
Flow Pd(II)-Catalysed Carbonylative Cyclisation in the Total Synthesis of Jaspine B

Abstract

1. Introduction
2. Results and Discussion
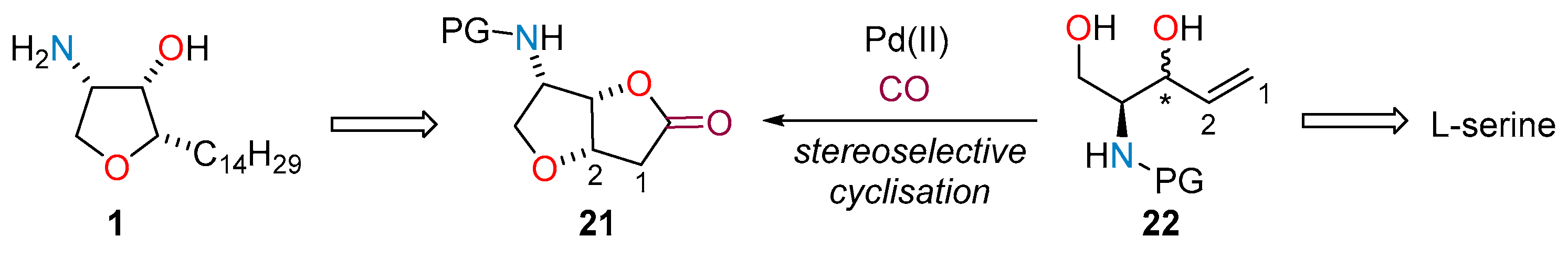
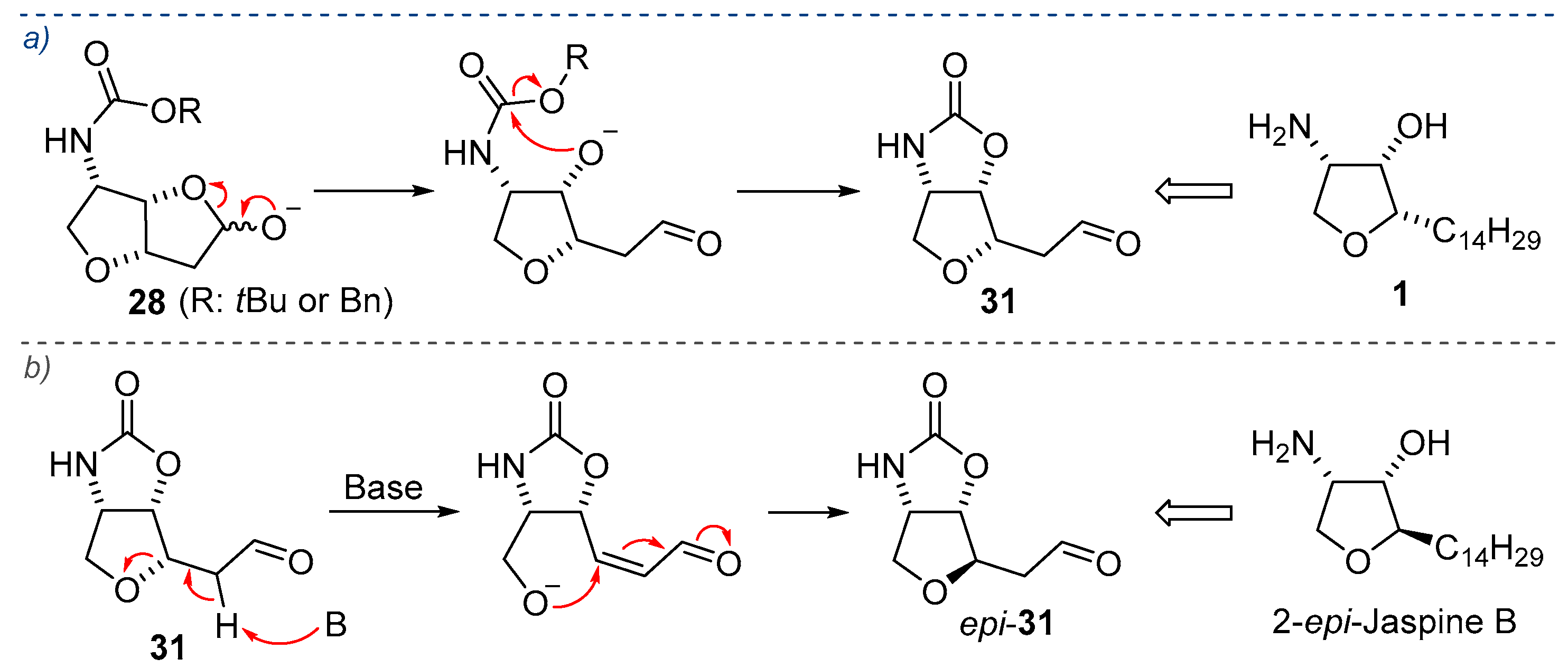
2.1. Total Synthesis of Jaspine B
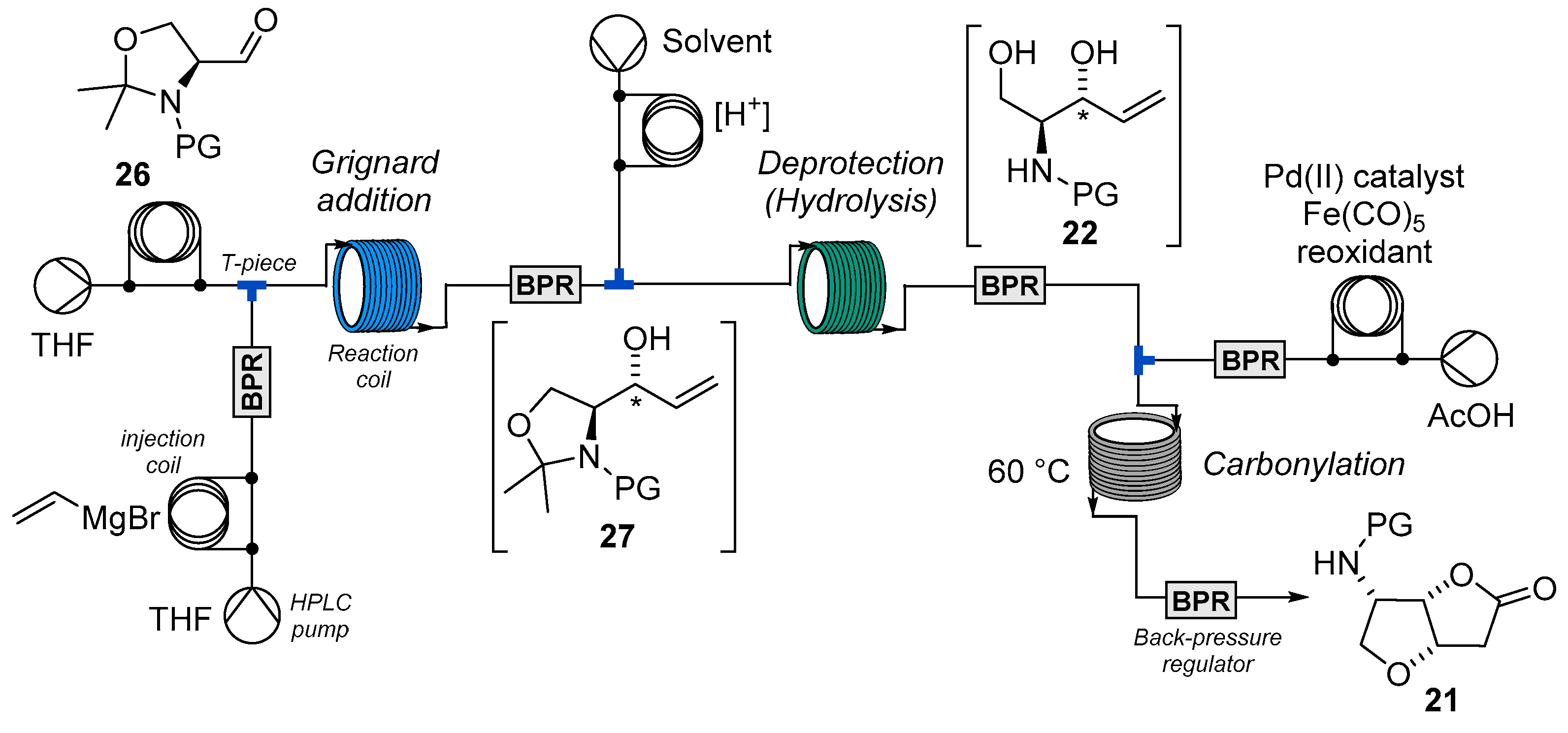
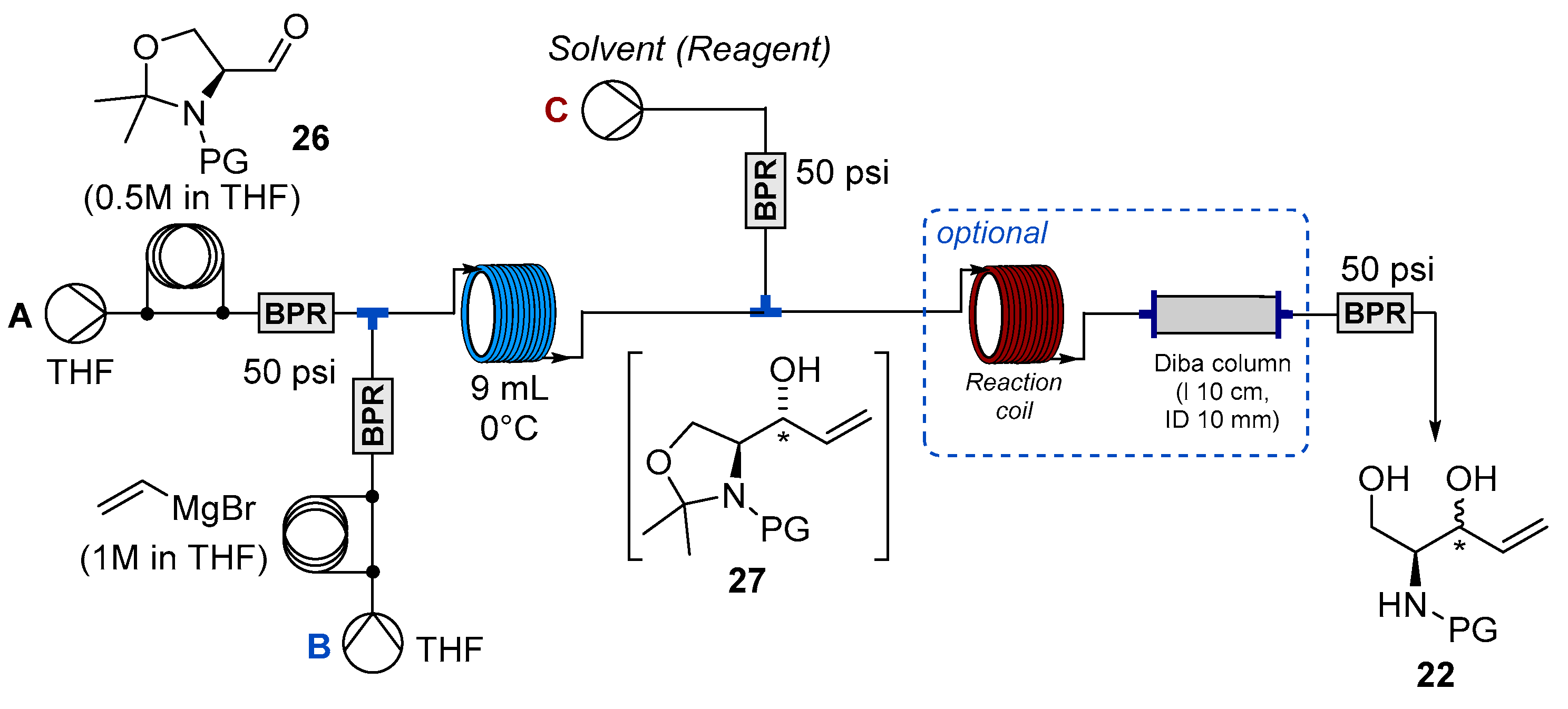
2.2. Flow Synthesis of the Key Intermediate 21 for the Preparation of Jaspine B 1
3. Experimental Section
3.1. Material and Methods
3.2. Representative Flow Procedures
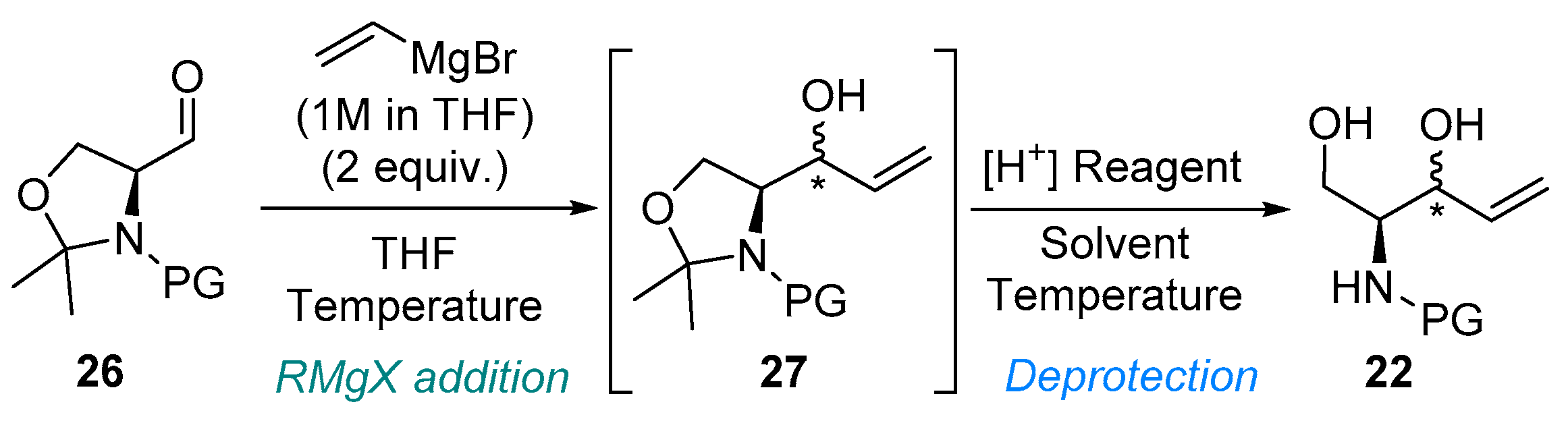
3.2.1. Grignard Reaction
3.2.2. Preparation of Unsaturated Aminodiols
3.2.3. Carbonylative Cyclisation Using pBQ/LiCl Reoxidation System
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koruda, I.; Musman, M.; Ohtani, I.I.; Ichiba, T.; Tanaka, J.; Gravalos, D.G.; Higa, T. Pachastrissamine, a Cytotoxic Anhydrophytosphingosine from a Marine Sponge, Pachastrissa sp. J. Nat. Prod. 2002, 65, 1505–1506. [Google Scholar] [CrossRef]
- Ledroit, V.; Debitus, C.; Lavaud, C.; Massiot, G. Jaspines A and B: Two new cytotoxic sphingosine derivatives from the marine sponge Jaspis sp. Tetrahedron Lett. 2003, 44, 225–228. [Google Scholar] [CrossRef]
- Canals, D.; Moemenoe, D.; Farias, G.; Llebaria, A.; Casa, J.; Delgado, A. Synthesis and biological properties of Pachastrissamine (jaspine B) and diastereoisomeric jaspines. Bioorg. Med. Chem. 2009, 17, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, P.; Ajay, S.; Meena, S.; Sinha, S.; Shaw, A.K. Stereoselective total synthesis of jaspine B (pachastrissamine) utilizing iodocyclization and an investigation of its cytotoxic activity. Tetrahedron Asymmetry 2013, 24, 903–908. [Google Scholar] [CrossRef]
- Salma, Y.; Lafont, E.; Therville, N.; Carpentier, S.; Bonnafé, M.-J.; Levade, T.; Génisson, Y.; Andrieu-Abadie, N. The natural marine anhydrophytosphingosine, Jaspine B, induces apoptosis in melanoma cells by interfering with ceramide metabolism. Biochem. Pharmacol. 2009, 78, 477–485. [Google Scholar] [CrossRef]
- Jayachitra, G.; Sudhakar, N.; Anchoori, R.K.; Rao, B.V.; Roy, S.; Banerjee, R. Stereoselective synthesis and biological studies of the C2 and C3 epimer and the enantiomer of Pachastrissamine (Jaspine B). Synthesis 2010, 1, 115–119. [Google Scholar]
