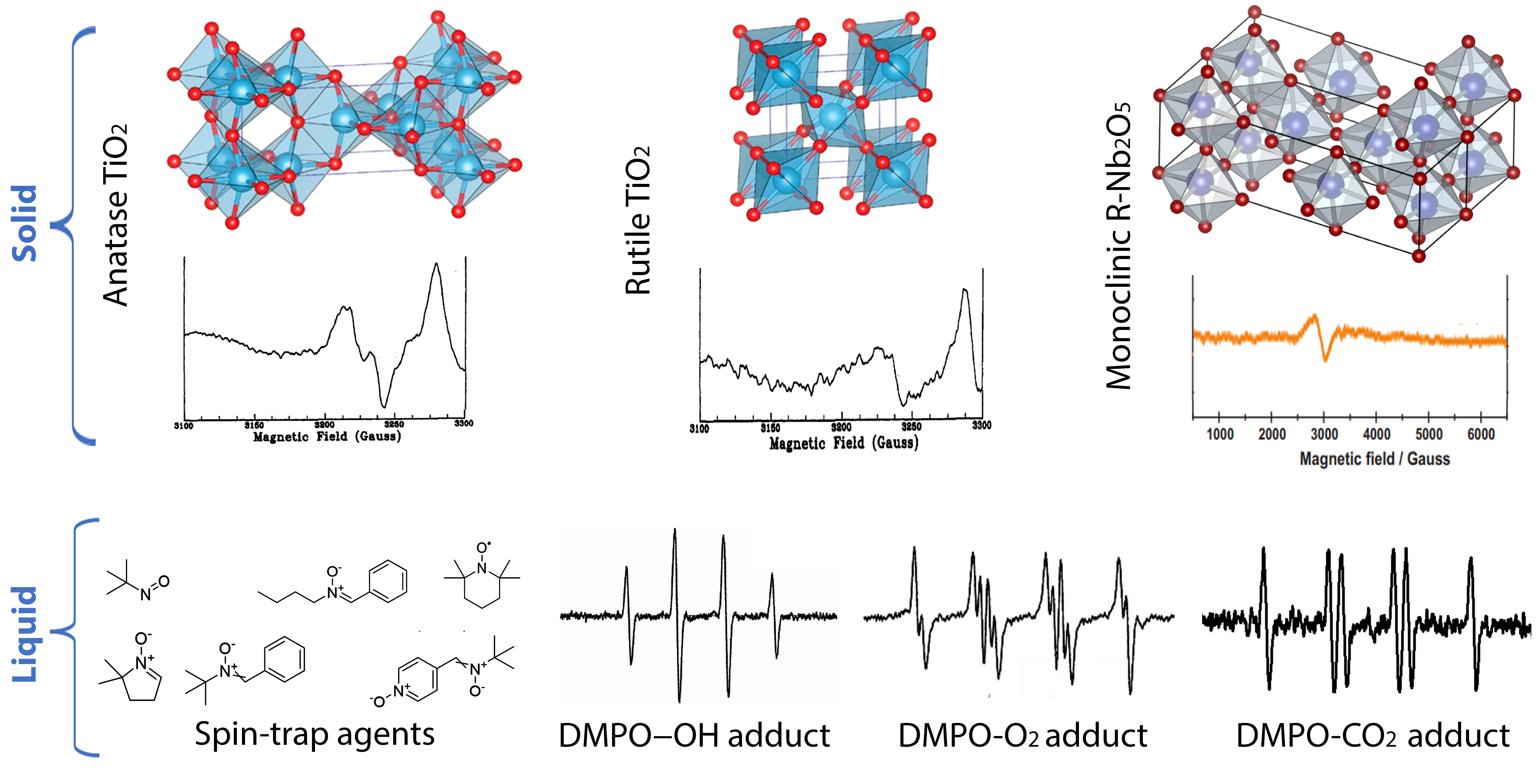
2. Titanium Dioxide Photocatalyst
Titanium dioxide (TiO
2) is a solid material used in large-scale practical applications, including photocatalysis. It has been used in many photocatalytic reactions, e.g., the mineralization of water and air pollutants, super hydrophilicity, and solar energy conversion [
44]. Titanium dioxide has three polymorphs, i.e., rutile (the thermodynamically stable phase), anatase, and brookite. These polymorphs are built up by distorted octahedral TiO
6 units, in which the oxygen atoms are surrounded by three Ti ions (OTi
3). Although some papers have reported promising activity for the brookite phase, the most investigated polymorphs are rutile and anatase They are differed from each other by their bandgap energy (3 and 3.2 eV for rutile and anatase, respectively), and by the more stable crystal faces (110 and 101 for rutile and anatase, respectively) [
1].TiO
2-based photocatalytic processes are induced by an initial excitation through the irradiation by a suitable light source, having an energy equal to or higher than the band-gap energy of the TiO
2 [
45].
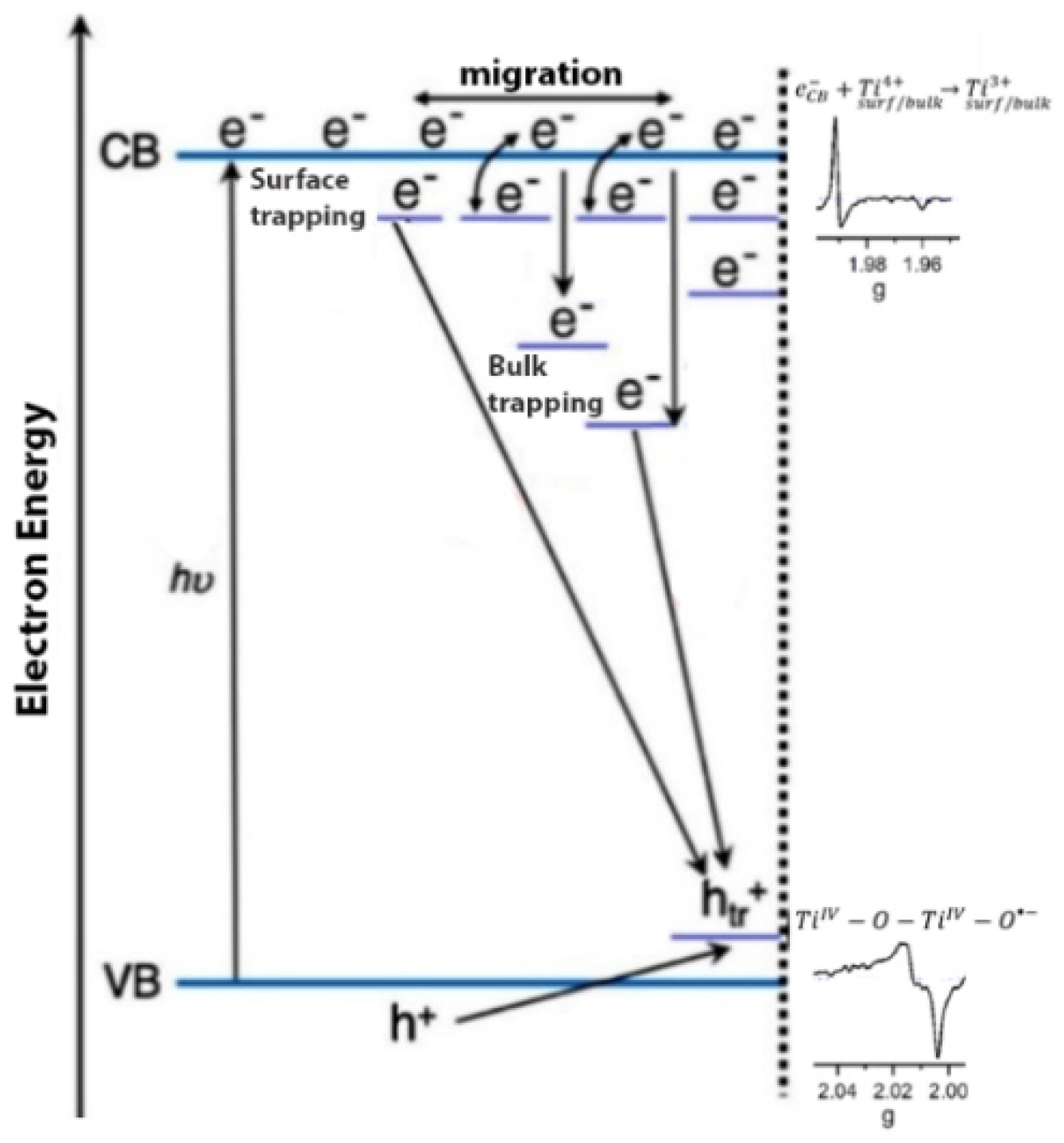
The irradiation of TiO
2 by a suitable light leads to a charge spatial separation, i.e., the promotion of an electron (e
−) in the conduction band (CB) and the formation of a hole, (h
+) in the valence band (VB). These charge carriers can be then migrated and trapped at the surface of TiO
2, before their transfer to the adsorbed species at the surface [
46]. The trapped electrons and trapped holes can also be promoted due to chemical modifications of TiO
2. While anatase has shown better photocatalytic activity than rutile, the mixed-phase Degussa P25 has demonstrated, in some cases, better activity due to the presence of an interface between the anatase and rutile, which improves the spatial separation of the photo-generated charge carriers [
47]. Nevertheless, the recombination of charge carriers, the nature and location of the charge traps, and the stability of the trapped charge carriers are important phenomena that affect the overall photocatalytic activity. The photocatalytic activity of TiO
2 is highly related to its surface properties and, therefore, it is of high importance to investigate TiO
2 from the surface science point of view. Titanium dioxide is considered a reducible metal oxide because it can lose oxygen, forming oxygen vacancies and excess electrons, as can be seen in Equation (17) [
48]. The insulating TiO
2 turns into an n-type semiconductor due to oxygen depletion, while the excess electrons are stabilized into the reduced solid and trapped as Ti
3+ centers. Thus, when excess electrons are photogenerated in TiO
2, they are usually stabilized by Ti cations with the formation of Ti
3+ ions.
is an empty vacancy in the Kröger–Vink notation.
The generation of Ti
3+ upon the irradiation of TiO
2 has been detected by Electron Energy Loss Spectroscopy (EELS) [
49], stopped-flow spectrophotometry [
50], laser-flush photolysis [
44], polarized optical spectroscopy [
51], photoelectron spectroscopy [
52], and Electron Paramagnetic Resonance spectroscopy (EPR) [
53]. EPR spectroscopy is considered as a sensitive technique to follow the surface defects and the radical formation at semiconductor surfaces, especially the paramagnetic species and the hyperfine interaction. EPR spectroscopy has been mainly employed to investigate the surface chemistry of polycrystalline TiO
2, i.e., the formation of Ti
3+ centers under irradiation or other conditions and its reactivity towards different adsorbed species. It is also very useful in clarifying the charge trapping and the surface charge transfer upon irradiating the surface of TiO
2.
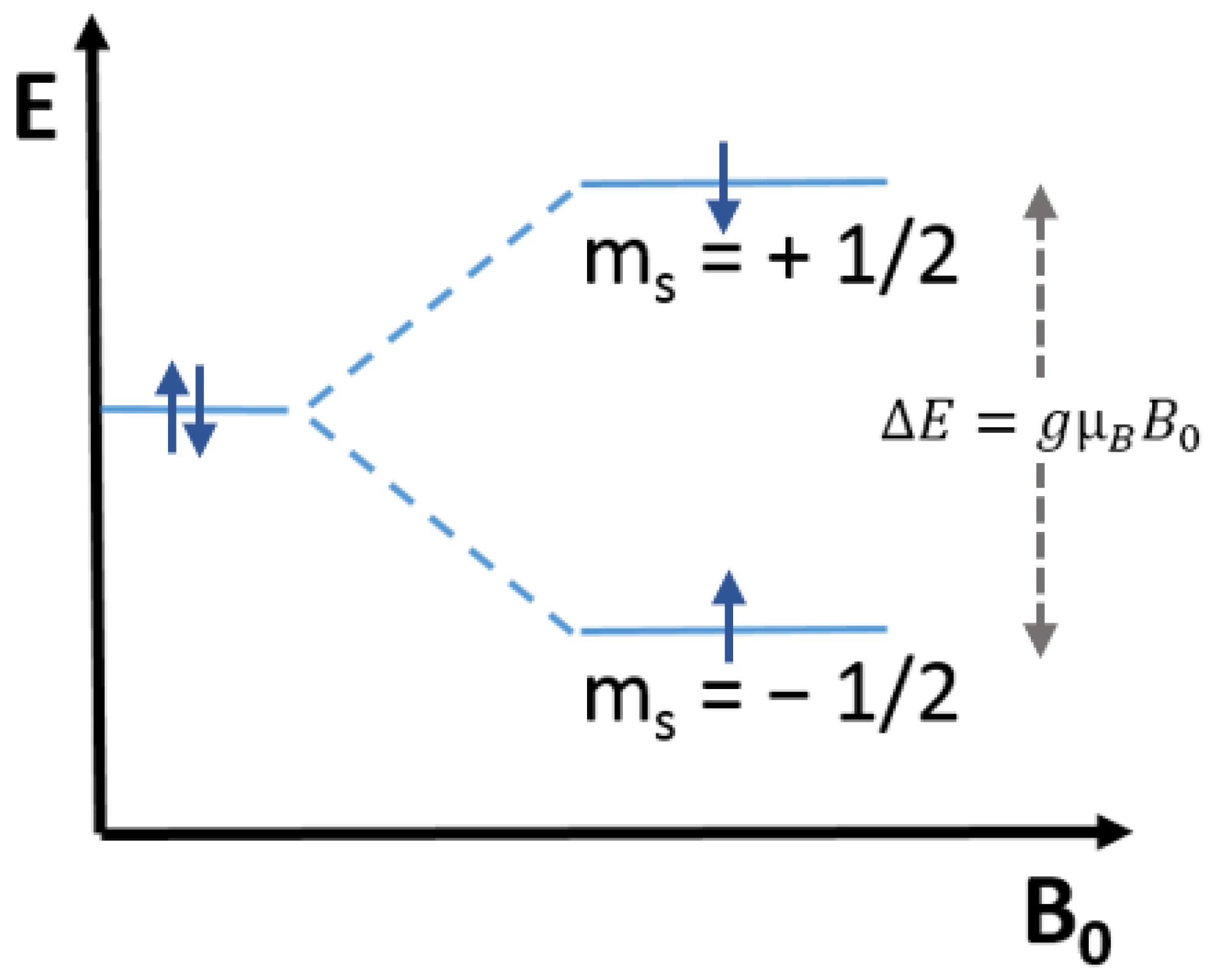
2.1. Trapped Electrons and Holes in TiO2: EPR and g Tensor
TiO
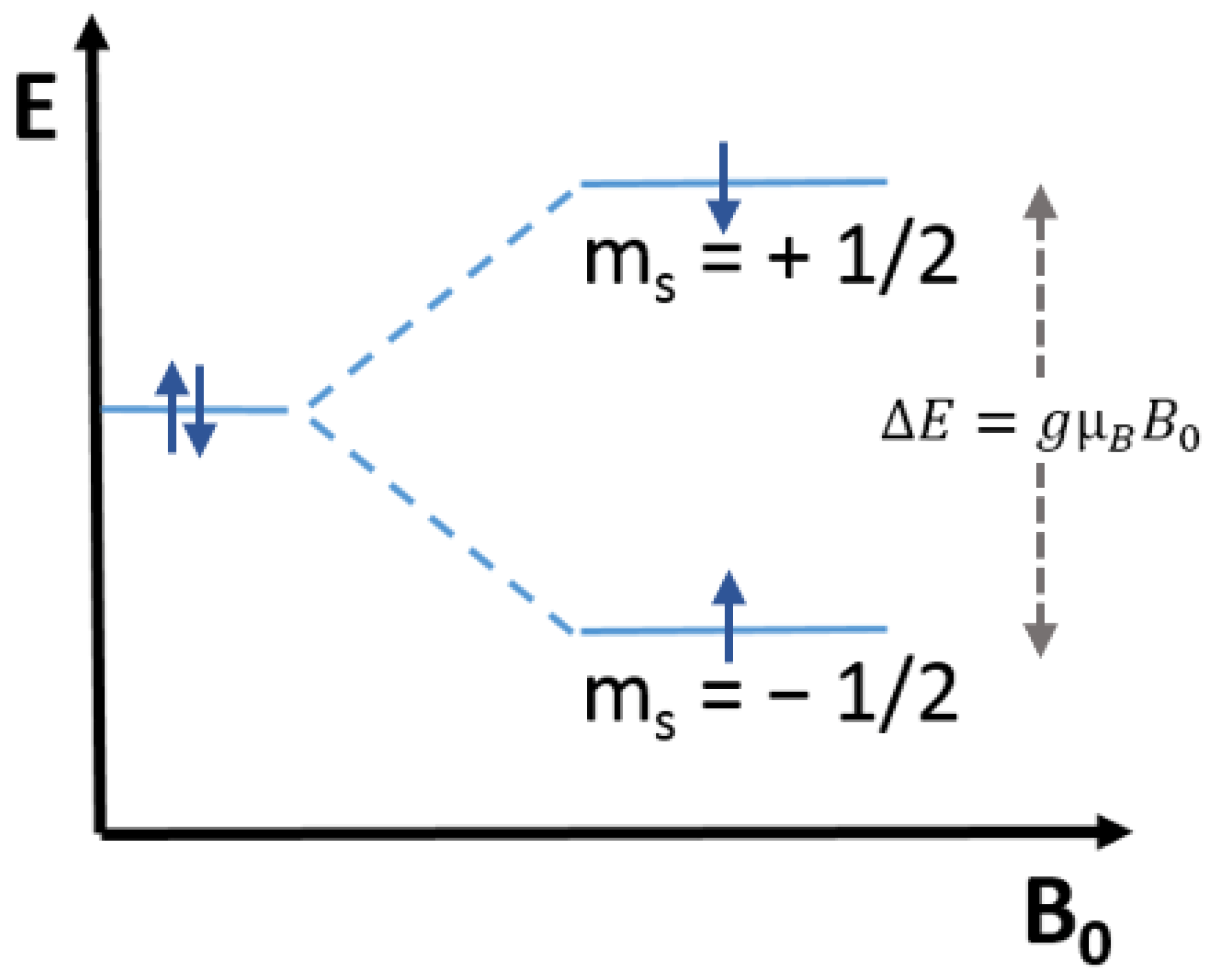
2 has been mainly investigated employing the Continuous Wave-EPR (CW-EPR) technique, which commonly uses X-band microwaves having 9.5 GHz frequency and generates the spectrum of the first derivative of the microwave absorption as a function of the magnetic field (Tesla or Gauss). Upon irradiation, the photogenerated electrons are trapped as Ti
3+ centers, which have paramagnetic properties (S = ½) having a 3d
1 configuration. When the metal ion is octahedrally coordinated, as in the case of all TiO
2 polymorphs, the free-ion ground state is split into two subgroups having three t
2g and two e
g orbitals separated by the energy term Δ
O. The degeneracy of the t
2g and e
g levels owing to a tetragonal (
Scheme 3A) or trigonal (
Scheme 3B) distortion leads to anisotropic g values [
39].
When the distortion compresses the tetragonal (D
4h), the d
xy orbital is the ground state, however, if the distortion elongates this tetragonal, the ground state is a degenerate d
xy, d
yz orbital. The g values for a Ti
3+ in a tetragonally distorted octahedral environment are:
where λ is the spin-orbit coupling constant for Ti
3+ (154 cm
−1), δ
1 and δ
2 are the energy separation between the d-orbitals (
Scheme 3A) [
39].
