
Prospective Screening of Cancer Syndromes in Patients with Mesenchymal Tumors

 , , , , ,
, , , , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Whole Genome and Transcriptome Analysis
2.3. Identification and Reporting of Germline Variants
2.4. Identification of Somatic Variants
2.5. Methylation
2.6. Statistical Analyses
3. Results
3.1. A Minority of Sarcoma Patients Have Tumor Predisposition Syndromes
3.2. Many Germline Variants Currently Have Limited Clinical Utility
3.3. Somatic Analysis Confirms Biallelic Inactivation and Establishes Sarcoma Syndromes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020; Volume 5. [Google Scholar]
- Burningham, Z.; Hashibe, M.; Spector, L.; Schiffman, J.D. The epidemiology of sarcoma. Clin. Sarcoma Res. 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Farid, M.; Ngeow, J. Sarcomas Associated with Genetic Cancer Predisposition Syndromes: A Review. Oncologist 2016, 21, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Yang, Q.; Friedman, J.M. Mortality in neurofibromatosis 1: An analysis using U.S. death certificates. Am. J. Hum. Genet. 2001, 68, 1110–1118. [Google Scholar] [CrossRef]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Kleinerman, R.A.; Schonfeld, S.J.; Tucker, M.A. Sarcomas in hereditary retinoblastoma. Clin. Sarcoma Res. 2012, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Öfverholm, I.; Wallander, K.; Haglund, C.; Chellappa, V.; Wejde, J.; Gellerbring, A.; Wirta, V.; Renevey, A.; Caceres, E.; Tsagkozis, P.; et al. Comprehensive Genomic Profiling Alters Clinical Diagnoses in a Significant Fraction of Tumors Suspicious of Sarcoma. Clin. Cancer Res. 2024, 30, 2647–2658. [Google Scholar] [CrossRef]
- Tesi, B.; Robinson, K.L.; Abel, F.; Díaz de Ståhl, T.; Orrsjö, S.; Poluha, A.; Hellberg, M.; Wessman, S.; Samuelsson, S.; Frisk, T.; et al. Diagnostic yield and clinical impact of germline sequencing in children with CNS and extracranial solid tumors-a nationwide, prospective Swedish study. Lancet Reg. Health Eur. 2024, 39, 100881. [Google Scholar] [CrossRef]
- MIP Piepline. Available online: https://github.com/Clinical-Genomics/MIP/blob/develop/documentation/README.md (accessed on 10 October 2023).
- Stranneheim, H.; Lagerstedt-Robinson, K.; Magnusson, M.; Kvarnung, M.; Nilsson, D.; Lesko, N.; Engvall, M.; Anderlid, B.M.; Arnell, H.; Johansson, C.B.; et al. Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021, 13, 40. [Google Scholar] [CrossRef]
- Balsamic Pipline. Available online: https://balsamic.readthedocs.io/en/v12.0.2/ (accessed on 22 December 2023).
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef]
- Wallander, K.; Thonberg, H.; Nilsson, D.; Tham, E. Massive parallel sequencing in individuals with multiple primary tumours reveals the benefit of re-analysis. Hered. Cancer Clin. Pract. 2021, 19, 46. [Google Scholar] [CrossRef]
- Gargano, M.A.; Matentzoglu, N.; Coleman, B.; Addo-Lartey, E.B.; Anagnostopoulos, A.V.; Anderton, J.; Avillach, P.; Bagley, A.M.; Bakštein, E.; Balhoff, J.P.; et al. The Human Phenotype Ontology in 2024: Phenotypes around the world. Nucleic Acids Res. 2024, 52, D1333–D1346. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.S.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Andeer, R.; Dalberg, M.; Laaksonen, M.; Magnusson, M.; Nilsson, D.; Rasi, C. Scout, Clinical DNA variant visualizer and browser, repository code. Available online: https://github.com/Clinical-Genomics/scout (accessed on 17 September 2020).
- Sondka, Z.; Dhir, N.B.; Carvalho-Silva, D.; Jupe, S.; Madhumita; McLaren, K.; Starkey, M.; Ward, S.; Wilding, J.; Ahmed, M.; et al. COSMIC: A curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 2024, 52, D1210–D1217. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Nationellt Vårdprogram för Bröstcancer (Regional Cancer Centra, Sweden). Available online: https://kunskapsbanken.cancercentrum.se/diagnoser/brostcancer/vardprogram/ (accessed on 17 September 2020).