- Xu, J.-M.; Zhang, E.; Shi, X.-J.; Wang, Y.-C.; Yu, B.; Jiao, W.-W.; Guo, Y.-Z.; Liu, H.-M. Synthesis and preliminary biological evaluation of 1,2,3-triazole-Jaspine B hybrids as potential cytotoxic agents. Eur. J. Med. Chem. 2014, 80, 593–604. [Google Scholar] [CrossRef]
- Martinková, M.; Mezeiová, E.; Fabišíková, M.; Gonda, J.; Pilátová, M.; Mojžiš, J. Total synthesis of pachastrissamine together with its 4-epi-congener via [3,3]-sigmatropic rearrangements and antiproliferative/cytotoxic evaluation. Carbohyde. Res. 2015, 402, 6–24. [Google Scholar] [CrossRef]
- Santos, C.; Fabing, I.; Saffon, N.; Ballereau, S.; Génisson, Y. Rapid access to jaspine B and its enantiomer. Tetrahedron 2013, 69, 7227–7233. [Google Scholar] [CrossRef]
- Jeon, H.; Bae, H.; Baek, D.; Kwak, Y.-S.; Kim, D.; Kim, S. Syntheses of sulfur and selenium analogues of pachastrissamine via double displacements of cyclic sulfate. Org. Biomol. Chem. 2011, 9, 7237–7242. [Google Scholar] [CrossRef]
- Rives, A.; Ladeira, S.; Levade, T.; Andrieu-Abadie, N.; Génisson, Y. Synthesis of Cytotoxic Aza Analogues of Jaspine B. J. Org. Chem. 2010, 75, 7920–7923. [Google Scholar] [CrossRef]
- Salma, Y.; Ballereau, S.; Maaliki, C.; Ladeira, S.; Andrieu-Abadie, N.; Génisson, Y. Flexible and enantioselective access to jaspine B and biologically active chain-modified analogues thereof. Org. Biomol. Chem. 2010, 8, 3227–3243. [Google Scholar] [CrossRef]
- Salma, Y.; Ballereau, S.; Ladeira, S.; Lepetit, C.; Chauvin, R.; Andrieu-Abadie, N. Single- and double-chained truncated jaspine B analogues: Asymmetric synthesis, biological evaluation and theoretical study of an unexpected 5-endo-dig process. Tetrahedron 2011, 67, 4253–4262. [Google Scholar] [CrossRef]
- Ballereau, S.; Andrieu-Abadie, N.; Saffon, N.; Génisson, Z. Synthesis and biological evaluation of aziridine-containing analogs of phytosphingosine. Tetrahedron 2011, 67, 2570–2578. [Google Scholar] [CrossRef]
- Pashikanti, A.; Ukani, R.; David, S.A.; Datta, A. Total Synthesis and Structure–Activity Relationship Studies of the Cytotoxic Anhydrophytosphingosine Jaspine B (Pachastrissamine). Synthesis 2017, 49, 2088–2100. [Google Scholar]
- Schmiedel, V.M.; Stefani, S.; Reissig, H.-U. Stereodivergent synthesis of jaspine B and its isomers using a carbohydrate-derived alkoxyallene as C3-building block. Beilstein. J. Org. Chem. 2013, 9, 2564–2569. [Google Scholar]
