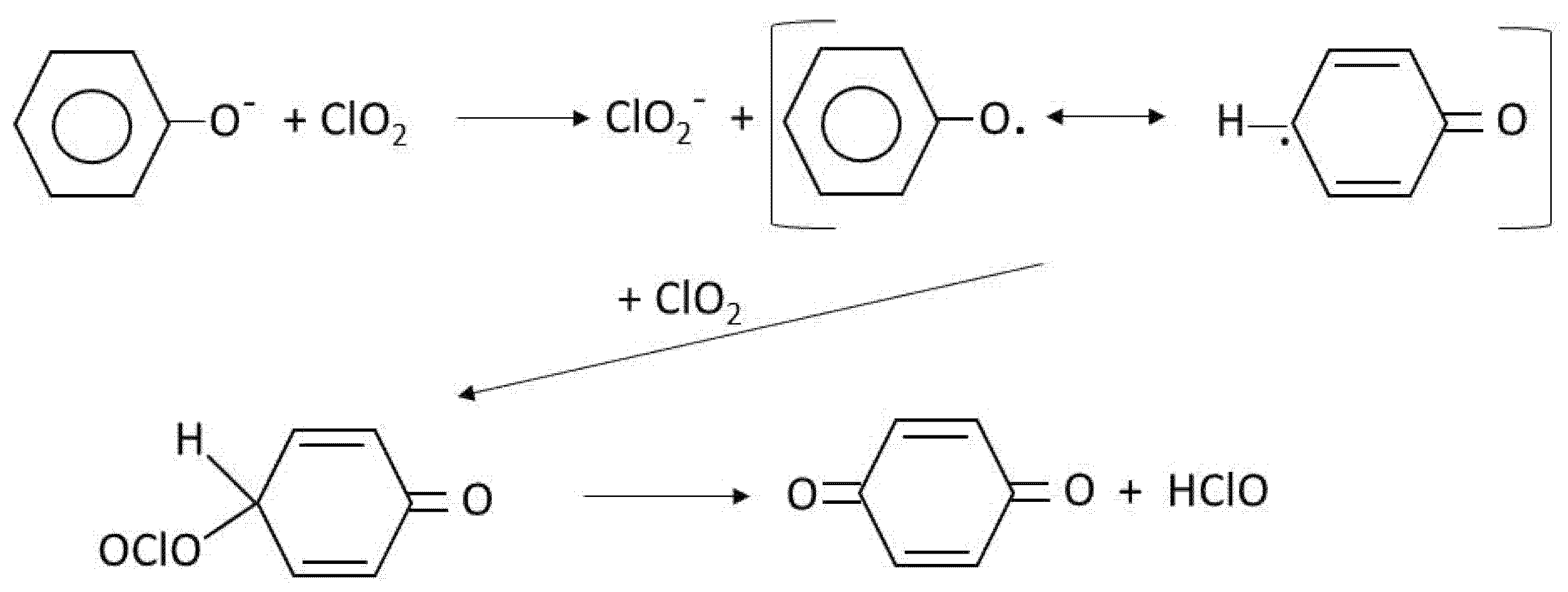
- Rav-Acha, C. Review Paper the Reactions of Chlorine Dioxide With Aquatic Organic Materials and. Water Res. 1984, 18, 1329–1341. [Google Scholar] [CrossRef]
- Wajon, J.E.; Rosenblatt, D.H.; Burrows, E.P. Oxidation of Phenol and Hydroquinone by Chlorine Dioxide. Environ. Sci. Technol. 1982, 16, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.G.; Gomez-Taylor, M. Occurrence of by-products of strong oxidants reacting with drinking water contaminants—Scope of the problem. Environ. Health Perspect. 1986, 69, 31–44. [Google Scholar] [CrossRef][Green Version]
- Song, K.; Mohseni, M.; Taghipour, F. Application of ultraviolet light-emitting diodes (UV-LEDs) for water disinfection: A review. Water Res. 2016, 94, 341–349. [Google Scholar] [CrossRef]
- Dalrymple, O.K.; Stefanakos, E.; Trotz, M.A.; Goswami, D.Y. A review of the mechanisms and modeling of photocatalytic disinfection. Appl. Catal. B Environ. 2010, 98, 27–38. [Google Scholar] [CrossRef]
- Fernández, P.; Blanco, J.; Sichel, C.; Malato, S. Water disinfection by solar photocatalysis using compound parabolic collectors. Catal. Today 2005, 101, 345–352. [Google Scholar] [CrossRef]
- Booshehri, A.Y.; Polo-Lopez, M.I.; Castro-Alférez, M.; He, P.; Xu, R.; Rong, W.; Malato, S.; Fernández-Ibáñez, P. Assessment of solar photocatalysis using Ag/BiVO4 at pilot solar Compound Parabolic Collector for inactivation of pathogens in well water and secondary effluents. Catal. Today 2017, 281, 124–134. [Google Scholar] [CrossRef]
- Madaeni, S.S. The application of membrane technology for water disinfection. Water Res. 1999, 33, 301–308. [Google Scholar] [CrossRef]
- Sun, X.; Liu, J.; Ji, L.; Wang, G.; Zhao, S.; Yoon, J.Y.; Chen, S. A review on hydrodynamic cavitation disinfection: The current state of knowledge. Sci. Total Environ. 2020, 737. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Mahendra, S.; Lyon, D.Y.; Brunet, L.; Liga, M.V.; Li, D.; Alvarez, P.J.J. Antimicrobial nanomaterials for water disinfection and microbial control: Potential applications and implications. Water Res. 2008, 42, 4591–4602. [Google Scholar] [CrossRef] [PubMed]
- Pina, A.S.; Batalha, Í.L.; Fernandes, C.S.M.; Aoki, M.A.; Roque, A.C.A. Exploring the potential of magnetic antimicrobial agents for water disinfection. Water Res. 2014, 66, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Emile Coleman, W.; Melton, R.G.; Kopfler, F.C.; Barone, K.A.; Aurand, T.A.; Jellison, M.G. Identification of Organic Compounds in a Mutagenic Extract of a Surface Drinking Water by a Computerized Gas Chromatography/Mass Spectrometry System (GC/MS/COM). Environ. Sci. Technol. 1980, 14, 576–588. [Google Scholar] [CrossRef]
- Glaze, W.H. Brogan & Partners Reaction Products of Ozone: A Review. Environ. Health Perspect. 1986, 69, 151–157. [Google Scholar]
- Werdehoff, K.S.; Singer, P.C. Chlorine Dioxide Effects on Thmfp, Toxfp, and the Formation of Inorganic By-Products. J. Am. Water Work. Assoc. 1987, 79, 107–113. [Google Scholar] [CrossRef]
- Richardson, S.D. Disinfection by-products and other emerging contaminants in drinking water. TrAC Trends Anal. Chem. 2003, 22, 666–684. [Google Scholar] [CrossRef]
- Lavonen, E.E.; Gonsior, M.; Tranvik, L.J.; Schmitt-Kopplin, P.; Köhler, S.J. Selective chlorination of natural organic matter: Identification of previously unknown disinfection byproducts. Environ. Sci. Technol. 2013, 47, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Bougeard, C.M.M.; Goslan, E.H.; Jefferson, B.; Parsons, S.A. Comparison of the disinfection by-product formation potential of treated waters exposed to chlorine and monochloramine. Water Res. 2010, 44, 729–740. [Google Scholar] [CrossRef]
- Le Roux, J.; Nihemaiti, M.; Croué, J.P. The role of aromatic precursors in the formation of haloacetamides by chloramination of dissolved organic matter. Water Res. 2016, 88, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Padhi, R.K.; Subramanian, S.; Satpathy, K.K. Formation, distribution, and speciation of DBPs (THMs, HAAs, ClO2−,andClO3−) during treatment of different source water with chlorine and chlorine dioxide. Chemosphere 2019, 218, 540–550. [Google Scholar] [CrossRef]
- Gan, W.; Huang, S.; Ge, Y.; Bond, T.; Westerhoff, P.; Zhai, J.; Yang, X. Chlorite formation during ClO2 oxidation of model compounds having various functional groups and humic substances. Water Res. 2019, 159, 348–357. [Google Scholar] [CrossRef]
- Postigo, C.; Zonja, B. Iodinated disinfection byproducts: Formation and concerns. Curr. Opin. Environ. Sci. Health 2019, 7, 19–25. [Google Scholar] [CrossRef]
- Richardson, S.D.; Plewa, M.J.; Wagner, E.D.; Schoeny, R.; DeMarini, D.M. Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research. Mutat. Res. Rev. Mutat. Res. 2007, 636, 178–242. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, Y.; Li, A.; Xu, B.; Xian, Q.; Shuang, C.; Shi, P.; Zhou, Q. Detection, formation and occurrence of 13 new polar phenolic chlorinated and brominated disinfection byproducts in drinking water. Water Res. 2017, 112, 129–136. [Google Scholar] [CrossRef]
- How, Z.T.; Kristiana, I.; Busetti, F.; Linge, K.L.; Joll, C.A. Organic chloramines in chlorine-based disinfected water systems: A critical review. J. Environ. Sci. 2017, 58, 2–18. [Google Scholar] [CrossRef]
- Mian, H.R.; Hu, G.; Hewage, K.; Rodriguez, M.J.; Sadiq, R. Prioritization of unregulated disinfection by-products in drinking water distribution systems for human health risk mitigation: A critical review. Water Res. 2018, 147, 112–131. [Google Scholar] [CrossRef]
- Kimura, S.Y.; Ortega-Hernandez, A. Formation mechanisms of disinfection byproducts: Recent developments. Curr. Opin. Environ. Sci. Heal. 2019, 7, 61–68. [Google Scholar] [CrossRef]
- Tardiff, R.G.; Garlson, G.P.; Simmon, V. Halogenated organics in tap water: A toxicological evaluation in Water Chlorination. Environmental Impact and Health Effects. Ann Arbor Sci. Ann Arbor Michigan. USA 1978, 1, 195–209. [Google Scholar]
- U.S. National Cancer institute. Report on the Carcinogenesis Bioassay of Chloroform (CAS No. 67-66-3); TR-000. NTIS Rpt No PB264018; U.S. National Cancer institute: Bethesda, MD, USA, 1976.