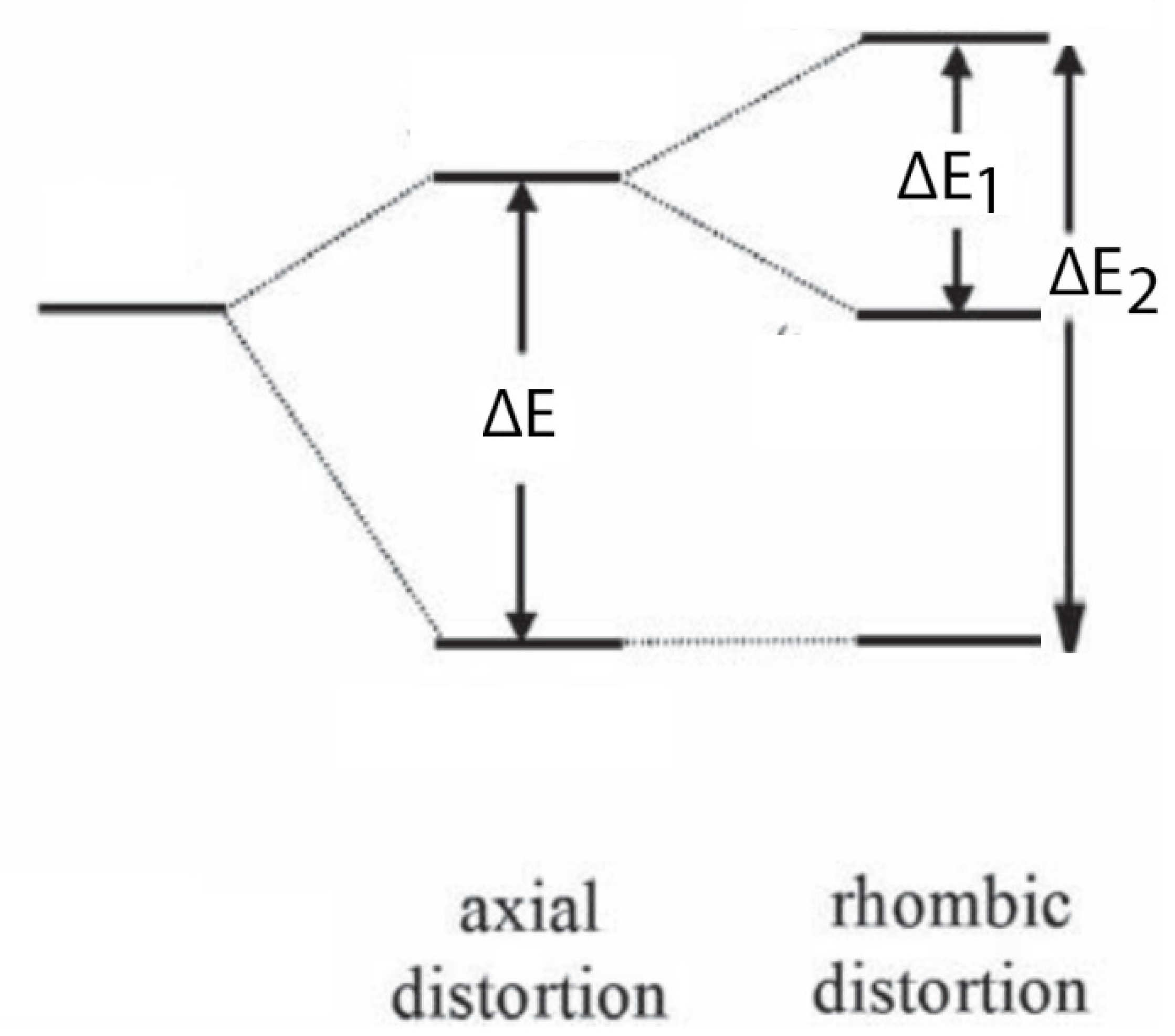
On the other hand, the photoexcitation of TiO
2 photocatalyst produces a paramagnetic center, i.e., O
− ion due to the hosting of the unpaired electron in the 2P orbital of the oxygen atom (O
2− + h
+ → O
−). In this case, the g tensor can be either axial (g
II = g
zz ≈ g
e; g
I = g
e + 2λ/ΔE) or rhombic (g
zz ≈ g
e; g
xx = g
e + 2λ/ΔE
1; g
yy = g
e + 2λ/ΔE
2) depending on the symmetry of the environment (
Scheme 4) [
39].
2.2. Charge Trapping Centers in Photoexcited TiO2
In-situ EPR investigations have been widely done especially for anatase, rutile, and mixed-phase P25 TiO2 photocatalysts under irradiation. These investigations were performed either for TiO2 suspensions in aqueous phases, or for dehydrated TiO2 powders at the solid-gas interfaces. In the former case, the presence of hydroxyl ions and water molecules on the surface of TiO2 stabilizes longer the charge carriers.
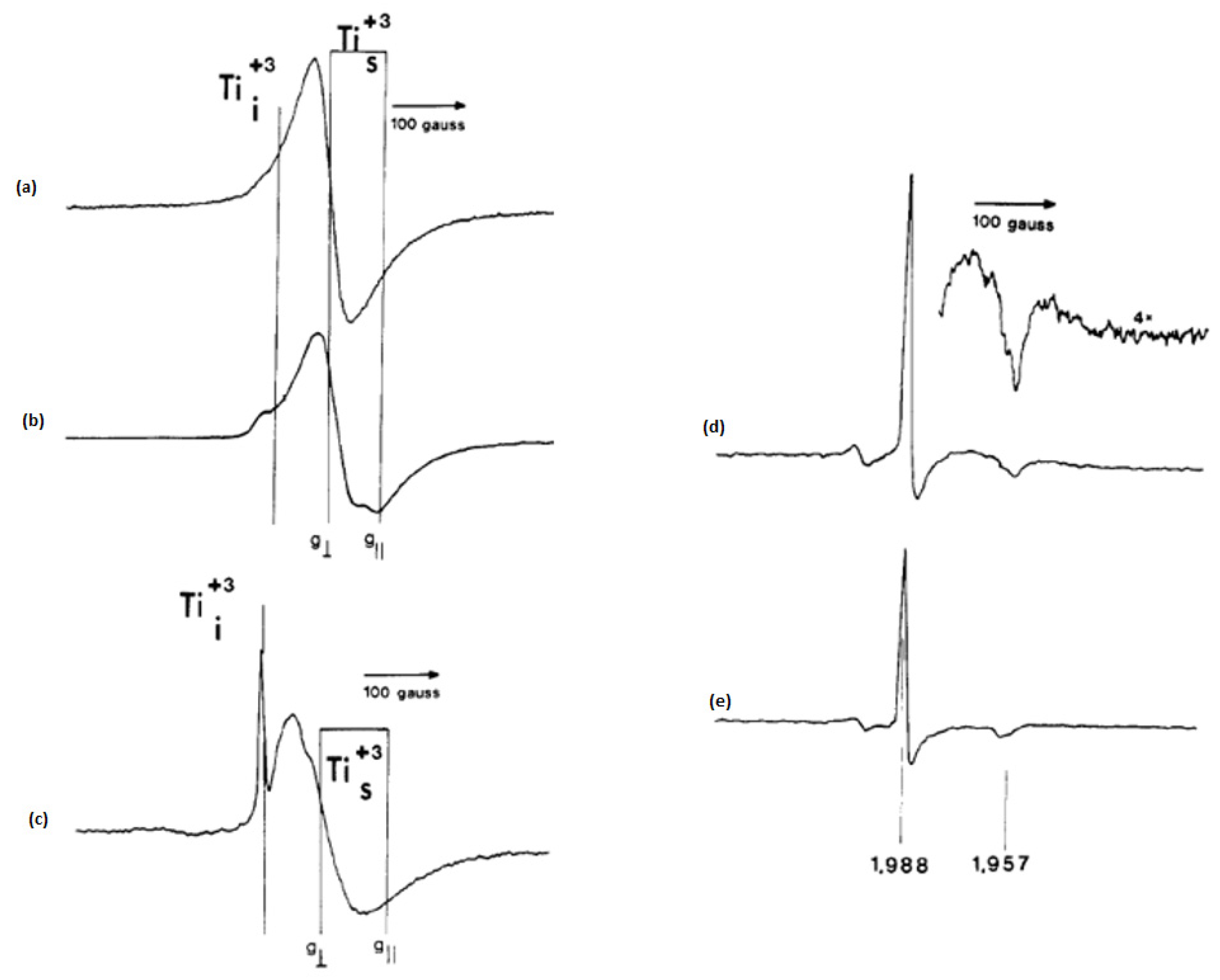
In their premier work to study the paramagnetic species on semiconducting photocatalysts, Howe and Grätzel [
25] have reported an EPR observation of trapped electrons in irradiated colloidal TiO
2. Although a blue coloration of TiO
2 suspension appeared upon the irradiation of degassed acidic solution of colloidal TiO
2 in the presence of PVA (polyvinyl alcohol), no EPR signals were detected at room temperature. However, the authors were able to detect such signals at 77 K after the irradiation at room temperature. They showed that the collected spectra consisted mainly of two overlapping signals: (i) a broad signal at g = 1.92, and (ii) a smaller and narrower feature at g = 1.988, which was more pronounced in lower concentrated solutions as presented in
Figure 1b. In alkaline solution, the broad signal at g = 1.93 was less intense and the signal at g = 1.988 was better resolved as the perpendicular component of an axially symmetric signal with the corresponding parallel component of g = 1.957 (
Figure 1c).
However, the authors noticed that a narrow axial signal was most prominent when the irradiation and the detection were carried out at 77K as can be seen in
Figure 1d. They clarified that in all signals the g values below 2 were due to the formation of Ti
3+ species and the insensitivity of the narrow line to changes in pH reveals that Ti
3+ species are located in the interior of colloid particles. They commented also that the values of the g-tensor components for the colloid Ti
3+ signals are significantly lower than those for Ti
3+ in powdered TiO
2. The authors have also found that the presence of a hole scavenger induced many electrons to be trapped at surface Ti
4+ sites, generating Ti
3+ species with distorted octahedral that responsible for the blue color and the broad EPR signals. Using a hole scavenger in the absence of oxygen, both surface and bulk trapped electrons were stable at room temperature. The irradiation of the suspension in presence of a hole scavenger at 77 K produced faster bulk trapped electrons than those trapped at the surface
Figure 1d,e. The authors suggested that the origin of the blue color in irradiated colloids was not due to electrons trapped at oxide vacancies because no EPR signals at g = 2.0 were observed. Hence, they distinguished the surface Ti
3+ species from the interstitial Ti
3+ species and showed that increasing the acidity and temperature of the suspension induces the trapping of electrons in surface sites.
In another publication, Howe and Grätzel [
55] did an EPR study of hydrated anatase, in which a high-vacuum sample cell was immersed in the liquid helium (4.2 K) or in the liquid nitrogen (77 K) and irradiated with a 450-W xenon lamp. Upon the irradiation at 4.2 K, hydrated anatase produced an EPR spectrum that consisted of two signals as shown in
Figure 2a: a low-field slightly nonaxial signal (signal B) and a high-field axial signal (signal A), which was disappeared in the presence of traces of residual oxygen. These signals were stable with time, however, they decayed rapidly when the lamp was turned off. On the other hand at 77 K, an order of magnitude less intense identical signals have appeared. In this case, signal B was only produced upon the irradiation in the presence of O
2 and was stable even in the dark (
Figure 2b). While the authors attributed the high-field signal A (g
1 = 1.990, g
2 = 1.990, g
3 = 1.960) to interstitial Ti
3+ cations, they assigned the low-field signal B (g
1 = 2.016, g
2 = 2.012, g
3 = 2.002) to the trapping of positive holes at lattice oxide ions. The trapped holes could be located in the subsurface layer as Ti
4+O
−Ti
4+OH since signal B was not broadened by the presence of O
2. The authors considered different possible pathways for the generation of these paramagnetic species. Upon the irradiation at 4.2 K, most of the charge carriers are trapped, i.e., electrons as Ti
3+ and holes at subsurface oxide ions, which are then quickly recombined each other after turning off the lamp, decaying the EPR signals. The adsorbed oxygen prevents recombination and stabilizes the trapped holes by scavenging the trapped electrons through a multielectron process. They concluded also that the existence of hydroxyl radicals on the surface appears to be transient and they were apparently not primary products of hole trapping.
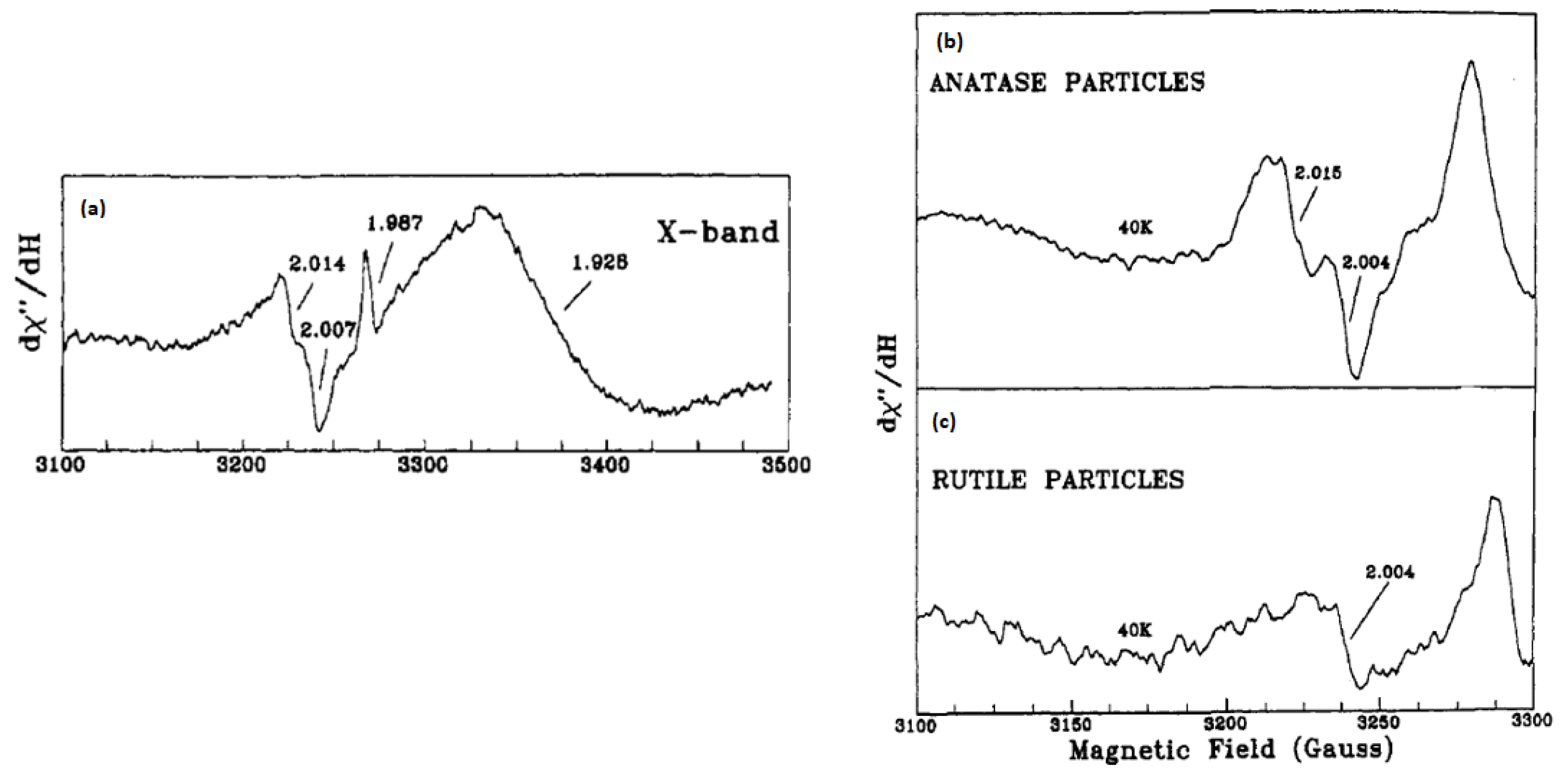
Micic et al. [
56] investigated the trapped holes on colloidal TiO
2 by EPR spectroscopy, in which they assigned the signals at g < 2 (see
Figure 3a) to the capture of electrons on by Ti
4+ ions. They showed a broad asymmetric EPR signal at g = 1.928 of Ti
3+ species at surface sites, in addition to a narrower EPR signal at g = 1.987 of trapped electrons at interior sites since it was not affected by the presence of electron scavengers. The authors clarified that as the photogenerated electrons can be located on Ti
4+, in the bulk lattice as (Ti
3+)
latt, at the surface as (Ti
3+)surf, and in the conduction band [e
−CB]. On the other hand, the signals with g = 2.007, g = 2.014, and the shoulder at about g = 2.020–2.028 were assigned to hole centers on the surface or subsurface layers because their intensities had been suppressed in the presence of polyvinyl alcohol or KI as hole scavengers. They explained that the surface of anatase TiO
2 is covered with hydroxide ions or water molecules with an average number of 5–15 OH
− groups per nm
2. Part of these hydroxide ions that are located on the (110) surface of TiO
2 can therefore trap the photogenerated holes. Such hydroxide ions are present as acidic Ti
4+-O(H)-Ti
4+ and basic Ti
4+-OH groups. Because the latter has a higher electron density, it is expected to be the primary hole trapping site. They showed also that the relative intensities of the EPR signals at g = 2.014 and 2.007 changed with pH. This was associated with the ionic form dominated by Ti
4+-OH groups (Equations (18) and (19)). The authors investigated as well the EPR signals of anatase and rutile TiO
2 powders suspended in water as presented in
Figure 3b,c. Same signals were obtained for anatase as for the colloidal TiO
2 but with lower intensities. The authors attributed that to the smaller surface area for suspended anatase TiO
2 powder. Nevertheless, a very weak signal was noticed for rutile powder that was similar to one of those found in anatase particles. The authors ruled out the OH radicals to be the species produced by hole trapping. They suggested instead an oxygen-centered radical, which can most probably be a hole trap on the basic OH-group having an energy level below the valance band of TiO
2 as illustrated in Equation (20). This was explained by the fact that such a group has a higher electron density. Such trapped holes can react through a direct transfer mechanism with chemical substrates adsorbed on the surface as in the case of methanol in the aqueous colloidal TiO
2 solution.