- Dhand, V.; Chang, S.; Britton, R. Total Synthesis of the Cytotoxic Anhydrophytosphingosine Pachastrissamine (Jaspine B). J. Org. Chem. 2013, 78, 8208–8213. [Google Scholar] [CrossRef]
- Enders, D.; Terteryan, V.; Paleček, J. Asymmetric Synthesis of Jaspine B (Pachastrissamine) via an Organocatalytic Aldol Reaction as Key Step. Synthesis 2008, 14, 2278–2282. [Google Scholar] [CrossRef]
- Urano, H.; Enomoto, M.; Kuwahara, S. Enantioselective Syntheses of Pachastrissamine and Jaspine A via hydroxylactonization of a Chiral Epoxy Ester. Biosci. 2010, 74, 152–157. [Google Scholar]
- Llaveria, J.; Díaz, Y.; Matheu, M.I.; Castillón, S. Eur. Enantioselective Synthesis of Jaspine B (Pachastrissamine) and Its C-2 and/or C-3 Epimers. J. Org. Chem. 2011, 1514–1519. [Google Scholar]
- Alnazer, H.; Castellan, T.; Salma, Y.; Génissom, Y.; Ballereau, S. Enantioselective Stereodivergent Synthesis of Jaspine B and 4-epi-Jaspine B from Axially Chiral Allenols. Synlett 2019, 30, 185–188. [Google Scholar]
- Yakura, T.; Sato, S.; Yoshimoto, Y. Enantioselective Synthesis of Pachastrissamine (Jaspin B) Using Dirhodium(II)-Catalyzed C–H Amination and Asymmetric Dihydroxylation as Key Steps. Chem. Pharm. Bull. 2007, 55, 1284–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abraham, E.; Candela-Lena, J.; Davies, S.G.; Georgiou, M.; Nicholson, R.L.; Roberts, P.M.; Russell, A.J.; Sánchez-Fernández, E.M.; Smith, A.D.; Thomson, J.E. Asymmetric synthesis of N,O,O,O-tetra-acetyl D-lyxo-phytosphingosine, jaspine B (pachastrissamine) and its C(2)-epimer. Tetrahedron Asymmetry 2007, 18, 2510–2513. [Google Scholar] [CrossRef]
- Abraham, E.; Brock, E.A.; Candela-Lena, J.; Davies, S.G.; Georgiou, M.; Nicholson, R.L.; Perkins, J.H.; Roberts, P.M.; Russell, A.J.; Sánchez-Fernández, E.M.; et al. Asymmetric synthesis of N,O,O,O-tetra-acetyl D-lyxo-phytosphingosine, jaspine B (pachastrissamine), 2-epi-jaspine B, and deoxoprosophyllinevialithium amide conjugate addition. Org. Biomol. Chem. 2008, 6, 1665–1673. [Google Scholar]
- Inuki, S.; Yoshimitsu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Ring-Construction/Stereoselective Functionalization Cascade: Total Synthesis of Pachastrissamine (Jaspine B) through Palladium-Catalyzed Bis-cyclization of Bromoallenes. Org. Lett. 2009, 11, 4478–4481. [Google Scholar] [CrossRef] [PubMed]