- Carvalho, N.A.; Santiago, K.M.; Maia, J.M.L.; Costa, F.D.; Formiga, M.N.; Soares, D.C.Q.; Paixão, D.; Mello, C.A.L.; Costa, C.; Rocha, J.; et al. Prevalence and clinical implications of germline pathogenic variants in cancer predisposing genes in young patients across sarcoma subtypes. J. Med. Genet. 2023, 61, 61–68. [Google Scholar] [CrossRef]
- Yap, T.A.; Ashok, A.; Stoll, J.; Mauer, E.; Nepomuceno, V.M.; Blackwell, K.L.; Garber, J.E.; Meric-Bernstam, F. Prevalence of Germline Findings Among Tumors From Cancer Types Lacking Hereditary Testing Guidelines. JAMA Netw. Open 2022, 5, e2213070. [Google Scholar] [CrossRef]
- Ballinger, M.L.; Pattnaik, S.; Mundra, P.A.; Zaheed, M.; Rath, E.; Priestley, P.; Baber, J.; Ray-Coquard, I.; Isambert, N.; Causeret, S.; et al. Heritable defects in telomere and mitotic function selectively predispose to sarcomas. Science 2023, 379, 253–260. [Google Scholar] [CrossRef]
- Thariat, J.; Chevalier, F.; Orbach, D.; Ollivier, L.; Marcy, P.Y.; Corradini, N.; Beddok, A.; Foray, N.; Bougeard, G. Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol. 2021, 22, e562–e574. [Google Scholar] [CrossRef]
- Therkildsen, C.; Jensen, L.H.; Rasmussen, M.; Bernstein, I. An Update on Immune Checkpoint Therapy for the Treatment of Lynch Syndrome. Clin. Exp. Gastroenterol. 2021, 14, 181–197. [Google Scholar] [CrossRef]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef]
- Fiala, E.M.; Jayakumaran, G.; Mauguen, A.; Kennedy, J.A.; Bouvier, N.; Kemel, Y.; Fleischut, M.H.; Maio, A.; Salo-Mullen, E.E.; Sheehan, M.; et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat. Cancer 2021, 2, 357–365. [Google Scholar] [CrossRef]
- Mutetwa, T.; Goudie, C.; Foulkes, W.D.; Polak, P. Companion Tumor Sequencing to Assess the Clinical Significance of Germline Sequencing in Children With Cancer. JAMA Netw. Open 2021, 4, e2135135. [Google Scholar] [CrossRef]