- King, W.D.; Marrett, L.D. Case-control study of bladder cancer and chlorination by-products in treated water (Ontario, Canada). Cancer Causes Control 1996, 7, 596–604. [Google Scholar] [CrossRef]
- Hildesheim, M.E.; Cantor, K.P.; Lynch, C.F.; Dosemeci, M.; Lubin, J.; Alavanja, M.; Craun, G. Drinking water source and chlorination byproducts II. Risk of colon and rectal cancers. Epidemiology 1998, 9, 29–35. [Google Scholar] [CrossRef]
- Boorman, G.A.; Dellarco, V.; Dunnick, J.K.; Chapin, R.E.; Hauchman, F.; Gardner, H.; Cox, M.; Sills, R.C.; Boorman, G.A.; Dellarco, V.; et al. Brogan & Partners Drinking Water Disinfection Byproducts: Review and Approach to Toxicity Evaluation Source: Environmental Health Perspectives, Vol. 107, Supplement 1: Reviews in Environmental Health, 1999 (Feb., 1999), pp. 207–217 Published by. Environ. Health Perspect. 1999, 107, 207–217. [Google Scholar] [PubMed]
- DeMarini, D.M. A review on the 40th anniversary of the first regulation of drinking water disinfection by-products. Environ. Mol. Mutagen. 2020, 61, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.D.; Plewa, M.J. CHO cell cytotoxicity and genotoxicity analyses of disinfection by-products: An updated review. J. Environ. Sci. 2017, 58, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Demarini, D.M.; Abu-Shakra, A.; Felton, C.F.; Patterson, K.S.; Shelton, M.L. Mutation spectra in salmonella of chlorinated, chloraminated, or ozonated drinking water extracts: Comparison to MX. Environ. Mol. Mutagen. 1995, 26, 270–285. [Google Scholar] [CrossRef]
- Villanueva, C.M.; Cantor, K.P.; Grimalt, J.O.; Malats, N.; Silverman, D.; Tardon, A.; Garcia-Closas, R.; Serra, C.; Carrato, A.; Castaño-Vinyals, G.; et al. Bladder cancer and exposure to water disinfection by-products through ingestion, bathing, showering, and swimming in pools. Am. J. Epidemiol. 2007, 165, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, J.; Plewa, M.J.; Wagner, E.D.; Nihemaiti, M.; Dad, A.; Croué, J.-P. Chloramination of wastewater effluent: Toxicity and formation of disinfection byproducts. J. Environ. Sci. 2017, 58, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.; Goslan, E.H.; Parsons, S.A.; Jefferson, B. Treatment of disinfection by-product precursors. Environ. Technol. 2011, 32, 1–25. [Google Scholar] [CrossRef]
- Sillanpää, M.; Ncibi, M.C.; Matilainen, A. Advanced oxidation processes for the removal of natural organic matter from drinking water sources: A comprehensive review. J. Environ. Manag. 2018, 208, 56–76. [Google Scholar] [CrossRef]
- Matilainen, A.; Sillanpää, M. Removal of natural organic matter from drinking water by advanced oxidation processes. Chemosphere 2010, 80, 351–365. [Google Scholar] [CrossRef]
- Glaze, W.H.; Peyton, G.R.; Lin, S.; Huang, R.Y.; Burleson, J.L. Destruction of pollutants in water with ozone in combination with ultraviolet radiation. II. Natural trihalomethane precursors. Environ. Sci. Technol. 1982, 16, 454–458. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, X.; Zhang, X.; Peng, S. A review of different drinking water treatments for natural organic matter removal. Water Sci. Technol. Water Supply 2015, 15, 442–455. [Google Scholar] [CrossRef]
- Miklos, D.B.; Remy, C.; Jekel, M.; Linden, K.G.; Drewes, J.E.; Hübner, U. Evaluation of advanced oxidation processes for water and wastewater treatment—A critical review. Water Res. 2018, 139, 118–131. [Google Scholar] [CrossRef]
- Hua, G.; Reckhow, D.A. Characterization of Disinfection Byproduct Precursors Based on Hydrophobicity and Molecular Size. Environ. Sci. Technol. 2007, 41, 3309–3315. [Google Scholar] [CrossRef] [PubMed]
- Lamsal, R.; Walsh, M.E.; Gagnon, G.A. Comparison of advanced oxidation processes for the removal of natural organic matter. Water Res. 2011, 45, 3263–3269. [Google Scholar] [CrossRef] [PubMed]
- De Vera, G.A.; Stalter, D.; Gernjak, W.; Weinberg, H.S.; Keller, J.; Farré, M.J. Towards reducing DBP formation potential of drinking water by favouring direct ozone over hydroxyl radical reactions during ozonation. Water Res. 2015, 87, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Petronijević, M.; Agbaba, J.; Ražić, S.; Molnar Jazić, J.; Tubić, A.; Watson, M.; Dalmacija, B. Fate of bromine-containing disinfection by-products precursors during ozone and ultraviolet-based advanced oxidation processes. Int. J. Environ. Sci. Technol. 2019, 16, 171–180. [Google Scholar] [CrossRef]
- Sakai, H.; Autin, O.; Parsons, S. Change in haloacetic acid formation potential during UV and UV/H2O2 treatment of model organic compounds. Chemosphere 2013, 92, 647–651. [Google Scholar] [CrossRef]
- Murray, C.A.; Parsons, S.A. Comparison of AOPs for the removal of natural organic matter: Performance and economic assessment. Water Sci. Technol. 2004, 49, 267–272. [Google Scholar] [CrossRef][Green Version]
- Moncayo-Lasso, A.; Pulgarin, C.; Benítez, N. Degradation of DBPs’ precursors in river water before and after slow sand filtration by photo-Fenton process at pH 5 in a solar CPC reactor. Water Res. 2008, 42, 4125–4132. [Google Scholar] [CrossRef]
- Moncayo-Lasso, A.; Rincon, A.G.; Pulgarin, C.; Benitez, N. Significant decrease of THMs generated during chlorination of river water by previous photo-Fenton treatment at near neutral pH. J. Photochem. Photobiol. A Chem. 2012, 229, 46–52. [Google Scholar] [CrossRef]
- Lee, S.; Ohgaki, S. Oxidative degradation of toc and thmfp by fluidized bed photocatalysis reactor. J. Environ. Sci. Heal. Part A 1999, 34, 1933–1944. [Google Scholar] [CrossRef]
- Liu, S.; Lim, M.; Fabris, R.; Chow, C.; Drikas, M.; Amal, R. TiO2 Photocatalysis of Natural Organic Matter in Surface Water: Impact on Trihalomethane and Haloacetic Acid Formation Potential. Environ. Sci. Technol. 2008, 42, 6218–6223. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lim, M.; Fabris, R.; Chow, C.; Chiang, K.; Drikas, M.; Amal, R. Removal of humic acid using TiO2 photocatalytic process—Fractionation and molecular weight characterisation studies. Chemosphere 2008, 72, 263–271. [Google Scholar] [CrossRef]
- Gerrity, D.; Mayer, B.; Ryu, H.; Crittenden, J.; Abbaszadegan, M. A comparison of pilot-scale photocatalysis and enhanced coagulation for disinfection byproduct mitigation. Water Res. 2009, 43, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.M.; Gordillo, M.V.; Rey, A.; Beltrán, F.J. Impact of TiO2/UVA photocatalysis on THM formation potential. Catal. Today 2018, 313, 167–174. [Google Scholar] [CrossRef]
- Murray, C.A.; Parsons, S.A. Preliminary laboratory investigation of disinfection by-product precursor removal using an advanced oxidation process. Water Environ. J. 2006, 20, 123–129. [Google Scholar] [CrossRef]
- Murray, C.A.; Goslan, E.H.; Parsons, S.A. TiO2/UV: Single stage drinking water treatment for NOM removal? J. Environ. Eng. Sci. 2007, 6, 311–317. [Google Scholar] [CrossRef]
- Kent, F.C.; Montreuil, K.R.; Brookman, R.M.; Sanderson, R.; Dahn, J.R.; Gagnon, G.A. Photocatalytic oxidation of DBP precursors using UV with suspended and fixed TiO2. Water Res. 2011, 45, 6173–6180. [Google Scholar] [CrossRef]
- Lu, J.; Dong, W.; Ji, Y.; Kong, D.; Huang, Q. Natural Organic Matter Exposed to Sulfate Radicals Increases Its Potential to Form Halogenated Disinfection Byproducts. Environ. Sci. Technol. 2016, 50, 5060–5067. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Kong, X.; Hou, S.; Zou, S.; Xu, X.; Huang, H.; Fang, J. DBP alteration from NOM and model compounds after UV/persulfate treatment with post chlorination. Water Res. 2019, 158, 237–245. [Google Scholar] [CrossRef]
- Wang, Z.; An, N.; Shao, Y.; Gao, N.; Du, E.; Xu, B. Experimental and simulation investigations of UV/persulfate treatment in presence of bromide: Effects on degradation kinetics, formation of brominated disinfection byproducts and bromate. Sep. Purif. Technol. 2020, 242. [Google Scholar] [CrossRef]
- Wang, L.; Ji, Y.; Lu, J.; Yin, X.; Zhou, Q.; Kong, D. Transformation of iodide and formation of iodinated by-products in heat activated persulfate oxidation process. Chemosphere 2017, 181, 400–408. [Google Scholar] [CrossRef]
- Pisarenko, A.N.; Stanford, B.D.; Snyder, S.A.; Rivera, S.B.; Boal, A.K. Investigation of the use of chlorine based advanced oxidation in surface water: Oxidation of natural organic matter and formation of disinfection byproducts. J. Adv. Oxid. Technol. 2013, 16, 137–150. [Google Scholar] [CrossRef]
- Wang, D.; Bolton, J.R.; Andrews, S.A.; Hofmann, R. Formation of disinfection by-products in the ultraviolet/chlorine advanced oxidation process. Sci. Total Environ. 2015, 518–519, 49–57. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, B.; Zhang, T.Y.; Hu, C.Y.; Tang, Y.L.; Dong, Z.Y.; Cao, T.C.; El-Din, M.G. Formation of disinfection by-products in a UV-activated mixed chlorine/chloramine system. J. Hazard. Mater. 2021, 407, 124373. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Al-Ani, Y.; Malik Ismael, Z.; Wu, X. Insights into the role of humic acid on Pd-catalytic electro-fenton transformation of toluene in groundwater. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Trellu, C.; Péchaud, Y.; Oturan, N.; Mousset, E.; Huguenot, D.; van Hullebusch, E.D.; Esposito, G.; Oturan, M.A. Comparative study on the removal of humic acids from drinking water by anodic oxidation and electro-Fenton processes: Mineralization efficiency and modelling. Appl. Catal. B Environ. 2016, 194, 32–41. [Google Scholar] [CrossRef]
- Mao, Y.; Guo, D.; Yao, W.; Wang, X.; Yang, H.; Xie, Y.F.; Komarneni, S.; Yu, G.; Wang, Y. Effects of conventional ozonation and electro-peroxone pretreatment of surface water on disinfection by-product formation during subsequent chlorination. Water Res. 2018, 130, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.K.; Daugherty, E.; Abbaszadegan, M. Evaluation of the relationship between bulk organic precursors and disinfection byproduct formation for advanced oxidation processes. Chemosphere 2015, 121, 39–46. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Q.; Song, H.; Zhang, J.; Wang, L.; Qi, J.; Liu, Y.; Ma, J. Comparative study about oxidation of trace N-nitrosamines by seven oxidation processes with a sensitivity improved determination method. Sep. Purif. Technol. 2020, 236, 116009. [Google Scholar] [CrossRef]
- Xu, B.; Chen, Z.; Qi, F.; Ma, J.; Wu, F. Inhibiting the regeneration of N-nitrosodimethylamine in drinking water by UV photolysis combined with ozonation. J. Hazard. Mater. 2009, 168, 108–114. [Google Scholar] [CrossRef]
- Lee, C.; Yoon, J.; Von Gunten, U. Oxidative degradation of N-nitrosodimethylamine by conventional ozonation and the advanced oxidation process ozone/hydrogen peroxide. Water Res. 2007, 41, 581–590. [Google Scholar] [CrossRef]
