Asymmetric Michael Addition of Malononitrile with Chalcones via Rosin-Derived Bifunctional Squaramide


Abstract
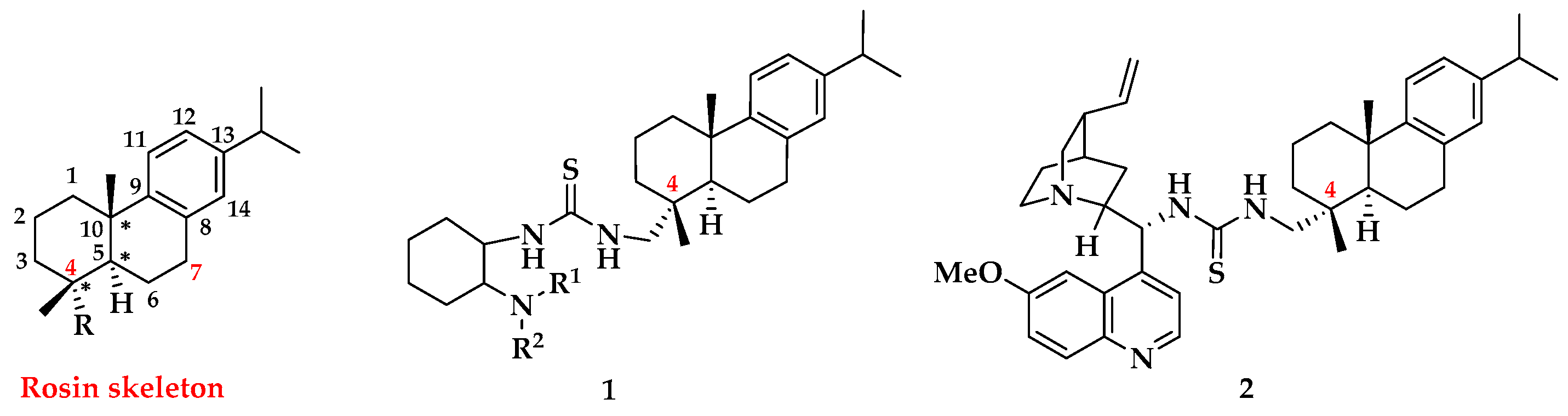
1. Introduction
2. Results and Discussion
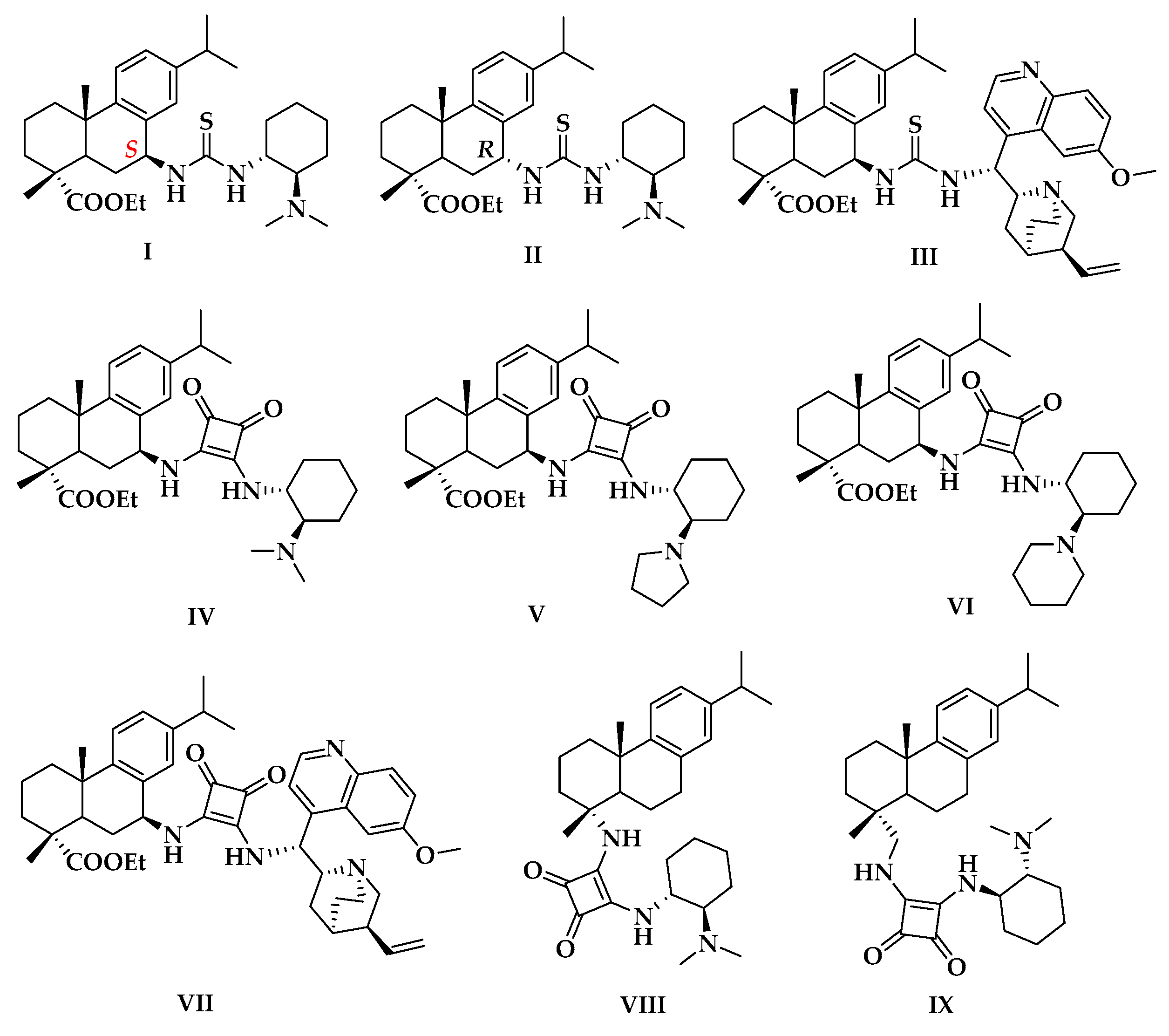
2.1. Screening of the Catalysts for the Asymmetirc Michael Addtion
2.2. Optimization of the Reaction Conditions
2.3. The Scope of the Asymmetric Michael Reaction
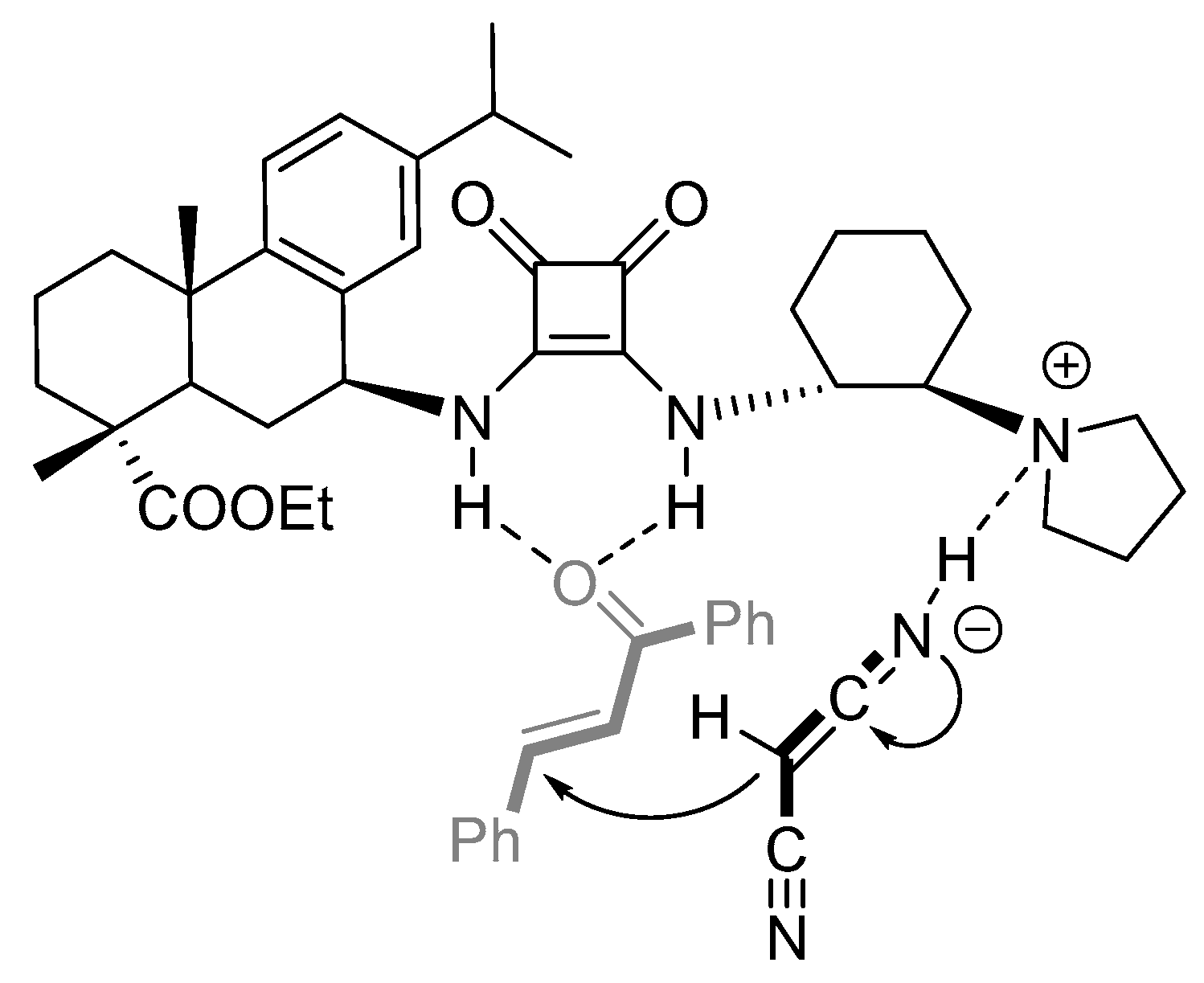
2.4. Plausible Transition-State Model of the Asymmetric Michael Reaction
3. Experimental Section
3.1. General Information
3.2. Typical Procedure for the Michael Addition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Conjugate additions of nitroalkanes to electron-poor alkenes: Recent results. Chem. Rev. 2005, 105, 933–971. [Google Scholar] [CrossRef] [PubMed]
- Almasi, D.; Alonso, D.A.; Nájera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent advances in asymmetric organocatalytic 1, 4-conjugate additions. Eur. J. Org. Chem. 2007, 11, 1701–1716. [Google Scholar] [CrossRef]
- Csaky, A.G.; Herran, G.D.L.; Murcia, M.C. Conjugate addition reactions of carbon nucleophiles to electron-deficient dienes. Chem. Soc. Rev. 2010, 39, 4080–4102. [Google Scholar] [CrossRef] [PubMed]
- Roca-Lopez, D.; Sadaba, D.; Delso, I.; Herrera, R.P.; Tejero, T.; Merino, P. Asymmetric organocatalytic synthesis of γ-nitrocarbonyl compounds through Michael and Domino reactions. Tetrahedron Asymmetry 2010, 21, 2561–2601. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Zheng, K.; Liu, X.; Feng, X. Recent advances in metal-catalyzed asymmetric 1, 4-conjugate addition (ACA) of nonorganometallic nucleophiles. Chem. Rev. 2018, 118, 7586–7656. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Enantioselective Michael additions to α, β-unsaturated imides catalyzed by a salen−Al complex. J. Am. Chem. Soc. 2003, 125, 11204–11205. [Google Scholar] [CrossRef]
- Taylor, M.S.; Zalatan, D.N.; Lerchner, A.M.; Jacobsen, E.N. Highly enantioselective conjugate additions to α, β-unsaturated ketones catalyzed by a (salen) Al complex. J. Am. Chem. Soc. 2005, 127, 1313–1317. [Google Scholar] [CrossRef]
