
Neural Regulation of Pancreatic Cancer: A Novel Target for Intervention


{kind=link}
Abstract
:1. Introduction
2. Nerve Fibers Are a Component of the Pancreatic Tumor Microenvironment
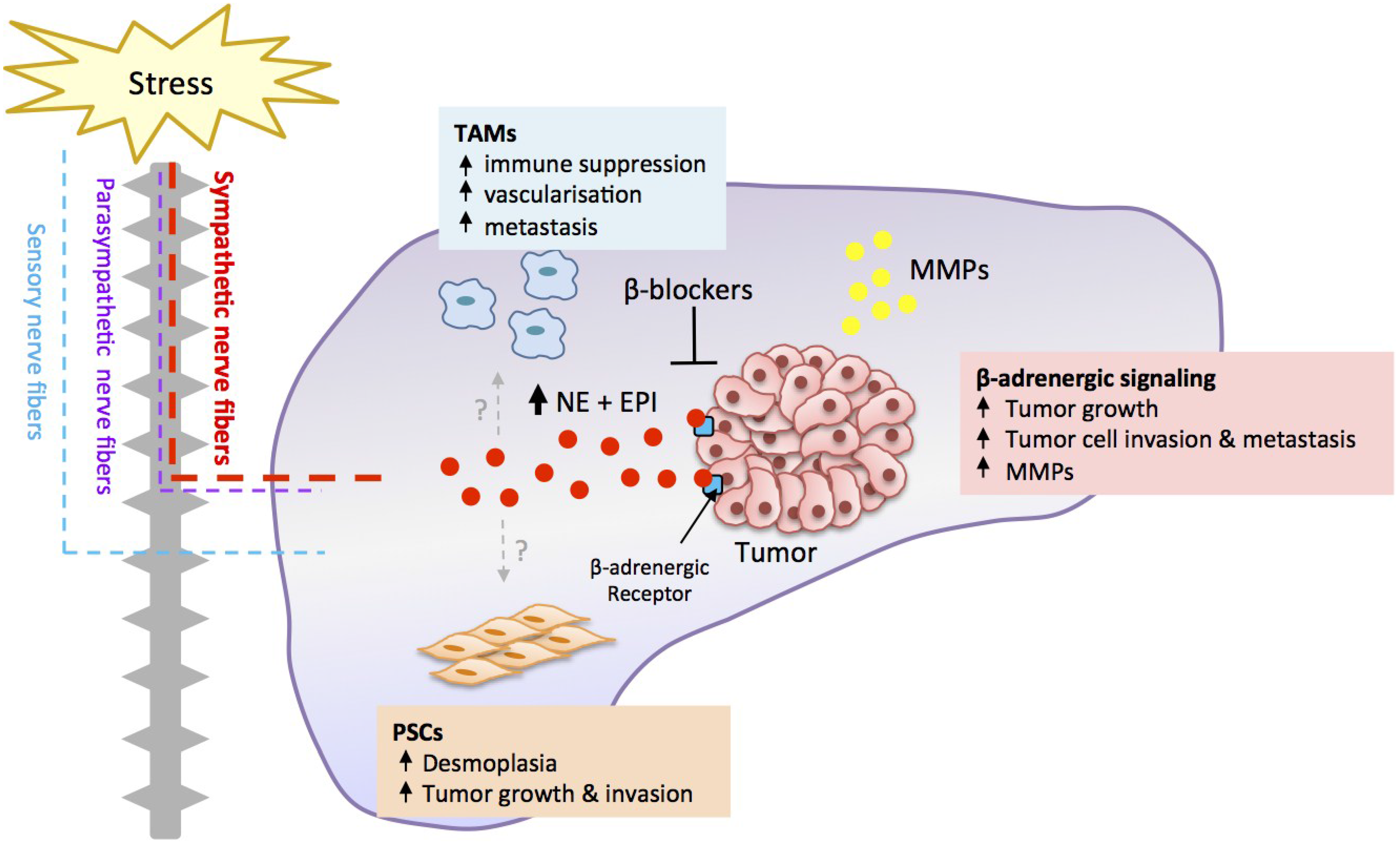
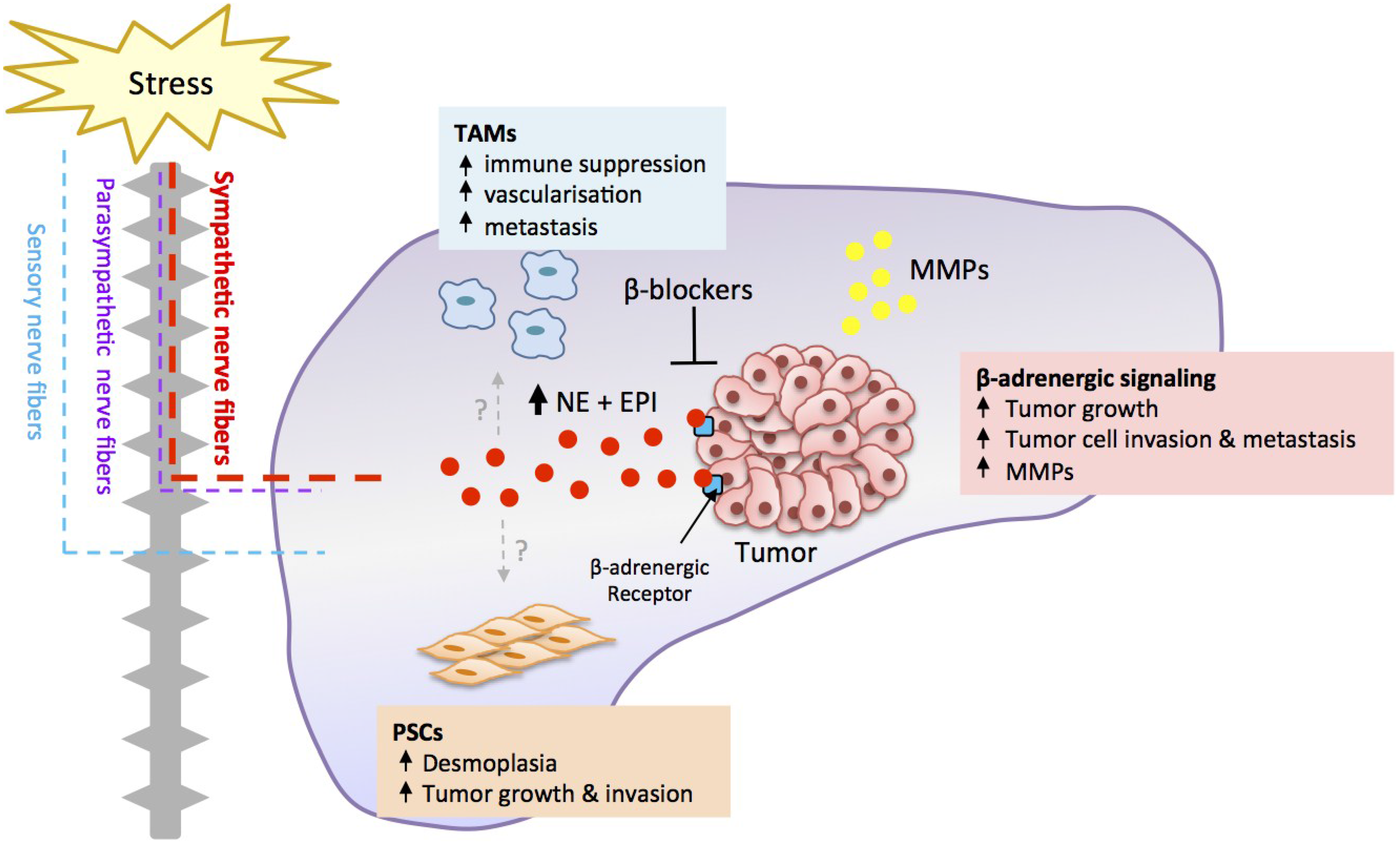
3. Orthotopic Preclinical Models Recapitulate Tumor-Stromal Interactions
4. Stress Regulates Pancreatic Cancer Progression
5. Stress Acts through a β-Adrenergic Signaling Pathway
6. β-Adrenergic Signaling Drives Tumor Cell Invasion
7. Stress Regulates Tumor Cell Proliferation and Apoptosis
8. Translation of β-Blockers for the Treatment of Pancreatic Cancer
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H. Progress in the knowledge and treatment of advanced pancreatic cancer: From benchside to bedside. Cancer Treat. Rev. 2014, 40, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Hirai, I.; Kimura, W.; Ozawa, K.; Kudo, S.; Suto, K.; Kuzu, H.; Fuse, A. Perineural invasion in pancreatic cancer. Pancreas 2002, 24, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Jiang, Y.; Jiang, Y.; Sun, Y.; Zhao, X. Expression of nerve growth factor and tyrosine kinase receptor a and correlation with perineural invasion in pancreatic cancer. J. Gastroenterol. Hepatol. 2008, 23, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, G.O.; Bergmann, F.; Kadihasanoglu, M.; Altintas, B.; Demir, I.E.; Hinz, U.; Muller, M.W.; Giese, T.; Buchler, M.W.; Giese, N.A.; et al. Pancreatic neuropathy and neuropathic pain—A comprehensive pathomorphological study of 546 cases. Gastroenterology 2009, 136, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Pour, P.M.; Bell, R.H.; Batra, S.K. Neural invasion in the staging of pancreatic cancer. Pancreas 2003, 26, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H. Anatomy of the pancreas and spleen. Surgery 2013, 31, 263–266. [Google Scholar] [CrossRef]
- Bond-Smith, G.; Banga, N.; Hammond, T.M.; Imber, C.J. Pancreatic adenocarcinoma. BMJ 2012, 344, e2476. [Google Scholar] [CrossRef] [PubMed]
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Moller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic stellate cells are an important source of mmp-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and cam assay. J. Cell Sci. 2007, 120, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Peng, S.J.; Chien, H.J. Imaging of the islet neural network. Diabetes Obes. Metab. 2014, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Diaz, R.; Abdulreda, M.H.; Formoso, A.L.; Gans, I.; Ricordi, C.; Berggren, P.O.; Caicedo, A. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab. 2011, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bockman, D.E. Nerves in the pancreas: What are they for? Am. J. Surg. 2007, 194, S61–S64. [Google Scholar] [CrossRef]
- Teff, K.L. Visceral nerves: Vagal and sympathetic innervation. JPEN J. Parenter. Enteral. Nutr. 2008, 32, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, S.; Olsson, C. Autonomic control of glands and secretion: A comparative view. Auton. Neurosci. 2011, 165, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B.; Ericson, L.E.; Lundquist, I.; Loren, I.; Sundler, F. Adrenergic innervation of pancreatic islets and modulation of insulin secretion by the sympatho-adrenal system. Cell Tissue Res. 1981, 216, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Kurose, T.; Seino, Y.; Nishi, S.; Tsuji, K.; Taminato, T.; Tsuda, K.; Imura, H. Mechanism of sympathetic neural regulation of insulin, somatostatin, and glucagon secretion. Am. J. Physiol. 1990, 258, E220–E227. [Google Scholar] [PubMed]