- Borja, N.A.; Silva-Smith, R.; Huang, M.; Parekh, D.J.; Sussman, D.; Tekin, M. Atypical ATMs: Broadening the phenotypic spectrum of ATM-associated hereditary cancer. Front. Oncol. 2023, 13, 1068110. [Google Scholar] [CrossRef]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef]
- Nurmi, A.; Muranen, T.A.; Pelttari, L.M.; Kiiski, J.I.; Heikkinen, T.; Lehto, S.; Kallioniemi, A.; Schleutker, J.; Bützow, R.; Blomqvist, C.; et al. Recurrent moderate-risk mutations in Finnish breast and ovarian cancer patients. Int. J. Cancer 2019, 145, 2692–2700. [Google Scholar] [CrossRef]
- Van Goethem, G.; Dermaut, B.; Löfgren, A.; Martin, J.J.; Van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001, 28, 211–212. [Google Scholar] [CrossRef]
- Lv, M.; Zhang, S.; Dong, Y.; Cao, L.; Guo, S. PolG Inhibits Gastric Cancer Glycolysis and Viability by Suppressing PKM2 Phosphorylation. Cancer Manag. Res. 2021, 13, 1559–1570. [Google Scholar] [CrossRef]
- Vyas, B.; Puri, R.D.; Namboodiri, N.; Nair, M.; Sharma, D.; Movva, S.; Saxena, R.; Bohora, S.; Aggarwal, N.; Vora, A.; et al. KCNQ1 mutations associated with Jervell and Lange-Nielsen syndrome and autosomal recessive Romano-Ward syndrome in India-expanding the spectrum of long QT syndrome type 1. Am. J. Med. Genet. A 2016, 170, 1510–1519. [Google Scholar] [CrossRef]
- Tommiska, J.; Känsäkoski, J.; Skibsbye, L.; Vaaralahti, K.; Liu, X.; Lodge, E.J.; Tang, C.; Yuan, L.; Fagerholm, R.; Kanters, J.K.; et al. Two missense mutations in KCNQ1 cause pituitary hormone deficiency and maternally inherited gingival fibromatosis. Nat. Commun. 2017, 8, 1289. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, C.; Meng, L.; Wang, Z.; Chen, D.; Peng, Y.; Yang, K.; Bian, Z. Activated KCNQ1 channel promotes fibrogenic response in hereditary gingival fibromatosis via clustering and activation of Ras. J. Periodontal Res. 2021, 56, 471–481. [Google Scholar] [CrossRef]