- Lv, J.; Li, Y.; Song, Y. Reinvestigation on the ozonation of N-nitrosodimethylamine: Influencing factors and degradation mechanism. Water Res. 2013, 47, 4993–5002. [Google Scholar] [CrossRef]
- Hama Aziz, K.H. Application of different advanced oxidation processes for the removal of chloroacetic acids using a planar falling film reactor. Chemosphere 2019, 228, 377–383. [Google Scholar] [CrossRef]
- Lovato, M.E.; Martín, C.A.; Cassano, A.E. Degradation of dichloroacetic acid in homogeneous aqueous media employing ozone and UVC radiation. Photochem. Photobiol. Sci. 2011, 10, 367–380. [Google Scholar] [CrossRef]
- Wang, K.; Guo, J.; Yang, M.; Junji, H.; Deng, R. Decomposition of two haloacetic acids in water using UV radiation, ozone and advanced oxidation processes. J. Hazard. Mater. 2009, 162, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Zazouli, M.A.; Kalankesh, L.R. Removal of precursors and disinfection by-products (DBPs) by membrane filtration from water; a review. J. Environ. Heal. Sci. Eng. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, Z.; Zhang, Z.; Shi, B.; Hu, C.; Lyu, L.; Zuo, P.; Metz, J.; Wang, H. Heterogeneous Fenton-like reaction followed by GAC filtration improved removal efficiency of NOM and DBPs without adjusting pH. Sep. Purif. Technol. 2021, 260, 118234. [Google Scholar] [CrossRef]
- Zhong, J.; Zhao, Y.; Ding, L.; Ji, H.; Ma, W.; Chen, C.; Zhao, J. Opposite photocatalytic oxidation behaviors of BiOCl and TiO2: Direct hole transfer vs. indirect OH oxidation. Appl. Catal. B Environ. 2019, 241, 514–520. [Google Scholar] [CrossRef]
- Moussavi, G.; Rezaei, M. Exploring the advanced oxidation/reduction processes in the VUV photoreactor for dechlorination and mineralization of trichloroacetic acid: Parametric experiments, degradation pathway and bioassessment. Chem. Eng. J. 2017, 328, 331–342. [Google Scholar] [CrossRef]
- Park, J.-A.; Nam, H.-L.; Choi, J.-W.; Ha, J.; Lee, S.-H. Oxidation of geosmin and 2-methylisoborneol by the photo-Fenton process: Kinetics, degradation intermediates, and the removal of microcystin-LR and trihalomethane from Nak-Dong River water, South Korea. Chem. Eng. J. 2017, 313, 345–354. [Google Scholar] [CrossRef]
- Aslani, H.; Nasseri, S.; Nabizadeh, R.; Mesdaghinia, A.; Alimohammadi, M.; Nazmara, S. Haloacetic acids degradation by an efficient Ferrate/UV process: Byproduct analysis, kinetic study, and application of response surface methodology for modeling and optimization. J. Environ. Manage. 2017, 203, 218–228. [Google Scholar] [CrossRef]
- Li, Y.; Kemper, J.M.; Datuin, G.; Akey, A.; Mitch, W.A.; Luthy, R.G. Reductive dehalogenation of disinfection byproducts by an activated carbon-based electrode system. Water Res. 2016, 98, 354–362. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, X.; Shang, H.; Li, X.; Li, W.; Li, J.; Xia, W.; Zhou, L.; Zhao, C. Degradation of trichloroacetic acid with an efficient Fenton assisted TiO2 photocatalytic hybrid process: Reaction kinetics, byproducts and mechanism. Chem. Eng. J. 2016, 289, 319–329. [Google Scholar] [CrossRef]
- Aslani, H.; Nabizadeh, R.; Nasseri, S.; Mesdaghinia, A.; Alimohammadi, M.; Mahvi, A.H.; Rastkari, N.; Nazmara, S. Application of response surface methodology for modeling and optimization of trichloroacetic acid and turbidity removal using potassium ferrate(VI). Desalin. Water Treat. 2016, 57, 25317–25328. [Google Scholar] [CrossRef]
- Zhao, B.; Li, X.; Li, W.; Yang, L.; Li, J.; Xia, W.; Zhou, L.; Wang, F.; Zhao, C. Degradation of trichloroacetic acid by an efficient Fenton/UV/TiO2 hybrid process and investigation of synergetic effect. Chem. Eng. J. 2015, 273, 527–533. [Google Scholar] [CrossRef]
- Alavi, N.; Tahvildarij, K. Removal of trihalomethanes in tehran drinking water by an advanced oxidation process. Nat. Environ. Pollut. Technol. 2015, 14, 211–216. [Google Scholar] [CrossRef]
- Park, B.; Cho, E.; Son, Y.; Khim, J. Distribution of electrical energy consumption for the efficient degradation control of THMs mixture in sonophotolytic process. Ultrason. Sonochem. 2014, 21, 1982–1987. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, X.M.; Yang, H.W.; Xie, Y.F. Haloacetic acid removal by sequential zero-valent iron reduction and biologically active carbon degradation. Chemosphere 2013, 90, 1563–1567. [Google Scholar] [CrossRef]
- Radjenović, J.; Farré, M.J.; Mu, Y.; Gernjak, W.; Keller, J. Reductive electrochemical remediation of emerging and regulated disinfection byproducts. Water Res. 2012, 46, 1705–1714. [Google Scholar] [CrossRef]
- Esclapez, M.D.; Tudela, I.; Díez-García, M.I.; Sáez, V.; Rehorek, A.; Bonete, P.; González-García, J. Towards the complete dechlorination of chloroacetic acids in water by sonoelectrochemical methods: Effect of the anodic material on the degradation of trichloroacetic acid and its by-products. Chem. Eng. J. 2012, 197, 231–241. [Google Scholar] [CrossRef]
- Wang, X.; Ning, P.; Liu, H.; Ma, J. Dechlorination of chloroacetic acids by Pd/Fe nanoparticles: Effect of drying method on metallic activity and the parameter optimization. Appl. Catal. B Environ. 2010, 94, 55–63. [Google Scholar] [CrossRef]
- Czili, H.; Horváth, A. Photodegradation of chloroacetic acids over bare and silver-deposited TiO2: Identification of species attacking model compounds, a mechanistic approach. Appl. Catal. B Environ. 2009, 89, 342–348. [Google Scholar] [CrossRef]
- Li, Y.P.; Cao, H.B.; Zhang, Y. Reductive dehalogenation of haloacetic acids by hemoglobin-loaded carbon nanotube electrode. Water Res. 2007, 41, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Shemer, H.; Narkis, N. Trihalomethanes aqueous solutions sono-oxidation. Water Res. 2005, 39, 2704–2710. [Google Scholar] [CrossRef] [PubMed]
- Shemer, H.; Narkis, N. Sonochemical removal of trihalomethanes from aqueous solutions. Ultrason. Sonochem. 2005, 12, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Shemer, H.; Narkis, N. Effects of aqueous solutions composition and acoustic intensity on THM compounds sonolysis. Environ. Eng. Sci. 2005, 22, 138–144. [Google Scholar] [CrossRef]
- Shemer, H.; Narkis, N. Mechanisms and inorganic byproducts of trihalomethane compounds sonodegradation. Environ. Sci. Technol. 2004, 38, 4856–4859. [Google Scholar] [CrossRef]
- Lifongo, L.L.; Bowden, D.J.; Brimblecombe, P. Photodegradation of haloacetic acids in water. Chemosphere 2004, 55, 467–476. [Google Scholar] [CrossRef]
- Zhang, L.; Arnold, W.A.; Hozalski, R.M. Kinetics of haloacetic acid reactions with Fe(O). Environ. Sci. Technol. 2004, 38, 6881–6889. [Google Scholar] [CrossRef]
- Wu, C.; Wei, D.; Fan, J.; Wang, L. Photosonochemical degradation of trichloroacetic acid in aqueous solution. Chemosphere 2001, 44, 1293–1297. [Google Scholar] [CrossRef]
- Hozalski, R.M.; Zhang, L.; Arnold, W.A. Reduction of haloacetic acids by FeO: Implications for treatment and fate. Environ. Sci. Technol. 2001, 35, 2258–2263. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.Z.; Tassos, S. Oxidation kinetics and mechanisms of trihalomethanes by Fenton’s reagent. Water Res. 1997, 31, 1117–1125. [Google Scholar] [CrossRef]
- Spangenberg, D.; Mbller, U. Photooxidation and Thermal Decomposition of Trichloroacetic Acid. Chemosphere 1996, 33, 43–49. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Ziółek, M.; Nawrocki, J. Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment. Appl. Catal. B Environ. 2003, 46, 639–669. [Google Scholar] [CrossRef]
- Hill, G.R. The Kinetics of the Oxidation of Cobaltous Ion by Ozone. J. Am. Chem. Soc. 1949, 71, 2434–2435. [Google Scholar] [CrossRef]
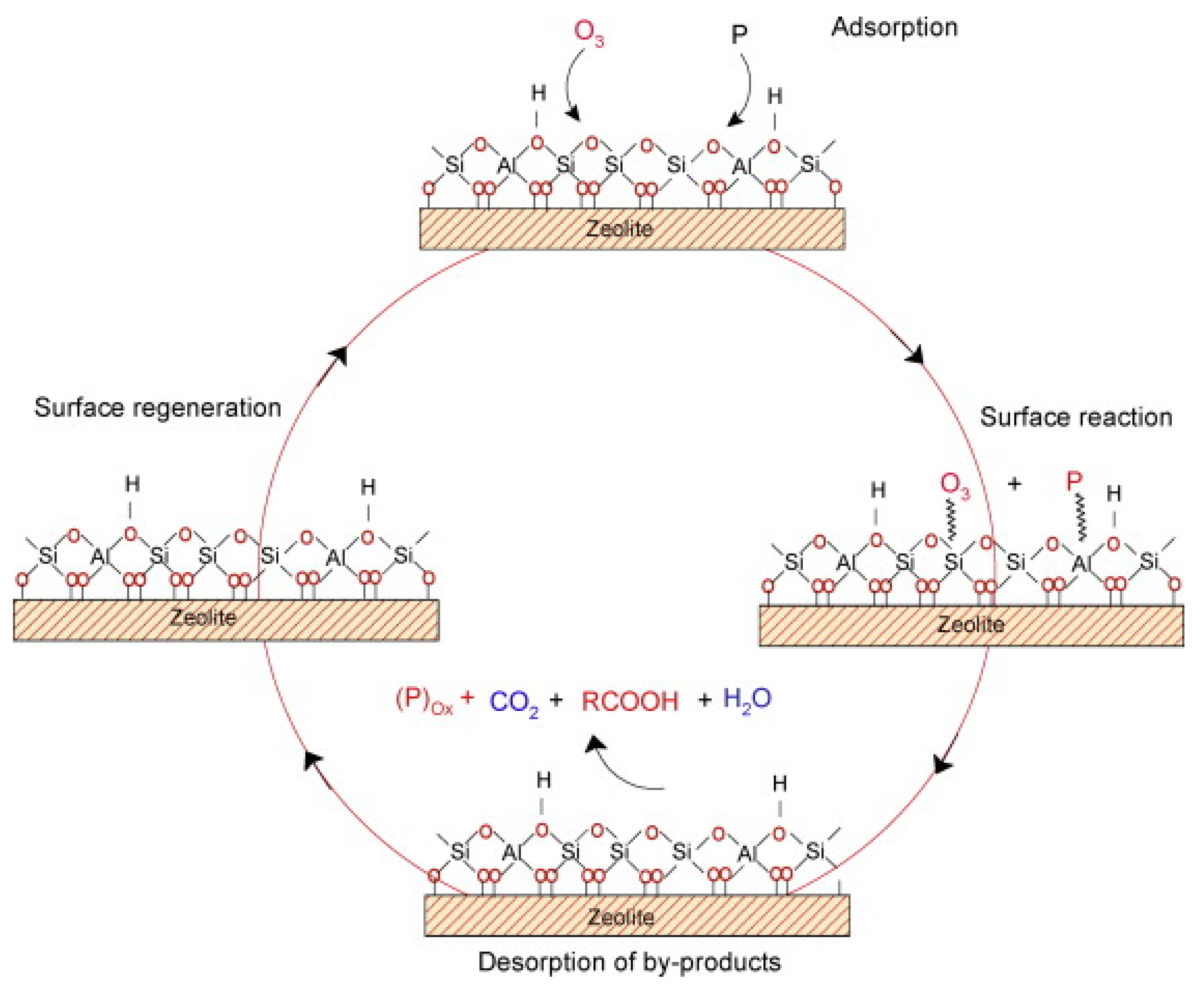
- Nawrocki, J.; Kasprzyk-Hordern, B. The efficiency and mechanisms of catalytic ozonation. Appl. Catal. B Environ. 2010, 99, 27–42. [Google Scholar] [CrossRef]
- Yu, D.; Wu, M.; Hu, Q.; Wang, L.; Lv, C.; Zhang, L. Iron-based metal-organic frameworks as novel platforms for catalytic ozonation of organic pollutant: Efficiency and mechanism. J. Hazard. Mater. 2019, 367, 456–464. [Google Scholar] [CrossRef]
- Yu, D.; Wang, L.; Yang, T.; Yang, G.; Wang, D.; Ni, H.; Wu, M. Tuning Lewis acidity of iron-based metal-organic frameworks for enhanced catalytic ozonation. Chem. Eng. J. 2021, 404, 127075. [Google Scholar] [CrossRef]
- Nawrocki, J. Catalytic ozonation in water: Controversies and questions. Discussion paper. Appl. Catal. B Environ. 2013, 142–143, 465–471. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H. Catalytic ozonation for water and wastewater treatment: Recent advances and perspective. Sci. Total Environ. 2020, 704, 135249. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Utrilla, J.; López-Ramón, M.V.; Sánchez-Polo, M.; Álvarez, M.Á.; Velo-Gala, I. Characteristics and behavior of different catalysts used for water decontamination in photooxidation and ozonation processes. Catalysts 2020, 10, 1485. [Google Scholar] [CrossRef]
- Yu, G.; Wang, Y.; Cao, H.; Zhao, H.; Xie, Y. Reactive Oxygen Species and Catalytic Active Sites in Heterogeneous Catalytic Ozonation for Water Purification. Environ. Sci. Technol. 2020, 54, 5931–5946. [Google Scholar] [CrossRef]
- Fogler, H.S. Elements of Chemical Reaction Engineering, 3rd ed.; Goodwin, B.M., Ed.; Prentice-Hall: Englewood-Cliffs, NJ, USA, 1999. [Google Scholar]
- Liu, Y.; Shen, J.; Chen, Z.; Yang, L.; Liu, Y.; Han, Y. Effects of amorphous-zinc-silicate-catalyzed ozonation on the degradation of p-chloronitrobenzene in drinking water. Appl. Catal. A Gen. 2011, 403, 112–118. [Google Scholar] [CrossRef]
- Andreozzi, R.; Insola, A.; Caprio, V.; Marotta, R.; Tufano, V. The use of manganese dioxide as a heterogeneous catalyst for oxalic acid ozonation in aqueous solution. Appl. Catal. A Gen. 1996, 138, 75–81. [Google Scholar] [CrossRef]
- Martins, R.C.; Quinta-Ferreira, R.M. Catalytic ozonation of phenolic acids over a Mn-Ce-O catalyst. Appl. Catal. B Environ. 2009, 90, 268–277. [Google Scholar] [CrossRef]
- Ikhlaq, A.; Brown, D.R.; Kasprzyk-Hordern, B. Catalytic ozonation for the removal of organic contaminants in water on ZSM-5 zeolites. Appl. Catal. B Environ. 2014, 154–155, 110–122. [Google Scholar] [CrossRef]
- Zhao, L.; Ma, J.; Sun, Z.; Zhai, X. Mechanism of influence of initial pH on the degradation of nitrobenzene in aqueous solution by ceramic honeycomb catalytic ozonation. Environ. Sci. Technol. 2008, 42, 4002–4007. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Ma, J.; Cui, Y.H.; Zhao, L.; Zhang, B.P. Factors affecting the catalytic activity of multi-walled carbon nanotube for ozonation of oxalic acid. Sep. Purif. Technol. 2011, 78, 147–153. [Google Scholar] [CrossRef]