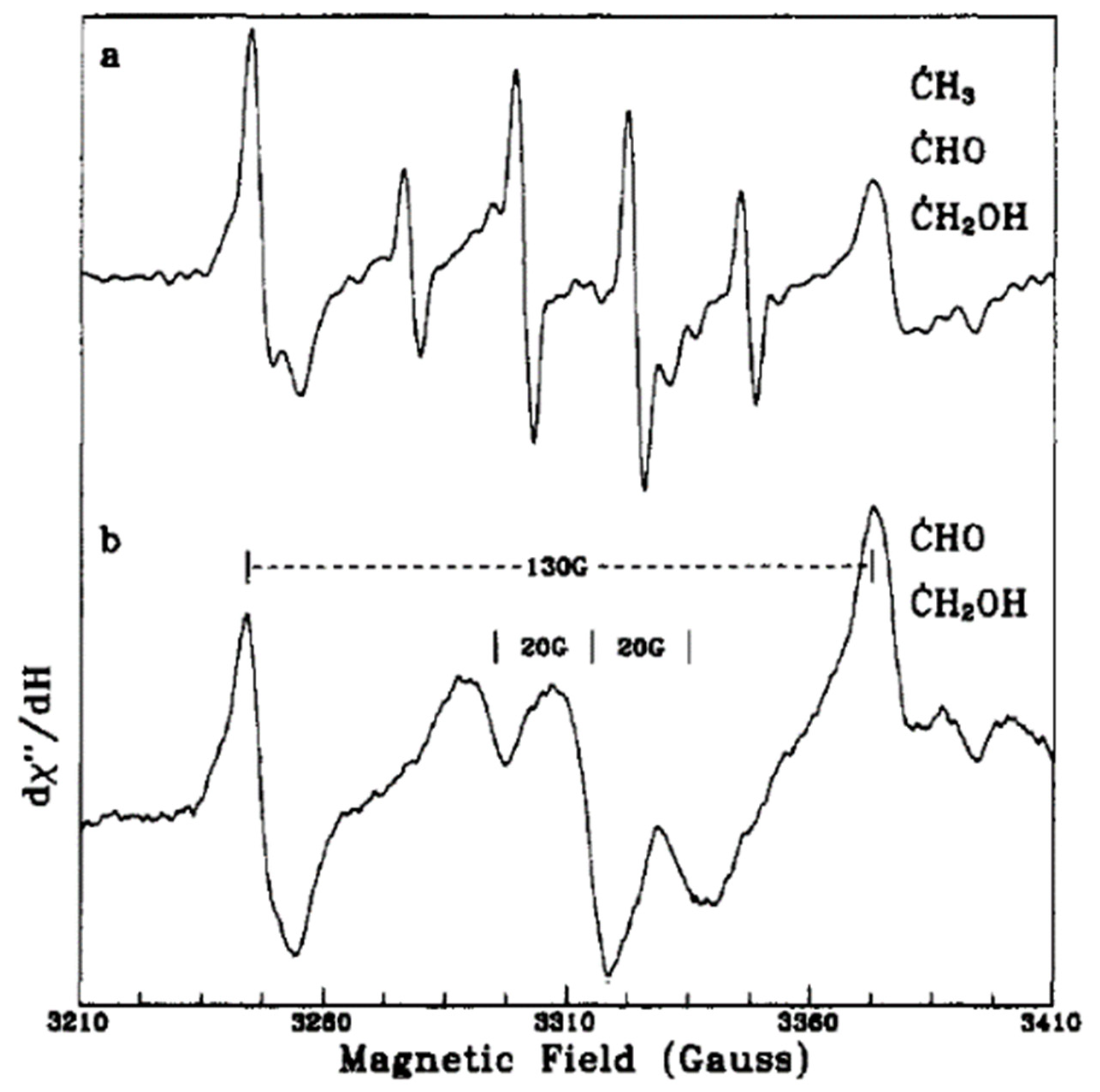
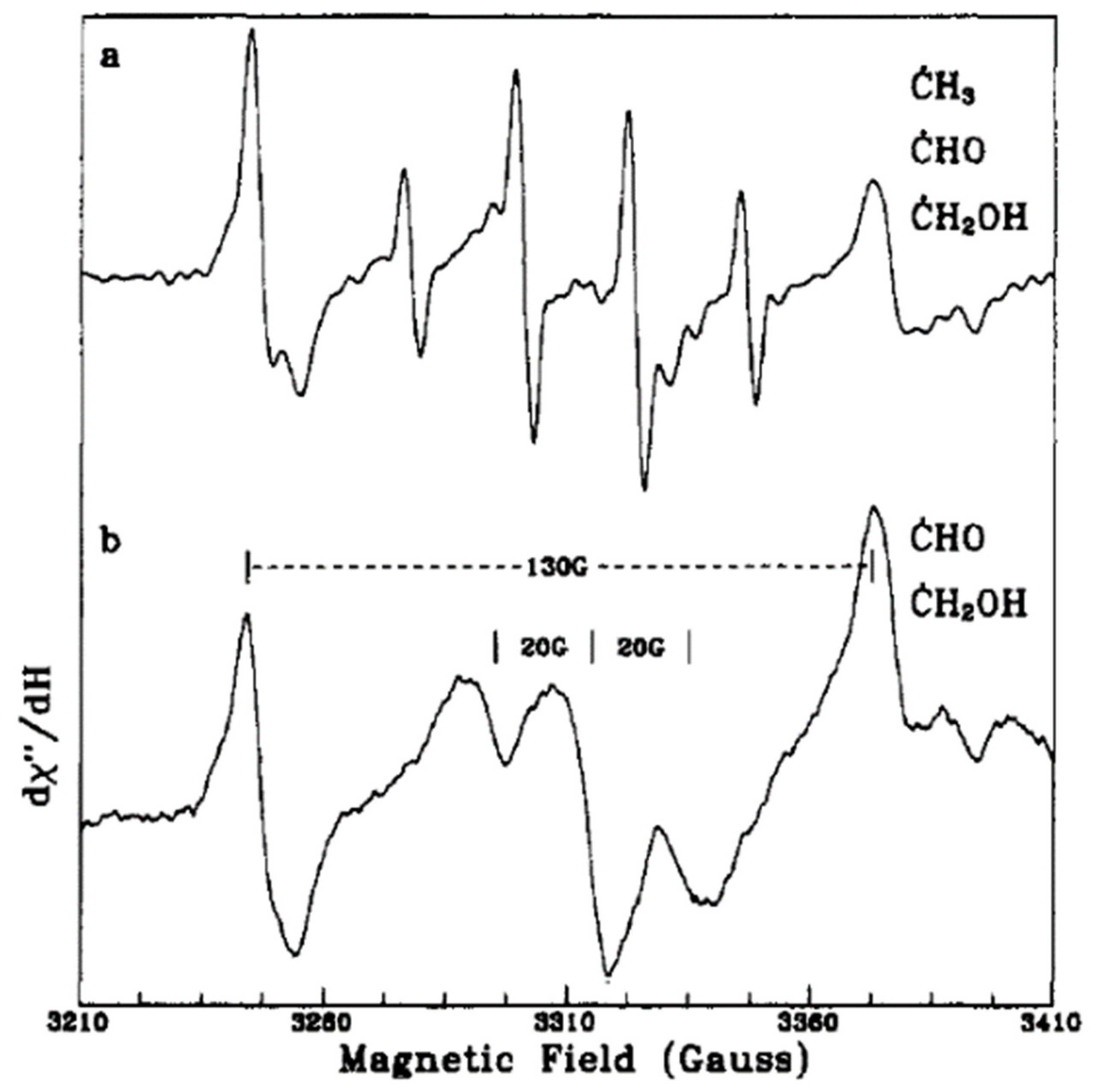
In another work, Micic et al. [
57] presented an EPR study for the hole transfer from irradiated TiO
2 colloids to methanol in an aqueous solution. Upon the excitation of degassed solutions of alkoxide TiO
2 colloids at different temperatures, they noticed three different signals around g = 2. At high laser pulse intensities, a quartet EPR signal (1:3:3:1) and a doublet signal with a 130-G separation appeared at 4.2 K as shown in
Figure 4a and were attributed to the methyl (CH
3) and formyl (CHO) radicals (Equations (21)–(24)), respectively. However, at 60 K (
Figure 4b), the EPR signal of the methyl radical disappeared, while the other signal can be related to the methanol radical [CH
3O(H)]. The authors suggested possible pathways for radical formation as per, Equations (25) and (26). On the other hand, the EPR signal for trapped holes on hydrous TiO
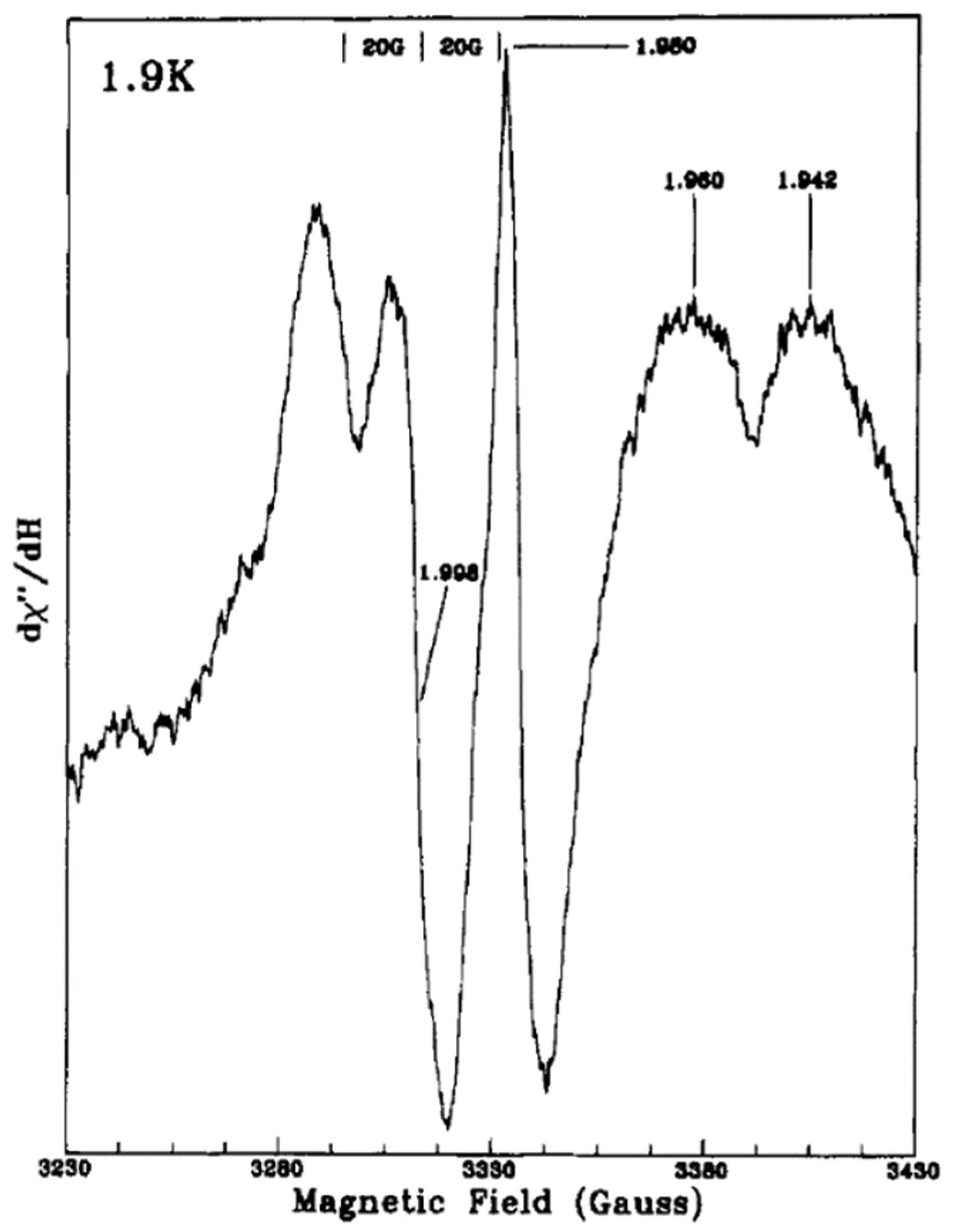
2 colloids showed resonances at g = 2.014 and g = 2.007 of O
●− radical with an additional signal at g = 1.989 of electron trapping in the bulk lattice. In this case, hydroxide ions on the surface can be the trap centers for photogenerated holes. Then they obtained at 1.9 K EPR signals of methanol radical (20-G splitting around g = 2) and Ti
3+ centers at (g = 1.980, g = 1.960, and g = 1.942) due to the addition of methanol to the aqueous TiO
2 solution as can be seen in
Figure 5, suggesting that
•CH
2OH radicals are the first hole trapping sites.
Coronado et al. [
58] investigated the surface characteristics of anatase TiO
2 under UV Irradiation using EPR spectroscopy. At room temperature, the EPR spectrum of amorphous TiO
2 gel showed a signal with g
1 = 2.023, g
2 = 2.009, and g
3 = 2.004, which was very close to those corresponding to Ti
4+-O
2− species. It showed a broader signal as well with g = 1.93 of Ti
3+ centers that were identical to those found in the spectra of colloidal TiO
2 after UV irradiation. On the other hand, the authors investigated the effect of the TiO
2 preparation method, i.e., hydrothermal and thermal, on EPR spectra. While the irradiated samples prepared by the hydrothermal method showed Ti
3+ centers, only weak signals associated with oxygenated radicals were observed for the samples prepared by the thermal method. They found that after UV irradiation of crystalized TiO
2 at 77 K under vacuum, the signal at g > 2.00 that corresponds to Ti
4+-O
2− species were overlapped. However, a narrow signal with
= 1.990 and g
II = 1.960 was observed, which could be related to the electrons stabilized in Ti cations located at crystallization defects or at surface sites (O
2−)
x-Ti
3+-(OH
−)
y. In another experiment, the authors examined the EPR spectra of irradiated samples after the adsorption of oxygen. As oxygen is considered as an electron scavenger competing effectively with recombination, a higher amount of radicals was formed in this case. The simulation of the spectra has revealed three or four overlapping signals according to the sample examined. They assigned the signal at g
1 = 2.024, g
2 = 2.013, and g
3 = 2.003 to photogenerated holes trapped by subsurface lattice oxygens, giving O
− species. They explained that the signal of Ti
3+ ions was located at the surface or within a few monolayers of the surface because of their rapid suppression upon the introduction of oxygen. Interestingly, Paranelli and coauthors have clarified the hole trapping sites’ nature in the anatase polymorph by coupling CW-EPR and ENDOR (pulse electron-nuclear double resonance) techniques [
59]. In their studies, the surface and subsurface holes in anatase were discriminated, in which the interaction of surface trapped holes with surface adsorbed water molecules was first measured using ENDOR. Water molecules play an important role in stabilizing the surface hole centers, where a complex environment composed by both surface hydroxyl groups and less close physisorbed water molecules are necessary.
Chiesa et al. [
39] used CW-EPR spectroscopy to study charge carrier trapping in anatase and rutile TiO
2 polymorphs, in which they irradiated fully dehydrated samples kept under vacuum. At 77 K, photogenerated electrons gave an EPR signal at g
II = 1.962 and g
I = 1.992, which had been assigned to trapped electrons on lattice titanium ions. However, they clarified that the trapped hole signal can be related to at least two akin species with similar but distinct parameters. These holes were trapped at the surface since they reacted with molecular hydrogen acting as a hole scavenger. One dominant species of trapped holes appeared at g
1 = 2.027, g
2 = 2.015, g
3 = 2.003.
Yan et al. [
60] studied the photoinduced electron-trapping states of anatase TiO
2 nanoparticles in two anaerobic systems, i.e., a proton-free system consisting of iodide ions in acetonitrile as a hole scavenger, and a protonated system consisting of methanol as a hole scavenger. Under UV illumination TiO
2 nanoparticles in the methanolic system exhibited a broad visible absorption peak at 725 nm and blue color of the electron-trapping state on TiO
2 nanoparticles (
Figure 6a). In contrast, low absorption from 400 to 600 nm was observed in the LiI/MeCN system related to the absorption spectra of I
3− that was formed due to the oxidation of I
− by photoinduced holes (
Figure 6b). EPR spectroscopy was then used to detect
●O
2− radicals produced under UV illumination from the reaction between O
2 and TiO
2 nanoparticles in different systems (O
2 + e
CB− →
●O
2−). The authors only detected
●O
2− signals in the methanol system, indicating that photoinduced electrons under the proton-free condition are not able to react with O
2 to produce
●O
2− radicals. They used then low-temperature (4 K) EPR spectroscopy to distinguish the different photoinduced electron-trapping states on TiO
2 nanoparticles. As shown in
Figure 6c, a rapid increase of a signal at g = 1.979 in systems containing methanol, ethanol, or acetonitrile was observed, in addition to a weak signal at g = 1.930 that appeared after 4 h of irradiation. While the first signal can be attributed to surface distorted four-coordinate tetrahedral Ti
4c3+ species, the latter can be assigned to anatase surface six-coordinate octahedral Ti
6c3+ species. The authors, thus, explained that the use of different electron donors in the presence of protons allowed the photoinduced electrons to be trapped mainly as surface Ti
4c3+ species with some surface Ti
6c3+ species. However, in LiI/MeCN proton-free electron donor system (
Figure 6d), only an increasing peak at g
┴ = 1.990 corresponds to the anatase lattice octahedral Ti
6c3+ state was observed. The authors, thus, explained that in the absence of protons, photoinduced electrons are trapped as interior interstitial Ti
6c3+ species in the lattice of the TiO
2 nanoparticles. The authors have finally concluded that the visible absorption signature at around 700 nm is dependent on the location of the trapped electrons, which can be controlled by protons’ content. Hence, the presence of protons is required to induce the conventional blue coloration of TiO
2 in order to stabilize trapped electrons as surface Ti
4c3+ species.
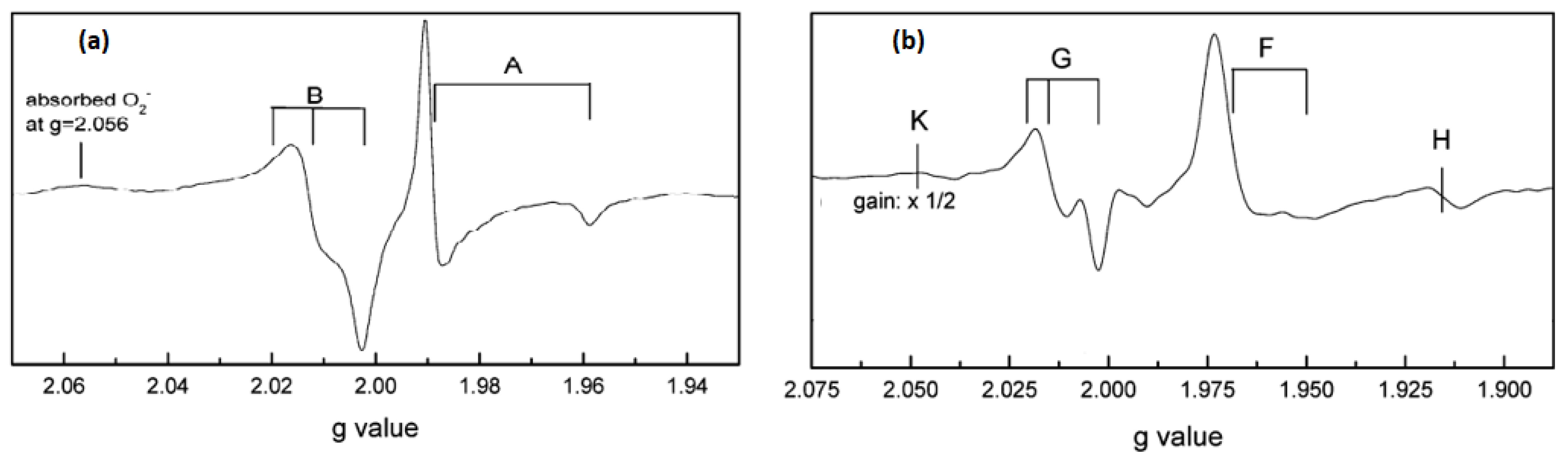
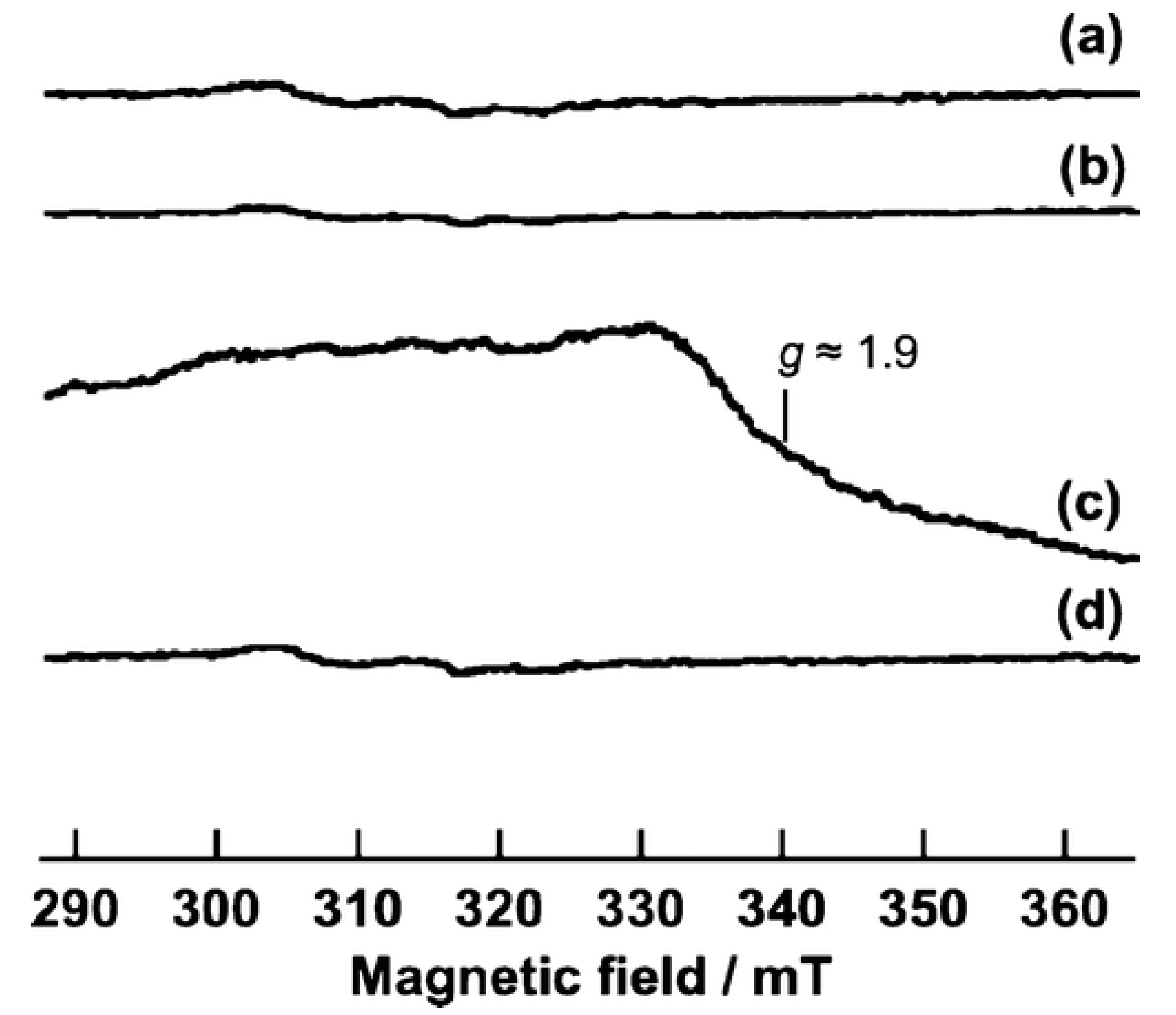
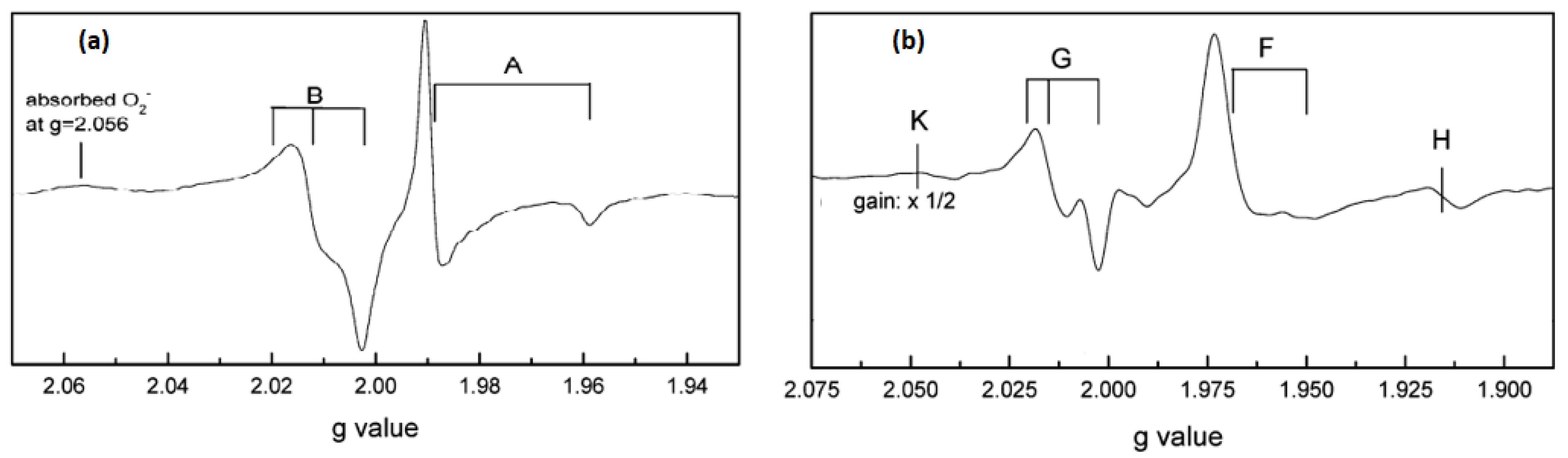
Kumar et al. [
61] studied in situ EPR for anatase (Hombikat UV100) and rutile TiO
2 nanoparticles under UV irradiation in air and vacuum conditions. The measurements were done at liquid helium (He) at a temperature of (4.2 K). EPR spectrum of anatase TiO
2 (
Figure 7a) showed two signals, i. e., trapped electrons (signal A) at g
1 = 2.016, g
2 = 2.012, g
3 = 2.002 and g
| = 1.958, g
⊥ = 1.988, and trapped holes (signal B) at g
1 = 2.016, g
2 = 2.012 and g
3 = 2.002. The authors assigned the sharp signal at g
⊥ and g
| to surface electron trapping sites, while the latter signal was related to photogenerated holes trapped at the lattice oxygen atoms located in the subsurface layer as Ti
4+O
•−Ti
4+OH
− radical. Based on their observations, they suggested a charge-transfer mechanism in anatase TiO
2. On the other hand, EPR signals of rutile TiO
2 illuminated at 4.2 K (
Figure 7b) were characterized by two sets of g values, g
⊥ = 1.969, g
| = 1.947 (signal F) and g
1 = 2.019, g
2 = 2.014, g
3 = 2.002 (signal G). The authors have assigned signals F and G to the surface electron traps as Ti
3+ and rutile hole trapping sites Ti
4+O
•−Ti
4+OH, respectively. They noticed also a third signal (H) at g = 1.91 that was related to vacancy-stabilized Ti
3+ in the lattice sites. The authors noticed also that the trapped electrons generated by irradiation at 4.2 K in anatase and rutile were stable below 85 K and vanished upon increasing temperature, while trapped holes are more stable up to ∼150 K. Nevertheless, heating to ambient temperature showed the disappearance of hole trapping sites due to electron-hole recombination.
