
Targeting Protein Kinase C Downstream of Growth Factor and Adhesion Signalling

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Protein Kinase C: Structure, Function and Activity

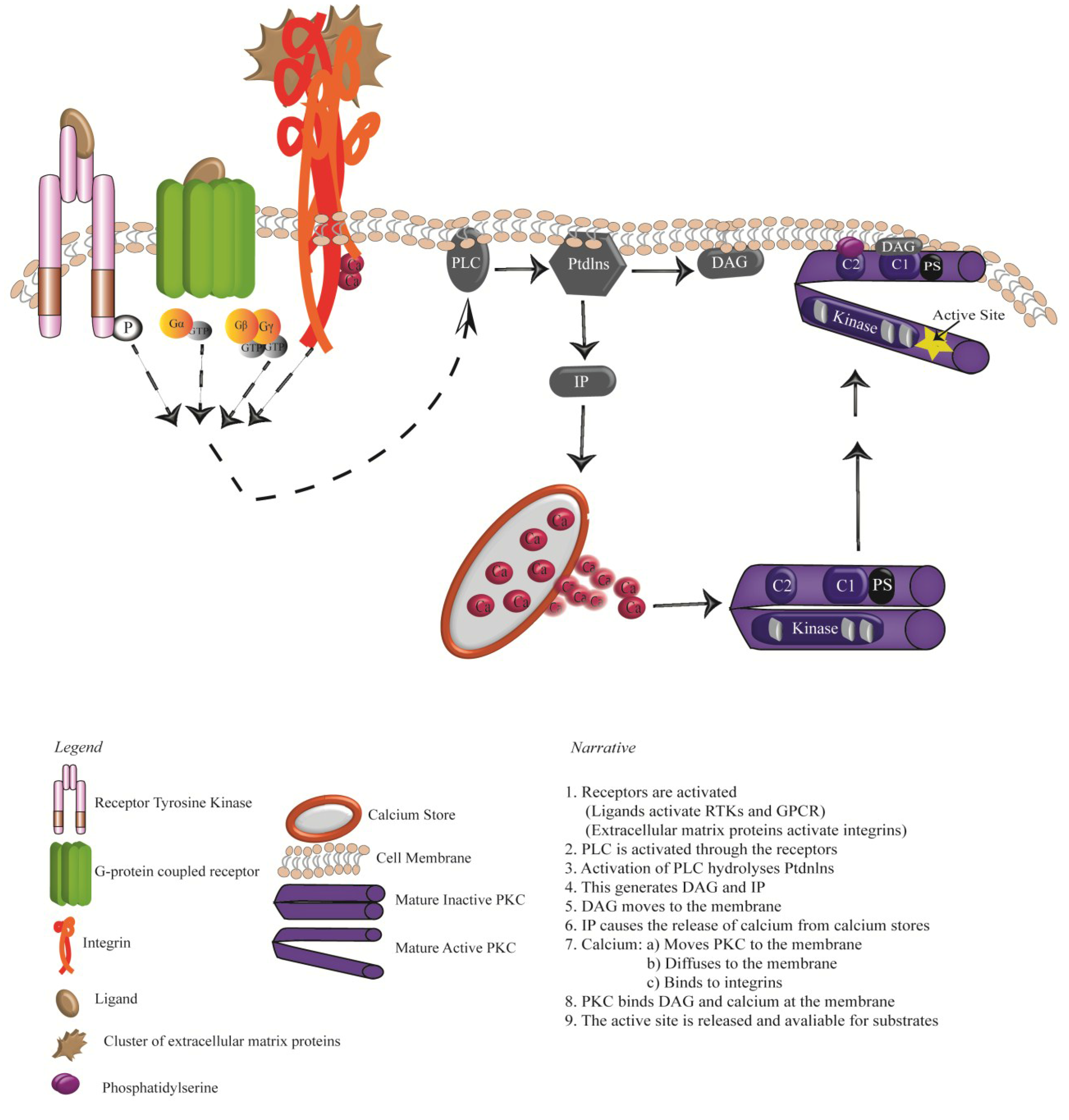
3. Activation of PKCs: Receptor Tyrosine Kinases, G-protein Coupled Receptors and Integrins
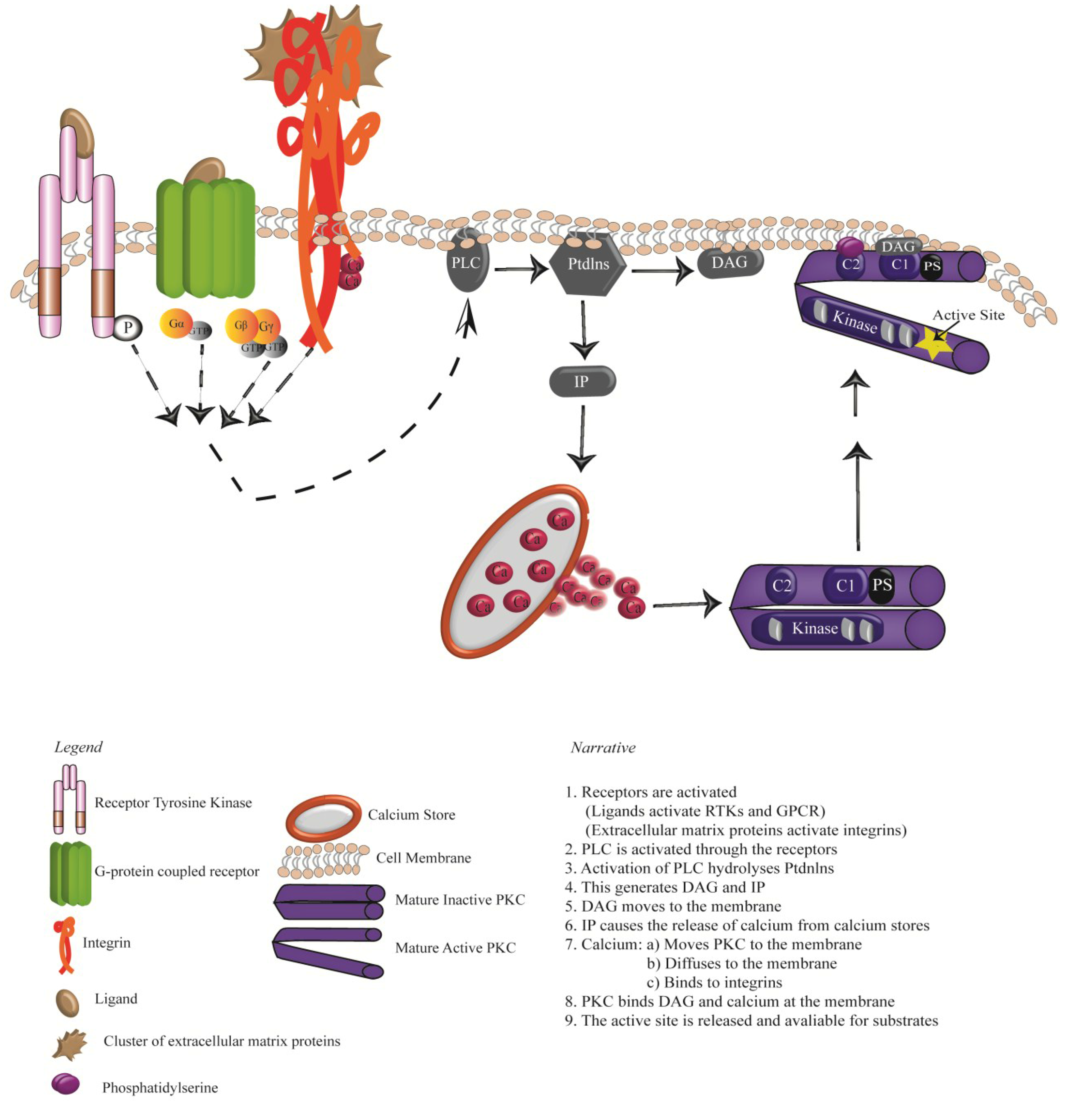
4. Oncogenic Signalling Downstream of RTKS, GPCRs and Integrins
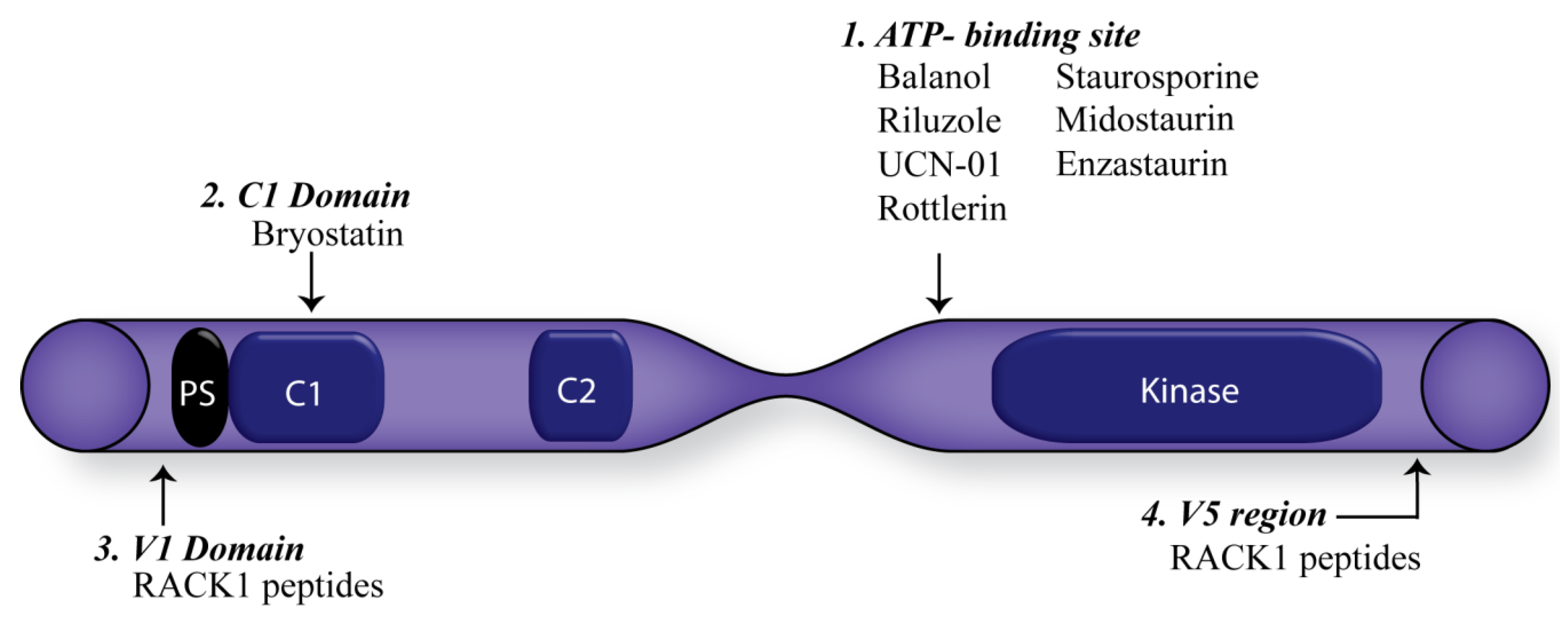
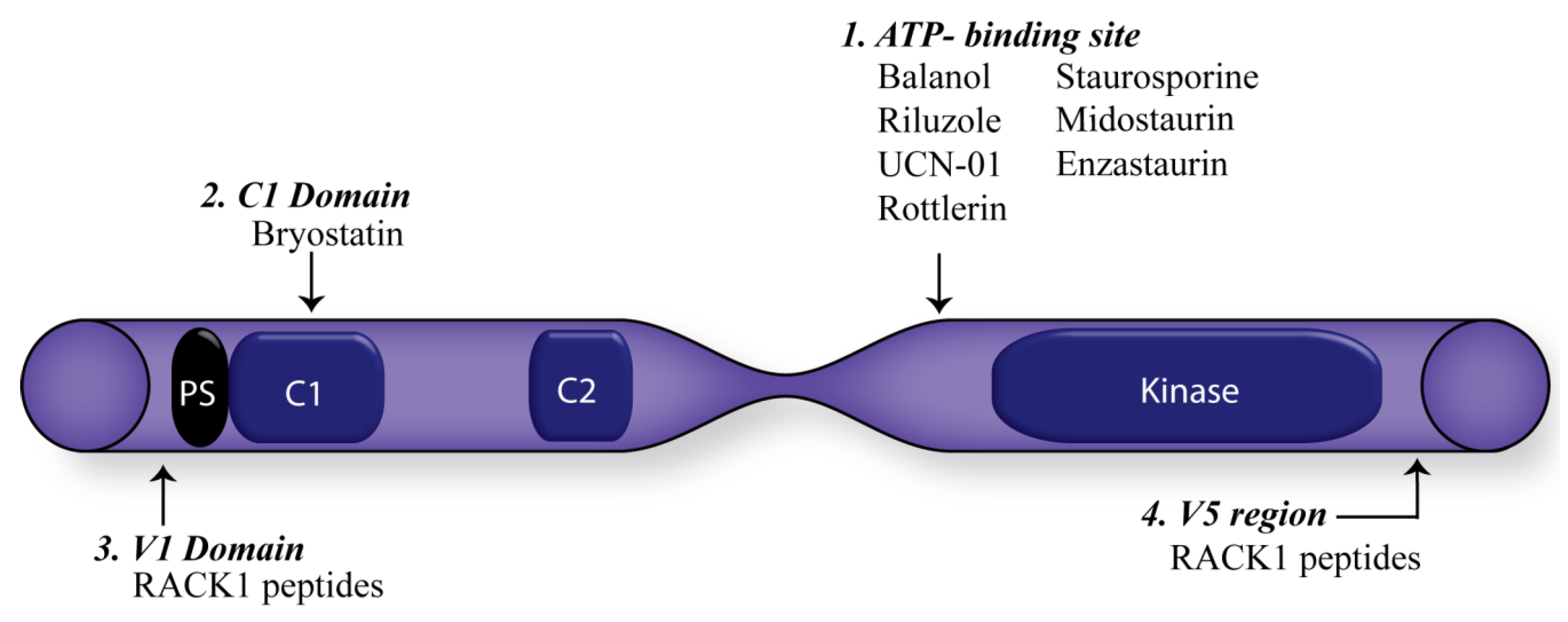
5. Consequences of Targeting PKC
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Mecham, R.P. Overview of extracellular matrix. In Current Protocols in Cell Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; Volume 10. [Google Scholar]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Scott, J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997, 278, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Sever, S.; Faul, C. Signal transduction in podocytes[mdash]spotlight on receptor tyrosine kinases. Nat. Rev. Nephrol. 2014, 10, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Lee, C.S.; Jang, J.-H.; Ghim, J.; Kim, Y.-J.; You, S.; Hwang, D.; Suh, P.-G.; Ryu, S.H. Phospholipase signalling networks in cancer. Nat. Rev. Cancer 2012, 12, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Plopper, G.E.; McNamee, H.P.; Dike, L.E.; Bojanowski, K.; Ingber, D.E. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 1995, 6, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Miyamoto, S. Integrin transmembrane signaling and cytoskeletal control. Curr. Opin.Cell Biol. 1995, 7, 681–689. [Google Scholar] [CrossRef]
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Ron, D.; Kiely, P.A. Rack1, a multifaceted scaffolding protein: Structure and function. Cell Commun. Signal 2011, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Dwane, S.; Durack, E.; O’Connor, R.; Kiely, P.A. Rack1 promotes neurite outgrowth by scaffolding agap2 to fak. Cell. Signal 2014, 26, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.A.; Baillie, G.S.; Barrett, R.; Buckley, D.A.; Adams, D.R.; Houslay, M.D.; O’Connor, R. Phosphorylation of rack1 on tyrosine 52 by c-abl is required for insulin-like growth factor i-mediated regulation of focal adhesion kinase. J. Biol. Chem. 2009, 284, 20263–20274. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.A.; Baillie, G.S.; Lynch, M.J.; Houslay, M.D.; O’Connor, R. Tyrosine 302 in rack1 is essential for insulin-like growth factor-i-mediated competitive binding of pp2a and beta1 integrin and for tumor cell proliferation and migration. J. Biol. Chem. 2008, 283, 22952–22961. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.A.; Leahy, M.; O’Gorman, D.; O’Connor, R. Rack1-mediated integration of adhesion and insulin-like growth factor I (IGF-I) signaling and cell migration are defective in cells expressing an IGF-I receptor mutated at tyrosines 1250 and 1251. J. Biol. Chem. 2005, 280, 7624–7633. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.A.; O’Gorman, D.; Luong, K.; Ron, D.; O’Connor, R. Insulin-like growth factor i controls a mutually exclusive association of rack1 with protein phosphatase 2a and β1 integrin to promote cell migration. Mol. Cell. Biol. 2006, 26, 4041–4051. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.A.; Sant, A.; O’Connor, R. Rack1 is an insulin-like growth factor 1 (IGF-1) receptor-interacting protein that can regulate IGF-1-mediated akt activation and protection from cell death. J. Biol. Chem. 2002, 277, 22581–22589. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, H.C.; Kiely, P.A.; O’Connor, R. Effects of rack1 on cell migration and IGF-I signalling in cardiomyoctes are not dependent on an association with the IGF-IR. Cell. Signal 2007, 19, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Webster, S.; Newton, P. The biology of protein kinase C. In Calcium Signaling; Islam, M.S., Ed.; Springer: Dordrecht, The Netherlands, 2012; Volume 740, pp. 639–661. [Google Scholar]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [Google Scholar] [CrossRef] [PubMed]