- Hisatomi, A.; Maruyama, H.; Orci, L.; Vasko, M.; Unger, R.H. Adrenergically mediated intrapancreatic control of the glucagon response to glucopenia in the isolated rat pancreas. J. Clin. Investig. 1985, 75, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Stopczynski, R.E.; Normolle, D.P.; Hartman, D.J.; Ying, H.; DeBerry, J.J.; Bielefeldt, K.; Rhim, A.D.; DePinho, R.A.; Albers, K.M.; Davis, B.M. Neuroplastic changes occur early in the development of pancreatic ductal adenocarcinoma. Cancer Res. 2014, 74, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, G.O.; Giese, N.A.; Erkan, M.; Kerscher, A.G.; Wente, M.N.; Giese, T.; Buchler, M.W.; Friess, H. The neurotrophic factor artemin promotes pancreatic cancer invasion. Ann. Surg. 2006, 244, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, G.O.; Schafer, K.H.; Kerscher, A.G.; Rauch, U.; Demir, I.E.; Kadihasanoglu, M.; Bohm, C.; Muller, M.W.; Buchler, M.W.; Giese, N.A.; et al. Nerve growth factor and artemin are paracrine mediators of pancreatic neuropathy in pancreatic adenocarcinoma. Ann. Surg. 2010, 251, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lu, K.Y. Neural invasion in pancreatic carcinoma. Hepatobiliary Pancreat. Dis. Int. 2002, 1, 469–476. [Google Scholar] [PubMed]
- Weiner, H. Perturbing the Organism: The Biology of Stressful Experience; University of Chicago Press: Chicago, IL, USA, 1992. [Google Scholar]
- Haass, M.; Kubler, W. Nicotine and sympathetic neurotransmission. Cardiovasc. Drugs Ther. 1997, 10, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, M.H.; Al-Wadei, H.A.; Schuller, H.M. Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors alpha3, alpha5, and alpha7. Mol. Cancer Res. 2012, 10, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, M.H.; Al-Wadei, H.A.; Schuller, H.M. Effects of chronic nicotine on the autocrine regulation of pancreatic cancer cells and pancreatic duct epithelial cells by stimulatory and inhibitory neurotransmitters. Carcinogenesis 2012, 33, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Loscalzo, M.; Trask, P.C.; Zabora, J.; Philip, E.J. Psychological distress in patients with pancreatic cancer—An understudied group. Psychooncology 2010, 19, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Zabora, J.; BrintzenhofeSzoc, K.; Curbow, B.; Hooker, C.; Piantadosi, S. The prevalence of psychological distress by cancer site. Psychooncology 2001, 10, 19–28. [Google Scholar] [CrossRef]
- Carlson, L.E.; Angen, M.; Cullum, J.; Goodey, E.; Koopmans, J.; Lamont, L.; MacRae, J.H.; Martin, M.; Pelletier, G.; Robinson, J.; et al. High levels of untreated distress and fatigue in cancer patients. Br. J. Cancer 2004, 90, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Wang, H.C.; Yuan, Z.; Huang, J.; Zheng, Q. Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via beta-adrenergic receptor-dependent activation of p38/mapk pathway. Hepato-Gastroenterology 2012, 59, 889–893. [Google Scholar] [PubMed]