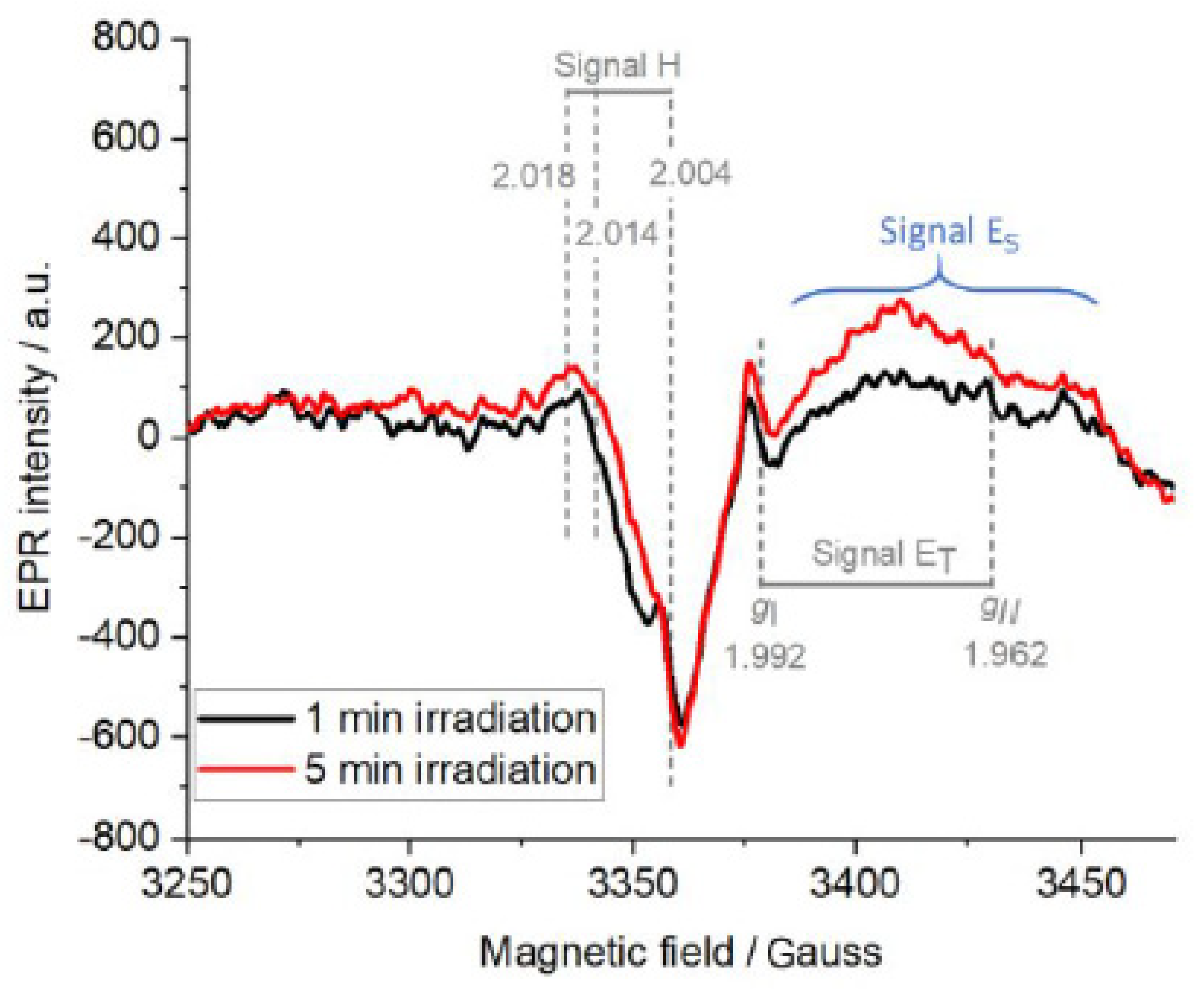
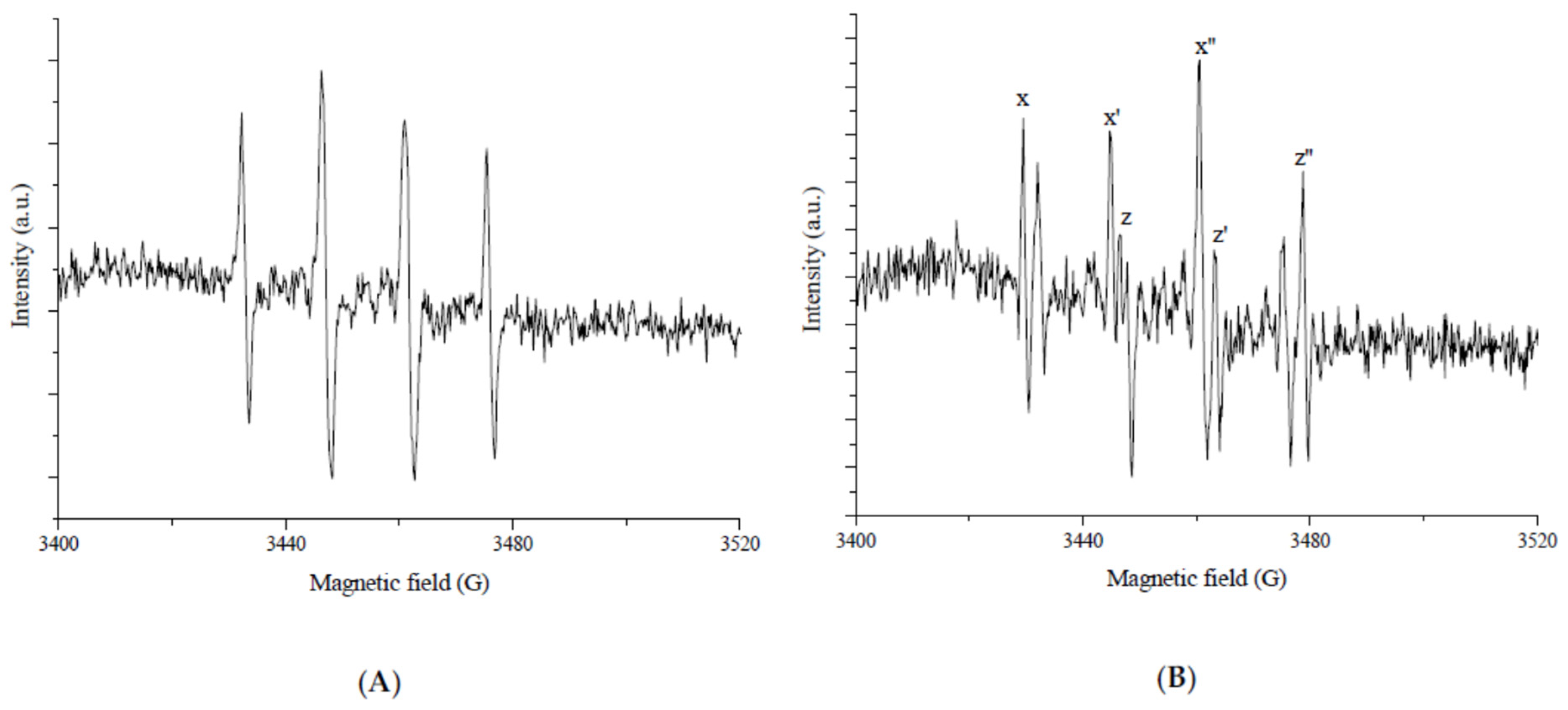
In order to investigate the role of platinum as an electron scavenger on the surface of TiO
2, AlSalka et al. [
21] investigated the paramagnetic centers produced upon the irradiation of self-prepared Pt/TiO
2 anatase powders via in situ EPR spectroscopy under an argon atmosphere at 77 K. In the presence of pre-adsorbed oxalic acid as a hole scavenger, the EPR spectra of irradiated Pt/TiO
2 showed two signals (
Figure 8), weak signal H (g ˃ 2) of trapped holes and signal ET (g ˂ 2) of trapped electrons.
The authors assigned signal H (g
x = 2.004, g
y = 2.014, g
z = 2.018) to the photogenerated holes trapped near the hydrated anatase surface at the oxygen atoms in the form of Ti
4+-O
−•-Ti
4+-OH-. In addition, they attributed signal ET (g
II = 1.962 and g
⊥ = 1.992) to the photogenerated electrons trapped in the bulk as Ti
3+ ions. The authors clarified that the appearance of such a signal of trapped electrons even in the presence of platinum nanoparticles attached to the surface of TiO
2 can only be interpreted as not all the photogenerated electrons were scavenged by the loaded Pt NPs. Hence, not all the electrons can be used, thereafter, in reduction reactions, i.e., the hydrogen evolution reaction. Interestingly, the authors noticed another broad and unfeatured signal (ES) centered at 3450 gausses with increasing intensity over the irradiation time. They have explained this signal to the injection of electrons from
•CO
2− radicals formed through the oxidation of oxalic acid by photogenerated holes, which can be then spread on the surface of TiO
2. Such a signal was also observed by Chiesa et al. [
39] due to the excess electrons near the surface of anatase TiO
2 through the injection of electrons in the solid. Micic et al. [
57] have also reported a broad asymmetric EPR signal at g = 1.981 of trapped electrons on the surface due to electron injection from methanol radicals into TiO
2 particles.
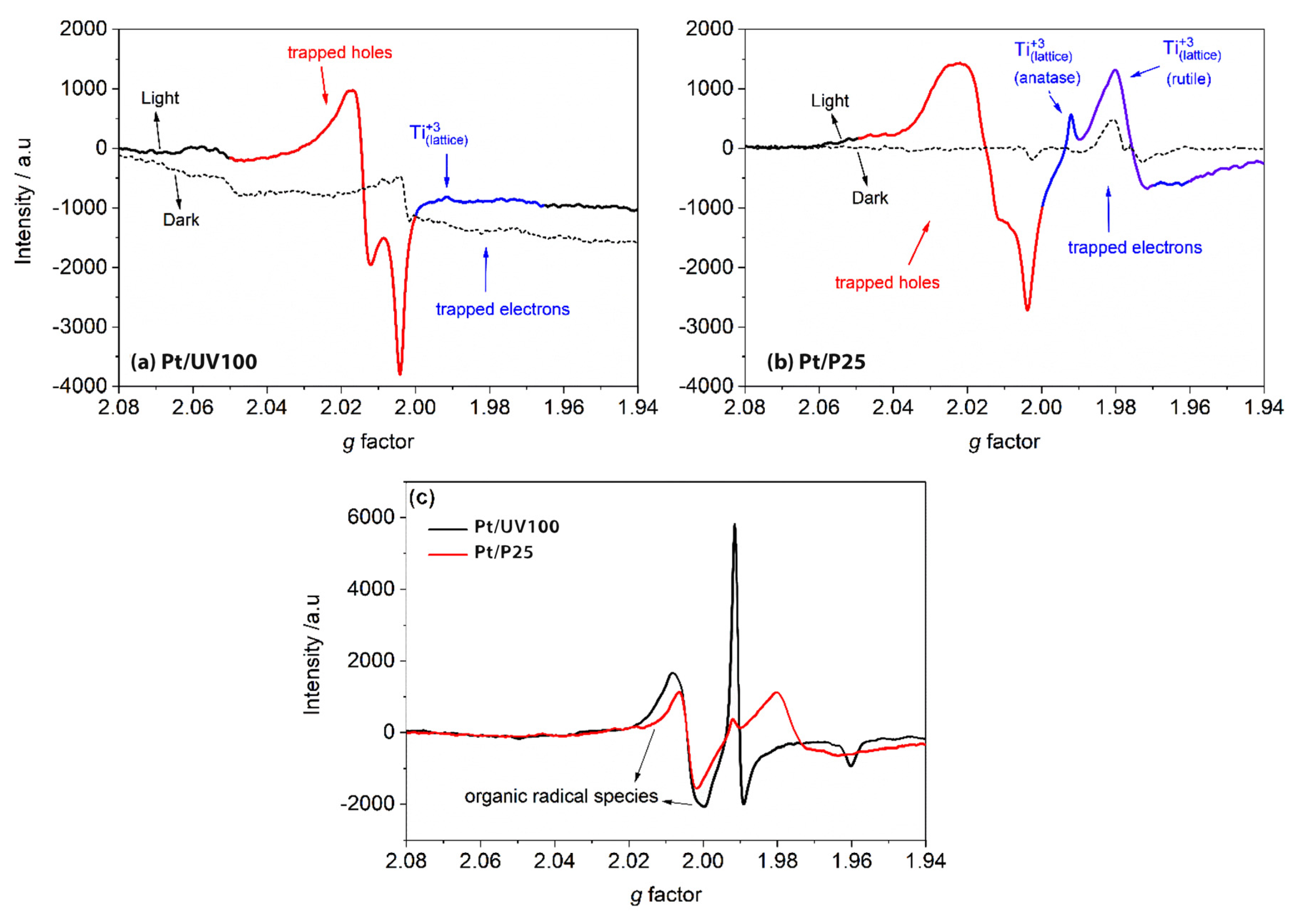
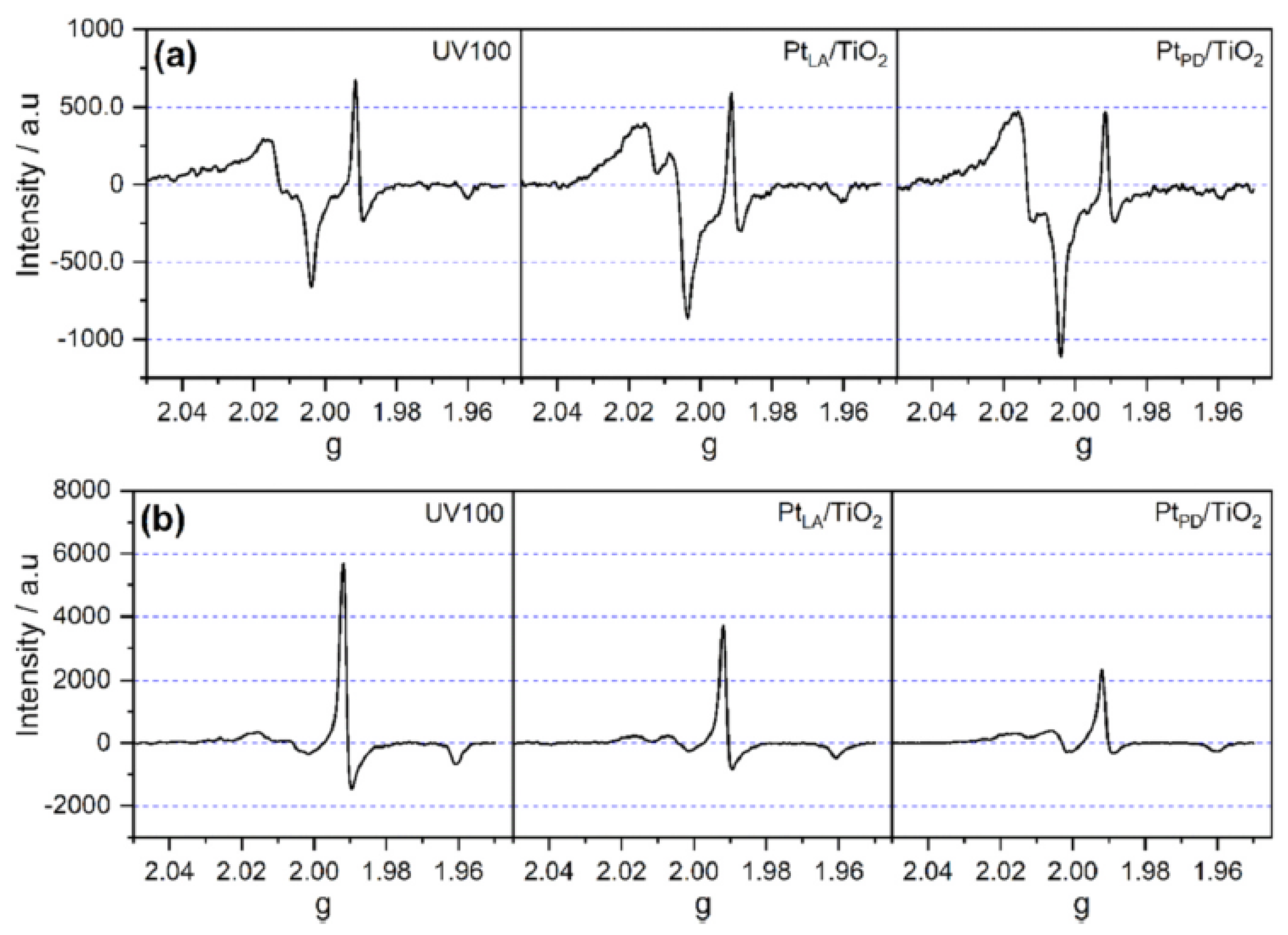
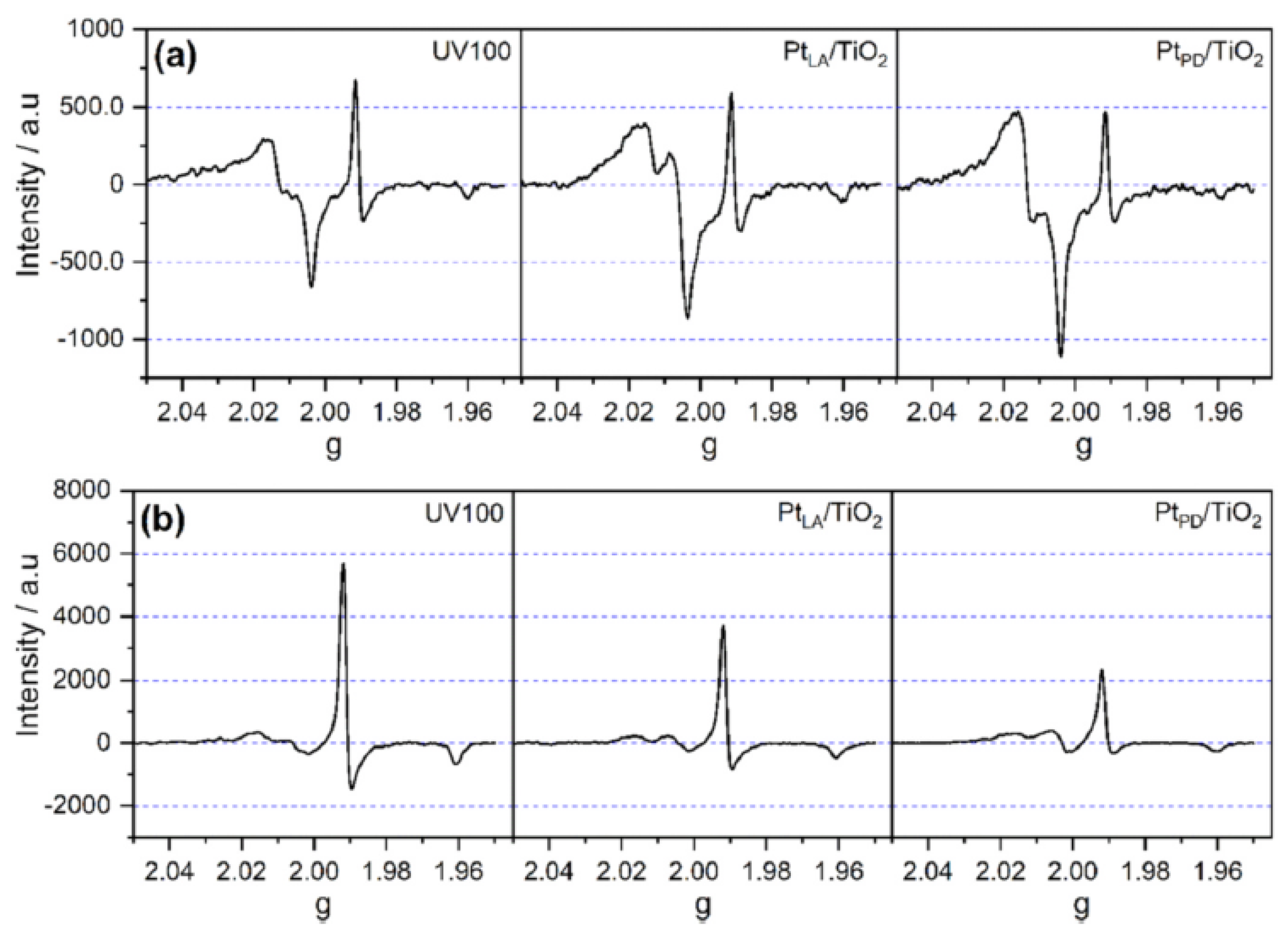
Al-Madanat et al. [
62] compared the EPR spectra of 1.0 wt% Pt-loaded P25 and UV100 TiO
2 in the N
2 atmosphere to investigate the effect of the photocatalyst nature and PtNPs on the photogenerated electron and hole paramagnetic species. Upon illumination, both materials showed two main features shown in
Figure 9a,b, i.e., the signals at g > 2.00 of the surface trapped holes and the signals at g < 2.00 of the trapped electrons. They explained that Pt-loaded UV100 showed the hole trapping site at g-tensor components g
x = 2.004, g
y = 2.015, and g
z = 2.019 that corresponds to the anatase oxygen site, while the small-signal at g
⊥ = 1.992 and the shoulder at g
II = 1.965 were related to the anatase lattice trapping electron site as Ti
+3. On the other hand, the signals of trapped holes in Pt-loaded P25 TiO
2 having g-tensor components g
x = 2.003, g
y = 2.019, and g
z = 2.026 were not completely resolved, considering a combination of trapped oxygen sites from anatase and rutile. However, the Ti
+3 signal from anatase can be observed clearly at the same g-tensor components assigned for Pt-loaded UV100. Moreover, two other signals at g
⊥ = 1.980 and gII = 1.945 were attributed to Ti
+3 sites for trapped electrons in the rutile lattice. The authors showed that around 20% higher relative intensity of trapped holes was noticed in Pt-loaded UV100, while Pt-loaded P25 had more intense signals of both trapped Ti
+3 sites. In this case and because no hole scavenger was used, the authors believed that UV100 exhibited better electron transfer to Pt islands than P25, inhibiting the charge carriers’ recombination and increasing the photocatalytic activity.