- Restivo, J.; Órfão, J.J.M.; Pereira, M.F.R.; Vanhaecke, E.; Rönning, M.; Iouranova, T.; Kiwi-Minsker, L.; Armenise, S.; Garcia-Bordejé, E. Catalytic ozonation of oxalic acid using carbon nanofibres on macrostructured supports. Water Sci. Technol. 2012, 65, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Orge, C.A.; Órfão, J.J.M.; Pereira, M.F.R. Composites of manganese oxide with carbon materials as catalysts for the ozonation of oxalic acid. J. Hazard. Mater. 2012, 213–214, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Orge, C.A.; Órfão, J.J.M.; Pereira, M.F.R. Carbon xerogels and ceria-carbon xerogel materials as catalysts in the ozonation of organic pollutants. Appl. Catal. B Environ. 2012, 126, 22–28. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, X.; Xie, Y.; Sun, H.; Wang, S. Nanocarbon-Based Catalytic Ozonation for Aqueous Oxidation: Engineering Defects for Active Sites and Tunable Reaction Pathways. ACS Catal. 2020, 10, 13383–13414. [Google Scholar] [CrossRef]
- Qu, X.; Zheng, J.; Zhang, Y. Catalytic ozonation of phenolic wastewater with activated carbon fiber in a fluid bed reactor. J. Colloid Interface Sci. 2007, 309, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Polo, M.; Leyva-Ramos, R.; Rivera-Utrilla, J. Kinetics of 1,3,6-naphthalenetrisulphonic acid ozonation in presence of activated carbon. Carbon N. Y. 2005, 43, 962–969. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Gómez-Serrano, V.; Álvarez, P.M.; Alvim-Ferraz, M.C.M.; Dias, J.M. Activated carbon modifications to enhance its water treatment applications. An overview. J. Hazard. Mater. 2011, 187, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Salla, J.S.; Padoin, N.; Amorim, S.M.; Li Puma, G.; Moreira, R.F.P.M. Humic acids adsorption and decomposition on Mn2O3 and α-Al2O3 nanoparticles in aqueous suspensions in the presence of ozone. J. Environ. Chem. Eng. 2020, 8, 102780. [Google Scholar] [CrossRef]
- Alver, A.; Kılıç, A. Catalytic ozonation by iron coated pumice for the degradation of natural organic matters. Catalysts 2018, 8, 219. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J. Catalytic Ozonation of Humic Acids by Ce–Ti Composite Catalysts. Kinet. Catal. 2017, 58, 734–740. [Google Scholar] [CrossRef]
- Gümüş, D.; Akbal, F. A comparative study of ozonation, iron coated zeolite catalyzed ozonation and granular activated carbon catalyzed ozonation of humic acid. Chemosphere 2017, 174, 218–231. [Google Scholar] [CrossRef]
- Turkay, O.; Inan, H.; Dimoglo, A. Experimental and theoretical study on catalytic ozonation of humic acid by ZnO catalyst. Sep. Sci. Technol. 2017, 52, 778–786. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, Z.; Chai, B.; Cheng, S.; Lu, X.; Bai, X. Heterogeneous catalytic ozonation of natural organic matter with goethite, cerium oxide and magnesium oxide. RSC Adv. 2016, 6, 14730–14740. [Google Scholar] [CrossRef]
- Wang, J.; Wang, G.; Yang, C.; Yang, S.; Huang, Q. Catalytic ozonation of organic compounds in water over the catalyst of RuO2/ZrO2-CeO2. Front. Environ. Sci. Eng. 2015, 9, 615–624. [Google Scholar] [CrossRef]
- Turkay, O.; Inan, H.; Dimoglo, A. Experimental study of humic acid degradation and theoretical modelling of catalytic ozonation. Environ. Sci. Pollut. Res. 2015, 22, 202–210. [Google Scholar] [CrossRef]
- Turkay, O.; Inan, H.; Dimoglo, A. Experimental and theoretical investigations of CuO-catalyzed ozonation of humic acid. Sep. Purif. Technol. 2014, 134, 110–116. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, C.; Wang, Y.; Hu, C. Inhibition of Nano-Metal Oxides on Bromate Formation during Ozonation Process. Ozone Sci. Eng. 2014, 36, 549–559. [Google Scholar] [CrossRef]
- Chen, K.C.; Wang, Y.H. The effects of Fe-Mn oxide and TiO2/α-Al2O3 on the formation of disinfection by-products in catalytic ozonation. Chem. Eng. J. 2014, 253, 84–92. [Google Scholar] [CrossRef]
- Wang, Y.H.; Chen, K.C. Removal of disinfection by-products from contaminated water using a synthetic goethite catalyst via catalytic ozonation and a biofiltration system. Int. J. Environ. Res. Public Health 2014, 11, 9325–9344. [Google Scholar] [CrossRef]
- Han, Q.; Wang, H.; Dong, W.; Liu, T.; Yin, Y. Suppression of bromate formation in ozonation process by using ferrate(VI): Batch study. Chem. Eng. J. 2014, 236, 110–120. [Google Scholar] [CrossRef]
- Wang, Y.H.; Chen, K.C.; Chen, C.R. Combined catalytic ozonation and membrane system for trihalomethane control. Catal. Today 2013, 216, 261–267. [Google Scholar] [CrossRef]
- Han, Q.; Wang, H.; Dong, W.; Liu, T.; Yin, Y. Formation and inhibition of bromate during ferrate(VI)—Ozone oxidation process. Sep. Purif. Technol. 2013, 118, 653–658. [Google Scholar] [CrossRef]
- Molnar, J.; Agbaba, J.; Dalmacija, B.; Klašnja, M.; Watson, M.; Kragulj, M. Effects of Ozonation and Catalytic Ozonation on the Removal of Natural Organic Matter from Groundwater. J. Environ. Eng. 2012, 138, 804–808. [Google Scholar] [CrossRef]
- Mortazavi, S.B.; Asgari, G.; Hashemian, S.J.; Moussavi, G. Degradation of humic acids through heterogeneous catalytic ozonation with bone charcoal. React. Kinet. Mech. Catal. 2010, 100, 471–485. [Google Scholar] [CrossRef]
- Chen, K.C.; Wang, Y.H.; Chang, Y.H. Using catalytic ozonation and biofiltration to decrease the formation of disinfection by-products. Desalination 2009, 249, 929–935. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Zhu, W.; He, X. Catalytic ozonation of dimethyl phthalate and chlorination disinfection by-product precursors over Ru/AC. J. Hazard. Mater. 2009, 166, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lu, J.; Ma, J.; Qiang, Z. Fluorescence spectroscopic characterization of DOM fractions isolated from a filtered river water after ozonation and catalytic ozonation. Chemosphere 2008, 71, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Alsheyab, M.A.; Muñoz, A.H. Comparative study of ozone and MnO2/O3 effects on the elimination of TOC and COD of raw water at the Valmayor station. Desalination 2007, 207, 179–183. [Google Scholar] [CrossRef]
- Karnik, B.S.; Davies, S.H.; Baumann, M.J.; Masten, S.J. Fabrication of catalytic membranes for the treatment of drinking water using combined ozonation and ultrafiltration. Environ. Sci. Technol. 2005, 39, 7656–7661. [Google Scholar] [CrossRef]
- Karnik, B.S.; Baumann, M.J.; Masten, S.J.; Davies, S.H. AFM and SEM characterization of iron oxide coated ceramic membranes. J. Mater. Sci. 2006, 41, 6861–6870. [Google Scholar] [CrossRef]
- Shioyama, M.; Kawanishi, T.; Yokoyama, S.; Nuno, M.; Yamamoto, T. Development of advanced ceramic membrane filtration system combined with ozonation and powdered activated carbon treatment. Water Sci. Technol. Water Supply 2001, 1, 91–96. [Google Scholar] [CrossRef]
- Gracia, R.; Cortés, S.; Sarasa, J.; Ormad, P.; Ovelleiro, J.L. Catalytic ozonation with supported titanium dioxide. The stability of catalyst in water. Ozone Sci. Eng. 2000, 22, 185–193. [Google Scholar] [CrossRef]
- Gracia, R.; Cortes, S.; Sarasa, J.; Ormad, P.; Ovelleiro, J.L. Heterogeneous catalytic ozonation with supported titanium dioxide in model and natural waters. Ozone Sci. Eng. 2000, 22, 461–471. [Google Scholar] [CrossRef]
- Volk, C.; Roche, P.; Joret, J.C.; Paillard, H. Comparison of the effect of ozone, ozone-hydrogen peroxide system and catalytic ozone on the biodegradable organic matter of a fulvic acid solution. Water Res. 1997, 31, 650–656. [Google Scholar] [CrossRef]
- Allemane, H.; Delouane, B.; Legube, B. Comparative Efficiency of Three Systems (Os, O3/H2O2 and O3/TiO2) for the Oxidation of Natural Organic Matter in Water. Ozone Sci. Eng. 1993, 15, 419–432. [Google Scholar] [CrossRef]
- Bai, Z.Y.; Wang, J.L.; Yang, Q. Catalytic ozonation of dimethyl phthalate by Ce-substituted goethite. Int. J. Environ. Sci. Technol. 2017, 14, 2379–2388. [Google Scholar] [CrossRef]
- Beltrán, F.J.; Pocostales, J.P.; Alvarez, P.M.; Oropesa, A. Diclofenac removal from water with ozone and activated carbon. J. Haz. Mater. 2009, 163, 768–776. [Google Scholar] [CrossRef]
- Beltrán, F.J.; Pocostales, J.P.; Alvarez, P.M.; Jaramillo, J. Mechanism and kinetic considerations of TOC removal from the powdered activated carbon ozonation of diclofenac aqueous solutions. J. Hazard. Mater. 2009, 169, 532–538. [Google Scholar] [CrossRef]
- Tomiyasu, H.; Fukutomi, H.; Gordon, G. Kinetics and Mechanism of Ozone Decomposition in Basic Aqueous Solution. Inorg. Chem. 1985, 24, 2962–2966. [Google Scholar] [CrossRef]
- Staehelin, J.; Buehler, R.E.; Hoigne, J. Ozone decomposition in water studied by pulse radiolysis. 2. Hydroxyl and hydrogen tetroxide (HO4) as chain intermediates. J. Phys. Chem. 1984, 88, 5999–6004. [Google Scholar] [CrossRef]
- Elovitz, M.S.; von Gunten, U. Hydroxyl Radical/Ozone Ratios During Ozonation Processes. I. The Rct Concept. Ozone Sci. Eng. 1999, 21, 239–260. [Google Scholar] [CrossRef]
- Yuan, R.; Zhou, B.; Zhang, X.; Guan, H. Photocatalytic degradation of humic acids using substrate-supported Fe3+-doped TiO2 nanotubes under UV/O3 for water purification. Environ. Sci. Pollut. Res. 2015, 22, 17955–17964. [Google Scholar] [CrossRef] [PubMed]
- Agustina, T.E.; Ang, H.M.; Vareek, V.K. A review of synergistic effect of photocatalysis and ozonation on wastewater treatment. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 264–273. [Google Scholar] [CrossRef]
- Mehrjouei, M.; Müller, S.; Möller, D. A review on photocatalytic ozonation used for the treatment of water and wastewater. Chem. Eng. J. 2015, 263, 209–219. [Google Scholar] [CrossRef]
- Mecha, A.C.; Chollom, M.N. Photocatalytic ozonation of wastewater: A review. Environ. Chem. Lett. 2020, 18, 1491–1507. [Google Scholar] [CrossRef]
- Beltrán, F.J.; Rey, A. Solar or UVA-Visible Photocatalytic Ozonation of Water Contaminants. Molecules 2017, 22, 177. [Google Scholar] [CrossRef]
- Yu, D.; Li, L.; Wu, M.; Crittenden, J.C. Enhanced photocatalytic ozonation of organic pollutants using an iron-based metal-organic framework. Appl. Catal. B Environ. 2019, 251, 66–75. [Google Scholar] [CrossRef]
- Cao, S.; Low, J.; Yu, J.; Jaroniec, M. Polymeric Photocatalysts Based on Graphitic Carbon Nitride. Adv. Mater. 2015, 27, 2150–2176. [Google Scholar] [CrossRef]
- Xiao, J.; Xie, Y.; Rabeah, J.; Brückner, A.; Cao, H. Visible-Light Photocatalytic Ozonation Using Graphitic C3N4 Catalysts: A Hydroxyl Radical Manufacturer for Wastewater Treatment. Acc. Chem. Res. 2020, 53, 1024–1033. [Google Scholar] [CrossRef]
- Kerc, A.; Bekbolet, M.; Saatci, A.M. Sequential Oxidation of Humic Acids by Ozonation and Photocatalysis. Ozone Sci. Eng. 2003, 25, 497–504. [Google Scholar] [CrossRef]