- Inuki, S.; Yoshimitsu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Ring-Construction/Stereoselective Functionalization Cascade: Total Synthesis of Pachastrissamine (Jaspine B) through Palladium-Catalyzed Bis-cyclization of Propargyl Chlorides and Carbonates. J. Org. Chem. 2010, 75, 3831–3842. [Google Scholar] [CrossRef]
- Yoshimitsu, Y.; Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Stereoselective Divergent Synthesis of Four Diastereomers of Pachastrissamine(Jaspine B). J. Org. Chem. 2010, 75, 3843–3846. [Google Scholar] [CrossRef]
- Passiniemi, M.; Koskinen, A.M.P. Asymmetric synthesis of Pachastrissamine (Jaspine B) and its diastereomers via η3-allylpalladium intermediates. Org. Biomol. Chem. 2011, 9, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, Y.; Miyagaki, J.; Oishi, S.; Fujii, N.; Ohno, H. Synthesis of pachastrissamine (jaspine B) and its derivatives by the late-stage introduction of the C-2 alkyl side-chains using olefin crossmetathesis. Tetrahedron 2013, 69, 4211–4220. [Google Scholar] [CrossRef]
- Jana, A.K.; Panda, G. Stereoselective synthesis of Jaspine B and its C2 epimer from Garner aldehyde. RSC Adv. 2013, 3, 16795–16801. [Google Scholar] [CrossRef]
- Yoshimitsu, Y.; Oishi, S.; Miyagaki, J.; Inuki, S.; Ohno, H.; Fujii, N. Pachastrissamine (jaspine B) and its stereoisomers inhibit sphingosine kinases and atypical protein kinase C. Bioorg. Med. Chem. 2011, 19, 5402–5408. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Jeon, H.; Baek, D.J.; Lee, D.; Kim, S. Stereochemically Reliable Syntheses of Pachastrissamine and Its 2-epi-Congener via Oxazolidinone Precursors from an Established Starting Material N-tert-Butoxycarbonyl-Protected Phytosphingosine. Synthesis 2012, 44, 3609–3612. [Google Scholar]
- Vichare, P.; Chattopadhyay, A. Nitrolaldol reaction of (R)-2,3-cyclohexylideneglyceraldehyde: A simple and stereoselective synthesis of the cytotoxic Pachastrissamine (Jaspine B). Tetrahedron Asymmetry 2010, 21, 1983–1987. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Matsunaga, K.; Masuda, T.; Kotsuki, H.; Nakano, K. Stereocontrolled synthesis of cytotoxic anhydrosphingosine pachastrissamine by using [3.3] sigmatropic rearrangement of allyl cyanate. Tetrahedron 2008, 64, 11313–11318. [Google Scholar] [CrossRef]
- Bhaket, P.; Morris, K.; Stauffer, C.S.; Datta, A. Total Synthesis of Cytotoxic Anhydrophytosphingosine Pachastrissamine (Jaspine B). Org. Lett. 2005, 7, 875–876. [Google Scholar] [CrossRef]