The authors have then made the experiment in the presence of naphthalene to investigate the naphthalene radical cation generated through the reaction via single-electron transfer with photogenerated holes. Both materials as can be seen in
Figure 9c produced similar EPR signals at g
x = 2.002, g
y = 2.006. However, Pt-loaded UV100 produced relatively higher signals of organic radicals and trapped electrons, which could be due to its better charge carrier separation.
Al-Madanat et al. [
63] have also investigated the effect of platinum loading method on TiO
2, i.e., through photodeposition or physical mixing after the laser ablation, on the e
─/h
+ populations under irradiation in N
2 or N
2-methanol atmosphere using EPR spectroscopy and commercial TiO
2 (anatase UV100). As shown in
Figure 10a, upon illumination, two groups of signals were recorded for all the materials used, the first at (g
x = 2.004, g
y = 2.015, and g
z = 2.019) and the second at (g
‖ = 1.992 and g
⊥ = 1.961) and (g
‖ = 1.961 and g
⊥ = 1.94–1.93), which were assigned to trapped holes at or near the surface and trapped electrons in the bulk and at the surface, respectively. In an inert atmosphere, Pt/TiO
2 prepared via the photodeposition method showed the strongest h
+ signal and the weakest e
─ signal, indicating a better electron transfer to Pt than in the other samples. However, this was also evidence of the non-complete scavenging of electrons by Pt. In the N
2-methanol atmosphere (
Figure 10b), a triple signal of the
•CH
2OH radical was produced with a simultaneous appearance of stronger signals of the trapped electrons attributed to the current doubling effect. Pt/TiO
2 prepared via the photodeposition showed again the smallest electron signal, confirming the best charge carrier separation among other samples, and thus the highest photocatalytic activity.
3. Niobium(V) Oxide Photocatalyst
Apart from the TiO
2-based photocatalysts, Nb
2O
5 is also a widely used semiconductor oxide due to its properties such as thermal and chemical stability, and appropriate electronic and morphological properties for photocatalytic applications [
64,
65]. Nb
2O
5 is a polymorphic material with different crystalline phases related to the preparation conditions, with their structures formed by different arrangements of NbO
6 octahedra [
66]. Its surface contains strong redox ability and both Lewis (LASs) and Brønsted acid sites (BASs), offering rich surface chemistry. In addition, the values of Nb
2O
5 bandgap energy are found to be between 3.0 and 3.4 eV, being suitable for photocatalytic reactions [
67]. Similar to TiO
2, under excitation by suitable wavelength energy, an electron (e
−) is promoted to the conduction band (CB) while a hole (h
+) is formed in the valence band (VB). Nb
2O
5 has not been yet so deeply investigated by EPR but, in general, it does not present so many characteristic signals as observed in the literature for TiO
2. Nevertheless, the EPR technique can also bring up much insights about the processes on the surface of the photocatalyst. Some reported works have shown the presence of new states as Nb(IV) centers or oxygen vacancies by using EPR measurements on doped Nb
2O
5 [
68,
69]
. Besides, Nb
2O
5 photocatalyst can also be combined with other materials, including TiO
2, to promote better efficiencies. One work regarding the heterojunction of TiO
2 and Nb
2O
5 was reported by Li and coworkers [
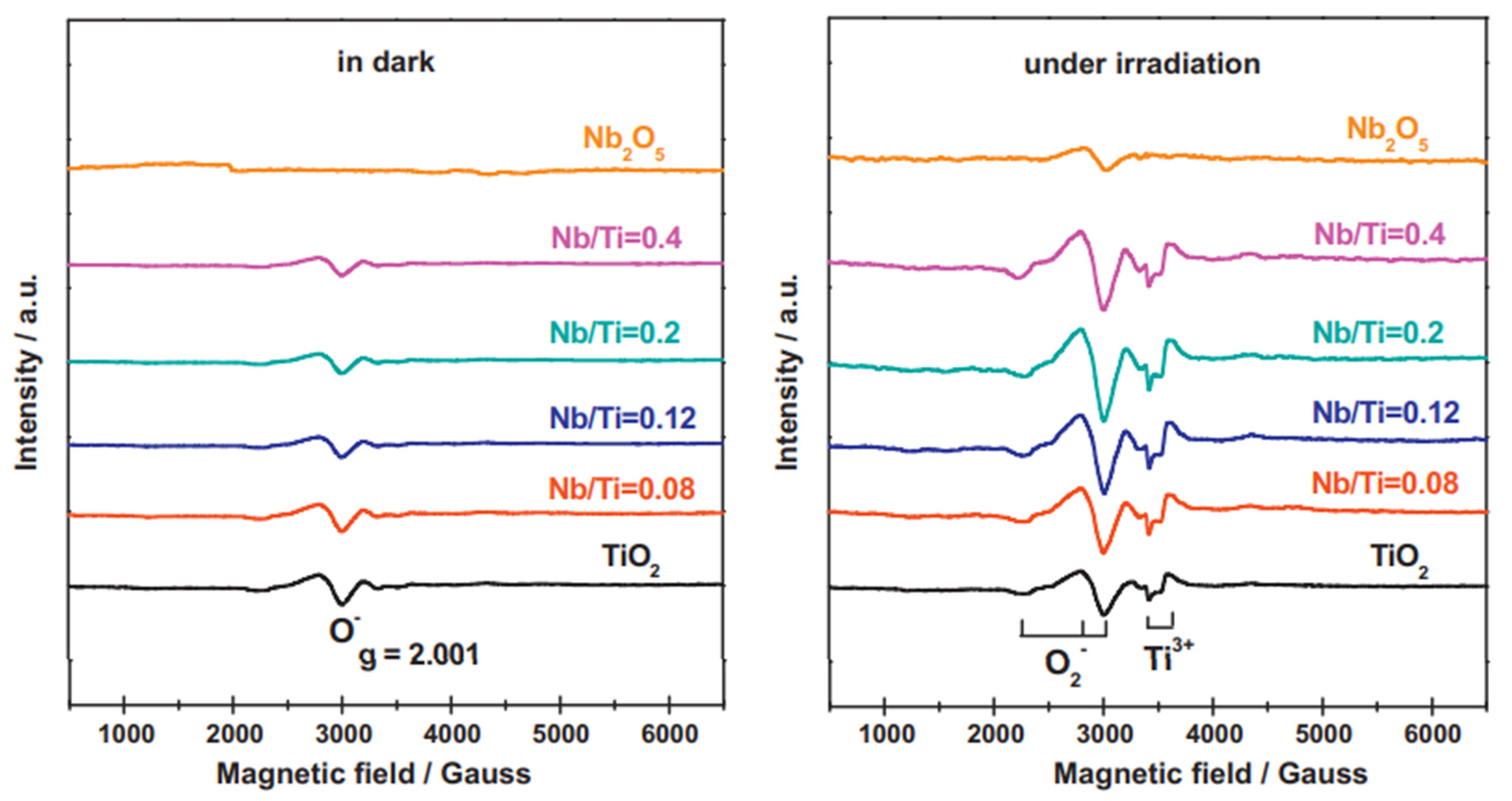
70]. The authors prepared ultra-fine niobium oxide nanoparticles on the surface of rutile TiO
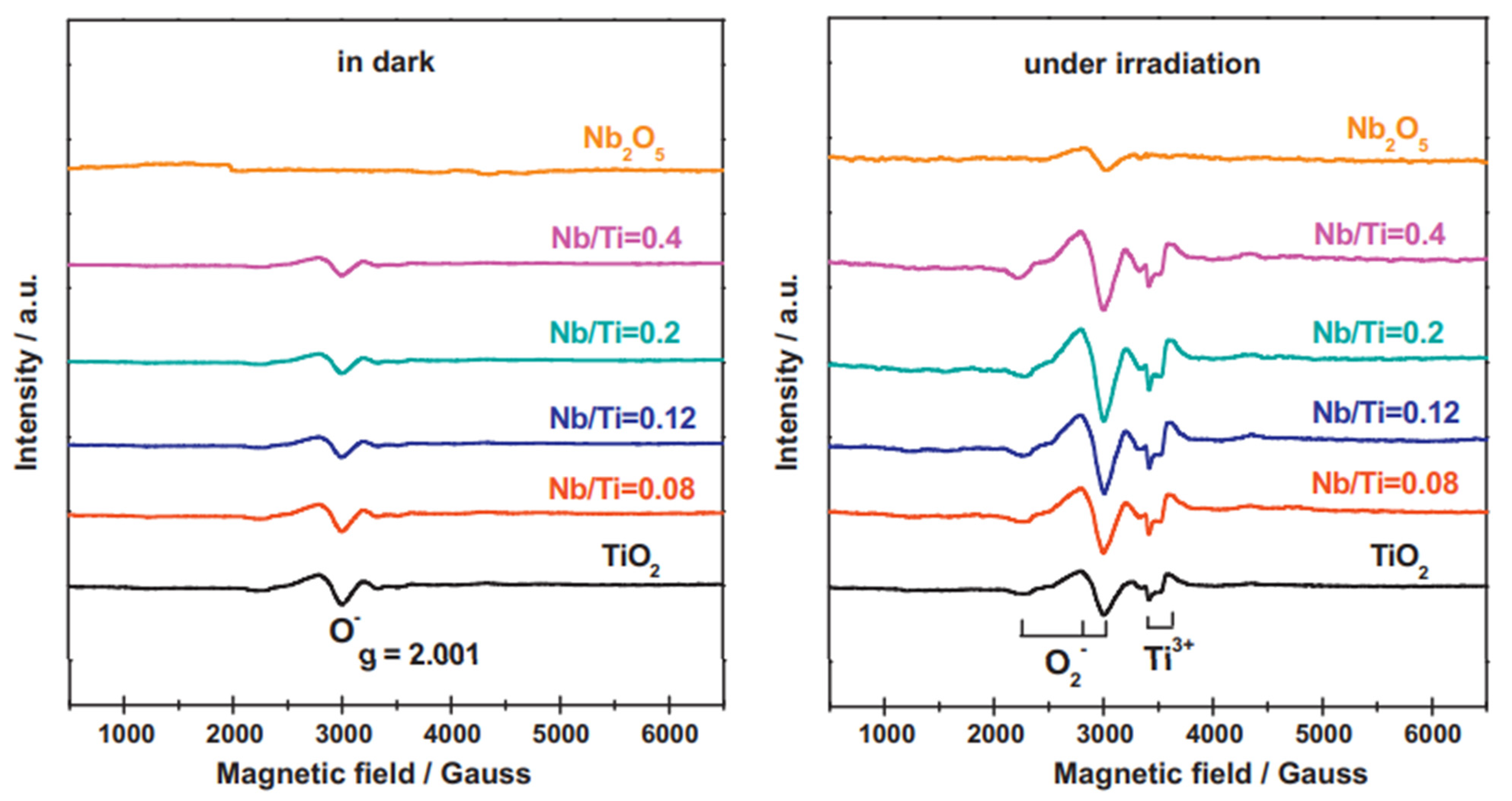
2 and evaluated their different proportions and activities for photocatalytic oxidation of phenylethanol and methanol photoreforming. They investigated the prepared materials employing EPR (
Figure 11). Under dark conditions, no signal was detected for pure Nb
2O
5 but one weak peak (g = 2.001) was observed for pure rutile TiO
2 and Nb
2O
5/TiO
2 composites which attributed to O
− formed from O
2 onto the surface. In addition, when the proportion of Nb
2O
5 in the Nb
2O
5/TiO
2 composites was increased, the intensity of this signal decreased, indicating that Nb
2O
5masked the defect sites of TiO
2, which seemed to work as nucleation centers for niobium oxide. When the samples were exposed to UV-vis irradiation, typical EPR signals assigned to superoxide species (g
1 = 2.002, g
2 = 2.009, g
3 = 2.028) were identified. The authors reported that more superoxide species were produced on Nb
2O
5/TiO
2 heterojunctions. Furthermore, two signals corresponding to the bulk Ti
3+ species (g = 1.982 and 1.978) were also found higher for the heterojunction. These facts evidence a better charge separation efficiency for the composite’s materials.
The group of Tanaka and coworkers performed many EPR studies towards the surface of Nb
2O
5 when applied for organics photooxidation. Firstly, they explore the photooxidation of alcohols at low temperatures and in organic solvent-free conditions by using niobium oxide in an oxygen atmosphere [
71,
72]. The prepared Nb
2O
5 was identified as TT phase (a kind of Nb
2O
5 phase) and a pseudohexagonal structure. A remarkable result observed by the author was the photooxidation of 1-pentanol under irradiation up to 480 nm, even though bare Nb
2O
5 is not able to absorb wavelengths higher than 390 nm. This fact could indicate that the photooxidation occurred by a different pathway rather than the expected electron transfer from the excited semiconductor. Based on EPR investigations, in
Figure 12, the mechanism for 1-pentanol photooxidation was then clarified. Firstly, when Nb
2O
5 was irradiated in presence of an excess of 1-pentanol (123 K), a broad ESR signal assigned to Nb
4+ (g = 1.9) was observed (
Figure 12c). In the presence of O
2, this signal disappeared, indicating that Nb
4+ was oxidized to Nb
5+ (
Figure 12d). Other EPR analyses showed a signal related to alkenyl radical (g = 2.006, A
H1 = 2.0 mT, A
H2 = 4.4 mT) which was not significantly affected by the addition of O
2. Besides, no oxygen anion radicals were detected, supporting the selectivity of the reaction. The mechanism of 1-pentanol to produce carbonyl compound was correlated to an electron transfer from the absorbed alcoholate to Nb
2O
5 forming Nb
4+ centers. The alcoholate was further converted to the carbonyl compound and the Nb
4+ sites are reoxidized to Nb
5+ by the molecular oxygen.
Similarly, the authors further studied the partial photooxidation of hydrocarbons by Nb
2O
5 and also compared its efficiency with TiO
2 [
73]. In principle, Nb
2O
5 showed higher selectivity than TiO
2 for some hydrocarbons such as cyclohexane and ethylbenzene. At this time, no significant activity was observed under visible light irradiation showing that the photooxidation mechanism involves the Nb
2O
5 excitation. The EPR analysis (77 K) at first showed the formation of O
2− species from absorbed O
2 by excited electrons on the oxide. Then, when ethylbenzene was adsorbed on Nb
2O
5 and exposed to irradiation, signals related both to ethylbenzyl radical (g = 2.003, A
H1 = 6.0 mT, A
H2 = 2.0 mT) and Nb
4+ centers (g = 1.933) were observed. These results could support the proposed mechanism in which the benzylic C–H bond of ethylbenzene was oxidized by the photogenerated hole while the excited electrons formed the Nb
4+ centers. When the sample was exposed to O
2, the signal for ethylbenzyl radical vanished and the oxygen radical was detected, indicating the formation of diamagnetic alkyl peroxide anion (ROO
−) or hydroperoxide (ROOH). Along with other analyses, the suggested mechanism was based on the oxidation of alkyl radical to alkyl hydroperoxide and then, after protonation, the alkyl hydroperoxide produced ketone and water. The higher selectivity found for Nb
2O
5 was related to the lower amount of oxygen radicals and also to the absence of O
3− species which are strong oxidizing radicals when compared to TiO
2.