- Bekbolet, M.; Uyguner, C.S.; Selcuk, H.; Rizzo, L.; Nikolaou, A.D.; Meriç, S.; Belgiorno, V. Application of oxidative removal of NOM to drinking water and formation of disinfection by-products. Desalination 2005, 176, 155–166. [Google Scholar] [CrossRef]
- Uyguner, C.S.; Suphandag, S.A.; Kerc, A.; Bekbolet, M. Evaluation of adsorption and coagulation characteristics of humic acids preceded by alternative advanced oxidation techniques. Desalination 2007, 210, 183–193. [Google Scholar] [CrossRef]
- Li, G.; Li, K.; Liu, A.; Yang, P.; Du, Y.; Zhu, M. 3D Flower-like β-MnO2/Reduced Graphene Oxide Nanocomposites for Catalytic Ozonation of Dichloroacetic Acid. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Shin, D.; Jang, M.; Cui, M.; Na, S.; Khim, J. Enhanced removal of dichloroacetonitrile from drinking water by the combination of solar-photocatalysis and ozonation. Chemosphere 2013, 93, 2901–2908. [Google Scholar] [CrossRef]
- Mehrjouei, M.; Müller, S.; Möller, D. Synergistic effect of the combination of immobilized TiO2, UVA and ozone on the decomposition of dichloroacetic acid. J. Environ. Sci. Health Part A Toxic/Hazardous Subst. Environ. Eng. 2012, 47, 1073–1081. [Google Scholar] [CrossRef]
- Gu, L.; Yu, X.; Xu, J.; Lv, L.; Wang, Q. Removal of dichloroacetic acid from drinking water by using adsorptive ozonation. Ecotoxicology 2011, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Chen, Z.; Zhao, S.; Wang, H.; Yang, L. Enhanced ozonation of dichloroacetic acid in aqueous solution using nanometer ZnO powders. J. Environ. Sci. 2010, 22, 1527–1533. [Google Scholar] [CrossRef]
- Yang, J.; Dong, Z.; Jiang, C.; Wang, C.; Liu, H. An overview of bromate formation in chemical oxidation processes: Occurrence, mechanism, influencing factors, risk assessment, and control strategies. Chemosphere 2019, 237, 124521. [Google Scholar] [CrossRef]
- Fischbacher, A.; Löppenberg, K.; von Sonntag, C.; Schmidt, T.C. A New Reaction Pathway for Bromite to Bromate in the Ozonation of Bromide. Environ. Sci. Technol. 2015, 49, 11714–11720. [Google Scholar] [CrossRef]
- Parrino, F.; Camera-Roda, G.; Loddo, V.; Palmisano, G.; Augugliaro, V. Combination of ozonation and photocatalysis for purification of aqueous effluents containing formic acid as probe pollutant and bromide ion. Water Res. 2014, 50, 189–199. [Google Scholar] [CrossRef][Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidant-Disinfectant | Oxidation Potential, V | Relative Oxidation Power 2 |
|---|---|---|
| Ozone | 2.07 | 1.00 |
| Hydrogen peroxide | 1.77 | 0.86 |
| Potassium permanganate | 1.49 | 0.72 |
| Hypochlorous acid | 1.49 | 0.72 |
| Chlorine | 1.36 | 0.66 |
| Hypobromous acid | 1.33 | 0.64 |
| Chlorine dioxide | 1.28 | 0.62 |
| Monochloramine | 1.16 | 0.56 |
| Pathogen | Chlorine | Chlorine Dioxide | Chloramine | Ozone |
|---|---|---|---|---|
| E. Coli | 0.034–0.05 | 0.4–0.75 | 95–180 | 0.02 |
| Rotavirus | 0.01–0.05 | 0.2–2.1 | 3810–6480 | 0.006–0.05 |
| G. Lambia Cyst | 47–150 | - | - | 0.5–0.6 |
| G. Muris | 30–630 | 7.2–18.5 | 1400 | 1.8–2.0 |
| NR-DBP Family Group | Representative Compound | ADC, µg L−1 |
|---|---|---|
| Halogenated compounds | ||
| THMs 4 | Iodoform | 0.2 2 |
| HAAs 4 | Bromochloracetic acid | <1 1 |
| Halonitromethanes | chloropicrin (trichloronitromethane) | 0.5 2 |
| Haloacetonitriles | dichloroacetontrile | 3.08 2 |
| Haloacetamides | dichloroacetamide | 1.62 2 |
| Haloamines | N-chloroaminoacetic acid 3 | - |
| Haloaldehydes | trichloroacetaldehyde | 3.67 2 |
| Haloketones | trichloropropanone | 3.55 2 |
| Halofuranones | MX: 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone | <1.0 1 |
| Haloquinones | 2,6-Dichloro-1,4-benzoquinone | <1.0 1 |
| Iodinated DBP (other than THM and HAACs) | Iodoacetaldehyde | <1.0 1 |
| Halogencyanide | Cyanogen chloride | 1.54 2 |
| Non halogenated compounds | ||
| Aldehydes | Formaldehyde | 3.46 2 |
| Ketones | Dimethylglyoxal | <1.0 1 |
| N-Nitrosamines | N-nitrosodimethylamine | 0.01 2 |
| Target DBPs | Processes Applied | Reactor and Experimental Conditions | Main Results | Ref. |
|---|---|---|---|---|
| 9 N-nitrosamines | O3 O3/H2O2 | 1 L glass beaker with magnetic stirring. 30 min degradation experiments; room temperature (21 °C); [N-nitrosamines] = 100 ng L−1; [H2O2] = 2 mg L−1; [O3] = 1.5 mg L−1 | O3/H2O2 process got the highest removal efficiency of 36.6% (NDMA)—91.4% (NDBA) among all the investigated methods. In comparison, ozonation merely removed less than 20% of N-nitrosamines (except NDphA for 29.3%). | [102] |
| Chloroacetic acids MCA DCA | O3/H2O2 O3 (pH = 11) | Planar falling film reactor. Seven UVA lamps (15W, 360 nm) fixed inside the reactor. Intensity UV light: 1 mW cm−2; water flow rate: 1 L min−1, volume 0.5 L; [CAA] = 1 mM; pH 3, pure gaseous oxygen rate of 10 L h−1, ozone gas with 130 ± 5 mg L−1 ozone at a power of 30 W. | Chloroacetic acids are highly resistant towards direct ozonation in the darkness as only about 2% degradation was observed after 90 min treatment (pH 3). However, increasing the pH of the solution to 11 shows a dramatic improvement in the degradation efficiency and by the combination of O3 with H2O2. | [106] |
| NDMA in ultrapure water and natural water (River Sanhaowu) | O3 (pH = 7.6) | Batch and continuous experiments. A sealed cylindrical reactor with a volume of 5 L. The reactor was stirred mildly and set in the dark at room temperature (24 ± 1 °C). Buffer solution (5 mM phosphate and 1 mM carbonate, prepared with ultrapure water) and pH 7.6). [O3]0 = 0.1 mM (4.8 mg L−1), [NDMA]0 = 400 ng L−1. | Ozonation was an efficient process for NDMA degradation. The removal efficiency was affected by initial NDMA concentration; higher NDMA dosing required higher ozone utilization. NDMA oxidation was favored at high ozone dosage and high pH. NDMA ozonation under various pH as well as hydroxyl radical (HO•) inhibition experiments verified that HO• generated from ozone dominated NDMA oxidation. | [105] |
| DCA in aqueous media. | O3 O3/UVC | The photo-reactor was a cylinder made of Teflon™ closed at both ends with two demountable, flat, circular windows made of quartz. Reactor length 5.2 cm, and the inner diameter 5.2 cm (VRirra = 110.4 cm3). Dissolved ozone concentrations of 1.46 to 2.1 × 10−7 mol cm−3. 15 and 40 W lamps. [DCA]o = 20, 40 and 50 ppm; pH 3.5 ± 0.1. | O3 or UVC by themselves did not result in appreciable decomposition of DCA. Conversely, the O3/UV combination can be considered a suitable process for degrading DCA in water. The combination of ozone and UVC radiation produces a significant amount of hydrogen peroxide as an important reaction by-product. | [107] |
| NDMA in distilled water. | O3/UVC | A cylindrical glass reactor with 700 mL valid sample bulk. Low-pressure Hg lamp (8W, emission at 253.7 nm). [NDMA]0 = 0.1 mmol L−1, pH 6.0, irradiation 1000 W cm−2, [O3]0 = 6.6 mg L−1. | UV irradiation and the UV/O3 combination are effective methods for NDMA removal from drinking water. The introduction of ozone into the UV process had little influence on the effectiveness of NDMA removal. However, it had a great influence on the formation of degradation products from NDMA. As the main products, DMA and NO2− decreased markedly in the UV/O3 process compared with UV irradiation. | [103] |
| DCA TCA | O3 O3/UVC O3/H2O2 O3/H2O2/UVC | A cylindrical stainless steel column (2 L vol.). Its diameter and height are 100 and 300 mm, respectively. Inside the reaction column is a quartz well containing a UV lamp with a diameter of 30 mm and a height of 300 mm—a 15-W low pressure mercury vapor lamp (254 nm). Ozone adding (mg min−1): 0.3 ± 0.06; [H2O2]o = 2.5 mg L−1; wavelength of UV lamp (nm): 254; power input of UV lamp (W): 15; reaction volume (L): 2; Initial DCA and TCA concentration (mg L−1): 2.0. | O3/UV showed to be more suitable for the decomposition of DCA and TCA in water among the six methods of oxidation. Decomposition of DCA was easier than TCA by AOPs. | [108] |
| NDMA in buffered deionized water and natural waters. | O3 O3/H2O2 | 500 mL glass bottle equipped with a dispenser. ([NDMA]0 = 1 µM, [O3]0 = 40 µM, ratio of [O3]0/[H2O2]0 = 2. 10 mM phosphate buffer. | In experiments with natural waters, NDMA could not be significantly oxidized during conventional ozonation. In the AOP O3/H2O2, ozone doses of 160–320 mM ([O3]0/[H2O2]0 = 2:1) were necessary for > 50% NDMA oxidation depending on the HO• scavenging rates of the natural waters. Bromate formation may be the limiting factor for NDMA oxidation during ozonation and ozone-based AOPs in bromide containing waters. | [104] |
| Target DBPs | Processes Applied | Reactor Configuration | Experimental Conditions | Ref. |
|---|---|---|---|---|
| THMs HAAs HANs HKs | Heterogeneous Fenton-like reaction + GAC filtration. | Pilot plant that includes mixing tank, mechanical flocculation tank, tube settler, sand filter column, intermediate water tank, heterogeneous Fenton column, active carbon column, water-producing water tank and automatic control device. | The design flow of single-set process is 2 m3 h−1. Poly aluminum chloride (PACL): 30 mg L−1; dosage of H2O2 is 0.15 mM (5 mg L−1). | [110] |
| MCA DCA TCA | UVA/TiO2; UVA/BiOCl. | 50 mL quartz reactor with a recycling water glass jacket. Light source a 500 W xenon lamp (simulated sunlight, containing 4% UV light). | T = 298 K, 40 mg BiOCl/TiO2 (P25) (1g L−1) dispersed in 40 mL model pollutant (20 mg L−1). Reaction time: 60 min. | [111] |
| TCA | Advanced oxidation/reduction processes (AORPs): the vacuum UV (VUV; 185 nm + 254 nm). | A tubular glass photoreactor. Internal diameter of 25 mm and a height of 400 mm. Working vol of 100 mL. 5.7 W dichromatic low-pressure mercury UV lamp emitting UV at two distinct wavelengths (185 and 254 nm). | 50 mg L−1 TCA, pH = 7; reaction time = 20 min. | [112] |
| THMs | Photo-Fenton process: UV/Fe/H2O2 | Photochemical reactor equipped with a 254 nm low-pressure mercury UV-C lamp. Stirred with magnetic bar. | The average fluence rate was 0.93 mW cm−2. T (25–31 °C). Reaction time: 60 min. CFe(II) = 2 mg L−1. CH2O2 = 20 mg L−1. pH = 7.2–7.4; CHCl3=163.6 μg L−1, CH2BrCl = 145.3 μg L−1, CHBr2Cl = 131.8 μg L−1, CHBr3 = 124.6 μg L−1. | [113] |
| DCA TCA | A combination of Ferrate [Fe(VI)] and UV irradiation. | Self-made photoreactor (effective volume 0.5 L) equipped with a cooling system and placed on a magnetic stirrer. A 75-W low-pressure mercury UV lamp (λmax = 365, 310, and 254 nm, respectively) with 0.225 mWcm−2 intensity placed in the center of the reactor. | Experimental design: HAAs 100–1000 μg L−1; pH: 3–9; Fe (VI) 10–40 mg L−1; time 5–60 min. | [114] |