- Bosco, R.; Melloni, E.; Celeghini, C.; Rimondi, E.; Vaccarezza, M.; Zauli, G. Fine tuning of protein kinase C (PKC) isoforms in cancer: Shortening the distance from the laboratory to the bedside. Mini Rev. Med. Chem. 2011, 11, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Nishizuka, Y. Protein kinase C isotypes and their specific functions: Prologue. J. Biochem. 2002, 132, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Fogh, B.S.; Multhaupt, H.A.B.; Couchman, J.R. Protein kinase C, focal adhesions and the regulation of cell migration. J. Histochem. Cytochem. 2014, 62, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.; Parker, P. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [PubMed]
- Martiny-Baron, G.; Fabbro, D. Classical PKC isoforms in cancer. Pharmacol. Res. 2007, 55, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Mongiorgi, S.; Cocco, L.; Follo, M.Y. Protein kinase C involvement in cell cycle modulation. Biochem. Soc. Trans. 2014, 42, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Defilippi, P.; Venturino, M.; Gulino, D.; Duperray, A.; Boquet, P.; Fiorentini, C.; Volpe, G.; Palmieri, M.; Silengo, L.; Tarone, G. Dissection of pathways implicated in integrin-mediated actin cytoskeleton assembly involvement of protein kinase C, RHO GTPASE, and tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 21726–21734. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, J.; Spong, S.; Sheppard, D. The integrin alphavbeta6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J. Cell Sci. 1998, 111, 2189–2195. [Google Scholar] [PubMed]
- Bordeleau, F.; Galarneau, L.; Gilbert, S.; Loranger, A.; Marceau, N. Keratin 8/18 modulation of protein kinase C-mediated integrin-dependent adhesion and migration of liver epithelial cells. Mol. Biol. Cell 2010, 21, 1698–1713. [Google Scholar] [CrossRef] [PubMed]
- Gonelli, A.; Mischiati, C.; Guerrini, R.; Voltan, R.; Salvadori, S.; Zauli, G. Perspectives of protein kinase C (PKC) inhibitors as anti-cancer agents. Mini Rev. Med. Chem. 2009, 9, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zong, C.S.; Hermanto, U.; Lopez-Bergami, P.; Ronai, Z.E.; Wang, L.-H. Rack1 recruits STAT3 specifically to insulin and insulin-like growth factor 1 receptors for activation, which is important for regulating anchorage-independent growth. Mol. Cell. Biol. 2006, 26, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Haimovich, B.; Kaneshiki, N.; Ji, P. Protein kinase C regulates tyrosine phosphorylation of PP125FAK in platelets adherent to fibrinogen. Blood 1996, 87, 152–161. [Google Scholar] [PubMed]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.M.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell. Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Violin, J.D.; Kunkel, M.T.; Skovsø, S.; Newton, A.C. Intramolecular conformational changes optimize protein kinase C signaling. Chem. Biol. 2014, 21, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Colón-González, F.; Kazanietz, M.G. C1 domains exposed: From diacylglycerol binding to protein–protein interactions. Biochim. Biophys. Acta 2006, 1761, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Cho, W. Membrane targeting by C1 and C2 domains. J. Biol. Chem. 2001, 276, 32407–32410. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, B.; Stahelin, R.V.; Digman, M.A.; Cho, W. Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J. Biol. Chem. 2003, 278, 46886–46894. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V. Ready, set, go! How protein kinase c manages dynamic signaling. Chem. Biol. 2014, 21, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Oancea, E.; Meyer, T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 1998, 95, 307–318. [Google Scholar] [CrossRef]