The group also showed an effective photoactivity by LMCT (ligand to metal charge transfer) transitions between small aromatic hydrocarbons and the surface of Nb
2O
5 [
74]. The material was able to selectively photooxidative hydrocarbons to carbonyl compounds under visible light irradiation. In this case, a pre-treatment was performed; the catalyst powder was first evacuated and then heated (573 K) in a reactor in situ, which showed enhanced activity for toluene photooxidation with high selectivity. In principle, the treatment did not affect the Nb
2O
5 structure as the surface area, crystal phase, and crystallinity, and just a very slight change was observed in the UV-vis spectra. In fact, the enhanced photoactivity was explained by the toluene adsorption on treated Nb
2O
5 which generated an additional visible light absorption. This transition was related to an LMCT from toluene to Nb
5+. Interestingly, the same phenomenon was not observed for TiO
2 even when the pre-treatment was applied. In the case of toluene as an electron donor, it was assumed that Nb
2O
5 had a higher affinity to the electrons in comparison to TiO
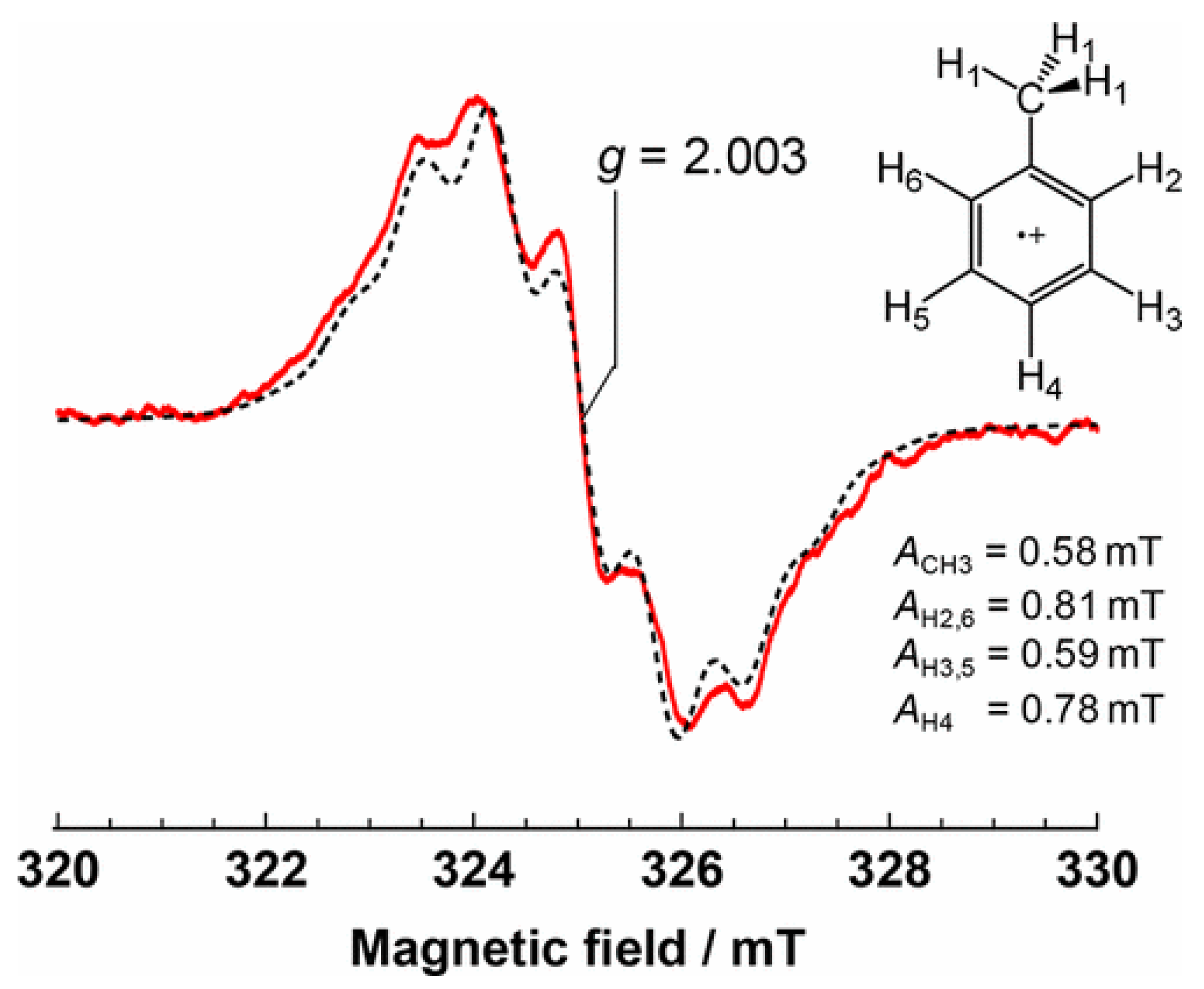
2. In the EPR measurements (77 K), the treated Nb
2O
5 in absence of toluene did not show any signal also under visible light irradiation. In contrast, when toluene was absorbed on Nb
2O
5 surface, a EPR signal was observed and assigned to toluene radical cation (g = 2.003, A
CH3 = 0.58 mT, A
H2,6 = 0.81 mT, A
H3,5 = 0.59 mT, and A
H4 = 0.78 mT), which was confirmed by a simulated spectrum as shown in
Figure 13. At this time, the authors did not observe the signal related to Nb
4+ formation. They suggested that the excited electrons would be delocalized in the conduction band of the oxide. When the sample was exposed to O
2 in the dark, the intensity of the toluene radical signal did not change; evidencing that O
2 itself could not oxidize toluene. Thus, the mechanism was based on the formation of superoxide radicals from the excited Nb
2O
5, which provoked the cleavage of the benzyl C−H bond of the toluene radical cation to form benzyl (Ph−CH
2•) and hydroperoxyl (
•OOH) radicals. In sequence, the benzyl radical was converted to benzylperoxy radical by the intercalation of adsorbed O
2. The main product of the toluene photooxidation was found to be benzaldehyde.
Other organic compounds also investigated by Tanaka’s group for selective photooxidation over Nb
2O
5 were amines [
75]. Similar to the previous works, the reaction to produce imines was active under visible light irradiation. This time, the mechanism was related to the adsorbed amine on Nb
2O
5 which gave rise to a donor level localized in the nitrogen from the amine species. In the EPR analysis (77 K), under visible light irradiation, the spectrum of Nb
2O
5 in presence of butylamine showed signals related to both the organic radical (g = 2.005) and Nb(IV) (g ∼ 1.92). Based on this result and some DFT calculations, it was explained that the photoactivation of the surface complex formed by the amine and Nb
2O
5 occurred via an electronic transfer from the N 2p orbital to the Nb 4d orbitals from the oxide conduction band. Thus, the amine oxidative dehydrogenation to form imine was based on the photoexcitation of the Nb
2O
5−amide surface complex which was active under visible light.
4. Spin Trapping in the Liquid Phase
After the pioneering works of Janzen et al. in 1969 [
76] and Lagercrantz in 1970 [
77], the free radical trapping technique “spin trapping” has been intensively introduced as a valuable tool in different fields, such as chemistry, physics, biology, and medicine for the detection and identification of the transient radicals formed in the reaction system [
78,
79,
80,
81,
82]. Since these formed radicals have a very short lifetime usually nanoseconds half-lives, the base of this method is to add a new diamagnetic reagent; spin trap agent, to the reaction medium which reacts with these radicals producing a new and more stable persistent paramagnetic species “spin adduct” that can be registered, qualified and quantified by the electron paramagnetic spectroscopy (EPR),
Scheme 5 [
76,
83].
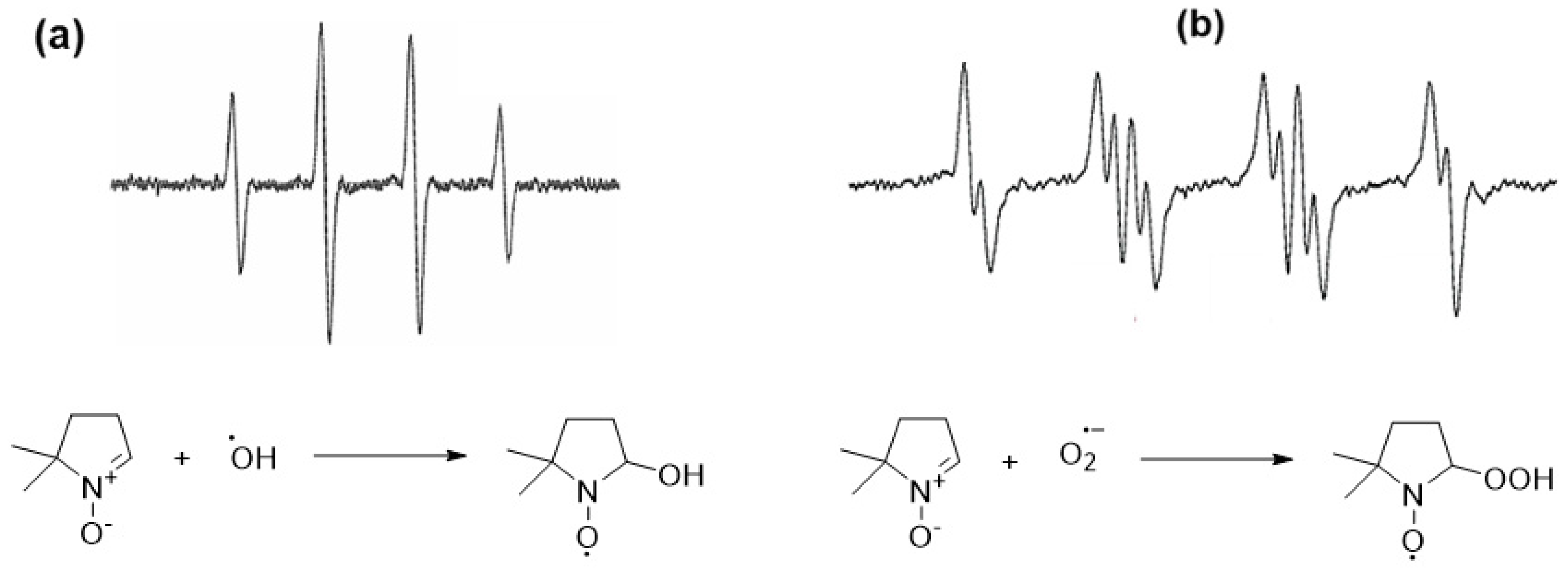
Nitroso and nitrone derivatives have been widely used as the main types of spin trapping agents (
Chart 1). The former is characterized by the faster trapping rate and trapping the carbon-center radical, while, the latter is more suitable for trapping the oxygen-centered radicals [
84,
85]. However, due to the limitations of the linear nitrone to facilitate the identification of the trapped radicals, new spin traps based on pyrroline N-oxide derivatives have been developed [
85,
86].
Recently, the spin trapping technique is widely employed in the photocatalytic reaction as a valuable tool to study the reaction mechanism [
21,
87]. Among several spin traps agents, such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), 4-hydroxy-5,5-dimethyl-2- trifluoromethylpyrroline-1-oxide (FDMPO), α-(4-pyridyl-1-oxide)-
N-
tert-butyl nitrone (POBN), and N-tert-butyl-a-phenylnitron (PBN), 5,5-Dimethyl-1-pyrrolin-N-oxid (DMPO) is the most used spin trap due to the well resolved and characteristic EPR spectra with the superoxide and hydroxyl radicals [
10,
86,
87,
88].
Figure 14 shows the EPR spectra for the DMPO-OH and DMPO-O
2 adducts after the reaction of the hydroxyl radicals and superoxide with DMPO under irradiation.
In the photocatalytic process over TiO
2, it has been frequently claimed that the hydroxyl radical is produced during the irradiation of an aqueous TiO
2 suspension through the decomposition of the water molecule by the photogenerated hole (Equations (27) and (28)). The hydroxyl radical was introduced as the most important reactive species “oxidant” generated during the photocatalysis process.
Besides, in the presence of molecular oxygen (O
2) in the photocatalytic system, it is likely that the adsorbed O
2 on TiO
2 surfaces will react with the conduction band photogenerated electron to produce superoxide radicals (perhydroxyl radical), according to Equations (29) and (30).
Jaeger and brad [
90] employed the spin trapping technique to detect the formed free radical species in situ during the irradiation of the aqueous suspension of TiO
2 and Pt-TiO
2 (anatase) in the presence of the spin traps PBN or POBN at room temperature. The authors did not observe any EPR signal in the dark, however, immediately after the illumination of the TiO
2 suspension with a light (>3.2 eV), the EPR signal was observed in the presence of both spin traps. The authors attributed the signal in the PBN\TiO
2 system to the formation of 4 different paramagnetic species. Two of them was assigned to the formation of the
and
(perhydroxyl radical). Although the authors are not certain from the origin and kind of the others two species, they are suggested that these species are formed due to the decomposition of t+he spin trap. Moreover, by using the spin trap POBN at different pH (4, and 7), they confirmed the formation of the
and
species. On the other hand, using the pristine TiO
2 instead of the Pt-TiO
2 leads to the formation the
and
, however, their concentrations are lower than that in the case of Pt-TiO
2.
Several research groups have been explored the photocatalytic activity, as well as the photocatalytic mechanism involved in the presence of different photocatalysts employing the EPR spin trapping techniques [
19,
21,
86,
87,
88,
91]. Fu et al. [
92] investigated the photocatalytic degradation of 4-chlorophenol (4-CP) using N-doped TiO
2 prepared by the high-temperature nitridation of commercial P25 (Degussa) and undoped P25-TiO
2 annealed with N
2 in the same process as a reference sample, under UV and visible light (λ > 420 nm). The authors observed that P25-TiO
2 exhibited higher activity for 4-CP decomposition than that of N-doped TiO
2, while, under visible light irradiation, P25-TiO
2 did not exhibit photocatalytic activity for degradation of the 4-CP degradation, since it was not visible-light active material. The degradation of 4-CP, however, was observed from N-doped TiO
2, suggesting that N doping for TiO
2 is an effective approach for achieving visible-light-driven photocatalysis.
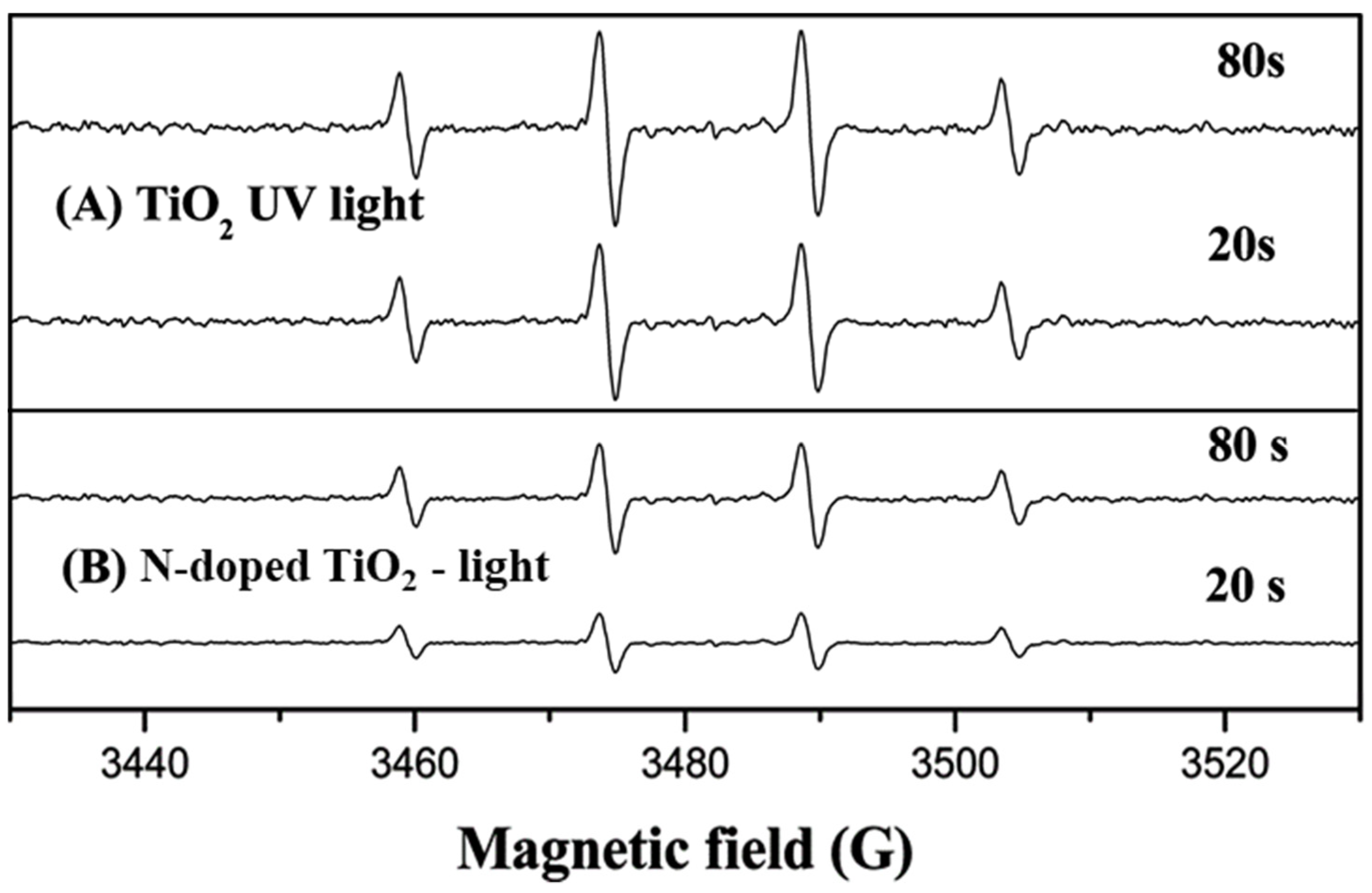
To explain the different activities between the studied materials the authors employ the ESR spin-trap technique using DMPO, to monitor the intermediate radicals and probe the nature of the reactive oxygen species generated during the UV and visible light irradiation in both systems. No EPR signals were observed by the authors in the dark in the presence of the catalysts or when 4-CP was absent (
Figure 15). Under UV irradiation, the signal of DMPO−OH adduct (quartet peaks with a 1:2:2:1 intensity) was observed after a 20 s of illumination, and its intensity further increased after 80 s of irradiation in the presence of both photocatalysts, however, the peak intensity of DMPO−OH adduct generated by N-doped TiO
2 was less than that of TiO
2, suggested that the lower production of
•OH.
In the previous experiments, although the formation of the superoxide radical (
) was expected through the scavenging of the conduction band electrons by O
2, the authors did not observe such signal. They explained that to the instability of this radical in the aqueous medium, where it reacts with the present proton (Equation (31)) to produce H
2O
2 and O
2 (
k = 6.6 × 10
3 M
−1 s
−1), due to the slow reactions between
•OOH/O
2•− and DMPO (
k = 10 M
−1 s
−1). Therefore, they performed the same experiment in an ethanolic medium to confirm the formation of DMPO-
/
, and they observed a similar trend to the
•OH production in both systems.