- Lee, H.-J.; Lim, C.; Hwang, S.; Jeong, B.-S.; Kim, S. Silver-Mediated exo-Selective Tandem Desilylative Bromination/Oxycyclization of Silyl-Protected Alkynes: Synthesis of 2-Bromomethylene-Tetrahydrofuran. Chem. Asian J. 2011, 6, 1943–1947. [Google Scholar] [CrossRef]
- Prasad, K.R.; Chandrakumar, A. Stereoselective Synthesis of Cytotoxic Anhydrophytosphingosine Pachastrissamine [Jaspine B]. J. Org. Chem. 2007, 72, 6312–6315. [Google Scholar] [CrossRef]
- Lee, T.; Lee, S.; Kwak, Y.S.; Kim, D.; Kim, S. Synthesis of Pachastrissamine from Phytosphingosine: A Comparison of Cyclic Sulfate vs an Epoxide Intermediate in Cyclization. Org. Lett. 2007, 9, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Van der Berg, R.J.B.H.N.; Boltje, T.J.; Verhagen, C.P.; Litjens, E.J.N.; Van der Marel, G.A.; Overkleeft, H.S. An Efficient Synthesis of the Natural Tetrahydrofuran Pachastrissamine Starting from D-ribo-Phytosphingosine. J. Org. Chem. 2006, 71, 836–839. [Google Scholar] [CrossRef]
- Fujiwara, T.; Liu, B.; Niu, W.; Hashimoto, K.; Nambu, H.; Yakura, T. Practical Synthesis of Pachastrissamine (Jaspine B), 2-epi-Pachastrissamine, and the 2-epi-Pyrrolidine Analogue. Chem. Pharm. Bull. 2016, 64, 179–188. [Google Scholar] [CrossRef]
- Du, Y.; Liu, J.; Linhardt, R.J. Stereoselective Synthesis of Cytotoxic Anhydrophytosphingosine Pachastrissamine (Jaspine B) from D-Xylose. J. Org. Chem. 2006, 71, 1251–1253. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reddy, L.V.R.; Reddy, P.V.; Shaw, A.K. An expedient route for the practical synthesis of pachastrissamine (jaspine B) starting from 3,4,6-tri-O-benzyl-D-galactal. Tetrahedron Asymmetry 2007, 18, 542–546. [Google Scholar] [CrossRef]
- Ribes, C.; Falomir, E.; Carda, M.; Marco, J.A. Stereoselective synthesis of pachastrissamine (jaspine B). Tetrahedron 2006, 62, 5421–5425. [Google Scholar] [CrossRef]
- Ramana, C.V.; Giri, A.G.; Suryawanshi, S.B.; Gonnade, R.G. Total synthesis of pachastrissamine (jaspine B) enantiomers from d-glucose. Tetrahedron Lett. 2007, 48, 265–268. [Google Scholar] [CrossRef]
- Rao, G.S.; Rao, B.V. A common strategy for the stereoselective synthesis of anhydrophytosphingosine pachastrissamine (jaspine B) and N,O,O,O-tetra-acetyl D-lyxo-phytosphingosine. Tetrahedron Lett. 2011, 52, 6076–6079. [Google Scholar]