- Hoash, Y.; Okino, T.; Takemoto, Y. Enantioselective Michael addition to α, β-unsaturated imides catalyzed by a bifunctional organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 4032–4035. [Google Scholar] [CrossRef]
- Inokuma, T.; Hoashi, Y.; Takemoto, Y. Thiourea-catalyzed asymmetric Michael addition of activated methylene compounds to α, β-unsaturated imides: Dual activation of imide by intra-and intermolecular hydrogen bonding. J. Am. Chem. Soc. 2006, 128, 9413–9419. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.W.; Huang, X.; Fan, L.P.; Xu, D.C.; Li, X.S.; Su, H.; Wen, Y.H. Efficient method for the synthesis of optically active 2-amino-2-chromene derivatives via one-pot tandem reactions. Adv. Synth. Catal. 2009, 351, 3077–3082. [Google Scholar] [CrossRef]
- Huang, X.; Li, P.; Li, X.-S.; Xu, D.-C.; Xie, J.-W. The organocatalytic two-step synthesis of diversely functionalized tricyclic tetrazoles. Org. Biomol. Chem. 2010, 8, 4527–4529. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-P.; Lou, C.-L.; Wang, J.-J.; Chen, C.-X.; Yan, M. Organocatalytic conjugate addition of malononitrile to conformationally restricted dienones. J. Org. Chem. 2011, 76, 3797–3804. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-M.; Wang, B.; Zhang, J.-M.; Yan, M. Asymmetric organocatalytic double-conjugate addition of malononitrile to dienones: Efficient synthesis of optically active cyclohexanones. Org. Lett. 2011, 13, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, W.; Du, D.-M. Efficient organocatalytic asymmetric synthesis of 2-amino-4H-chromene-3-carbonitrile derivatives. Tetrahedron Asymmetry 2012, 23, 339–344. [Google Scholar] [CrossRef]
- Arai, T.; Oka, I.; Morihata, T.; Awata, A.; Masu, H. A neutral, chiral, bis (imidazolidine)-derived NCN-type palladium pincer complex with catalytic activity. Chem. Eur. J. 2013, 19, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.R.; Gayen, P.; Panda, S.; Ghorai, P. Enantioselective, organocatalytic, dissymmetric 1, 4- and 1, 2-addition of malononitrile to a keto-bisenone followed by an oxa-Michael addition cascade. Org. Lett. 2019, 21, 5793–5797. [Google Scholar] [CrossRef]