| 4HANs 9THMs 4 HAcA 4 HAcAm Chloropicrin 500 nmol each | Reductive electrolysis. | Norit GAC (0.4 g) placed in a 1.5 cm × 4 cm cylinder constructed from sheet graphite to serve as the working cathode. It was then transferred to the cathodic chamber of an electrolysis cell. | A constant potential of −1000 mV vs. SHE was applied to the cathode, while cathodic chamber was continuously stirred with a Teflon-lined magnetic stir bar. | [115] |
| TCA | TiO2 photocatalytic process combined with Fenton reagent. | Self-made cylindrical reactor with cool water recycling cloth and a UV light tube. The UV light (kmax = 254 nm) 50 W low pressure mercury lamp placed in the center of the reactor and equipped with a protective quartz tube. | Irradiation intensity was about 35 Mw cm−2; TCAA aqueous solution (150 mL) containing TiO2. Flow rate of 40 mL min−1 of O2. [TCA] = 0.01 mmol L−1, UV irradiation intensity= 35 mW cm−2, [TiO2] = 1.0 g L−1, [Fe2+] = 0.1 mmol L−1, [H2O2] = 1.8 mmol L−1, natural pH = 6. | [116] |
| TCA | Ferrate(VI) a multipurpose chemical, is used as coagulant and oxidant. | Self-made photoreactor (effective volume 0.5 L) equipped with a cooling system and placed on a magnetic stirrer. | Initial pH of solution (3–9), ferrate (VI) dosage (1–10 mg L−1), contact time (5–60 min), trichloroacetic acid (100–1000 μg L−1), and initial turbidity (1–10 NTU). | [117] |
| TCA | Fenton with TiO2 photocatalytic oxidation. | Self-made photoreactor equipped with cool water recycling and a UV light tube. 50 W low-pressure mercury lamp placed in the center of the reactor and equipped with a protective quartz tube. | Irradiation intensity was about 35 mW cm−2. TCA aqueous solution (150 mL, 2 mg L−1). [TiO2] = 1.0 g L−1, [Fe2+]0 = 5.6 mg L−1, m(Fe2+):m(H2O2) = 1:10, intensity (UV) = 35 mW cm−2, flow rate (O2) = 40 mL min−1. pH = 5.8 (without adjustment). | [118] |
| THMs EPA method 551/1 | UV/ZnO/H2O2 | A photoreactor equipped with 4 UV lamps in the reactor corners. | 5 mL of concentrated solution (30%) H2O2; 0.5 g of ZnO in 100 mL of drinking water samples with constant reaction time (1hr) and UV irradiation. | [119] |
| 4THMs | Sonophotolytic degradation. | Rectangular shape of stainless-steel reactor (L100, W100, H250) and ultraviolet lamps (4). Transducer was located in a bottom of the reactor. | The applied ultrasonic frequency was 500 kHz and the electrical powers were 0–52.55 W. Electrical power of each lamp was 10.5 W. 10 mg L−1 of THMs mixture (1.5 L). | [120] |
| TCA | A sequential Fe0 (zero valent iron) and BAC column system. | Fe0 column and a BAC column made of glass, 30 cm in length and 3 cm in id. | 1.2 µM TCA; pH of the feed water was approximately 6.0. The feed was initially treated by the Fe0 column (BET was 1.3 m2 g−1) and then the BAC column. | [121] |
| 17 DBPs (i.e., halomethanes, haloacetonitriles, halopropanones, chloral hydrate, and trichloronitromethane) at low concentration (µg L−1) | Electrochemical reduction using a resin impregnated graphite cathode. | Flow-through electrochemical reactor consisting of two polycarbonate frames (internal dimensions of 20 × 5 × 1.2 cm). | The cathode potential from −700 to −900 mV vs. SHE. | [122] |
| TCA | High-frequency sonoelectrochemical methods. | A sonoreactor (0.5 L of volume) consisting of a cylindrical flask equipped with a cooling jacket where the electrodes (18 cm2 on each side) were placed. | 0.5 mM TCAA aqueous solution; 850 kHz ultrasonic irradiation, titanium cathode, and a platinized titanium anode. | [123] |
| MCA DCA TCA | Iron-based bimetallic particles: Two kinds of dry Pd/Fe nanoparticles (Pd/Fe−1 and Pd/Fe−2). | A series of glass vials (60 mL). The vials were sealed with Teflon-lined rubber septa and aluminium cap. | Pd content = 0.1 wt%, Pd/Fe loading = 3 g L−1, initial concentration of chloroacetic acid = 20 mg L−1, and reaction time = 180 min. | [124] |
| 4THMs | Photocatalysis with TiO2 (slurry). | Photo-Cat Lab consists of an air compressor for oxygenation of the system; eight 75-watt, low-pressure, mercury arc bulbs in series; and a submicron-pore-size ceramic membrane filter that produces TiO2-free effluent. | Batch configuration. Process flow rate of 25 L min−1; initial system volume of approximately 16 L. the average intensity of the UV bulbs was approximately 7.0 Mw cm−2. 400 mg L−1 TiO2. | [86] |
| MCA DCA TCA | Photocatalytic degradation over various bare and silver-deposited Degussa P25 TiO2 particles. | A medium scale (V = 2.5 dm3) photochemical reactor. | Irradiation performed under anaerobic and aerobic conditions. Flow rate of gases (air and Ar) was 40 dm3 h−1; light source (40 W, λmax = 350 nm), initial concentration of TiO2 (rutile, anatase, P25) was 1 g dm−3. initial concentration of MCA, DCA, and TCA was adjusted to be 1 mM. | [125] |
| DCA TCA | Electrochemical treatment (dehalogenation). | Packed-bed flow reactor. The reactor was composed of two glass compartments separated by a cationic exchange membrane. | Initial concentration of HAAs 10.5 mM. Flow rate: 1 mL min−1; electrolysis potential: −0.200–−0.400 V (vs. SCE). | [126] |
| 4THMs | Ultrasonic (US) irradiation, hydrogen peroxide (H2O2), Fenton’s oxidation, US/H2O2 and US/H2O2/Fe2 | 200 mL conical closed glass reactor kept in a temperature- controlled bath | THMs solute mixture, at 10 mg L−1 for each compound, in deionized water. Initial pH adjusted to 3.5 by 1N H2SO4. 250–500 mg L−1 H2O2 and 20–40 mg L−1 Fe(2+).90 min reaction. | [127] |
| 4THMs Iodoform (CHI3) | Sonodegradation | 200 mL conical closed glass reactor kept in a temperature- controlled bath | 100 mL aqueous solution at 25 °C. frequency of 20 kHz; acoustic intensity 3.75 W cm−2; initial pH 5.4–5.8. | [128] |
| 4 THMs CHI3 | Sonodegradation | 200 mL conical closed glass reactor kept in a temperature- controlled bath. | Ultrasonic frequency of 20 kHz. presence of inorganic components. increase the ultrasonic intensity, from 0.9 to 7.0 W cm−2. the power density, from 0.123 to 0.368 W mL−1. | [129] |
| 4THMs CHI3 | Ultrasonic irradiation | 200-mL conical closed glass reactor kept in a temperature-controlled bath. | Ultrasonic irradiation 20 kHz. Acoustic intensity 3.75 W cm−2, and the power density 0.184 W mL−1. initial pH 5.4–5.8 without buffer addition. Initial conc. of THMs 10 mg L−1. | [130] |
| MCA DCA TCA | Photodegradation in the presence of titanium dioxide suspensions. | Reaction vessel 350-mm long quick fit condenser tube. Connections to a thermostatic water bath. Two mercury vapor greenhouse lamps were used to mimic sunlight radiation (λ > 400 nm). | 250 cm3 of approximately 0.1 mol dm−3 solutions of the HAAs. 0.1 g of finely powdered titanium dioxide was added as a photocatalyst. | [131] |
| MCA DCA TCA MBA (bromoacetic acid) | Iron Fe (0) particles. | Batch experiments. 125 mL serum bottles. | Aqueous HAA solution buffered at pH 7.5 with 50 mM deoxygenated MOPS. 0.3 g of iron and rotated at 45 rpm. Different Initial HAA Concentrations: 15–405 μM. | [132] |
| TCA | Ultraviolet (UV) photolysis, ultrasound (US) sonolysis and their combination. | Quarz reactor immersed in a thermostathic bath. Two UV lamps located on either side of the sample reactor. | TCA (2.89 × 10−4 M). Vreacción 36 mL. pH 3.5; T = 30 °C. | [133] |
| HAAs: TCA TBA (tribromoacetic acid) CDBA (chlorodibromoacetic acid) BDCA (bromodichloroacetic acid) | Reduction with zerovalent iron (Fe0). | Batch experiments (glass serum bottle). | 0.5 g of Fe(0) in 36 mL of DI water. Desired conc. (100–200 µM). RT up to 94 h. No pH buffer employed. Initial pH values ranged 3.62 to 4.14 and final rose to 5.60 to 6.23. | [134] |
| 4THMs | Fentons Reagent. | The reactors consisted of 54 mL Pyrex test tubes filled to capacity with stock THM solution, plus Fenton’s reagent, and sealed with septum-fitted screw caps. | pH = 3.5 [THM]o = 37.2 µg L−1 each [H202] = 3.7 Mm; [Fe 2+] = 0.19 mM. | [135] |
| TCA | Photocatalysis with TiO2 and thermal decomposition. | Photocatalysis: The reactor equipped with a suprasil-glass adapter for the UV-lamp. Reaction vessel has inlet and outlet ports of Teflon tubes for bubbling air and N2. The apparatus was closed. Thermal: A double-walled thermostated jar of glass was used. | 0.5 g TiO2 were suspended in 500 mL aqueous solution containing 10 mmol (20 mM) of trichloroacetic acid. 20 W-Hg low pressure lamp. | [136] |
| Target Water | Catalyst and Main Properties | Reactor and Experimental Conditions | Main Results | Ref. |
|---|---|---|---|---|
| Humic acid | α-Al2O3 SBET = 45.8 m2 g−1 Pore volume 0.162 cm3 g−1 pHpzc = 4.2 Crystallite size 41.7 nm Mn2O3 SBET = 15.6 m2 g−1 Pore volume 0.008 cm3 g−1 pHpzc = 5.9 Crystallite size 15.9 nm XPS before and after ozonation indicates the change in the oxidation state of Mn and Al in both materials and the hydroxylation of the surface. | Semi-batch column reactor H = 68 cm, d = 8 cm V = 1 L Ozone flow rate = 0.063 m3 h−1 CO3l = 4–8 mg L−1 Catalyst loading 0.1–0.5 g L−1 CHA = 50 mg L−1 T = 25 °C pH = 5.5 | Al2O3 nanocatalysts showed better performance than Mn2O3 in HA removal using lower ozone dosage due to favorable surface charge. Mn2O3 decomposes O3 faster tan Al2O3. Adsorption of HA contributes to its catalytic ozonation. Higher adsorption capacity of Al2O3. Some deactivation observed in 4 consecutive runs. No post-chlorination. No DBPs were analyzed. | [160] |
| Humic acid | Fe coated pumice Prepared from natural pumice with FeCl3 impregnation. BET, TEM, XRD, DLS, FTIR and pHpzc XRD: α-FeOOH Particle size 200–250 nm. SBET = 10.56 m2 g−1 pHpzc = 7.13 | Semi-batch column reactor V = 1 L Ozone dose 0.333 g min−1 CO3ge = 6–12 mg L−1 Catalyst loading 25–100 mg L−1 CHA = 10 mg L−1 T = 22 °C pH = 3–10, 6.72 | Improved efficiency of catalytic ozonation (80% DOC removal vs. 20% ozonation). Contributions: *Surface adsorption 21.3% *HO• radicals 66.2% *Sole ozonation 12.5% Scavenger experiments with t-BuOH and phosphate confirmed the role of hydroxyl radicals in solution. Iron leaching < 13 μg L−1 No post-chlorination. No DBPs were analyzed. | [161] |
| Humic acid | Ce-Ti composites Sol-gel synthesis Ce/Ti = 0.2–1.0 Calcination at 600 °C XRD showed cubic fluorite CeO2 structure. Crystallite size decreased with Ce content, increasing SBET (not reported). | Semi-batch column reactor (h = 500 mm; D = 60 mm) V = 1 L Q = 0.5 L min−1 30 min reaction time CO3ge = 16.91 mg L−1 Catalyst loading 0.3 g L−1 CHA = 30 mg L−1 COD = 260 mg L−1 pH = 6.91 | The highest efficiency was found for Ti-Ce (1/0.8) composition due to its high surface area and low pHpzc. Increased the ozone efficiency by 62%. Apparent first order rate constants for homogeneous (0.054 g L−1 min−1) vs. heterogeneous (0.067 g L−1 min−1). Less of 50% heterogeneous contribution. The distribution of molecular weights tends to lower values in catalytic ozonation. No post-chlorination. No DBPs were analyzed. | [162] |