- Than, B.L.; Goos, J.A.; Sarver, A.L.; O’Sullivan, M.G.; Rod, A.; Starr, T.K.; Fijneman, R.J.; Meijer, G.A.; Zhao, L.; Zhang, Y.; et al. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene 2014, 33, 3861–3868. [Google Scholar] [CrossRef]
- Rapetti-Mauss, R.; Berenguier, C.; Allegrini, B.; Soriani, O. Interplay Between Ion Channels and the Wnt/β-Catenin Signaling Pathway in Cancers. Front. Pharmacol. 2020, 11, 525020. [Google Scholar] [CrossRef]
- van der Tuin, K.; Tops, C.M.J.; Adank, M.A.; Cobben, J.M.; Hamdy, N.A.T.; Jongmans, M.C.; Menko, F.H.; van Nesselrooij, B.P.M.; Netea-Maier, R.T.; Oosterwijk, J.C.; et al. CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 4534–4540. [Google Scholar] [CrossRef]
- Poumeaud, F.; Valentin, T.; Vande Perre, P.; Jaffrelot, M.; Bonnet, D.; Chibon, F.; Chevreau, C.; Selves, J.; Guimbaud, R.; Fares, N. Special features of sarcomas developed in patients with Lynch syndrome: A systematic review. Crit. Rev. Oncol. Hematol. 2023, 188, 104055. [Google Scholar] [CrossRef]
- de Angelis de Carvalho, N.; Niitsuma, B.N.; Kozak, V.N.; Costa, F.D.; de Macedo, M.P.; Kupper, B.E.C.; Silva, M.L.G.; Formiga, M.N.; Volc, S.M.; Aguiar Junior, S.; et al. Clinical and Molecular Assessment of Patients with Lynch Syndrome and Sarcomas Underpinning the Association with MSH2 Germline Pathogenic Variants. Cancers 2020, 12, 1848. [Google Scholar] [CrossRef]
- Naslund-Koch, C.; Nordestgaard, B.G.; Bojesen, S.E. Increased Risk for Other Cancers in Addition to Breast Cancer for CHEK2(star)1100delC Heterozygotes Estimated From the Copenhagen General Population Study. J. Clin. Oncol. 2016, 34, 1208–1216. [Google Scholar] [CrossRef]