- Zhang, D.; Ma, Q.; Shen, S.; Hu, H. Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: The study of beta-adrenoceptor antagonist's anticancer effect in pancreatic cancer cell. Pancreas 2009, 38, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Ma, Q.; Li, J.; Wang, Z.; Shan, T.; Li, W.; Xu, Q.; Xie, K. Interaction of the sympathetic nerve with pancreatic cancer cells promotes perineural invasion through the activation of stat3 signaling. Mol. Cancer Ther. 2013, 12, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Ma, Q.Y.; Wang, L.C.; Hu, H.T.; Li, J.H.; Zhang, D.; Zhang, M. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol. Rep. 2009, 22, 825–830. [Google Scholar] [PubMed]
- Shan, T.; Ma, J.; Ma, Q.; Guo, K.; Guo, J.; Li, X.; Li, W.; Liu, J.; Huang, C.; Wang, F.; et al. Beta2-ar-hif-1alpha: A novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis. Curr. Mol. Med. 2013, 13, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Al-Wadei, H.A.; Ullah, M.F.; Plummer, H.K., 3rd. Regulation of pancreatic cancer by neuropsychological stress responses: A novel target for intervention. Carcinogenesis 2012, 33, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Fidler, I.J.; Coombes, K.R. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res. 2007, 67, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Borgstrom, P.; Oh, P.; Czarny, M.; Racine, B.; Schnitzer, J.E. Co-implanting orthotopic tissue creates stroma microenvironment enhancing growth and angiogenesis of multiple tumors. F1000 Res. 2013, 2, 129. [Google Scholar] [CrossRef] [PubMed]
- Wilmanns, C.; Fan, D.; O’Brian, C.A.; Bucana, C.D.; Fidler, I.J. Orthotopic and ectopic organ environments differentially influence the sensitivity of murine colon carcinoma cells to doxorubicin and 5-fluorouracil. Int. J. Cancer 1992, 52, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Radinsky, R.; Fan, D.; Tsan, R.; Bucana, C.D.; Wilmanns, C.; Fidler, I.J. Organ-specific modulation of steady-state mdr gene expression and drug resistance in murine colon cancer cells. J. Natl. Cancer Inst. 1994, 86, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Graves, E.E.; Vilalta, M.; Cecic, I.K.; Erler, J.T.; Tran, P.T.; Felsher, D.; Sayles, L.; Sweet-Cordero, A.; Le, Q.T.; Giaccia, A.J. Hypoxia in models of lung cancer: Implications for targeted therapeutics. Clin. Cancer Res. 2010, 16, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.S.; Poon, P.C.; Owen, S.C.; Shoichet, M.S. Blood vessel hyperpermeability and pathophysiology in human tumour xenograft models of breast cancer: A comparison of ectopic and orthotopic tumours. BMC Cancer 2012, 12, 579. [Google Scholar] [CrossRef] [PubMed]
- Westwood, J.A.; Potdevin Hunnam, T.C.; Pegram, H.J.; Hicks, R.J.; Darcy, P.K.; Kershaw, M.H. Routes of delivery for cpg and anti-cd137 for the treatment of orthotopic kidney tumors in mice. PLoS ONE 2014, 9, e95847. [Google Scholar] [CrossRef] [PubMed]