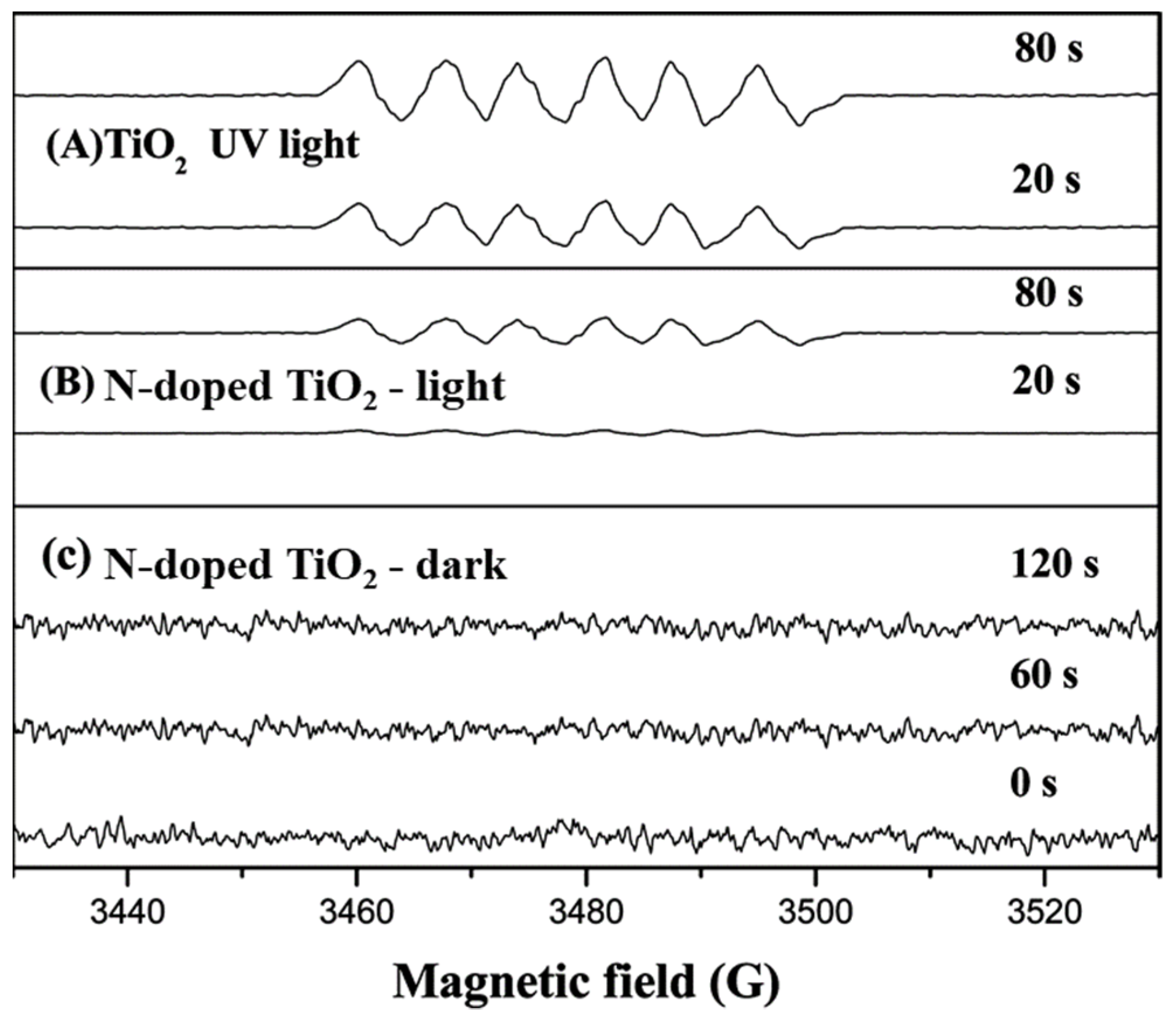
On the other hand, when 4-CP/N doped TiO
2 suspension in H
2O and ethanolic solution irradiated with visible light, both DMPO-OH and DMPO-OOH adducts were registered, respectively, and the EPR signal intensity was enhanced gradually with increasing illumination time (
Figure 16). However, the signal intensities were found weaker than those under UV light. Based on these results, the authors concluded that the photocatalytic degradation of 4-CP over N-doped TiO
2 under UV or visible light occurs in a similar mechanism, mainly by the radical reaction, which is similar to TiO
2 under UV light irradiation. According to the EPR results, the authors considering that the formation of DMPO-OH/OOH adducts give direct evidence that (
•OH and O
2•−) are the main active species responsible for the photodegradation of 4-CP, strongly suggesting that the photocatalytic reaction of organic compounds over N-doped TiO
2 proceed via surface intermediates of oxygen reduction or water oxidation (indirect path), not via direct reactions with holes trapped at the N-induced midgap level.
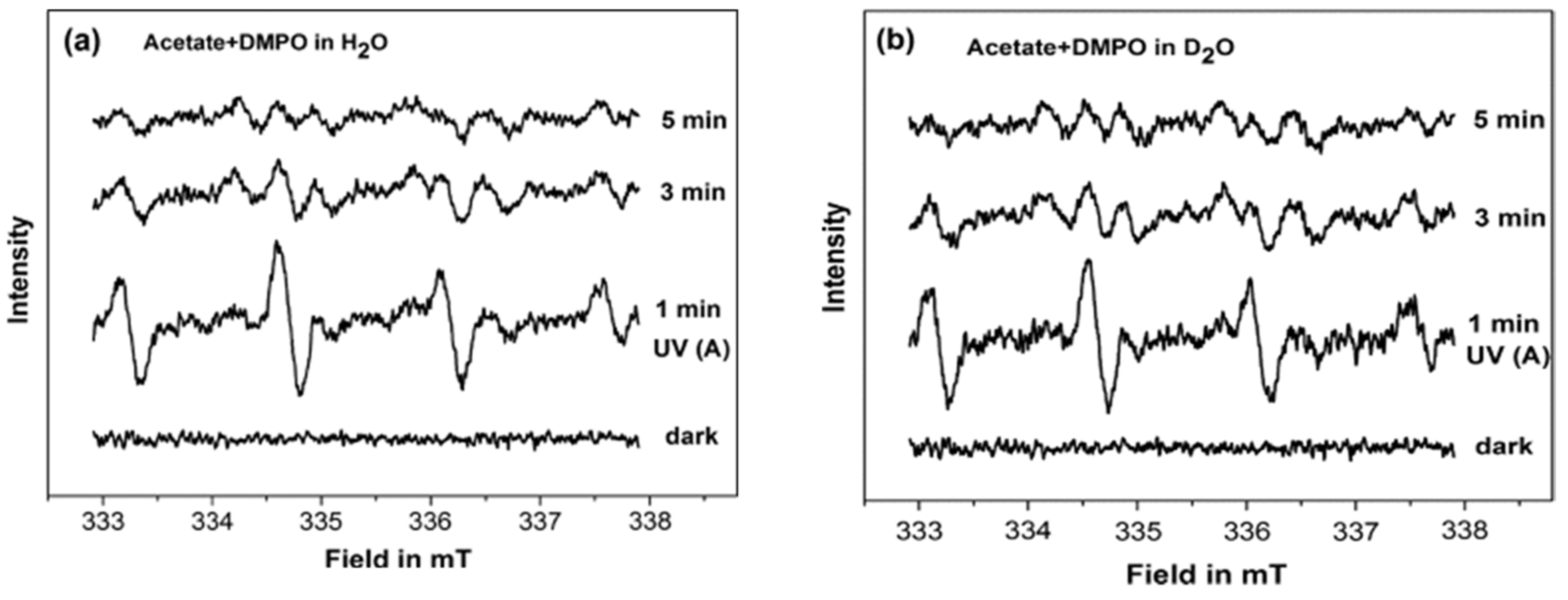
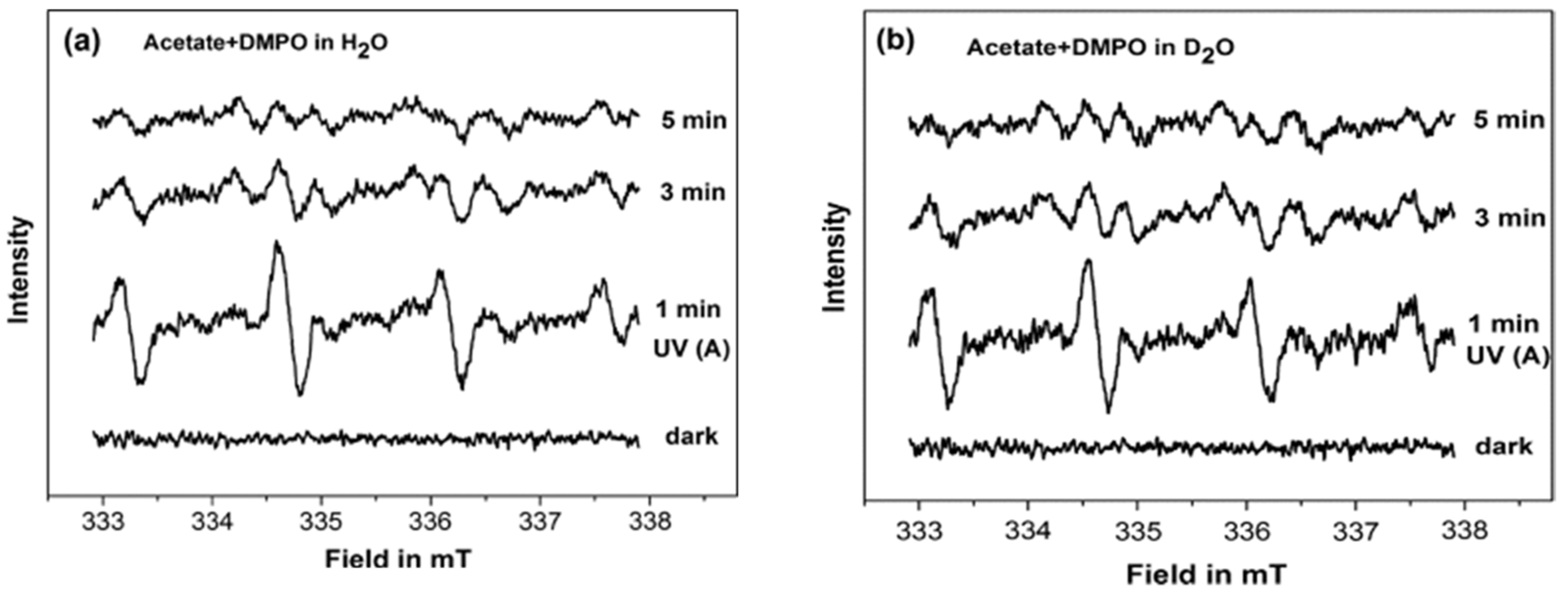
To better understand the interfacial interactions between the acetate and the TiO
2 at different pH, Belhadj et al. [
88] investigated the adsorption and the photocatalytic degradation of acetate on anatase surface (UV100) by combining the EPR and attenuated total reflection Fourier transform infrared (ATR-FTIR) techniques. In this study, the authors employed DMPO as a spin trap to probe the formed reactive oxygen species during the photocatalytic degradation of acetate in H
2O and D
2O under aerobic conditions at different pH. At pH 6.0 in H
2O and pD 6.4 in D
2O, similar EPR signals were registered under the UV(A) irradiation in the presence of DMPO (
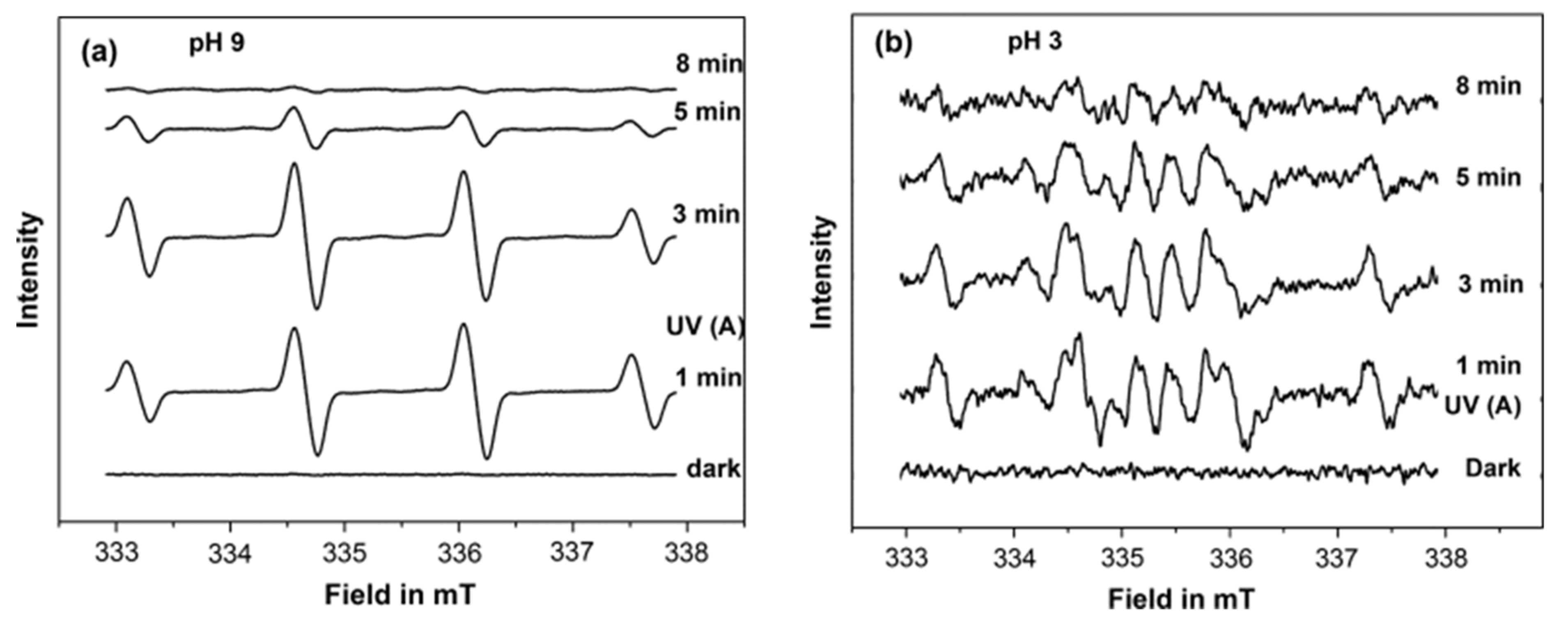
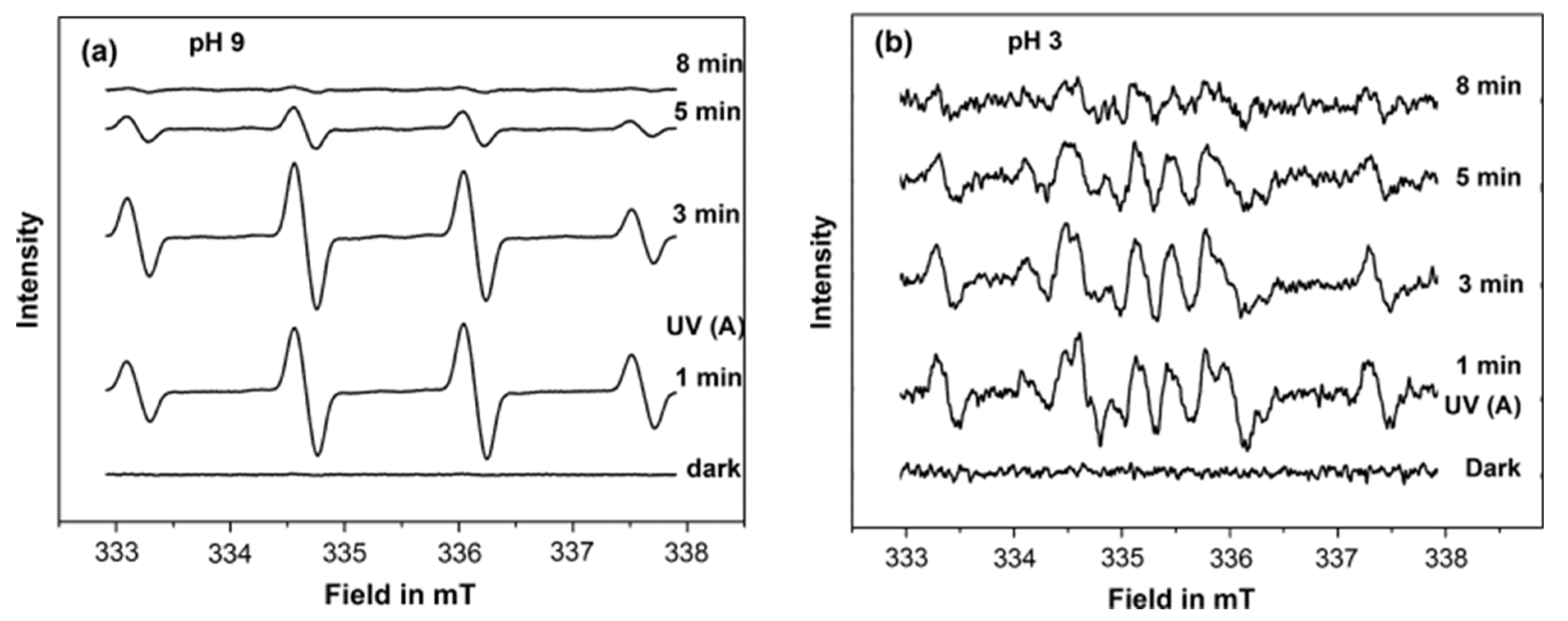
Figure 17), which was attributed to the formation of DMPO-OH and DMPO-OD adducts, respectively. While, at pH 9 in H
2O, a stronger signal for DMPO-OH was only registered (
Figure 18a). In contrast, a different EPR signal containing several peaks was observed at pH 3 (
Figure 18b). A negligible DMPO-OH signal was formed at pH 3 compared to that at pH 9. The authors attributed this signal to the formation of different DMPO adducts; i.e., DMPO-OH, DMPO-OOH, and DMO-OCH
3. Based on these results and the ATR-FTIR result, the authors suggested that the photocatalytic degradation of acetate at pH 9 mainly occurred by indirect oxidation by the
attack, as it is being predominately formed by oxidation of the adsorbed hydroxyl ions on the TiO
2 surfaces. On the other hand, the formation and the increase of DMPO-OCH
3 adduct signal during the irradiation at pH 3 suggested that the photooxidation of acetate occurs mainly through direct oxidation by the hole (h
+). These results show the existence of different radical intermediates at different pH, which would provide new insight into the mechanism of the oxidation of acetate at different pH.
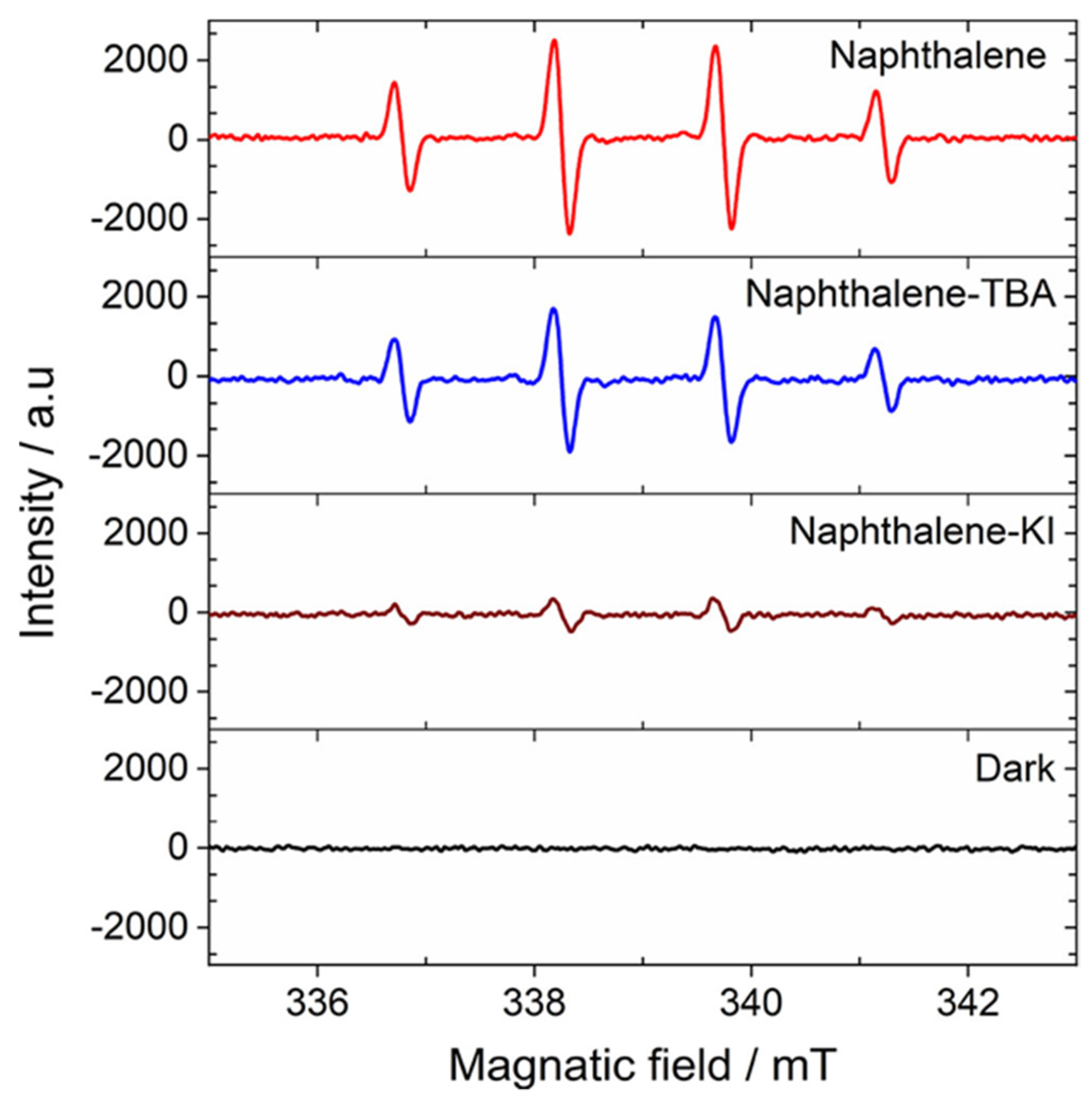
Although the EPR spin trapping has been always considered as a useful technique to confirm the involvement of the hydroxyl radical in the photocatalytic process, however, this technique can be also used to confirm the opposite. Al-Madanat et al. [
87] employed this technique during the study of the photocatalytic mechanism of naphthalene reforming over Pt-TiO
2 (UV100, anatase) to obtain additional insights regarding the role of
in the photocatalytic reforming process. In this study, the authors performed the photocatalytic reforming reaction of naphthalene in the presence of two scavengers, potassium iodide (KI) as a hole scavenger and 2-methylpropan-2-ol ((CH
3)
3COH, TBA) as a hydroxyl radical (
) scavenge to determine the involvement of different active species in the photoreforming process. They found that the conversion of naphthalene is completely inhibited in the presence of KI. while, the addition of TBA does not affect the photocatalytic process, which suggested that the presence of free hydroxyl radicals (
) has a very limited contribution in the naphthalene conversion. To prove that, they performed the photoreforming naphthalene in the presence of either TBA or KI and DMPO. The authors observed the formation of DMPO−OH adduct in all the detected samples, as shown in
Figure 19.