- Bychkovsky, B.L.; Agaoglu, N.B.; Horton, C.; Zhou, J.; Yussuf, A.; Hemyari, P.; Richardson, M.E.; Young, C.; LaDuca, H.; McGuinness, D.L.; et al. Differences in Cancer Phenotypes Among Frequent CHEK2 Variants and Implications for Clinical Care-Checking CHEK2. JAMA Oncol. 2022, 8, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Abdelghani, E.; Schieffer, K.M.; Cottrell, C.E.; Audino, A.; Zajo, K.; Shah, N. CHEK2 Alterations in Pediatric Malignancy: A Single-Institution Experience. Cancers 2023, 15, 1649. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Olivier, M.; Bergemann, T.L.; Hainaut, P. Sarcomas in TP53 germline mutation carriers: A review of the IARC TP53 database. Cancer 2012, 118, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, R.A.; Tucker, M.A.; Abramson, D.H.; Seddon, J.M.; Tarone, R.E.; Fraumeni, J.F., Jr. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J. Natl. Cancer Inst. 2007, 99, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef]
- Mandelker, D.; Marra, A.; Mehta, N.; Selenica, P.; Yelskaya, Z.; Yang, C.; Somar, J.; Mehine, M.; Misyura, M.; Basturk, O.; et al. Expanded genetic testing of GIST patients identifies high proportion of non-syndromic patients with germline alterations. NPJ Precis. Oncol. 2023, 7, 1. [Google Scholar] [CrossRef]
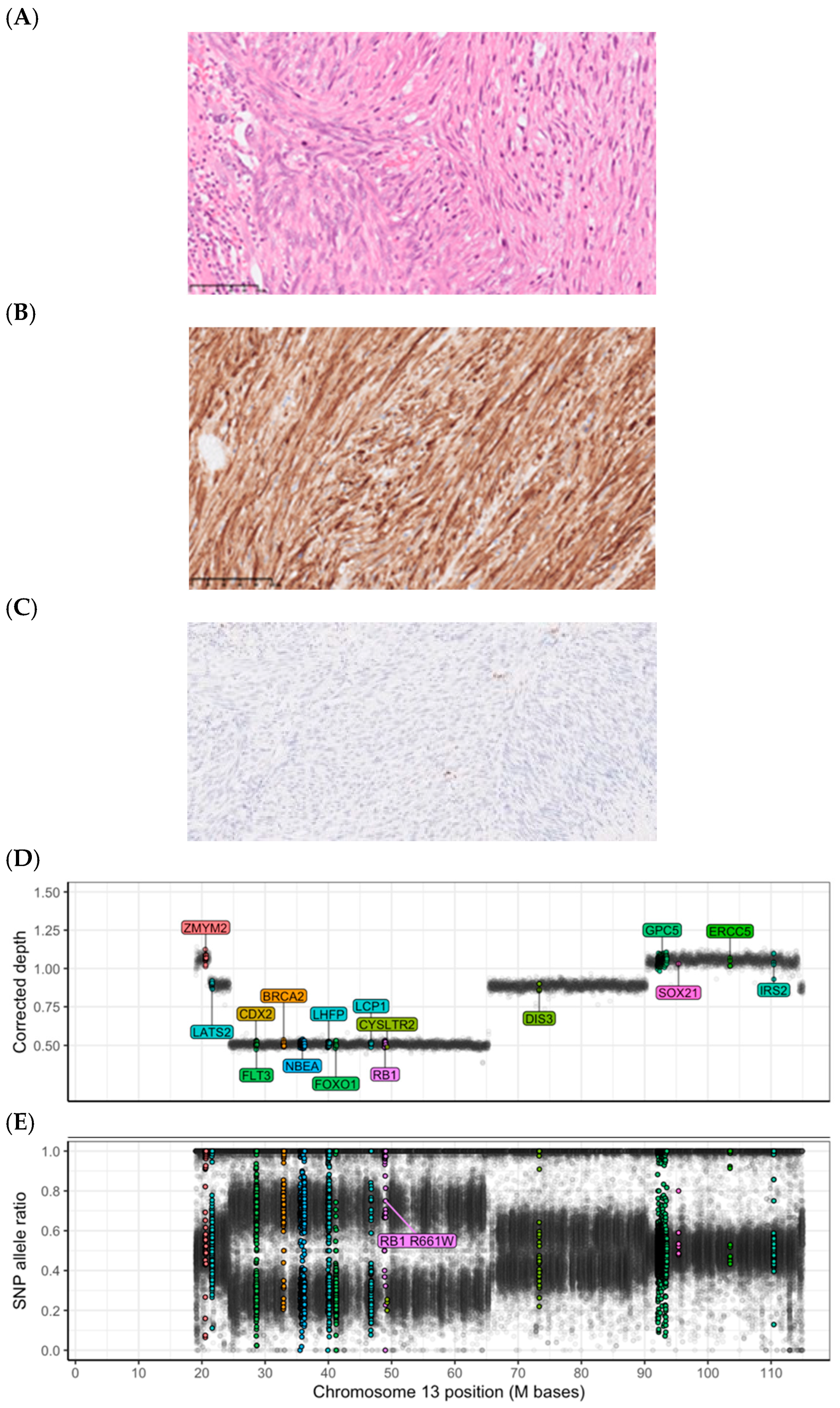
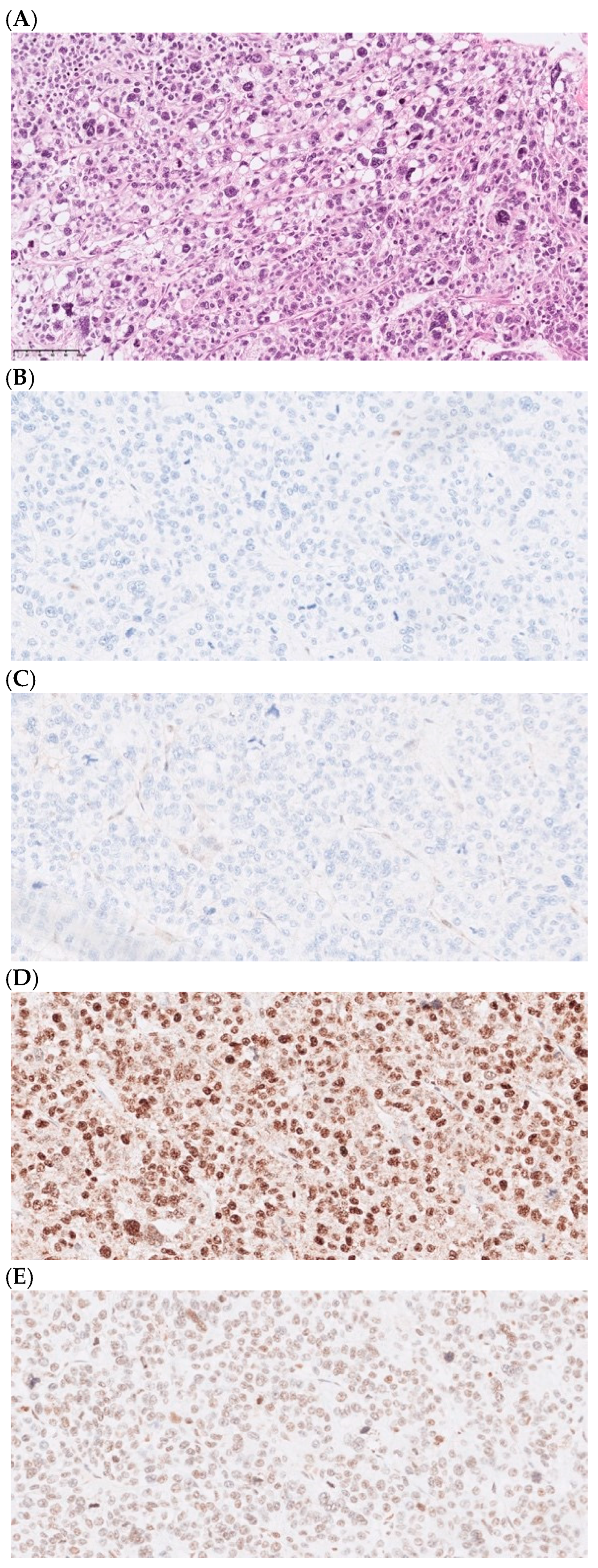
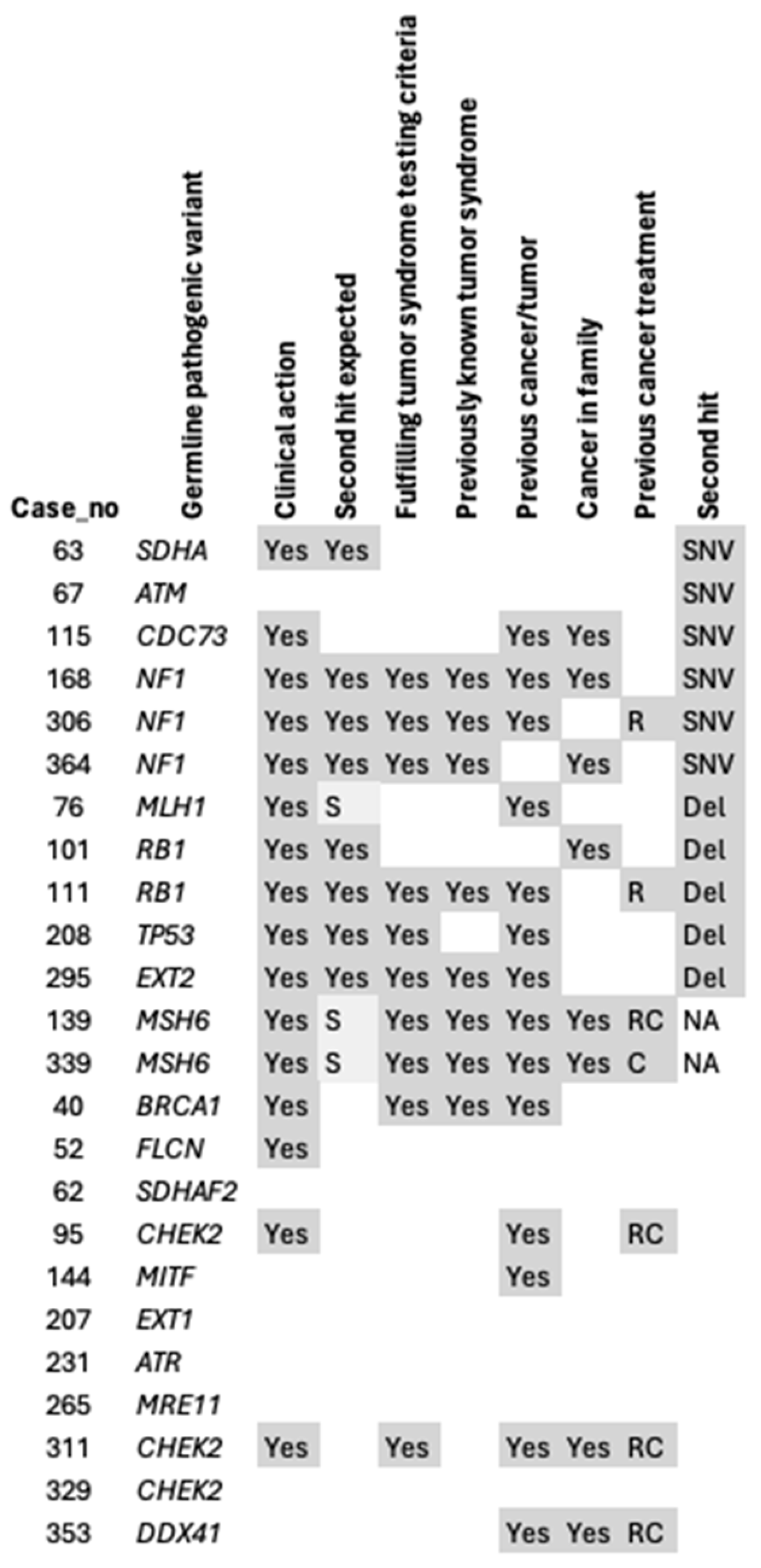
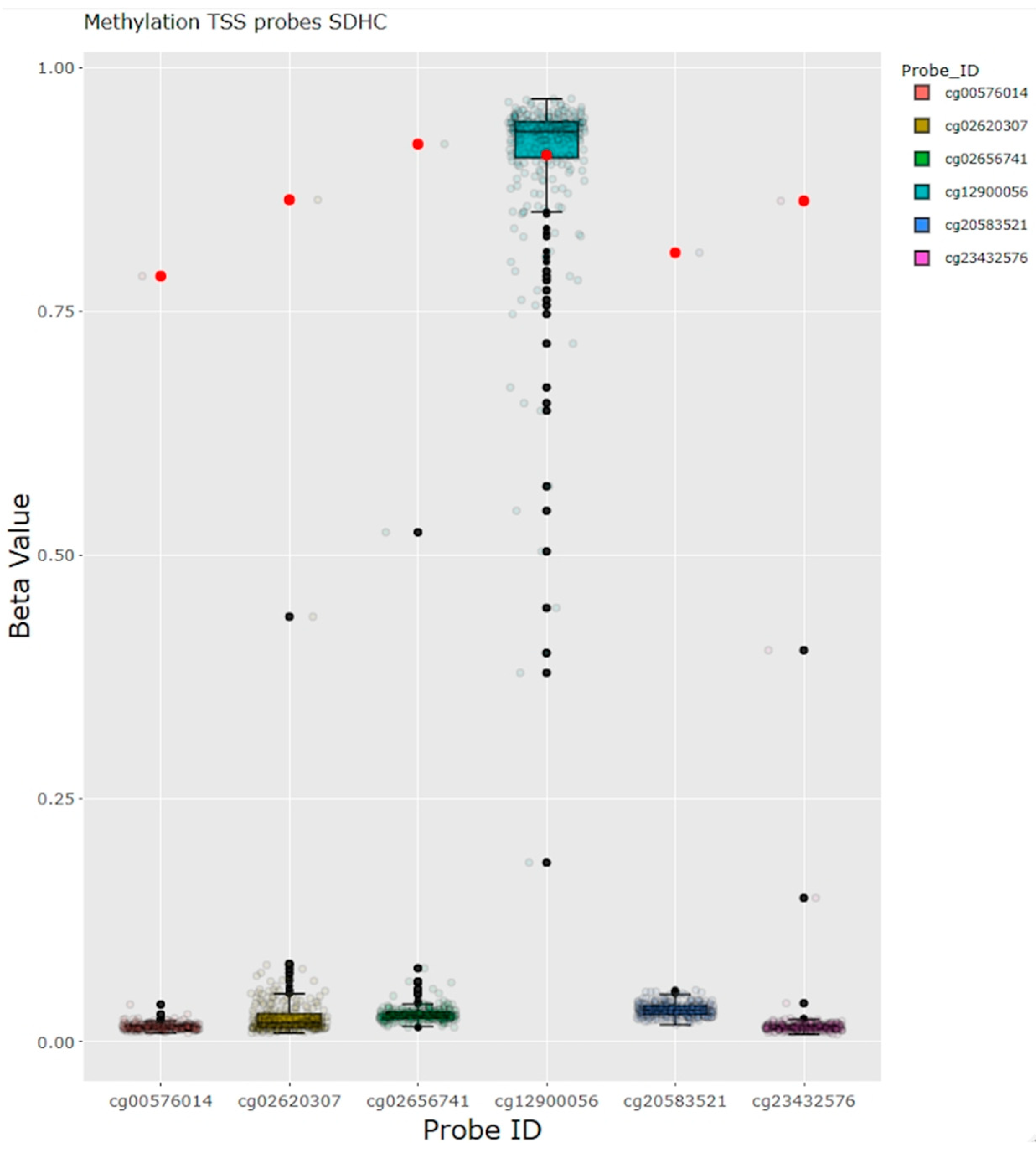

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case_No | Histological Diagnosis Including Sequencing Results | Germline Finding | Clinical Impact A | Pedigree | Previous Cancer Treatment | Co-Morbidity | Tumor Syndrome Previously Known | Fulfilling Tumor Syndrome Criteria B | Second Hit |
|---|---|---|---|---|---|---|---|---|---|
| 40 | Leiomyoma | BRCA1, NM_007294, c.406del, p.Arg136AspfsTer27 | Clinical action | No known cancer | No | OvC 55 y | Yes | Yes | No second hit |
| 52 | Osteosarcoma, parosteal | FLCN, NM_144606, c.779+1G>T | Clinical action | No known cancer | No | No | No | No | No second hit |
| 62 | GIST | SDHAF2, NM_017841, c.37-1G>C | Risk factor | No known cancer | No | HT | No | No | No second hit |