- Kwon, Y.; Song, J.; Bae, H.; Kim, W.J.; Lee, J.Y.; Han, G.H.; Lee, S.K.; Kim, S. Synthesis and Biological Evaluation of Carbocyclic Analogues of Pachastrissamine. Mar. Drugs 2015, 13, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Lopatka, P.; Markovič, M.; Koóš, P.; Ley, S.L.; Gracza, T. Continuous Pd-Catalyzed Carbonylative Cyclization Using Iron Pentacarbonyl as a CO Source. J. Org. Chem. 2019, 84, 14394–14406. [Google Scholar] [CrossRef] [PubMed]
- Markovič, M.; Koóš, P.; Gracza, T. A Short Asymmetric Synthesis of Sauropunols A–D. Synthesis 2017, 49, 2939–2942. [Google Scholar]
- Markoviš, M.; Koóš, P.; Čarný, T.; Sokoliová, S.; Boháčiková, N.; Moncoľ, J.; Gracza, T. Total Synthesis, Configuration Assignment, and Cytotoxic Activity Evaluation of Protulactone A. J. Nat. Prod. 2017, 80, 1631–1638. [Google Scholar] [CrossRef]
- Lopatka, P.; Koóš, P.; Markovič, M.; Gracza, T. Asymmetric Formal Synthesis of (+)-Pyrenolide D. Synthesis 2014, 46, 817–821. [Google Scholar]
- Koóš, P.; Špánik, I.; Gracza, T. Asymmetric intramolecular Pd (II)-catalysed amidocarbonylation of unsaturated amino alcohols. Tetrahedron Asymmetry 2009, 20, 2720–2723. [Google Scholar] [CrossRef]
- Kapitán, P.; Gracza, T. Stereocontrolled oxycarbonylation of 4-benzyloxyhepta-1,6-diene-3,5-diols promoted by chiral palladium (II) complexes. Tetrahedron Asymmetry 2008, 19, 38–44. [Google Scholar] [CrossRef]
- McKillop, A.; Taylor, R.; Watson, R.; Lewis, N. An Improved Procedure for the Preparation of the Garner Aldehyde and Its Use for the Synthesis of N-Protected 1-Halo-2(R)-amino-3-butanes. Synthesis 1994, 1, 31–33. [Google Scholar] [CrossRef]
- Ojima, I.; Vidal, S.E. Rhodium-Catalyzed Cyclohydrocarbonylation: Application to the Synthesis of (+)-Prosopinine and (−)-Deoxoprosophylline. J. Org. Chem. 1998, 63, 7999–8003. [Google Scholar] [CrossRef]
- Ghosh, A.; Chattopadhyay, S.K. A diversity oriented synthesis of D-erythro-sphingosine and siblings. Tetrahedron Asymmetry 2017, 28, 1139–1143. [Google Scholar] [CrossRef]
- Ghosal, P.; Shaw, A.K. An efficient total synthesis of the anticancer agent (+)-spisulosine (ES-285) from Garner’s aldehyde. Tetrahedron Lett. 2010, 51, 4140–4142. [Google Scholar] [CrossRef]
- Singh, P.; Panda, G. Intramolecular 5-endo-trig aminopalladation of β -hydroxy-γ -alkenylamine: Efficient route to apyrrolidine ring and its application for the synthesis of (-)-8,8a-di-epi-swainsonine. RSC Adv. 2014, 4, 2161–2166. [Google Scholar] [CrossRef]