- Shi, J.; Wang, M.; He, L.; Zheng, K.; Liu, X.; Lin, L.; Feng, X. Enantioselective Michael addition of malononitrile to chalcones catalyzed by a simple quinine–Al (OiPr)3 complex: A simple method for the synthesis of a chiral 4H-pyran derivative. Chem. Commun. 2009, 31, 4711–4713. [Google Scholar] [CrossRef]
- Li, X.; Ma, Y.; Xing, Z.; Tang, N.; Zhu, J.; Deng, J. The asymmetric addition of malononitrile to α, β-unsaturated ketones catalyzed by RuCl2 [(R, R)-DPEN] (PPh3)2 as the precatalyst. Tetrahedron Lett. 2014, 55, 3868–3872. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Duan, W.; Wang, W. Organocatalytic enantioselective conjugate additions to enones. J. Am. Chem. Soc. 2006, 128, 12652–12653. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cun, L.; Lian, C.; Zhong, L.; Chen, Y.; Liao, J.; Zhu, J.; Deng, J. Highly enantioselective Michael addition of malononitrile to α, β-unsaturated ketones. Org. Biomol. Chem. 2008, 6, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Perfetto, A.; Lattanzi, A. Back to natural cinchona alkaloids: Highly enantioselective Michael addition of malononitrile to enones. Adv. Synth. Catal. 2009, 351, 3067–3071. [Google Scholar] [CrossRef]
- Russo, A.; Capobianco, A.; Perfetto, A.; Lattanzi, A.; Peluso, A. Enantioselective conjugate addition of malononitrile to chalcones promoted by α, α-L-diaryl prolinols: Noncovalent versus covalent catalysis? Eur. J. Org. Chem. 2011, 10, 1922–1931. [Google Scholar] [CrossRef]
- Yang, W.; Jia, Y.; Du, D.-M. Squaramide-catalyzed enantioselective Michael addition of malononitrile to chalcones. Org. Biomol. Chem. 2012, 10, 332–338. [Google Scholar] [CrossRef]
- Molleti, N.; Rana, N.K.; Singh, V.K. Highly enantioselective conjugate addition of malononitrile to 2-enoylpyridines with bifunctional organocatalyst. Org. Lett. 2012, 14, 4322–4325. [Google Scholar] [CrossRef]