- Graves, E.E.; Maity, A.; Le, Q.T. The tumor microenvironment in non-small-cell lung cancer. Semin. Radiat. Oncol. 2010, 20, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Morikawa, K.; Fabra, A.; Bucana, C.D.; Fidler, I.J. Influence of organ environment on extracellular matrix degradative activity and metastasis of human colon carcinoma cells. J. Natl. Cancer Inst. 1990, 82, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Jiang, L.; Wang, X.Q.; Pan, W.; She, F.F.; Chen, Y.L. Establishment of and comparison between orthotopic xenograft and subcutaneous xenograft models of gallbladder carcinoma. Asian Pac. J. Cancer Prev. 2014, 15, 3747–3752. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, R.A.; Dinney, C.P.; Gohji, K.; Ordonez, N.G.; Killion, J.J.; Fidler, I.J. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J. Natl. Cancer Inst. 1992, 84, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Greene, G.F.; Kitadai, Y.; Pettaway, C.A.; von Eschenbach, A.C.; Bucana, C.D.; Fidler, I.J. Correlation of metastasis-related gene expression with metastatic potential in human prostate carcinoma cells implanted in nude mice using an in situ messenger rna hybridization technique. Am. J. Pathol. 1997, 150, 1571–1582. [Google Scholar] [PubMed]
- Singh, R.K.; Bucana, C.D.; Gutman, M.; Fan, D.; Wilson, M.R.; Fidler, I.J. Organ site-dependent expression of basic fibroblast growth factor in human renal cell carcinoma cells. Am. J. Pathol. 1994, 145, 365–374. [Google Scholar] [PubMed]
- Tan, M.H.; Chu, T.M. Characterization of the tumorigenic and metastatic properties of a human pancreatic tumor-cell line (aspc-1) implanted orthotopically into nude-mice. Tumour Biol. 1985, 6, 89–98. [Google Scholar] [PubMed]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Marincola, F.M.; Drucker, B.J.; Siao, D.Y.; Hough, K.L.; Holder, W.D., Jr. The nude mouse as a model for the study of human pancreatic cancer. J. Surg. Res. 1989, 47, 520–529. [Google Scholar] [CrossRef]
- Fu, X.; Guadagni, F.; Hoffman, R.M. A metastatic nude-mouse model of human pancreatic cancer constructed orthotopically with histologically intact patient specimens. Proc. Natl. Acad. Sci. USA 1992, 89, 5645–5649. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Yang, M.; Nardin, S.; Wang, X.; Jiang, P.; Baranov, E.; Moossa, A.R.; Hoffman, R.M. Chronologically-specific metastatic targeting of human pancreatic tumors in orthotopic models. Clin. Exp. Metastasis 2000, 18, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.S.; Kaushal, S.; Kono, Y.; Cao, H.S.; Hoffman, R.M.; Bouvet, M. Complementarity of ultrasound and fluorescence imaging in an orthotopic mouse model of pancreatic cancer. BMC Cancer 2009, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.G.; Kim-Fuchs, C.; Angst, E.; Sloan, E.K. Bioluminescent orthotopic model of pancreatic cancer progression. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Merk, J.; Rolff, J.; Becker, M.; Leschber, G.; Fichtner, I. Patient-derived xenografts of non-small-cell lung cancer: A pre-clinical model to evaluate adjuvant chemotherapy? Eur. J. Cardiothorac. Surg. 2009, 36, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.Y.; Lee, W.Y.; Jung, S.; Hong, H.K.; Nam, D.H.; Park, Y.A.; Huh, J.W.; Yun, S.H.; Kim, H.C.; Chun, H.K.; et al. Correlation between tumor engraftment in patient-derived xenograft models and clinical outcomes in colorectal cancer patients. Oncotarget 2015, 6, 16059–16068. [Google Scholar]
- Gupta, J.; Igea, A.; Papaioannou, M.; Lopez-Casas, P.P.; Llonch, E.; Hidalgo, M.; Gorgoulis, V.G.; Nebreda, A.R. Pharmacological inhibition of p38 mapk reduces tumor growth in patient-derived xenografts from colon tumors. Oncotarget 2015, 6, 8539–8551. [Google Scholar] [PubMed]