- Abraham, E.; Davies, S.G.; Roberts, P.M.; Russell, A.J.; Thomson, J.E. Jaspine B (pachastrissamine) and 2-epi-jaspine B:synthesis and structural assignment. Tetrahedron Asymmetry 2008, 19, 1027. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Preocess Res. Dev. 2016, 1, 2–25. [Google Scholar] [CrossRef]
- Brandão, P.; Pineiro, M.; Pinho e Melo, T.M. Flow Chemistry: Towards A More Sustainable Heterocyclic Synthesis. Eur. J. Org. Chem. 2019, 43, 7188–7217. [Google Scholar] [CrossRef]
- Levesque, F.; Seeberger, F.H. Continuous-Flow Synthesis of the Anti-Malaria Drug Artemisinin. Angew. Chem. Int. Ed. 2012, 51, 1706–1709. [Google Scholar] [CrossRef]
- Lau, S.-H.; Galván, A.; Merchant, R.R.; Battilocchio, C.; Souto, J.A.; Berry, M.B.; Ley, S.V. Machines vs Malaria: A Flow-Based Preparation of the Drug Candidate OZ439. Org. Lett. 2015, 17, 3218–3221. [Google Scholar] [CrossRef] [PubMed]
- LaPorte, T.L.; Spangler, L.; Hamedi, M.; Lobben, P.; Chan, S.H.; Mushlehiddinoglu, J.; Wang, S.S.Y. Development of a Continuous Plug Flow Process for Preparation of a Key Intermediate for Brivanib Alaninate. Org. Process Res. Dev. 2014, 18, 1492–1502. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Griffiths-Jones, C.M.; Ley, S.V.; Tranmer, G.K. Preparation of the Neolignan Natural Product Grossamide by a Continuous-Flow Process. Synlett 2006, 3, 427–430. [Google Scholar] [CrossRef]
- Correia, C.A.; Gilmore, K.; McQuade, D.T.; Seeberger, P.H. A Cocise Flow Synthesis of Efavirenz. Angew. Chem. Int. Ed. 2015, 54, 4945–4948. [Google Scholar] [CrossRef] [PubMed]
- Tsubogo, T.; Oyamada, H.; Kobayashi, S. Multistep continuous-flow synthesis of (R)- and (S)-rolipram using heterogeneous catalysts. Nature 2015, 520, 329–332. [Google Scholar] [CrossRef]
- Hartwig, J.; Ceylan, S.; Kupracz, L.; Coutable, L.; Kirschning, A. Heating under High-Frequency Inductive Conditions: Application to the Continuous Synthesis of the Neurolepticum Olanzapine (Zyprexa). Angew. Chem. Int. Ed. 2013, 52, 9813–9817. [Google Scholar] [CrossRef] [PubMed]
- Babjak, M.; Markovič, M.; Kandríková, B.; Gracza, T. Homogeneous Cyclocarbonylation of Alkenols with Iron Pentacarbonyl. Synthesis 2014, 46, 809–816. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | RMgX Addition | Deprotection | Product | ||||
|---|---|---|---|---|---|---|---|---|