- Newton, A. Regulation of the abc kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, A.; Macieira, S.; Velarde, M.; Bädeker, M.; Benda, C.; Jestel, A.; Brandstetter, H.; Neuefeind, T.; Blaesse, M. Crystal structure of the catalytic domain of human atypical protein kinase C-IOTA reveals interaction mode of phosphorylation site in turn motif. J. Mol. Biol. 2005, 352, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Matsubayashi, H.; Amano, T.; Shirai, Y.; Saito, N.; Sakai, N. Phosphorylation of PKC activation loop plays an important role in receptor-mediated translocation of PKC. Genes Cells 2005, 10, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Dutil, E.M.; Newton, A.C. Dual role of pseudosubstrate in the coordinated regulation of protein kinase C by phosphorylation and diacylglycerol. J. Biol. Chem. 2000, 275, 10697–10701. [Google Scholar] [CrossRef] [PubMed]
- Hauge, C.; Antal, T.L.; Hirschberg, D.; Doehn, U.; Thorup, K.; Idrissova, L.; Hansen, K.; Jensen, O.N.; Jørgensen, T.J.; Biondi, R.M. Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 2007, 26, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Behn-Krappa, A.; Newton, A.C. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr. Biol. 1999, 9, 728–737. [Google Scholar] [CrossRef]
- Oliva, J.L.; Griner, E.M.; Kazanietz, M.G. PKC isozymes and diacylglycerol-regulated proteins as effectors of growth factor receptors. Growth Factors 2005, 23, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; DeGudicibus, S.J.; Stacey, D.W. Requirement for C-RAS proteins during viral oncogene transformation. Nature 1986, 320, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Margolis, B.; Rhee, S.; Felder, S.; Mervic, M.; Lyall, R.; Levitzki, A.; Ullrich, A.; Zilberstein, A.; Schlessinger, J. EGF induces tyrosine phosphorylation of phospholipase C-II: A potential mechanism for EGF receptor signaling. Cell 1989, 57, 1101–1107. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem. Sci. 1994, 19, 459–463. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Domin, J.; Waterfield, M.D. Using structure to define the function of phosphoinositide 3-kinase family members. FEBS Lett. 1997, 410, 91–95. [Google Scholar] [CrossRef]
- White, M.F. The IRS-signalling system: A network of docking proteins that mediate insulin action. Mol. Cell. Biochem. 1998, 182, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.L.; Katan, M. Structural views of phosphoinositide-specific phospholipase C: Signalling the way ahead. Structure 1996, 4, 1387–1394. [Google Scholar] [CrossRef]
- Todderud, G.; Wahl, M.I.; Rhee, S.G.; Carpenter, G. Stimulation of phospholipase C-γ 1 membrane association by epidermal growth factor. Science 1990, 249, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.G.; Marie, J.; Wilson, E.; Ives, H.E.; Escobedo, J.; Rosario, M.D.; Mirda, D.; Williams, L.T. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature 1992, 358, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, S.; Wahl, M.I.; Wedegaertner, P.B.; Kim, J.; Rhee, S.G.; Carpenter, G.; Kim, J. Selectivity of phospholipase C phosphorylation by the epidermal growth factor receptor, the insulin receptor, and their cytoplasmic domains. Proc. Natl. Acad. Sci. USA 1990, 87, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Moriya, S.; Kazlauskas, A.; Akimoto, K.; Hirai, S.; Mizuno, K.; Takenawa, T.; Fukui, Y.; Watanabe, Y.; Ozaki, S.; Ohno, S. Platelet-derived growth factor activates protein kinase C ε through redundant and independent signaling pathways involving phospholipase C γ or phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Larose, L.; Gish, G.; Shoelson, S.; Pawson, T. Identification of residues in the beta platelet-derived growth factor receptor that confer specificity for binding to phospholipase C-γ 1. Oncogene 1993, 8, 2493–2499. [Google Scholar] [PubMed]