| Humic acid | Fe coated Zeolite (ICZ) Zeolite clinoptilolite Synthesis by impregnation and precipitation at basic pH, dried at 105 °C. SEM and XRD analyses showed 2.132% Fe and the ICZ morphology. Granular activated carbon (GAC, from Merck) Dp = 1.5 mm SBET = 971.7 m2 g−1 | Semi-batch stirred reactor V = 2 L 1 h reaction time CO3l = 10 mg L−1 (monitored) Q = 2 L min−1 Catalyst loading 0.75 g L−1 CHA = 30 mg L−1 DOC = 8.52 mg L−1 pH = 6.50–11 | DOC removal by ozone alone was 21.4% and increased up to 62% for ICZ-O3 and 48.1% for GAC-O3. The efficiency of the process was also tested with different humic acid fractions (<100 and <50 kDa). Fe leaching was analyzed near 60 µg L−1 at the beginning of the reaction. Kinetics was studied and showed the efficiency of both catalysts. Better results with ICZ in general. No post-chlorination. No DBPs were analyzed. | [163] |
| Humic acid | ZnO (Sigma Aldrich) 99.9% Density 5,61 g cm−3 Particle size < 5 µm | Semi-batch stirred reactor V = 2 L Inner diameter 12 cm Ozone dose 0.190 mg min−1 2 h reaction time Catalyst loading = 0.25 g L−1 DOC0 = 5.86 mg L−1 pH = 5.33 Ambient temperature | The degradation of HA by catalytic ozonation was much more effective (60% mineralization) than ozonation alone (30%). DFT modelling showed O3 disproportionation on the ZnO surface to form reactive oxygen species. No post-chlorination. No DBPs were analyzed. | [164] |
| NOM River water (Harbin Mo Panshan, Harbin, China) | FeOOH-goethite pHpzc = 6.8 SBET = 97 m2 g−1 Average pore size 23.2 nm CeO2 pHpzc = 6.7 SBET = 116 m2 g−1 Average pore size 12.6 nm MgO pHpzc = 11.1 SBET = 105 m2 g−1 Average pore size 16.7 nm SEM and XRD analyses to confirm structure and morphology. | Ozonation in semi-continuous mode T = 20 °C V = 1 L Catalyst loading 100 mg L−1 Qg = 150 mL min−1 DOC0 = 2.68 mg L−1 pH = 7.53 tert-BuOH as HO• scavenger | UV254 removal was mainly from direct ozonation. CeO2 was the best system in UV254 removal (69%). FeOOH (27.24%) and MgO (18.66%) better in mineralization than O3 alone (10.7%) and O3/CeO2 (2.24%). Adsorption plays a negative effect in mineralization. Fractionation of DOC. Catalytic stability was high, with very low ions leaching (up to 11 µg L−1) and SEM, XRD of used catalysts similar than for fresh samples. No post-chlorination. No DBPs analysis. | [165] |
| Oxalic acid (OA) Dimethyl phthalate (DMP) and NOM surface water (Jingmi Cannel, Beijing, China) | RuO2/ZrO2-CeO2 pHpzc = 6.0 Powder (less than 4 um) and pelletized (2 mm) 0.5 wt.% Ru SBET = 170 m2 g−1 By XRD: Cubic CeO2, RuO2 detected. RuO2/Al2O3 0.5–5 mm 0.1 wt.% Ru SBET = 113–183 m2 g−1 RuO2/AC Particle size 6–10 mesh 1–1.5 mm 0.5 wt.% Ru AC ash content 7.8 wt.% (mainly SiO2, Al2O3 and Fe2O3) SBET = 945.2 m2 g−1 | Cylinder reactor, bubble column D = 5 cm L = 120 cm V = 1 L Semi-batch for catalytic activity: Cat. Dosage = 2 g L−1 Ozone dosage 116 mg h−1 Qg = 400 mL min−1 Continuous operation for stability tests: Qw = 20 mL min−1 Hydraulic time 60 min Experiment time 48 h Cat. Dosage = 40 g TOC0 = 2.25–3.20 mg L−1 pH = 7.2–8.5 T = 15 °C Chlorination: pH = 7 Excess of chlorine T = 25 °C, 7 days | High efficiency in catalytic ozonation for OA and DMP, adsorption of intermediates. NOM mineralization but high adsorption capacity is observed. RuO2/ZrO2-CeO2 had higher stability than Ru/AC and Ru/Al2O3. Post-chlorination and DBPs. HAAFPs removal improved from 38–57%. THMFPs from 50–64% but O3 alone reacts fast with THMs precursors. | [166] |
| Humic acid and NOM Porsuk River (Turkey) | Degussa P-25 TiO2 80/20 anatase/rutile SBET 50 m2 g−1 Crystal size 21 nm. | Semi-batch stirred reactor V = 2 L Inner diameter 12 cm 2 h reaction time DOC (HA) = 5.8 mg L−1 DOC (NOM) = 3.76 mg L−1 pH = 5.5–6.0 Catalyst dose 0.25–1 g L−1 Ambient temperature O3 dose 0.190 mg min−1 | DOC removal of humic acid was highly improved by catalytic ozonation (from 30% to 70%). The process was less effective with NOM. Adsorption plays an important role. By DFT modelling, HO• are supposed to be formed from adsorbed O3 and its decomposition onto TiO2 surface in the presence of H2O. No post-chlorination. No DBPs were analyzed. | [167] |
| Humic acid | CuO (nanopowder, Sigma-Aldrich) SBET = 0.64 m2 g−1 Pore volume = 0.002 cm3 g−1 Monoclinic structure. | Semi-batch stirred reactor V = 2 L Inner diameter 12 cm Ozone dose 0.190 mg min−1 60 min reaction time DOC = 5.86 mg L−1 pH = 5.5–6 T = 23 °C Catalyst dose 0.25 g L−1 | CuO presented catalytic activity in the ozonation of humic acid, with 80% of DOC removal in a short time. It was fond that adsorption, desorption, oxidation and chelating reactions took place in solution. The DFT approach proposed that HO• radicals formed in the catalyst surface initiate the heterogeneous catalytic ozonation reaction. No post-chlorination. No DBPs were analyzed. | [168] |
| Bromide in UP water Humic acid | TiO2 P25 Anatase/rutile 80/20 Nano-SnO2 Tetragonal pase Dcrystal) 9.7 nm | Cylindrical bubble column (d = 8 cm, h = 40 cm) V = 2 L Vr = 1.5 L Br- = 0.40 mg L−1 CO3 liquid = 3.38 mg L−1 pH = 6 Ccat = 100 mg L−1 T = 26 °C CHumic acid = 0–3.0 mg L−1 | Analysis of the BrO3− formed by O3 and catalytic O3 The presence of both catalysts reduces the formation of BrO3− during ozonation. Inhibition of BrO3− formation with increasing humic acid concentration. No other DBPs analyzed. | [169] |
| NOM surface water (Wu-Lo River) pCBA | Fe-Mn oxide 23.0 % Fe 8.17% Mn 68.77% O SBET = 262.0 m2 g−1 pHpzc = 5.9 Acidic groups = 181 ueq g−1 Basic groups = 636 TiO2-αAl2O3 7.69% Ti 29.29% Al 63.02% O SBET = 14.5 m2 g−1 pHpzc = 8.3 Acidic groups = 2074 ueq g−1 Basic groups = 0.71 ueq g−1 | Fluidized bed reactor Semi-continuous operation D = 6 cm L = 38 cm Gas flow rate 100 mL min−1 Vr = 1.04 L Reaction time 60 min O3 concentration 2.5 mg L−1 Catalyst loading 1.25 g L−1 DOC0 = 8.22–11.26 mg L−1 pH = 8.15–8.57 THMFP = 216–266 μg L−1 HAAFP = 509–553 μg L−1 T = 20 °C Chlorination: According to SM 2350 Residual chlorine 0.2–1 mg L−1, pH 7, 48 h contact time. | Kinetics of O3 decomposition, Rct Fe-Mn catalyst presented high hydroxyl radical exposure. Fe-Mn catalyst showed a best performance in O3 decomposition, DOC (30%) and UV254 (85%) removal. Post-chlorination and DBPs analyses demonstrated that catalytic ozonation reduced THMs (70%) and HAA9 (75%) formation. | [170] |
| NOM-surface water polluted with DOC from domestic and agricultural effluents (Wu-Lo River, Pintung, Taiwan) | α-FeOOH goethite Precipitation synthesis SBET = 61.9 m2 g−1 SEM morphology micro needles. | Fluidized bed reactor Semi-continuous operation D = 6 cm L = 38 cm Gas flow rate 50 mL min−1 Vr = 1.04 L Reaction time 60 min O3 concentration 2.5 mg L−1 Catalyst loading 0.5–1.5 g L−1 DOC0 = 9.21 mg L-1 pH = 7.6 T = 20 °C Chlorination: According to SM 2350 Residual chlorine 0.2−1 mg L−1, pH 7, 48 h contact time. | Higher efficiency in DOC removal by catalytic ozonation. Post-chlorination and DBPs No BrO3− detected, THMs and 9HAAs highly reduced by catalytic ozonation + biofiltration. The bromine incorporation factor of THMs and HAAs increases with catalyst dosage. | [171] |
| Humic acid Bromide | Ferrate (VI) | Homogeneous ozonation Flasks, Vr = 0.5 L Ferrate dose 0.1–5 mg L−1 CBr- = 100–1500 µg L−1 DOC0 = 0.1–10 mg L−1 CO3 = 1.5–4 mg L−1 T = 5–40 °C pH = 3–11 | Ferrate (VI) reduced BrO3− during catalytic ozonation. Humic acid content decreased bromate formation. No post-chlorination. No other DBPs were analyzed. | [172] |
| Humic substances in phosphate buffer. | Degussa P-25 TiO2 80/20 anatase/rutile SBET 50 m2 g−1 | Catalytic ozonation/Ultrafiltration Reactor membrane module and mixing tank. Injection of O3 in a Y-type mixer. Gas flow rate 50–150 mL min-1 CO3 gas = 2.5–10 mg L−1 TiO2 loading 0–5 g L−1 DOC0 = 8 mg L−1 pH = 2 (O3 decomposition) Chlorination: SM 2350 (residual chlorine 0.5–2 mg L−1 after 48 h, pH 7). | Reduction of THMs formed during post-chlorination after catalytic ozonation. Positive effect of the TiO2 loading. Membrane permeate recovered 93% by catalytic ozonation vs. 78% in ozonation alone. | [173] |
| Bromide | Ferrate (VI) | Homogeneous ozonation Flasks, Vr = 0.5 L Ferrate dose 1–5 mg L−1 CBr- = 100–1500 μg L−1 CO3 = 0–5 mg L−1 T = 5–40 °C pH = 3–11 | Ferrate (VI) reduced BrO3− during catalytic ozonation No post-chlorination No other DBPs were analyzed. | [174] |
| NOM from groundwater (Banat, Serbia) | Aeroxide P25 TiO2 80/20 anatase/rutile SBET = 50 m2 g−1 | Ozonation: Reactor bubble glass column D = 85 mm V = 2 L Vr = 1.5 L Gas flow rate 8 L h−1 Contact time 3–25 min O3 dosage 0.4–3.0 mg O3/mg DOC Catalyst dose 1 mg L−1 DOC0 = 9.85 mg L−1 pH = 7.46 Chlorination: SM 2350 (residual chlorine 0.5–2 mg L−1 after 48 h, pH 7). | Removal THMFP 48% (no improvement observed compared to ozonation) Removal HAAFP 44% Less brominated species and haloacetonitrile after catalytic ozonation. | [175] |
| Humic substances | Bone charcoal (BC) Prepared at 600 °C for 4 h (bone from cattle and sheep). XRD revealed calcium phosphate (hydroxyapatite form) and amorphous carbon. XRF: CaO 92.9%; P2O5 3%; MgO 0.6%; SiO2 0.095%; MnO 0.008%. Boehm titration: Surface acidic groups 0.71 meq g−1 Surface basic groups 0.33 meq g−1 SBET = 121 m2 g−1 pHpzc = 8.5 | Semi-batch operation V = 1 L Catalyst 2 g C HS = 15–100 mg L−1 Ozone 0.5 mg L−1 min−1 T = 15–40 °C pH = 2–12 | The catalyst improves the reaction rate of HS degradation by 1.43- and 1.56- fold compared to ozonation. Kinetics and mechanism analysis. Kinetic studies showed that the rate greatly increased in the presence of BC. The reaction rate is related to BC dosage, HS concentration, pH, temperature. The presence of t-BuOH confirmed the main degradation route by hydroxyl radicals. Ea = 10 kJ mol−1 Diffusion controlled reaction. No chlorination. No DBPs were analyzed. | [176] |
| NOM from surface water (Dong-Gang River, Pingtung, Taiwan) | TiO2/αAl2O3 No characterization studies reported | Fluidized bed reactor Continuous operation Gas flow rate 100 mL min−1 Water flow rate 50 mL min−1 O3 concentration 2.5 mg L−1 Catalyst loading 0–25 g DOC0 = 2.8–4.7 mg L−1 pH = 7.82–8.15 T = 20 °C Chlorination: According to SM 2350 Residual chlorine 0.2–1 mg L−1, pH 7, 48 h contact time. | The combined catalytic ozonation significant reduced DOC up to 51.4% and DBPFP up to c.a. 50% (THMs and HAA6 analyses). The amount of catalyst had a positive effect in the process. | [177] |