- Yan, L.; Wang, H.; Xiong, F.; Tao, Y.; Wu, Y.; Chen, F. Chloramphenicol base chemistry. Part 11: Chloramphenicol base-derived thiourea-catalyzed enantioselective Michael addition of malononitrile to α, β-unsaturated ketones. Tetrahedron Asymmetry 2017, 28, 921–929. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, Y.; Wu, L.; Zhang, G.; Liu, X.; Zhang, H.; Fu, D.; Wang, R. Doubly stereocontrolled asymmetric aza-Henry reaction with in situ generation of N-Boc-imines catalyzed by novel rosin-derived amine thiourea catalysts. Adv. Synth. Catal. 2009, 351, 2096–2100. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, Y.; Liu, X.; Zhang, G.; Lai, L.; Wu, L.; Zhang, J.; Wang, R. Enantio and diastereoselective asymmetric addition of 1, 3-dicarbonyl compounds to nitroalkenes in a doubly stereocontrolled manner catalyzed by bifunctional rosin-derived amine thiourea catalysts. J. Org. Chem. 2009, 74, 5562–5567. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, Y.; Chan, A.S.C.; Wang, R. Highly enantioselective synthesis of γ-nitro heteroaromatic ketones in a doubly stereocontrolled manner catalyzed by bifunctional thiourea catalysts based on dehydroabietic amine: A doubly stereocontrolled approach to pyrrolidine carboxylic acids. Org. Lett. 2009, 11, 153–156. [Google Scholar] [CrossRef]
- Jiang, X.; Fu, D.; Zhang, G.; Cao, Y.; Liu, L.; Song, J.; Wang, R. Highly diastereo-and enantioselective Mannich reaction of lactones with N-Boc-aldimines catalyzed by bifunctional rosin-derived amine thiourea catalysts. Chem. Commun. 2010, 46, 4294–4296. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, G.; Fu, D.; Cao, Y.; Shen, F.; Wang, R. Direct organocatalytic asymmetric Aldol reaction of α-isothiocyanato imides to α-ketoesters under low ligand loading: A doubly stereocontrolled approach to cyclic thiocarbamates bearing chiral quaternary stereocenters. Org. Lett. 2010, 12, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Cao, Y.; Wang, Y.; Liu, L.; Shen, F.; Wang, R. A unique