- Akimoto, K.; Takahashi, R.; Moriya, S.; Nishioka, N.; Takayanagi, J.; Kimura, K.; Fukui, Y.; Osada, S.I.; Mizuno, K.; Hirai, S.I.; et al. EGF or PDGF receptors activate atypical pkclambda through phosphatidylinositol 3-kinase. EMBO J. 1996, 15, 788–798. [Google Scholar] [PubMed]
- Brodie, C.; Bogi, K.; Acs, P.; Lazarovici, P.; Petrovics, G.; Anderson, W.B.; Blumberg, P.M. Protein kinase C-epsilon plays a role in neurite outgrowth in response to epidermal growth factor and nerve growth factor in PC12 cells. Cell Growth Differ. 1999, 10, 183–191. [Google Scholar] [PubMed]
- Greco, S.; Muscella, A.; Elia, M.G.; Salvatore, P.; Storelli, C.; Mazzotta, A.; Manca, C.; Marsigliante, S. Angiotensin II activates extracellular signal regulated kinases via protein kinase C and epidermal growth factor receptor in breast cancer cells. J. Cell. Physiol. 2003, 196, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Burry, R.W. PKC activators (phorbol ester or bryostatin) stimulate outgrowth of ngf-dependent neurites in a subline of PC12 cells. J. Neurosc. Res. 1998, 53, 214–222. [Google Scholar] [CrossRef]
- Welsh, J.B.; Gill, G.N.; Rosenfeld, M.G.; Wells, A. A negative feedback loop attenuates EGF-induced morphological changes. J. Cell Biol. 1991, 114, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Morrison, P.; Takishima, K.; Rosner, M.R. Role of threonine residues in regulation of the epidermal growth factor receptor by protein kinase C and mitogen-activated protein kinase. J. Biol. Chem. 1993, 268, 15536–15543. [Google Scholar] [PubMed]
- Lund, K.; Lazar, C.S.; Chen, W.; Walsh, B.; Welsh, J.; Herbst, J.; Walton, G.; Rosenfeld, M.; Gill, G.; Wiley, H. Phosphorylation of the epidermal growth factor receptor at threonine 654 inhibits ligand-induced internalization and down-regulation. J. Biol. Chem. 1990, 265, 20517–20523. [Google Scholar] [PubMed]
- Cochet, C.; Gill, G.N.; Meisenhelder, J.; Cooper, J.A.; Hunter, T. C-kinase phosphorylates the epidermal growth factor receptor and reduces its epidermal growth factor-stimulated tyrosine protein kinase activity. J. Biol. Chem. 1984, 259, 2553–2558. [Google Scholar] [PubMed]
- Chen, P.; Xie, H.; Wells, A. Mitogenic signaling from the EGF receptor is attenuated by a phospholipase C-γ/protein kinase C feedback mechanism. Mol. Biol. Cell. 1996, 7, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Hackel, P.O.; Zwick, E.; Prenzel, N.; Ullrich, A. Epidermal growth factor receptors: Critical mediators of multiple receptor pathways. Curr. Opin. Cell Biol. 1999, 11, 184–189. [Google Scholar] [CrossRef]
- Chen, P.; Xie, H.; Sekar, M.C.; Gupta, K.; Wells, A. Epidermal growth factor receptor-mediated cell motility: Phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 1994, 127, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Zicha, D.; Parker, P.J. Protein kinase C controls microtubule-based traffic but not proteasomal degradation of C-MET. J. Biol. Chem. 2003, 278, 28921–28929. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Zicha, D.; Parker, P.J. PKC controls HGF-dependent C-MET traffic, signalling and cell migration. EMBO J. 2004, 23, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Davy, A.; Robbins, S.M.; Yong, V.W. Differential activation of ERKS to focal adhesions by PKC epsilon is required for PMA-induced adhesion and migration of human glioma cells. Oncogene 2001, 20, 7398–7407. [Google Scholar] [CrossRef] [PubMed]
- Hashigasako, A.; Machide, M.; Nakamura, T.; Matsumoto, K.; Nakamura, T. Bi-directional regulation of SER-985 phosphorylation of C-met via protein kinase c and protein phosphatase 2a involves C-met activation and cellular responsiveness to hepatocyte growth factor. J. Biol. Chem. 2004, 279, 26445–26452. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Vazquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. pharmacol. 2006, 147, S46–S55. [Google Scholar] [CrossRef] [PubMed]