| 63 | GIST, wild-type | SDHA, NM_004168, c.223C>T, p.Arg75Ter | Clinical action | No known cancer | No | DCMP, dementia | No | No | SNV: SDHA, NM_004168, c.896G>A, p.Gly299Asp |
| 67 | Angiosarcoma | ATM, NM_000051, c.7570G>C, p.Ala2524Pro | Risk factor | No known cancer | No | HT | No | No | SNV: ATM,NM_000051, c.5188C>T, p.Arg1730Ter |
| 76 | Pleomorphic liposarcoma | MLH1, NM_000249, c.546-2A>G | Clinical action | No known cancer | No | CRC 52 y, kidney failure, HT | No | No | Deletion MLH1 |
| 95 | Liposarcoma, dedifferentiated | CHEK2,NM_001005735, c.1229del, p.Thr410MetfsTer15 C | Clinical action | No known cancer | RT, chemo breast | BrC 65, HT | No | No | No second hit |
| 101 | Leiomyosarcoma | RB1,NM_000321, c.1981C>T, p.Arg661Trp | Clinical action | Brother’s son suspected RB 4y, mother UtC 50y. | No | No | No | No | Deletion RB1 |
| 111 | Leiomyosarcoma | RB1, NM_000321, c.184C>T, p.Gln62Ter | Clinical action | No known cancer | RT OS | RB bilateral 6 months, OS legs multiple during childhood, endometriosis | Yes | Yes | Deletion RB1 |
| 115 | Adenosarcoma, sarcomatous overgrowth | CDC73, NM_024529, c.664C>T, p.Arg222Ter | Clinical action | Mother and sister LuC | No | Hypothyreosis, lung embolus | No | No | SNV: CDC73, NM_024529, c.25C>T, p.Arg9Ter |
| 139 | Radiation-induced sarcoma | MSH6,NM_000179, c.2851_2858del, p.Leu951IlefsTer12 | Clinical action | Mother OvC, son testis cancer, daughter BC, daughter CxC | RT, chemo breast | BrC 44 y, UtC 48 y | Yes | Yes | No verified second hit |
| 144 | GIST | MITF, NM_198159, c.1255G>A, p.Glu419Lys | Risk factor | No known cancer | No | UtC 75 | No | No | No second hit |
| 168 | MPNST | NF1, NM_000267, c.1721+3A>C | Clinical action | Sister brain tumor | No | GIST small intestine | Yes | Yes | SNV: NF1, NM_000267, c.565A>T, p.Lys189Ter |