approach to the concise synthesis of highly optically active spirooxazolines and the discovery of a more potent oxindole-type phytoalexin analogue. J. Am. Chem. Soc. 2010, 132, 15328–15333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Y.; Jiang, X.; Yan, W.; Wang, R. Highly enantioslective synthesis of multisubstituted polyfunctional dihydropyrrole via an organocatalytic tandem Michael/cyclization sequence. Org. Lett. 2011, 13, 3806–3809. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, X.; Liu, L.; Shen, F.; Zhang, F.; Wang, R. Enantioselective Michael/cyclization reaction sequence: Scaffold-inspired synthesis of spirooxindoles with multiple stereocenters. Angew. Chem. Int. Ed. 2011, 50, 9124–9127. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, L.; Xing, Y.; Wang, L.; Wang, S.; Chen, Z.; Wang, R. Highly enantioselective Friedel–Crafts alkylation reaction catalyzed by rosin-derived tertiary amine–thiourea: Synthesis of modified chromanes with anticancer potency. Chem. Commun. 2012, 48, 446–448. [Google Scholar] [CrossRef]
- Zhu, H.; Jiang, X.; Li, X.; Hou, C.; Jiang, Y.; Hou, K.; Wang, R.; Li, Y. Highly enantioselective synthesis of N-protected β-amino malonates catalyzed by magnetically separable heterogeneous rosin-derived amino thiourea catalysts: A stereocontrolled approach to β-amino acids. ChemCatChem 2013, 5, 2187–2190. [Google Scholar] [CrossRef]
- Storer, R.I.; Aciro, C.; Jones, L.H. Squaramides: Physical properties, synthesis and applications. Chem. Soc. Rev. 2011, 40, 2330–2346. [Google Scholar] [CrossRef]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from molecular recognition to bifunctional organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional amine-squaramides: Powerful hydrogen-bonding organocatalysts for asymmetric domino/cascade reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Held, F.E.; Tsogoeva, S.B. Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: Recent advances. Catal. Sci. Technol. 2016, 6, 645–667. [Google Scholar] [CrossRef]