- Dupré, D.J.; Robitaille, M.; Rebois, R.V.; Hébert, T.E. The role of Gβγ subunits in the organization, assembly, and function of gpcr signaling complexes. Annu. Rev. pharmacol. Toxicol. 2009, 49, 31–56. [Google Scholar] [CrossRef] [PubMed]
- Kolanus, W.; Seed, B. Integrins and inside-out signal transduction: Converging signals from PKC and PIP 3. Curr. Opin. Cell Biol. 1997, 9, 725–731. [Google Scholar] [CrossRef]
- Parekh, D.B.; Ziegler, W.; Parker, P.J. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000, 19, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Disatnik, M.H.; Hernandez-Sotomayor, S.M.; Jones, G.; Carpenter, G.; Mochly-Rosen, D. Phospholipase C-gamma 1 binding to intracellular receptors for activated protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.H.; Du, X.; Plow, E.F. Inside-out integrin signalling. Curr. Opin. Cell Biol. 1992, 4, 766–771. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. Cross-regulation of signaling by itam-associated receptors. Nat. Immunol. 2009, 10, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Couchman, J.R. Protein kinase C involvement in focal adhesion formation. J. Cell Sci. 1992, 101, 277–290. [Google Scholar] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Kazanietz, M.G. New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 1999, 13, 1658–1676. [Google Scholar] [PubMed]
- Barr, L.F.; Mabry, M.; Nelkin, B.D.; Tyler, G.; May, W.S.; Baylin, S.B. C-MYC gene-induced alterations in protein kinase C expression: A possible mechanism facilitating MYC-RAS gene complementation. Cancer Res. 1991, 51, 5514–5519. [Google Scholar] [PubMed]
- Han, E.K.-H.; Cacace, A.M.; Sgambato, A.; Weinstein, I.B. Altered expression of cyclins and C-FOS in R6 cells that overproduce PKCε. Carcinogenesis 1995, 16, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Mans, D.R.A.; Regner, A.; Schwartsmann, G. Targeting protein kinase C: New therapeutic opportunities against high-grade malignant gliomas? Oncologist 2002, 7, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hornia, A.; Jiang, Y.W.; Zang, Q.; Ohno, S.; Foster, D.A. Tumor promotion by depleting cells of protein kinase C delta. Mol. Cell. Biol. 1997, 17, 3418–3428. [Google Scholar] [PubMed]
- Lu, Z.; Liu, D.; Hornia, A.; Devonish, W.; Pagano, M.; Foster, D.A. Activation of protein kinase C triggers its ubiquitination and degradation. Mol. Cell. Biol. 1998, 18, 839–845. [Google Scholar] [PubMed]
- Gould, C.M.; Newton, A.C. The life and death of protein kinase c. Curr. Drug Targets 2008, 9, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, J.; Aaltonen, V.; Peltonen, J. Protein kinase C (PKC) family in cancer progression. Cancer Lett. 2006, 235, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Langzam, L.; Koren, R.; Gal, R.; Kugel, V.; Paz, A.; Farkas, A.; Sampson, S.R. Patterns of protein kinase C isoenzyme expression in transitional cell carcinoma of bladder. Relation to degree of malignancy. Am. J. Clin. Pathol. 2001, 116, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Czifra, G.; Tallai, B.; Nemeth, T.; Kovacs, I.; Kovacs, L.; Biro, T. Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur. Urol. 2004, 46, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Koren, R.; Ben Meir, D.; Langzam, L.; Dekel, Y.; Konichezky, M.; Baniel, J.; Livne, P.M.; Gal, R.; Sampson, S.R. Expression of protein kinase C isoenzymes in benign hyperplasia and carcinoma of prostate. Oncol. Rep. 2004, 11, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Fournier, D.B.; Chisamore, M.; Lurain, J.R.; Rademaker, A.W.; Jordan, V.C.; Tonetti, D.A. Protein kinase C α expression is inversely related to er status in endometrial carcinoma: Possible role in AP-1-mediated proliferation of ER-negative endometrial cancer. Gynecol. Oncol. 2001, 81, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Tsai, M.T.; Su, W.W.; Chen, Y.L.; Wu, T.T.; Hsieh, Y.S.; Huang, C.Y.; Yeh, K.T.; Liu, J.Y. Expression of protein kinase C alpha in biopsies and surgical specimens of human hepatocellular carcinoma. Chin. J. Physiol. 2005, 48, 139–143. [Google Scholar] [PubMed]