| Dimethyl phthalate (DMP) and NOM in surface water (Jingmi Cannel, China) | Ru/AC Particle size 6–10 mesh 1–1.5 mm 0.5 wt.% Ru AC ash content 7.8 wt.% (mainly SiO2, Al2O3 and Fe2O3) SBET = 945.2 m2 g−1 | Cylinder reactor, bubble column D = 5 cm L = 120 cm V = 1 L Semi-batch for catalytic activity: Cat. Dosage = 2 g L−1 Ozone dosage 118 mg h−1 Q 300 mL min−1 Continuous operation for stability tests: Qw = 20 mL min−1 Cat. Dosage = 40 g COD0 = 2.25–3.29 mg L−1 pH = 7.2–8.5 T = 25 °C Chlorination: pH = 7 Excess of chlorine T = 25 °C, 7 days | Ru/AC active in catalytic ozonation improved DMP mineralization. Stable for 42 h with no Ru leaching and 75% DOC removal. In natural water, Ru/AC-O3 was better than O3 alone for DOC removal (40% vs. 10%) and DBPFP reduction (60% vs. 40%); THMFP and HAAFP analyzed. | [178] |
| NOM (Songhua River, Harbin, China) | FeOOH-goethite pHpzc = 7.0 SBET = 68.4 m2 g−1 Average pore size 24 nm Surface OH density = 0.5 mmol g−1 CeO2 pHpzc = 6.6 SBET = 117 m2 g−1 Average pore size 7.9 nm Surface OH density = 0.4 mmol g−1 D = 0.075–0.3 mm | Ozonized water (20 mL; 10 mg O3 L−1) VT = 40 mL DOC = 10 mg L−1 O3/DOC = 1 10 min reaction time pH = 7 T = 18 °C Fractionation of NOM with XAD-8 and XAD-4 amberlite resins: HOA: Hydrophobic acid HON: Hydrophobic neutral HIA: Hydrophilic acid HIB: Hydrophilic base Fluorescence analyses. | Ozonation decrease the aromaticity of humic-like structures and increases the generation of carboxylic groups. Catalytic ozonation improve the destruction of humic-like structures. Ozonation of HOA and HIA yields by-products with low aromaticity and low molecular weight. Catalytic ozonation enhances the formation of these by-products from HIA and improves the destruction of highly polycyclic aromatic structures. No post-chlorination. No DBPs were analyzed. | [179] |
| Humic and fulvic acid in distilled water | MnO2 No characterization studies reported. | Cylinder reactor (bubble column, semi-batch operation, total recirculation of water) D = 7 cm L = 1.5 m V = 5 L O3 dose = 47 mg L−1 in 30 min TOC0 = 28–38 mg L−1 pH = 8.9 T = 19 °C | TOC removal 79% in catalytic ozonation vs. 67% in single ozonation. No post-chlorination. No DBPs were analyzed. | [180] |
| NOM-surface water (Lake Lansing, Michigan USA) | Fe2O3 over ceramic membrane (Al2O3/ZrO2/TiO2) Average Fe2O3 particles 4–6 nm (TEM) | Conditions of ozone-membrane system: Water recirculation rate = 2.75 L min−1 T = 20 °C O3 flow rate: 100 mL min−1 Transmembrane pressure = 0.5 bar O3 gas concentration = 2.5 mg L−1 Ozone injection before membrane module DOC0 = 8.6–11.6 mg L−1 pH = 7.7–8.6 Chlorination SM 2350 (residual chlorine 0.5 mg L−1 after 48 h, pH 7). | Decomposition of O3 in the membrane surface. DOC reduced > 85% Decrease of THMFP (90%), HAAFP (85%) and also the concentration of aldehydes, ketones and ketoacids. | [181] [182] |
| DOC in river water (Japan) | Powdered AC (membrane as separator) | Ozonation: Reactor characteristics not specified. Column before membrane module O3 dose = 1–2 mg L−1 AC dose 10 mg L−1 DOC0 = 2.4 mg L−1 Q water = 5.5 m3 day−1 Chlorination conditions not specified. | Combination of PAC and O3 improved the performance of the subsequent membrane system. DOC reduced 59% T-THMFP reduced 75% | [183] |
| Humic acid solution NOM from Ebro River (Zaragoza, Spain) | TiO2 (anatase Probus, SBET = 8.5 m2 g−1) 2.5 wt.% supported by adsorption on: Attapulgyte (clay, SBET = 104 m2 g−1) α-Al2O3 (SBET = 164 m2 g−1) Silica gel (SBET = 362 m2 g−1) Calcination T = 350–600 °C Granular form, d = 2–4 and 4–6 mm. TiO2/Al2O3 catalyst characterization: SBET = 132 m2 g−1 Pore volume = 0.33 cm2 g−1 Pore size = 4.4 nm (12%); 20.56 um (88%). Anatase phase by XRD | Glass fixed bed reactor Semi-batch mode Humic acid experiments: V = 650 mL O3 = 405 mg O3 h−1 30 min contact time C HA = 5.34 mg TOC L−1 pH = 7.2 Catalyst loading: 2.5–10 g L−1 NOM experiments: V = 2 L O3 = 396.4 mg O3 h−1 Contact time 2–4 min. Catalyst loading: 2.5 g L−1 TOC0 = 4.46 mg L−1 pH = 7.94 Stability tests (4 consecutive runs): C Humic acid = 6.65 mg L−1 TOC pH = 7.62 Catalyst loading = 2.5 g L−1 V = 650 mL O3 405 mg O3 h−1 30 min contact time | TOC and UV254 removal were slightly greater in the presence of a catalyst (60% vs. 32% in O3 for TOC). The best results were obtained with TiO2/Al2O3 calcined at 500 °C with a concentration of 2.5 g L−1 of catalyst and particle diameter 2–4 mm. The importance of adsorption is highlighted. Higher doses of O3 have a positive effect in catalytic ozonation. Stability tests with the best catalyst showed a good stability with similar TOC, UV254 and pH results after the 4 experiments. No post-chlorination. No DBPs were analyzed. | [184] [185] |
| Fulvic acid extracted from surface water (Cebron, France) | Commercial TiO2 over Al2O3 beads Dbeads = 1.5–2.5 mm No characterization provided | Ozonation: Reactor: Conical glass flask 1.3 L V reaction = 1 L Contact time = 10 min O3 dose = 2–6.5 mg L−1 (homogeneous experiments) CCAT = 10 g L−1 DOC0 = 2.84 mg L−1 H2O2/O3 ratio = 0.35 mg H2O2/mg O3 pH = 7 Chlorination: Cl2 dose = 2 mg L−1 Contact time 1 h pH = 7 | Catalytic ozonation increased ozone consumption. Higher mineralization (24%) compared with O3 and H2O2/O3 systems. Chlorine demand was minimized in catalytic ozonation. No DBPs were analyzed. | [186] |
| Model compounds: Fulvic acid Protein disaccharide | Granular catalyst TiO2/Al2O3 GAT 925 SI D = 4 mm For comparison: BST: TiO2 stick compacted APF: Granular TiO2/Clay | Tested TiO2/O3 and TiO2/O3/H2O2 Bubble column Semi-continuous reactor with recirculation of the aqueous solution Total height 2 m D = 40 mm Porous glass to support the catalyst in the middle Q = 2.4–2.8 L h−1 CO3g = 60–90 mg L−1 pH = 8 TOC0 = 12 mg L−1 H2O2/O3 = 0.4 g/g Catalyst loading 30 g L−1 Room temperature | Cumulative effect of adsorption and ozonation. At high ozone dosage TOC abatement by catalytic ozonation is observed (80% vs. 50% in O3). APF was not stable under the conditions tested due to mechanic friction. No chlorination. No DBPs were analyzed. | [187] |
| Target DBPs | Catalyst and Main Properties | Reactor and Experimental Conditions | Main Results | Ref. |
|---|---|---|---|---|
| MCA DCA Deionized water | TiO₂ photocatalyst coated on the commercial product “Pilkington ActiveTM glass” (PAGs). | Planar falling film reactor. Seven UVA lamps (15 W, 360 nm) fixed inside the reactor. Intensity UV light: 1 mW cm−2; water flow rate: 1 L min−1, volume 0.5 L; [CAA] = 1 mM; pH 3, pure gaseous oxygen rate of 10 L h−1, ozone gas with 130 ± 5 mg L−1 ozone at a power of 30 W. | Single ozonation was ineffective method for the removal of chloroacetic acids. However, a significant enhancement in the degradation and mineralization efficiencies was observed by the combination of ozone with photocatalysis. | [106] |
| DCA | FMOG: Flower-like nanocomposite (FMOG) consisted of pure β -MnO2 (PMO) and reduced graphene oxide nanosheets (RGO). Atomic ratio between Mn and O element is ca. 1:2.1. | 500 mL glass conical flask. 110 min; flow rate of O3: 4.47 mmoL min−1, T = 20 °C, concentration of catalyst: 50 mg L−1, and pH = 4.4. [DCAA]0 = 100 mg L−1. | FMOG displayed higher catalytic performance compared with ozonation, and PMO catalytic ozonation. | [205] |
| DCAcN | Titanium dioxide (Degussa, P-25, anatase/rutile = 75/25%). | Bench and outdoor photocatalytic ozonation systems consisted of the combination of CPC and ozone reactor. Metal halide lamps of different powers (100, 250, and 400 W); UV-solar, 300–800 nm. pH 6.5, 20 ± 1 C, TiO2 dose 0.4 g L−1, ozone dose 1 g L−1 h−1, UV intensity 33.8 W m−2. pH: 3, 6.5, and 10. | Compared to the single process, the UV-solar/TiO2/O3 process had the highest DCAN-removal rate. Optimal conditions: pH neutral; 1 g L−1 TiO2 and 1.13 g L−1 h−1 ozone; T = 20 °C; UV-solar intensity: 4.6–25 W m−2. | [206] |
| DCA | Immobilized TiO2 in the form of a well-known commercial product called “Pilkington ActiveTM glass” (PAG) sheet with a contact area of 30 × 5 cm2. | A planar reactor: Polymethylmethacrylate box (160 cm3 vol) covered by an optical window made from the same material. Reactor connected to an ozonation chamber (500 cm3 vol). 400 mL of dichloroacetic acid solution. 30 W lamp (300–420 nm and a maximum at about 360 nm). T = 25 °C, pH = 3, [DCA]o = 1 Mm. O3 input concentration: 135 ± 5 mg L−1. | (PAG/O3/UVA) showed highly modified oxidation properties in the decontamination of DCA in aqueous solutions. Kinetics of first order reactions with respect to dichloroacetic acid were found. Higher initial concentrations of DCA and higher temperatures increased the initial degradation rate. More than 90% of DCA was removed. | [207] |
| DCA in simulated water: deionized water with different amounts of humic acid (HA). | Natural bentonite, composed primarily of Ca2+-montmorillonite. | Cylindrical reactors with an inner diameter of 60 mm and a length of 500 mm. Gas flow rate 20 mL min−1; ozone gaseous concentrations of 0.90 mg L−1; 298 K; 2 L simulated water: Deionized water containing 1 mg L−1 DCA or different amounts of HA, pH adjusted to 6.0.; Fe3+ dosage: 5 mg L−1. | Under the combined effects of adsorption, ozonation and catalytic oxidation, high DCA removal is obtained. | [208] |
| DCA | Nanometer ZnO powder: Size 90 nm, and surface area (11 m2 g−1). | Batch experiments. A glass flat-bottomed flask with the inside diameter of 45 cm and the volume of 1.2 L. T = 20 ± 1 °C; initial pH 6.88; [DCA]o = 100 μg L−1; initial ozone concentration 1.96 mg L−1; catalyst dosage 100 mg L−1. | ZnO as catalyst in water significantly improved the ozonation removal of DCA. The degradation efficiency of DCA increased with the increasing pH of solution, catalyst dosage and ozone dosage. The presence of t-BuOH had a negative effect on catalytic ozonation of DCA. | [209] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltrán, F.J.; Rey, A.; Gimeno, O. The Role of Catalytic Ozonation Processes on the Elimination of DBPs and Their Precursors in Drinking Water Treatment. Catalysts 2021, 11, 521. https://doi.org/10.3390/catal11040521
Beltrán FJ, Rey A, Gimeno O. The Role of Catalytic Ozonation Processes on the Elimination of DBPs and Their Precursors in Drinking Water Treatment. Catalysts. 2021; 11(4):521. https://doi.org/10.3390/catal11040521
Chicago/Turabian StyleBeltrán, Fernando J., Ana Rey, and Olga Gimeno. 2021. "The Role of Catalytic Ozonation Processes on the Elimination of DBPs and Their Precursors in Drinking Water Treatment" Catalysts 11, no. 4: 521. https://doi.org/10.3390/catal11040521
APA StyleBeltrán, F. J., Rey, A., & Gimeno, O. (2021). The Role of Catalytic Ozonation Processes on the Elimination of DBPs and Their Precursors in Drinking Water Treatment. Catalysts, 11(4), 521. https://doi.org/10.3390/catal11040521