- Zhao, B.-L.; Li, J.-H.; Du, D.-M. Squaramide-catalyzed asymmetric reactions. Chem. Rec. 2017, 17, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Mika, L.T.; Csámpai, A.; Holczbauer, T.; Kardosa, G.; Soós, T. Mechanistic investigations of a bifunctional squaramide organocatalyst in asymmetric Michael reaction and observation of stereoselective retro-Michael reaction. RSC Adv. 2015, 5, 95079–95086. [Google Scholar] [CrossRef]
- Huang, W.-J.; Chen, Q.; Lin, N.; Long, X.-W.; Pan, W.-G.; Xiong, Y.-S.; Weng, J.; Lu, G. Asymmetric synthesis of trifluoromethylsubstituted 3, 3′-pyrrolidinyl-dispirooxindoles through organocatalytic 1, 3-dipolar cycloaddition reactions. Org. Chem. Front. 2017, 4, 472–482. [Google Scholar] [CrossRef]
- Lin, N.; Long, X.-W.; Chen, Q.; Zhu, W.; Wang, B.; Chen, K.; Jiang, C.; Weng, J.; Lu, G. Highly efficient construction of chiral dispirocyclic oxindole/thiobutyrolactam/chromanone complexes through Michael/cyclization cascade reactions with a rosin-based squaramide catalyst. Tetrahedron 2018, 74, 3734–3741. [Google Scholar] [CrossRef]
- Jiang, L.; Long, X.; Huang, W.; Lin, N.; Jiang, C.; Lu, G. Synthesis and characterization of chiral ethyl 7-amino-dehydroabietate. Fine Chem. 2014, 31, 807–811. [Google Scholar]
- Yue, L.; Du, W.; Liu, Y.-K.; Chen, Y.-C. Organocatalytic asymmetric direct Michael addition of aromatic ketones to alkylidenemalononitriles. Tetrahedron Lett. 2008, 49, 3881–3884. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Yield (%) 2 | ee (%) 3 | Config. 4 |
|---|---|---|---|---|
| 1 | I | 88 | 76 | R |
| 2 | II | 31 | 26 | S |
| 3 | III | 36.8 | 18 | R |
| 4 | IV | >99 | 80 | R |
| 5 | V | >99 | 90 | R |
| 6 | VI | 85 | 83 | R |
| 7 | VII | 20 | 23 | S |
| 8 | VIII | 74 | 56 | R |
| 9 | IX | 93 | 75 | R |

| Entry | x | Solvent | Yield (%) 2 | ee (%) 3 |
|---|---|---|---|---|
| 1 | 10 | CH2Cl2 | 99 | 90 |
| 2 | 10 | CHCl3 | 99 | 90 |
| 3 | 10 | CH2ClCH2Cl | 95 | 90 |
| 4 | 10 | MeOH | 88 | 0 |
| 5 | 10 | THF | 32 | 60 |
| 6 | 10 | Et2O | 21 | 59 |
| 7 | 10 | toluene | 35 | 79 |