- Neill, G.W.; Ghali, L.R.; Green, J.L.; Ikram, M.S.; Philpott, M.P.; Quinn, A.G. Loss of protein kinase calpha expression may enhance the tumorigenic potential of GLI1 in basal cell carcinoma. Cancer Res. 2003, 63, 4692–4697. [Google Scholar] [PubMed]
- Kahl-Rainer, P.; Karner-Hanusch, J.; Weiss, W.; Marian, B. Five of six protein kinase C isoenzymes present in normal mucosa show reduced protein levels during tumor development in the human colon. Carcinogenesis 1994, 15, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Kerfoot, C.; Huang, W.; Rotenberg, S.A. Immunohistochemical analysis of advanced human breast carcinomas reveals downregulation of protein kinase C α. J. Histochem. Cytochem. 2004, 52, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Ways, D.K.; Kukoly, C.A.; deVente, J.; Hooker, J.L.; Bryant, W.O.; Posekany, K.J.; Fletcher, D.J.; Cook, P.P.; Parker, P.J. MCF-7 breast cancer cells transfected with protein kinase C-α exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. J. Clin. Investig. 1995, 95, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Lahn, M.; Kohler, G.; Sundell, K.; Su, C.; Li, S.; Paterson, B.M.; Bumol, T.F. Protein kinase C α expression in breast and ovarian cancer. Oncology 2004, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, J.; Aaltonen, V.; Koskela, S.; Lehenkari, P.; Laato, M.; Peltonen, J. Protein kinase C α/β inhibitor GO6976 promotes formation of cell junctions and inhibits invasion of urinary bladder carcinoma cells. Cancer Res. 2004, 64, 5693–5701. [Google Scholar] [CrossRef] [PubMed]
- Masur, K.; Lang, K.; Niggemann, B.; Zanker, K.S.; Entschladen, F. High PKC α and low E-cadherin expression contribute to high migratory activity of colon carcinoma cells. Mol. Biol. Cell 2001, 12, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, S. Protein kinase C alpha (PKCα): Regulation and biological function. J. Biochem. 2002, 132, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.; Shima, D.; Squire, A.; Bastiaens, P.I.; Gschmeissner, S.; Humphries, M.J.; Parker, P.J. Pkcalpha regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 1999, 18, 3909–3923. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitz, I.; Toker, A.; Mercurio, A.M. Protein kinase C-dependent mobilization of the α6β4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J. Cell. Biol. 1999, 146, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, P.; Winstanley, J.; Pearson, J.; Bishop, H.; Garrod, D. Protein kinase C α expression in normal breast, ductal carcinoma in situ and invasive ductal carcinoma. Eur. J. Cancer 2004, 40, 2269–2273. [Google Scholar] [CrossRef] [PubMed]
- Gilhooly, E.M.; Morse-Gaudio, M.; Bianchi, L.; Reinhart, L.; Rose, D.P.; Connolly, J.M.; Reed, J.A.; Albino, A.P. Loss of expression of protein kinase C β is a common phenomenon in human malignant melanoma: A result of transformation or differentiation? Melanoma Res. 2001, 11, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Weinstein, I.B. Protein kinase c beta enhances growth and expression of cyclin D1 in human breast cancer cells. Cancer Res. 2006, 66, 11399–11408. [Google Scholar] [CrossRef] [PubMed]