- Schuller, A.G.; Barry, E.R.; Jones, R.D.; Henry, R.E.; Frigault, M.M.; Beran, G.; Linsenmayer, D.; Hattersley, M.; Smith, A.; Wilson, J.; et al. The met inhibitor azd6094 (savolitinib, hmpl-504) induces regression in papillary renal cell carcinoma patient-derived xenograft models. Clin. Cancer Res. 2015, 21, 2811–2819. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Lindberg, J.M.; Adair, S.J.; Newhook, T.E.; Cowan, C.R.; Stokes, J.B.; Borgman, C.A.; Stelow, E.B.; Lowrey, B.T.; Chopivsky, M.E.; et al. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the mek inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/her2 inhibitor lapatinib. Neoplasia 2013, 15, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.; Morizono, K.; Karanikolas, B.D.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef] [PubMed]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R., Jr.; Danial, N.; et al. Behavioral stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Lamkin, D.M.; Sloan, E.K.; Patel, A.J.; Chiang, B.S.; Pimentel, M.A.; Ma, J.C.; Arevalo, J.M.; Morizono, K.; Cole, S.W. Chronic stress enhances progression of acute lymphoblastic leukemia via beta-adrenergic signaling. Brain Behav. Immun. 2012, 26, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; Hollande, F.; Sloan, E.K. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun. 2014, 40, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W.; Carroll, A.R.; Spannuth, W.A.; Deavers, M.T.; Allen, J.K.; et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Investig. 2010, 120, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Hishinuma, S.; Ogata, Y.; Tomikawa, M.; Ozawa, I.; Hirabayashi, K.; Igarashi, S. Patterns of recurrence after curative resection of pancreatic cancer, based on autopsy findings. J. Gastrointest. Surg. 2006, 10, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Reiser, C.; Hinz, U.; Bachmann, J.; Debus, J.; Jaeger, D.; Friess, H.; Buchler, M.W. Surgery for recurrent pancreatic ductal adenocarcinoma. Ann. Surg. 2007, 245, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Armaiz-Pena, G.N.; Gonzalez-Villasana, V.; Nagaraja, A.S.; Rodriguez-Aguayo, C.; Sadaoui, N.C.; Stone, R.L.; Matsuo, K.; Dalton, H.J.; Previs, R.A.; Jennings, N.B.; et al. Adrenergic regulation of monocyte chemotactic protein 1 leads to enhanced macrophage recruitment and ovarian carcinoma growth. Oncotarget 2015, 6, 4266–4273. [Google Scholar] [PubMed]
- Weddle, D.L.; Tithoff, P.; Williams, M.; Schuller, H.M. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 2001, 22, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Askari, M.D.; Tsao, M.S.; Schuller, H.M. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of egf receptors. J. Cancer Res. Clin. Oncol. 2005, 131, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ma, Q.Y.; Hu, H.T.; Zhang, M. Beta2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting creb, nfkappab and ap-1. Cancer Biol. Ther. 2010, 10, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ma, Q.; Wang, Z.; Zhang, M.; Guo, K.; Wang, F.; Wu, E. Beta2-adrenoceptor blockage induces g1/s phase arrest and apoptosis in pancreatic cancer cells via ras/akt/nfkappab pathway. Mol. Cancer 2011, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.P.; Luo, K.; Lv, Z.W.; Huang, J. Beta-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepato-Gastroenterology 2012, 59, 584–588. [Google Scholar] [PubMed]