| T (°C) | Rxn Time | [H+] Reagent (Equivalent) | Solvent | T (°C) | Rxn Time | Yield (%) a | ||
| 1 | 26-Boc | −78 | 2 h | pTSA.H2O (0.1) | MeOH | 40 °C | 1.5 h | 75 |
| 2 | 26-Boc | 0 | 2 h | Amberlyst 15 (3.2) b | MeOH | 40 °C | 0.5 h | 46 c |
| 3 | 26-Boc | 0 | 2 h | - | AcOH | 70 °C | 0.5 h | 35 c |
| 4 | 26-Cbz | −78 | 1.5 h | - | AcOH/H2O (4/1) | r.t. | 3 h | 59 |
| 5 | 26-Cbz | −78 | 1.5 h | pTSA.H2O (0.1) | MeOH | r.t. | 3 h | 87 |
| 6 | 26-Cbz | 0 | 1.5 h | Amberlyst 15 (3.2) b | MeOH | 40 °C | 0.5 h | 73 |
| Entry | Substrate 26-PG | STREAM | Optional Equipment | Yield b (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| A a | B | C | |||||||
| Inj. Coil (mL) | Flow Rate (mL/min.) | Inj. Coil (mL) | Flow Rate (mL/min.) | Solvent | Flow Rate (mL/min.) | ||||
| 1 | Boc | 2 | 0.3 | 2.3 | 0.3 | - | - | 84 c | |
| 2 | Cbz | 2 | 0.25 | 2.3 | 0.25 | - | - | 66 c | |
| 3 | Boc | 2 | 0.3 | 2.3 | 0.3 | MeOH | 0.6 | 1.7g (Amberlyst) | 34 d |
| 4 | Cbz | 2 | 0.25 | 2.3 | 0.25 | MeOH | 0.5 | 1.7g (Amberlyst) | 49 |
| 5 | Boc | 17.3 | 0.25 | 18.4 | 0.25 | pTSA in MeOH (1.1M) | 0.25 | reaction coil (10 mL, 50 °C) | 62 |
| 6 | Cbz | 17.3 | 0.25 | 18.4 | 0.25 | pTSA in MeOH (1.1M) | 0.25 | reaction coil (10 mL, 50 °C) | 82 |
| Entry | Product | Typical Conditions | New Conditions | |
|---|---|---|---|---|
| Batch Yield a,b (%) | Flow Yield a,c (%) | Flow Yield a,d (%) | ||
| 1 | 21-Cbz | 75 | 76 | 71 |
| 2 | 21-Boc | 75 | 64 | 72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopatka, P.; Gavenda, M.; Markovič, M.; Koóš, P.; Gracza, T. Flow Pd(II)-Catalysed Carbonylative Cyclisation in the Total Synthesis of Jaspine B. Catalysts 2021, 11, 1513. https://doi.org/10.3390/catal11121513
Lopatka P, Gavenda M, Markovič M, Koóš P, Gracza T. Flow Pd(II)-Catalysed Carbonylative Cyclisation in the Total Synthesis of Jaspine B. Catalysts. 2021; 11(12):1513. https://doi.org/10.3390/catal11121513
Chicago/Turabian StyleLopatka, Pavol, Michal Gavenda, Martin Markovič, Peter Koóš, and Tibor Gracza. 2021. "Flow Pd(II)-Catalysed Carbonylative Cyclisation in the Total Synthesis of Jaspine B" Catalysts 11, no. 12: 1513. https://doi.org/10.3390/catal11121513
APA StyleLopatka, P., Gavenda, M., Markovič, M., Koóš, P., & Gracza, T. (2021). Flow Pd(II)-Catalysed Carbonylative Cyclisation in the Total Synthesis of Jaspine B. Catalysts, 11(12), 1513. https://doi.org/10.3390/catal11121513