- Urtreger, A.J.; Kazanietz, M.G.; Bal de Kier Joffé, E.D. Contribution of individual PKC isoforms to breast cancer progression. IUBMB Life 2012, 64, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Grossoni, V.C.; Todaro, L.B.; Kazanietz, M.G.; de Kier Joffé, E.D.B.; Urtreger, A.J. Opposite effects of protein kinase c beta1 (PKCβ1) and PKCε in the metastatic potential of a breast cancer murine model. Breast Cancer Res.Treat. 2009, 118, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Gökmen-Polar, Y.; Murray, N.R.; Velasco, M.A.; Gatalica, Z.; Fields, A.P. Elevated protein kinase C βII is an early promotive event in colon carcinogenesis. Cancer Res. 2001, 61, 1375–1381. [Google Scholar] [PubMed]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E. The protein kinase Cβ-selective inhibitor, enzastaurin (ly317615. HCL), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-W.; Kim, S.G.; Kim, H.-P.; Kwon, E.; You, J.; Choi, H.-J.; Park, J.-H.; Kang, B.-C.; Im, S.-A.; Kim, T.-Y. Enzastaurin, a protein kinase C β inhibitor, suppresses signaling through the ribosomal S6 kinase and bad pathways and induces apoptosis in human gastric cancer cells. Cancer Res. 2008, 68, 1916–1926. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Kuriyama, S.; Ways, D.K.; Yoshii, J.; Miyamoto, Y.; Kawata, M.; Ikenaka, Y.; Tsujinoue, H.; Nakatani, T.; Shibuya, M. Protein kinase c lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res. 1999, 59, 4413–4418. [Google Scholar] [PubMed]
- Xia, P.; Aiello, L.P.; Ishii, H.; Jiang, Z.Y.; Park, D.J.; Robinson, G.S.; Takagi, H.; Newsome, W.P.; Jirousek, M.R.; King, G.L. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J. Clin. Investig. 1996, 98, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, K.; Hojo, H.; Abe, M. Characterization of expression of protein kinase c isozymes in human b-cell lymphoma: Relationship between its expression and prognosis. Pathol. Int. 2004, 54, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, E.; Adam, A.; de Kier Joffe, E.B.; Aguirre-Ghiso, J.A. Immortalized mammary epithelial cells overexpressing protein kinase C γ acquire a malignant phenotype and become tumorigenic in vivo. Mol. Cancer Res. 2003, 1, 776–787. [Google Scholar] [PubMed]
- Swannie, H.; Kaye, S. Protein kinase c inhibitors. Curr. Oncol. Rep. 2002, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.O.; Karliner, J.S.; Mochly-Rosen, D. A selective ε-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997, 272, 30945–30951. [Google Scholar] [CrossRef] [PubMed]
- Leskow, F.C.; Krasnapolski, M.A.; Urtreger, A.J. The pros and cons of targeting protein kinase C (PKC) in the management of cancer patients. Curr. Pharm. Biotechnol. 2011, 12, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Andrejauskas-Buchdunger, E.; Regenass, U. Differential inhibition of the epidermal growth factor-, platelet-derived growth factor-, and protein kinase C-mediated signal transduction pathways by the staurosporine derivative CGP 41251. Cancer Res. 1992, 52, 5353–5358. [Google Scholar] [PubMed]
- Tenzer, A.; Zingg, D.; Rocha, S.; Hemmings, B.; Fabbro, D.; Glanzmann, C.; Schubiger, P.A.; Bodis, S.; Pruschy, M. The phosphatidylinositide 3ʹ-kinase/akt survival pathway is a target for the anticancer and radiosensitizing agent PKC412, an inhibitor of protein kinase c. Cancer Res. 2001, 61, 8203–8210. [Google Scholar] [PubMed]
- Faul, M.M.; Gillig, J.R.; Jirousek, M.R.; Ballas, L.M.; Schotten, T.; Kahl, A.; Mohr, M. Acyclic n-(azacycloalkyl) bisindolylmaleimides: Isozyme selective inhibitors of PKCβ. Bioorg. Med. Chem. Lett. 2003, 13, 1857–1859. [Google Scholar] [CrossRef]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of su11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Kreisl, T.N.; Kotliarova, S.; Butman, J.A.; Albert, P.S.; Kim, L.; Musib, L.; Thornton, D.; Fine, H.A. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro-oncology 2010, 12, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Kaye, S.B.; Schwartsmann, G. Laboratory and phase I studies of new cancer drugs. Curr. Opin. Oncol. 1992, 4, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Pavlick, A.; Wu, J.; Roberts, J.; Rosenthal, M.; Hamilton, A.; Wadler, S.; Farrell, K.; Carr, M.; Fry, D.; Murgo, A.; et al. Phase I study of bryostatin 1, a protein kinase C modulator, preceding cisplatin in patients with refractory non-hematologic tumors. Cancer Chemother. Pharmacol. 