- Kondo, H.; Takeuchi, S.; Togari, A. Beta-adrenergic signaling stimulates osteoclastogenesis via reactive oxygen species. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E507–E515. [Google Scholar] [CrossRef] [PubMed]
- Stiles, J.; Amaya, C.; Pham, R.; Rowntree, R.K.; Lacaze, M.; Mulne, A.; Bischoff, J.; Kokta, V.; Boucheron, L.E.; Mitchell, D.C.; et al. Propranolol treatment of infantile hemangioma endothelial cells: A molecular analysis. Exp. Ther. Med. 2012, 4, 594–604. [Google Scholar] [PubMed]
- Turner, N.A.; Porter, K.E.; Smith, W.H.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef]
- Lamyel, F.; Warnken-Uhlich, M.; Seemann, W.K.; Mohr, K.; Kostenis, E.; Ahmedat, A.S.; Smit, M.; Gosens, R.; Meurs, H.; Miller-Larsson, A.; et al. The beta2-subtype of adrenoceptors mediates inhibition of pro-fibrotic events in human lung fibroblasts. Naunyn Schmied. Arch. Pharmacol. 2011, 384, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gaba b receptor is a novel drug target for pancreatic cancer. Cancer 2008, 112, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Munoz, S.A.; Zijlstra, A.; Yang, X.; Sterling, J.A.; et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol. 2012, 10, e1001363. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Bloomston, M.; Zervos, E.E.; Rosemurgy, A.S., 2nd. Matrix metalloproteinases and their role in pancreatic cancer: A review of preclinical studies and clinical trials. Ann. Surg. Oncol. 2002, 9, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Lutgendorf, S.K.; Lamkin, D.M.; Jennings, N.B.; Arevalo, J.M.; Penedo, F.; DeGeest, K.; Langley, R.R.; Lucci, J.A., 3rd; Cole, S.W.; Lubaroff, D.M.; et al. Biobehavioral influences on matrix metalloproteinase expression in ovarian carcinoma. Clin. Cancer Res. 2008, 14, 6839–6846. [Google Scholar] [CrossRef] [PubMed]
- Maatta, M.; Soini, Y.; Liakka, A.; Autio-Harmainen, H. Differential expression of matrix metalloproteinase (mmp)-2, mmp-9, and membrane type 1-mmp in hepatocellular and pancreatic adenocarcinoma: Implications for tumor progression and clinical prognosis. Clin. Cancer Res. 2000, 6, 2726–2734. [Google Scholar] [PubMed]
- Durlik, M.; Gardian, K. Metalloproteinase 2 and 9 activity in the development of pancreatic cancer. Pol. Prz. Chir. 2012, 84, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Izeboud, C.A.; Mocking, J.A.; Monshouwer, M.; van Miert, A.S.; Witkamp, R.F. Participation of beta-adrenergic receptors on macrophages in modulation of lps-induced cytokine release. J. Recept. Signal Transduct. Res. 1999, 19, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Nemeth, Z.H.; Szabo, C.; Zsilla, G.; Salzman, A.L.; Vizi, E.S. Isoproterenol inhibits il-10, tnf-alpha, and nitric oxide production in raw 264.7 macrophages. Brain Res. Bull. 1998, 45, 183–187. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.S.; Albo, D.; Kimsey, T.F.; White, S.L.; Wang, T.N. Macrophage inflammatory protein-3alpha promotes pancreatic cancer cell invasion. J. Surg. Res. 2005, 123, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Kestler, H.A.; Holzmann, K.; Ellenrieder, V.; Schneiderhan, W.; Siech, M.; Adler, G.; Bachem, M.G.; Gress, T.M. Transcriptome analysis of human hepatic and pancreatic stellate cells: Organ-specific variations of a common transcriptional phenotype. J. Mol. Med. (Berl.) 2005, 83, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Oben, J.A.; Yang, S.; Lin, H.; Ono, M.; Diehl, A.M. Norepinephrine and neuropeptide y promote proliferation and collagen gene expression of hepatic myofibroblastic stellate cells. Biochem. Biophys. Res. Commun. 2003, 302, 685–690. [Google Scholar] [CrossRef]