However, the authors observed that in the presence of TBA the adduct signal shows a decrease of 35%, confirming -scavenging properties of TBA. Nevertheless, this lower quantity of available radicals do not impact degradation of naphthalene as the authors mentioned before according to the scavenger experiments, providing evidence against a degradation mechanism initiated by the attack of . On the other hand, the addition of KI to the system practically nullifies the adduct formation, as could be expected from the efficient hole consumption by this scavenger. Therefore, according to these results, the authors excluded the involvement of the in the degradation of naphthalene during the photocatalytic process.
The EPR spin trapping technique was also used to study the mechanism of the photocatalytic reaction in the presence of other reactive species than the
and
radicals such as
[
93,
94] and organic radicals [
22,
23,
95]. Besides, it was also used to confirm the activity differences between the employing photocatalysts [
21,
96]. Alsalka et al. [
21] studied the photocatalytic reforming of oxalic acid employing self-prepared TiO
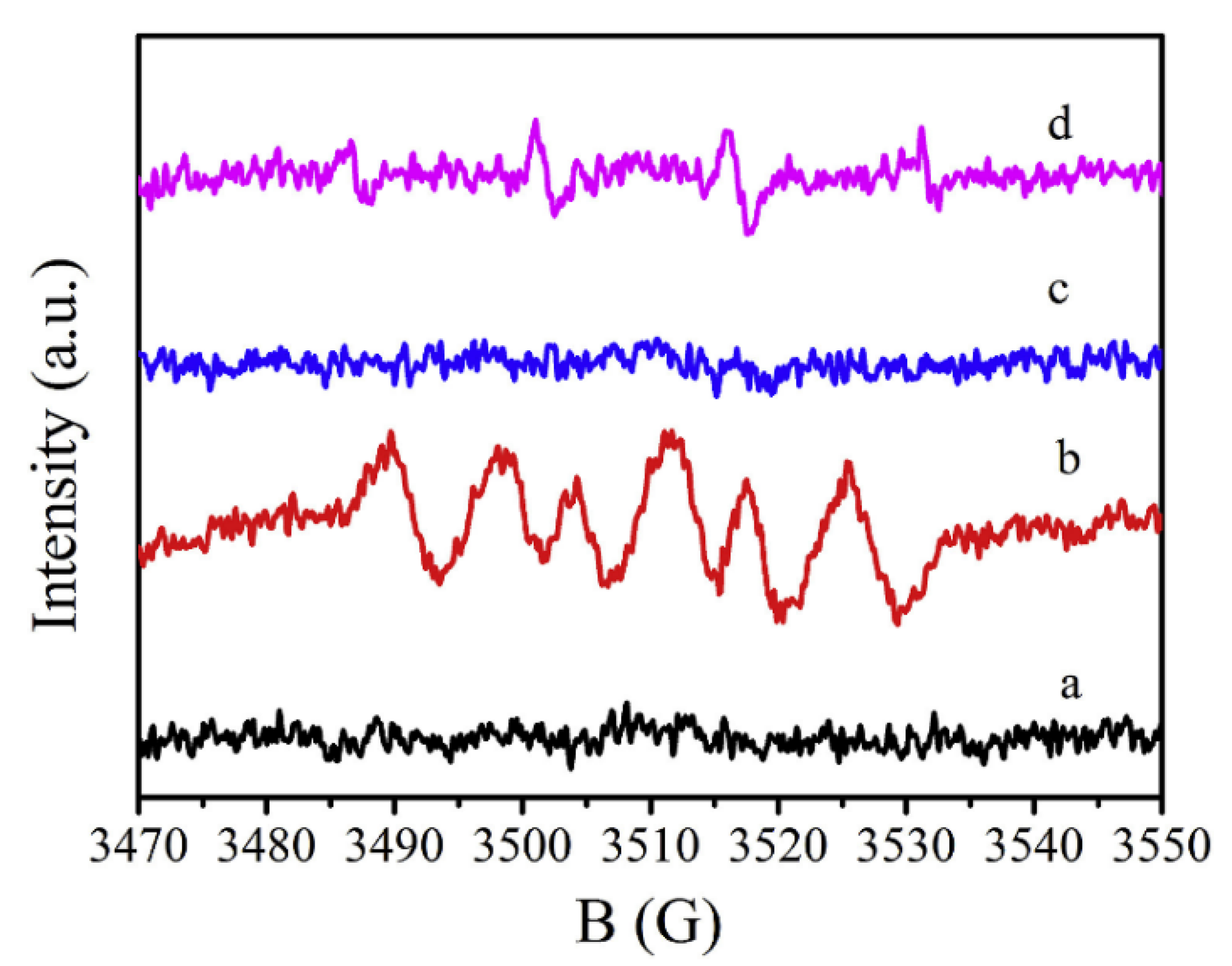
2 photocatalysts loaded with different noble metals (Pt and/or Au). By using DMPO as a spin-trapping agent, the authors traced the nature of the photogenerated species during the oxalic acid degradation under anaerobic conditions. The registered EPR spectra under irradiation that present in
Figure 20 showed that all the photocatalysts (bare and modified) exhibited the same spectra, however, the intensities of the signals were different. In this study, it was found that the higher amount of photocatalyzed H
2 formed by employing Pt-TiO
2 as a photocatalyst, which become in agreement with the presented results in
Figure 20 as the sample that contains the Pt-TiO
2 (
Figure 20c) exhibit the stronger signal.
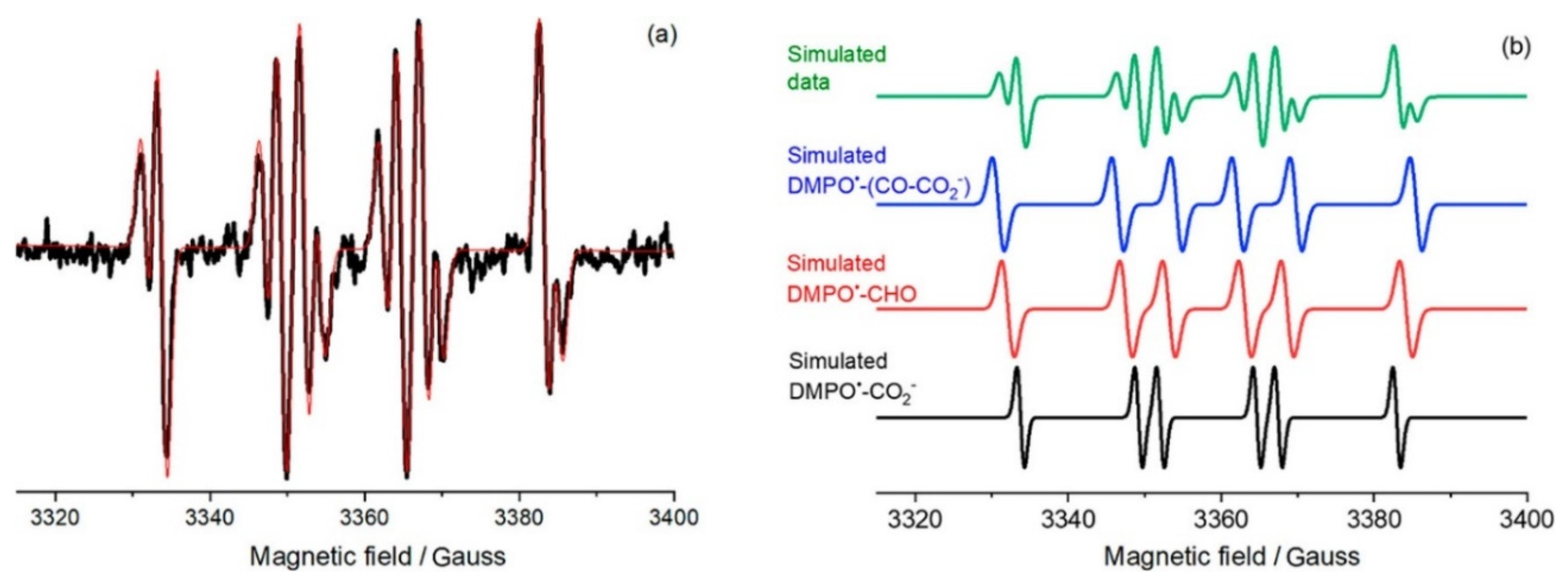
Moreover, the authors suggested that the complex EPR signal (
Figure 20) was formed due to the overlapping of different DMPO spin adducts produced during the photoreforming of oxalic acid. By simulating this spectrum (
Figure 21), they claimed that
,
), and
were formed during this process. Although the authors did not observe the formation of
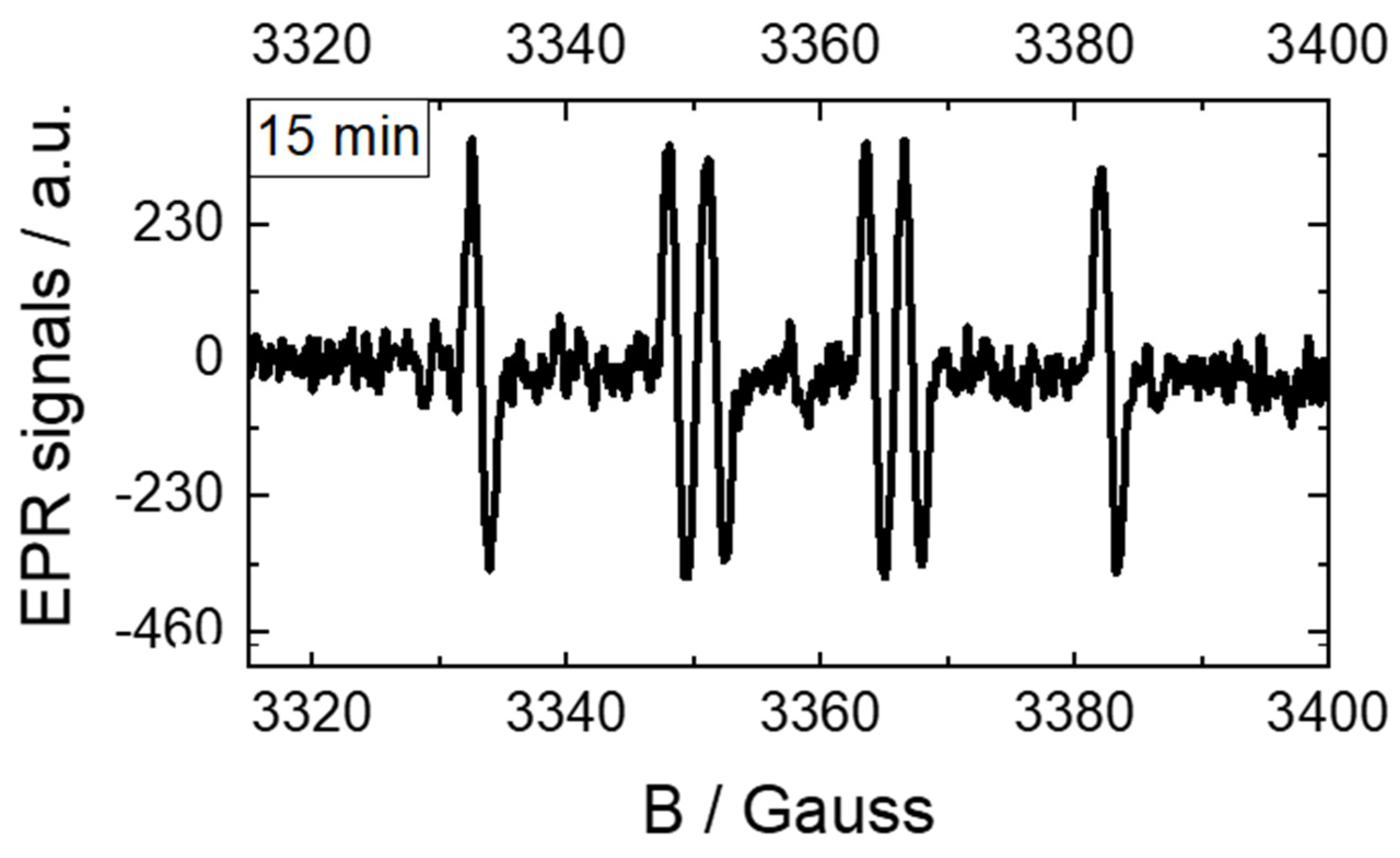
, however, they could not exclude their contribution in the photocatalytic process. However, the authors in another publication [
97] performed EPR spin-trap experiments as described before but with the presence of KI as a second hole scavenger in the addition to oxalic acid. They noticed that DMPO
•-CO
2-spin adduct is the lonely signal detected in the presence of KI with much lower EPR intensities as presented in
Figure 22. They explained this result by suggesting different pathways including the inhibition of by-products formation and lower
•CO
2− radicals’ production when KI was added to the suspension.
Although the TiO
2 has a thermodynamically feasible band structure for the water splitting (for both proton reduction and water oxidation under UV light irradiation) to H
2 and O
2, vast studies of the photocatalytic water splitting at TiO
2-based nanomaterials have shown that H
2 but not O
2 evolved during the photocatalytic process. To explain this phenomenon, Li et al., [
98] studied the photocatalytic overall water splitting over three different phases of TiO
2, namely, anatase, rutile, and brookite. The authors found that the overall water splitting can only take place on rutile, while, it becomes feasible on anatase and brookite under prolonged UV irradiation. To explain these results, the authors employed the EPR spin-trap technique using DMPO as a spin trapping agent to probe the reactive oxygen species derived on the surface of different phases of TiO
2 under UV light irradiation and Ar atmosphere. Under dark, no signal was detected for all photocatalysts, while, they obtained different signals for the three TiO
2 phases under light irradiation. Both anatase and brookite showed the same EPR signal consisting of four characteristic peaks with standard ratios of intensities 1:2:2:1, ascribed to
•DMPO-OH adduct. On the other hand, the seven-line EPR signal that was detected for rutile TiO
2 ascribed to
•DMPO-X generated from the oxidation of DMPO by peroxide, which can be easily decomposed to produce O
2 on rutile as the authors speculated. In the case of anatase and brookite TiO
2, the formed
•OH radical may be strongly absorbed on the surface and its amount increases with the prolonged UV irradiation. Thus, at saturation of absorption, the coupling of the formed
•OH radicals leads to evolving O
2 on the surface of the TiO
2.
Moreover, the spin trapping technique was not only used to detect and monitor the formed reactive species such as the oxygen and nitrogen reactive radicals [
94,
99] during the photocatalytic degradation process, but also is employed within the mechanistic pathway to monitor and/or to be involved in controlling the experimental conditions during the photocatalysis organic synthesis [
82,
100,
101,
102]. The involvement of TEMPO derivative radical within a mechanistic pathway of the synthesis reaction is assumed to act as a selective redox mediator involved in reactions of the generated reactive oxygen species. Balayeva et al. [
101] studied the visible-light-induced dehydrogenation of N-heterocycles such as tetra-hydroquinolines, tetrahydroisoquinolines, and indolines compounds on the surface of TiO
2 in an aerated system yielding the corresponding heteroarenes compounds. In this study, the authors found that 4-amino-TEMPO exhibits a beneficial role, as it improved the yield and increased the selectivity of the dehydrogenation reaction. According to the authors, the low conversion and selectivity in the absence of 4-amino-TEMPO were attributed to the formation of H
2O
2/TiO
2 surface complexes (Equation (32)), thus, preventing the formation of the N-heterocycle−TiO
2 surface complex, which is necessary for the efficient electron transfer from the excited organic moiety to the TiO
2 conduction band. However, in presence of the TEMPO (paramagnetic), it will be oxidized to TEMPO
+ (not paramagnetic) and reduces the O
2 to
, Equation (33). The formed cation further reacts with H
2O
2/TiO
2 surface complex generated superoxide radical
, Equations (34) and (35). To confirm their hypothesis, the authors employed the EPR technique in this reaction to monitor the 4-amino-TEMPO signal under visible light illumination in this system. They found that the 4-amino-TEMPO signal slightly decreases upon illumination, which suggests that the cation 4-amino-TEMPO
+ is formed upon visible-light illumination.
Furthermore, it is highly important to highlight that the fate of the photocatalytic reaction depends on the experimental conditions. In this regard, Mazzanti et al. [
102] demonstrated that the photocatalytic reaction of the azo dyes (methyl orange and acid orange 7) on the surface of the TiO
2 can be switched from the degradation path to the reductive path by introducing the sodium formate in the system. This change in the reaction conditions leads to the transformation of the azo dyes into useful products. In the absence of the sodium formate, the photocatalytic degradation of the azo dyes via the photogenerated hydroxyl radical leading to the formation of several independent pieces as secondary pollutants. The formation of the
radical in this system was confirmed by photoexcitation of deaerated azo dye/TiO
2 suspensions in the presence of the spin trapping agent DMPO, which leads to the formation of a quartet EPR signal ascribable to the formation of
•DMPO-OH adduct (
Figure 23).
However, the presence of the formate ion anion in the system leads to scavenge the photogenerated holes, thereby inhibiting the hydroxyl radical formation. Therefore, protons derived from the oxidation of formate by the photogenerated valance band holes and promoted electrons in the TiO2 conduction band lead to the reductive cleavage of N=N bonds yielding reduced valuable intermediates such as sulfanilic acid. The EPR signal that formed in this system in the presence of formate ions showed a triplet of doublets signal (•DMPO-) attributed to the formation of from oxidation of formate ions.
Some interesting mechanism studies were also performed in the literature using EPR measurements of Nb
2O
5 in suspension. Jiao and coworkers recently published a noteworthy case where simulated natural environment conditions were applied for photocatalytic studies [
103]. They investigate some materials such as plastic bags, containers, and wrap films for food, and some of their components to be photoconverted by Nb
2O
5 without any sacrificial agents. The oxide was prepared in a way to obtain Nb
2O
5 layers composed of a single-unit-cell thickness, in order to maximize the available surface. The photoreaction was found to produce CO
2 which was further selectively photoreduced to CH
3COOH. By in situ EPR analysis, the oxide suspension evidenced the formation of DMPO-OH
• and DMPO-O
2•− using pure water and methanol as the solvent, respectively. This result indicated that Nb
2O
5 photoexcited holes could oxidize H
2O into
•OH radicals and meanwhile the photogenerated electrons could reduce O
2 into O
2•−, H
2O
2 and H
2O. Along with further studies and other techniques, the mechanism was based on the oxidative C–C bond cleavage by O
2 and
•OH radical to form CO
2 which formed in sequence CH
3COOH by reductive photoinduced C–C coupling of
•COOH intermediates.
The same radicals have been reported in the work of Chen and coworkers [
104]. The authors have prepared Nb
2O
5 nanospheres by the hydrothermal method which were identified with the orthorhombic phase. The samples showed good photoactivity for Rhodamine B degradation under visible light. When EPR analysis was performed using a dispersion with Nb
2O
5 (
Figure 24), DMPO, and the dye under visible light irradiation, the generation of
•OH was observed in water suspension whereas O
2•− radicals were found in the presence of methanol. The higher intensity for the latter one could indicate that it would be the main active radical for the photodegradation of Rhodamine B. This fact was confirmed by photoluminescence experiments using different scavengers. Finally, the mechanism was explained based on a photosensitization for the photocatalytic oxidation and degradation of the dye on Nb
2O
5 under visible light.
Niobium oxide is also well known for H
2 evolution application and in many cases; deposited platinum is applied as a co-catalyst. In this sense, Zhang and coworkers have shown the performance of mesoporous Pt–Nb
2O
5 photocatalysts evaluating two different procedures, a sol-gel one-step method and by impregnation [
105]. The materials were applied to H
2 generation in a methanolic solution under UV irradiation in which the sample prepared by one-step method resulted in a higher rate (9.79 mmol h
−1). The photocatalytic activity mechanism, in relation to the generated radicals and their whole reaction, was deeply investigated by EPR studies. In principle, the same kinds of radicals mentioned before were found also in this case. In presence of methanol, both O
2•− and CH
2OH
• were detected for the tested samples under irradiation. Their intensities were higher for the one-step prepared sample, followed by the one produced by impregnation and then, the bare Nb
2O
5. When the analysis was run in water,
•OH radicals were identified with intensities following the same trend as for the other radicals. According to these results, it was possible to conclude that the efficiency of photogenerated charge carriers transfer from the photocatalyst has been enhanced by Pt nanoparticles, especially when applying the one-step method.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}