| 8 4 | 10 | CH2Cl2 | 50 | 91 |
| 9 5 | 10 | CH2Cl2 | 15 | 92 |
| 10 | 5 | CH2Cl2 | 93 | 90 |
| 11 | 1 | CH2Cl2 | 75 | 90 |
| 12 | 0.5 | CH2Cl2 | 75 | 90 |
| 13 | 0.3 | CH2Cl2 | 70 | 90 |
| 14 | 0.1 | CH2Cl2 | 29 | 90 |
| 15 6 | 0.3 | CH2Cl2 | 87 | 90 |

| Entry | R1 | R2 | Product | Yield (%) 2 | ee (%) 3 |
|---|---|---|---|---|---|
| 1 | C6H5 | C6H5 | 4a | 87 | 90 |
| 2 | 4-MeC6H4 | C6H5 | 4b | 97 | 86 |
| 3 | 4-OMeC6H4 | C6H5 | 4c | 92 | 87 |
| 4 | 4-FC6H4 | C6H5 | 4d | 94 | 90 |
| 5 | 4-ClC6H4 | C6H5 | 4e | 72 | 85 |
| 6 | 4-BrC6H4 | C6H5 | 4f | 45 | 85 |
| 7 | 3-ClC6H4 | C6H5 | 4g | 72 | 90 |
| 8 | 3-OMeC6H4 | C6H5 | 4h | 65 | 85 |
| 9 | 2-OMeC6H4 | C6H5 | 4i | 52 | 35 |
| 10 | C6H5 | 4-MeC6H4 | 4j | 64 | 88 |
| 11 | C6H5 | 4-OMeC6H4 | 4k | 76 | 80 |
| 12 | C6H5 | 4-FC6H4 | 4l | 99 | 80 |
| 13 | C6H5 | 4-ClC6H4 | 4m | 57 | 80 |
| 14 | C6H5 | 4-BrC6H4 | 4n | 41 | 79 |
| 15 | 4-MeC6H4 | 4-MeC6H4 | 4o | 70 | 86 |
| 16 | C6H5 | pyridin-2-yl | 4p | 55 | 22 |
| 17 | 1-Naphthyl | C6H5 | 4q | 50 | 39 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, N.; Wei, Q.-X.; Jiang, L.-H.; Deng, Y.-Q.; Zhang, Z.-W.; Chen, Q. Asymmetric Michael Addition of Malononitrile with Chalcones via Rosin-Derived Bifunctional Squaramide. Catalysts 2020, 10, 14. https://doi.org/10.3390/catal10010014
Lin N, Wei Q-X, Jiang L-H, Deng Y-Q, Zhang Z-W, Chen Q. Asymmetric Michael Addition of Malononitrile with Chalcones via Rosin-Derived Bifunctional Squaramide. Catalysts. 2020; 10(1):14. https://doi.org/10.3390/catal10010014
Chicago/Turabian StyleLin, Ning, Qiu-Xiang Wei, Li-Hua Jiang, Yan-Qiu Deng, Zhen-Wei Zhang, and Qing Chen. 2020. "Asymmetric Michael Addition of Malononitrile with Chalcones via Rosin-Derived Bifunctional Squaramide" Catalysts 10, no. 1: 14. https://doi.org/10.3390/catal10010014
APA StyleLin, N., Wei, Q.-X., Jiang, L.-H., Deng, Y.-Q., Zhang, Z.-W., & Chen, Q. (2020). Asymmetric Michael Addition of Malononitrile with Chalcones via Rosin-Derived Bifunctional Squaramide. Catalysts, 10(1), 14. https://doi.org/10.3390/catal10010014