2009, 64, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Souroujon, M.C.; Mochly-Rosen, D. Peptide modulators of protein–protein interactions in intracellular signaling. Nat. Biotechnol. 1998, 16, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Choi, Y.-L.; Vallentin, A.; Hunrichs, B.S.; Hellerstein, M.K.; Peehl, D.M.; Mochly-Rosen, D. Centrosomal PKCβII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008, 68, 6831–6839. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Koyanagi, T.; Palaniyandi, S.S.; Fajardo, G.; Churchill, E.N.; Budas, G.; Disatnik, M.-H.; Bernstein, D.; Brum, P.C.; Mochly-Rosen, D. Pharmacological inhibition of βIIPKC is cardioprotective in late-stage hypertrophy. J. Mol. Cell. Cardiol. 2011, 51, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [PubMed]
- O’Brian, C.A.; Liskamp, R.M.; Solomon, D.H.; Weinstein, I.B. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985, 45, 2462–2465. [Google Scholar] [PubMed]
- Kikkawa, U.; Takai, Y.; Tanaka, Y.; Miyake, R.; Nishizuka, Y. Protein kinase C as a possible receptor protein of tumor-promoting phorbol esters. J. Biol. Chem. 1983, 258, 11442–11445. [Google Scholar] [PubMed]
- Blobe, G.C.; Obeid, L.M.; Hannun, Y.A. Regulation of protein kinase C and role in cancer biology. Cancer Metastasis Rev. 1994, 13, 411–431. [Google Scholar] [CrossRef] [PubMed]
- Reyland, M. Protein kinase cdelta and apoptosis. Biochem. Soc. Trans. 2007, 35, 1001-04. [Google Scholar] [PubMed]
- Symonds, J.M.; Ohm, A.M.; Carter, C.J.; Heasley, L.E.; Boyle, T.A.; Franklin, W.A.; Reyland, M.E. Protein kinase C δ is a downstream effector of oncogenic k-ras in lung tumors. Cancer Res. 2011, 71, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.V.; Grossoni, V.C.; Urtreger, A.J.; Yang, C.; Colombo, L.L.; Morandi, A.; Pallotta, M.G.; Kazanietz, M.G.; de Kier Joffé, E.D.B.; Puricelli, L.L. PKC delta (PKCδ) promotes tumoral progression of human ductal pancreatic cancer. Pancreas 2010, 39, e31–e41. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ulloa, L.B.; Hernandez-Maqueda, J.G.; Santoyo-Ramos, P.; Castaneda-Patlan, M.C.; Robles-Flores, M. Protein kinase C ζ is a positive modulator of canonical wnt signaling pathway in tumoral colon cell lines. Carcinogenesis 2011, 32, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tao, Y.; Duran, A.; Llado, V.; Galvez, A.; Barger, J.F.; Castilla, E.A.; Chen, J.; Yajima, T.; Porollo, A. Control of nutrient stress-induced metabolic reprogramming by PKCζ in tumorigenesis. Cell 2013, 152, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase c family: Are we there yet? Nat. Rev. Cancer 2007, 7, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B. Cancer-associated protein kinase c mutations reveal kinase’s role as tumor suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cao, F.; Wang, Y.; Meng, F.; Zhang, Y.; Zhong, D.; Zhou, Q. The Protein Kinase C (PKC) inhibitors combined with chemotherapy in the treatment of advanced non-small cell lung cancer: Meta-analysis of randomized controlled trials. Clin. Trans. Oncol. 2015, 17, 371–377. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowling, C.M.; Kiely, P.A. Targeting Protein Kinase C Downstream of Growth Factor and Adhesion Signalling. Cancers 2015, 7, 1271-1291. https://doi.org/10.3390/cancers7030836
Dowling CM, Kiely PA. Targeting Protein Kinase C Downstream of Growth Factor and Adhesion Signalling. Cancers. 2015; 7(3):1271-1291. https://doi.org/10.3390/cancers7030836
Chicago/Turabian StyleDowling, Catríona M., and Patrick A. Kiely. 2015. "Targeting Protein Kinase C Downstream of Growth Factor and Adhesion Signalling" Cancers 7, no. 3: 1271-1291. https://doi.org/10.3390/cancers7030836
APA StyleDowling, C. M., & Kiely, P. A. (2015). Targeting Protein Kinase C Downstream of Growth Factor and Adhesion Signalling. Cancers, 7(3), 1271-1291. https://doi.org/10.3390/cancers7030836