- Sastry, K.S.; Karpova, Y.; Prokopovich, S.; Smith, A.J.; Essau, B.; Gersappe, A.; Carson, J.P.; Weber, M.J.; Register, T.C.; Chen, Y.Q.; et al. Epinephrine protects cancer cells from apoptosis via activation of camp-dependent protein kinase and bad phosphorylation. J. Biol. Chem. 2007, 282, 14094–14100. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.W.; Reed, C.B.; Kokolus, K.M.; Pitoniak, R.; Utley, A.; Bucsek, M.J.; Ma, W.W.; Repasky, E.A.; Hylander, B.L. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat. Commun. 2015, 6, 6426. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, K.; Xiao, X.; Zheng, S.; Xu, T.; Chen, S. Effects of propranolol on the proliferation and apoptosis of hemangioma-derived endothelial cells. J. Pediatr. Surg. 2012, 47, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.K.; Wolter, N.E.; Blanch, A.; Partridge, T.; Cheng, L.; Morgenstern, D.A.; Podkowa, M.; Kaplan, D.R.; Irwin, M.S. Anti-tumor activity of the beta-adrenergic receptor antagonist propranolol in neuroblastoma. Oncotarget 2014, 5, 161–172. [Google Scholar] [PubMed]
- Wrobel, L.J.; Le Gal, F.A. Inhibition of human melanoma growth by a non-cardioselective beta-blocker. J. Investig. Dermatol. 2015, 135, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Che, X.; Zhao, W.; Zhang, D.; Bi, T.; Wang, G. The beta-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor kappab signaling. Oncol. Rep. 2010, 24, 1669–1676. [Google Scholar] [PubMed]
- Storniolo, A.M.; Enas, N.H.; Brown, C.A.; Voi, M.; Rothenberg, M.L.; Schilsky, R. An investigational new drug treatment program for patients with gemcitabine: Results for over 3000 patients with pancreatic carcinoma. Cancer 1999, 85, 1261–1268. [Google Scholar] [CrossRef]
- Rougier, P.; Riess, H.; Manges, R.; Karasek, P.; Humblet, Y.; Barone, C.; Santoro, A.; Assadourian, S.; Hatteville, L.; Philip, P.A. Randomised, placebo-controlled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. Eur. J. Cancer 2013, 49, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, R.; Bodoky, G.; Ruhstaller, T.; Glimelius, B.; Bajetta, E.; Schuller, J.; Saletti, P.; Bauer, J.; Figer, A.; Pestalozzi, B.; et al. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: A randomized, multicenter, phase iii trial of the swiss group for clinical cancer research and the central european cooperative oncology group. J. Clin. Oncol. 2007, 25, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.; Storniolo, A.M. Assessing clinical benefit in the treatment of pancreas cancer: Gemcitabine compared to 5-fluorouracil. Eur. J. Cancer 1997, 33, S18–S22. [Google Scholar] [CrossRef]