| 207 | DFSP, fibrosarcoma | EXT1, NM_000127, c.1018C>T, p.Arg340Cys | Risk factor | No known cancer | No | No | No | No | No second hit |
| 208 | Leiomyosarcoma | TP53, NM_001126112, c.503del, p.His168ProfsTer2 | Clinical action | No known cancer | Interferone | OS 16 y, LMS 35 y | No | Yes | Deletion TP53 |
| 231 | Liposarcoma (DDLPS) | ATR, NM_001184, c.6836dupp.Asn2279LysfsTer4 | Risk factor | No known cancer | No | No | No | No | No second hit |
| 265 | Leiomyoma | MRE11, NM_005591, c.1090C>T, p.Arg364Ter | Risk factor | No known cancer | No | No | No | No | No second hit |
| 295 | Secondary peripheral chondrosarcoma | EXT2, NM_000401, c.441C>G, p.Tyr147Ter | Clinical action | No known cancer | No | CHS | Yes | Yes | Deletion EXT2 |
| 306 | MPNST | NF1, NM_000267, c.4974_4977del, p.Tyr1659ThrfsTer17 | Clinical action | No known cancer | RT brain | Intracranial sarcoma 23 y, Scwannoma 24 y, Café-au-laît spots | Yes | Yes | SNV: NF1, NM_000267, c.365_371del, p.His122LeufsTer41 |
| 311 | Synovial chondromatosis | CHEK2, NM_001005735, c.1229del, p.Thr410MetfsTer15 C | Clinical action | Sister breast cancer | RT, chemo breast, Tamoxifene | BrC 59 y | No | Yes | No second hit |
| 329 | Low-grade parosteal OS | CHEK2,NM_001005735, c.1229del, p.Thr410MetfsTer15 C | Risk factor | No known cancer | No | No | No | No | No second hit |
| 339 | Soft tissue sarcoma paravertebral D | MSH6, NM_000179, c.3261del, p.Phe1088SerfsTer2 | Clinical action | Sister OvC, mother BC 59y + UrC 59y + CRC 67y + UtC 53y, maternal grandmother UtC | Chemo | CRC 54 y | Yes | Yes | NA |
| 353 | Leiomyosarcoma | DDX41, NM_016222, c.415_418dup, p.Asp140GlyfsTer2 | Risk factor | Father CRC | RT, chemo kidney | Wilm’s tumor kidney and lung, cholecystectomy, myoma | No | No | No second hit |
| 364 | GIST, wild-type | NF1, NM_000267, c.6792C>A, p.Tyr2264Ter | Clinical action | Son molecularly verified NF1, no known cancer. | No | Café-au-laît spots | Yes | Yes | SNV: NF1, NM_000267, c.3723_3730dup, p.Val1244GlufsTer25 |
| No Germline Finding A | Germline Finding B | |||
|---|---|---|---|---|
| Number of Cases | Fraction of Total | Number of Cases | Fraction of Total | |
| Soft tissue sarcoma, high-grade | 120 | 42% | 11 | 46% |
| Soft tissue sarcoma, low-grade | 33 | 11% | 1 | 4% |
| Soft tissue, benign | 40 | 14% | 1 | 4% |
| Bone sarcoma, high-grade | 13 | 5% | 0 | 0% |
| Bone sarcoma, low-grade | 7 | 2% | 3 | 13% |
| Bone, benign | 5 | 2% | 0 | 0% |
| GIST | 36 | 13% | 4 | 17% |
| Gynaecological tract | 34 | 12% | 4 | 17% |
| Sum | 288 | 24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Öfverholm, I.; Lin, Y.; Mondini, J.; Hardingz, J.; Bränström, R.; Tsagkozis, P.; Wirta, V.; Gellerbring, A.; Lindberg, J.; Chellappa, V.; et al. Prospective Screening of Cancer Syndromes in Patients with Mesenchymal Tumors. Cancers 2024, 16, 3816. https://doi.org/10.3390/cancers16223816
Öfverholm I, Lin Y, Mondini J, Hardingz J, Bränström R, Tsagkozis P, Wirta V, Gellerbring A, Lindberg J, Chellappa V, et al. Prospective Screening of Cancer Syndromes in Patients with Mesenchymal Tumors. Cancers. 2024; 16(22):3816. https://doi.org/10.3390/cancers16223816
Chicago/Turabian StyleÖfverholm, Ingegerd, Yingbo Lin, Julia Mondini, John Hardingz, Robert Bränström, Panagiotis Tsagkozis, Valtteri Wirta, Anna Gellerbring, Johan Lindberg, Venkatesh Chellappa, and et al. 2024. "Prospective Screening of Cancer Syndromes in Patients with Mesenchymal Tumors" Cancers 16, no. 22: 3816. https://doi.org/10.3390/cancers16223816
APA StyleÖfverholm, I., Lin, Y., Mondini, J., Hardingz, J., Bränström, R., Tsagkozis, P., Wirta, V., Gellerbring, A., Lindberg, J., Chellappa, V., Mayrhofer, M., Haglund, C., Haglund de Flon, F., & Wallander, K. (2024). Prospective Screening of Cancer Syndromes in Patients with Mesenchymal Tumors. Cancers, 16(22), 3816. https://doi.org/10.3390/cancers16223816