- Zhou, R.Y.; Shanas, R.; Nelson, M.A.; Bhattacharyya, A.; Shi, J.Q. Increased expression of the heterogeneous nuclear ribonucleoprotein k in pancreatic cancer and its association with the mutant p53. Int. J. Cancer 2010, 126, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.M.; Carey, I.M.; Owen, C.G.; Harris, T.; Dewilde, S.; Cook, D.G. Does beta-adrenoceptor blocker therapy improve cancer survival? Findings from a population-based retrospective cohort study. Br. J. Clin. Pharmacol. 2011, 72, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, G.S.; Babu, K.G.; Bondarde, S.A.; Biswas, G.; Ranade, A.; Parikh, P.M.; Bascomb, N.F.; Malhotra, H. Effect of coadministered beta blocker and cox-2 inhibitor to patients with pancreatic cancer prior to receiving albumin-bound (nab) paclitaxel. J. Clin. Oncol. 2015, 33, Abstrat 302. [Google Scholar]
- Powe, D.G.; Voss, M.J.; Zanker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010, 1, 628–638. [Google Scholar] [PubMed]
- Melhem-Bertrandt, A.; Chavez-Macgregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Munzone, E.; Rotmensz, N.; Cipolla, C.; de Giorgi, V.; Santillo, B.; Zanelotti, A.; Adamoli, L.; Colleoni, M.; Viale, G.; et al. Therapeutic effect of beta-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res. Treat. 2013, 140, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Barron, T.I.; Connolly, R.M.; Sharp, L.; Bennett, K.; Visvanathan, K. Beta blockers and breast cancer mortality: A population- based study. J. Clin. Oncol. 2011, 29, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Grytli, H.H.; Fagerland, M.W.; Fossa, S.D.; Tasken, K.A.; Haheim, L.L. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate 2013, 73, 250–260. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Grazzini, M.; Gandini, S.; Benemei, S.; Lotti, T.; Marchionni, N.; Geppetti, P. Treatment with beta-blockers and reduced disease progression in patients with thick melanoma. Arch. Int. Med. 2011, 171, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Lemeshow, S.; Sorensen, H.T.; Phillips, G.; Yang, E.V.; Antonsen, S.; Riis, A.H.; Lesinski, G.B.; Jackson, R.; Glaser, R. Beta-blockers and survival among danish patients with malignant melanoma: A population-based cohort study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.S.; Karlan, B.Y.; Li, A.J. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol. Oncol. 2012, 127, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O'Reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Neeman, E.; Sharon, E.; Ben-Eliyahu, S. Exploiting the critical perioperative period to improve long-term cancer outcomes. Nat. Rev. Clin. Oncol. 2015, 12, 213–226. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, A.; Kim-Fuchs, C.; Le, C.P.; Hollande, F.; Sloan, E.K. Neural Regulation of Pancreatic Cancer: A Novel Target for Intervention. Cancers 2015, 7, 1292-1312. https://doi.org/10.3390/cancers7030838
Chang A, Kim-Fuchs C, Le CP, Hollande F, Sloan EK. Neural Regulation of Pancreatic Cancer: A Novel Target for Intervention. Cancers. 2015; 7(3):1292-1312. https://doi.org/10.3390/cancers7030838
Chicago/Turabian StyleChang, Aeson, Corina Kim-Fuchs, Caroline P. Le, Frédéric Hollande, and Erica K. Sloan. 2015. "Neural Regulation of Pancreatic Cancer: A Novel Target for Intervention" Cancers 7, no. 3: 1292-1312. https://doi.org/10.3390/cancers7030838
APA StyleChang, A., Kim-Fuchs, C., Le, C. P., Hollande, F., & Sloan, E. K. (2015). Neural Regulation of Pancreatic Cancer: A Novel Target for Intervention. Cancers, 7(3), 1292-1312. https://doi.org/10.3390/cancers7030838