
Synergistic Antitumor Activity of Talazoparib and Temozolomide in Malignant Rhabdoid Tumors

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Proliferation and Cytotoxicity Assay
2.3. Clonogenic Potential Analysis
2.4. Flow Cytometry Cell Cycle Assay
2.5. Apoptotic Cell Death Analysis
2.6. Confocal Imaging of DNA Damage Response Proteins
2.7. Protein Extraction and Western Blotting
2.8. Immunohistochemistry Analysis
2.9. DNA Damage Antibody Array
2.10. In Vivo Efficacy Study
2.11. Statistical Analyses
2.12. RNA Sequencing and Bioinformatics Analysis
3. Results
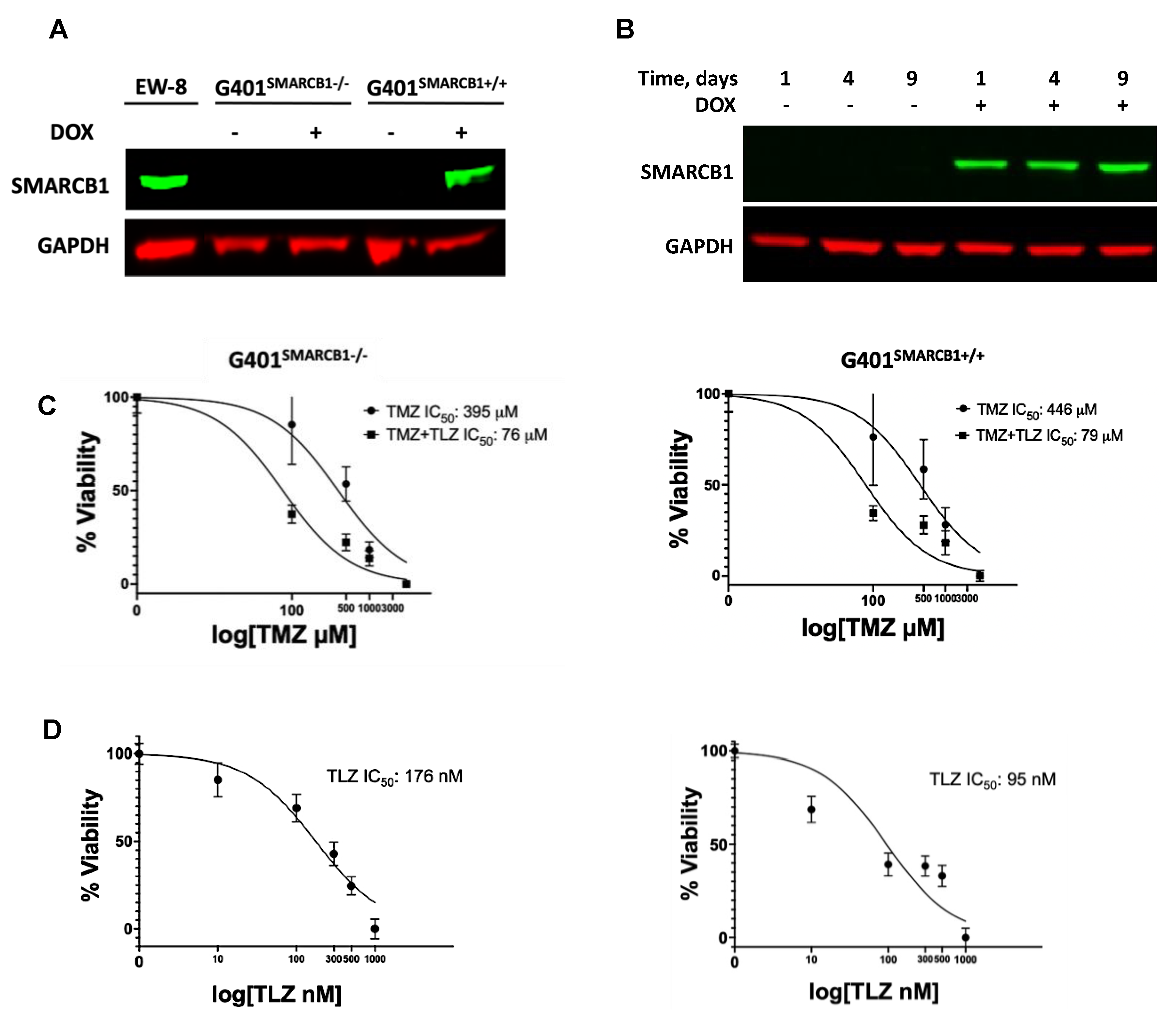
3.1. In Vitro Evaluation of the Impact of TLZ and TMZ Treatments on the Sensitization of MRT Cells
3.2. Evaluation of MRT Cellular Proliferation and Clonogenic Potential in Cells Treated with TLZ and TMZ
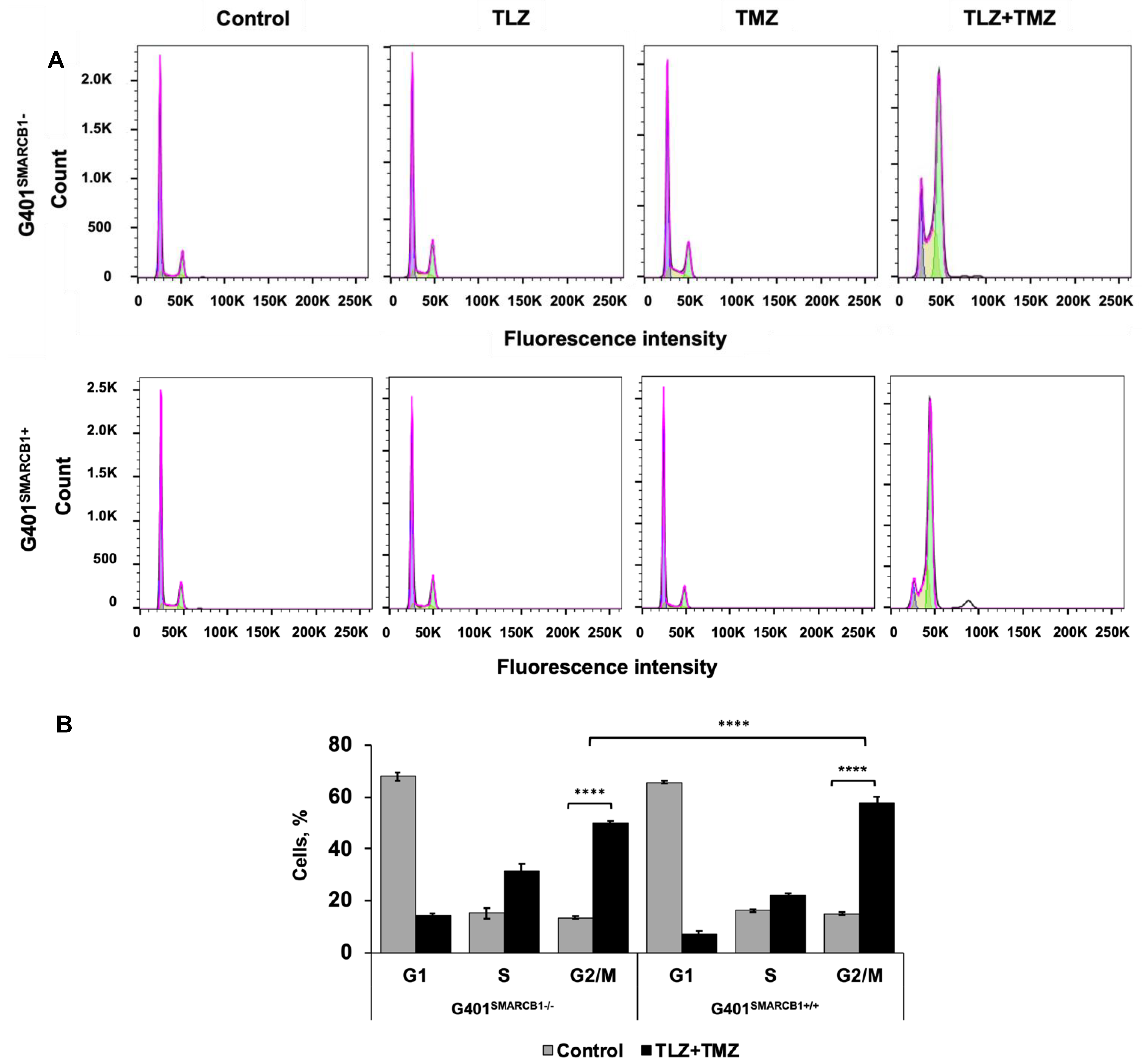
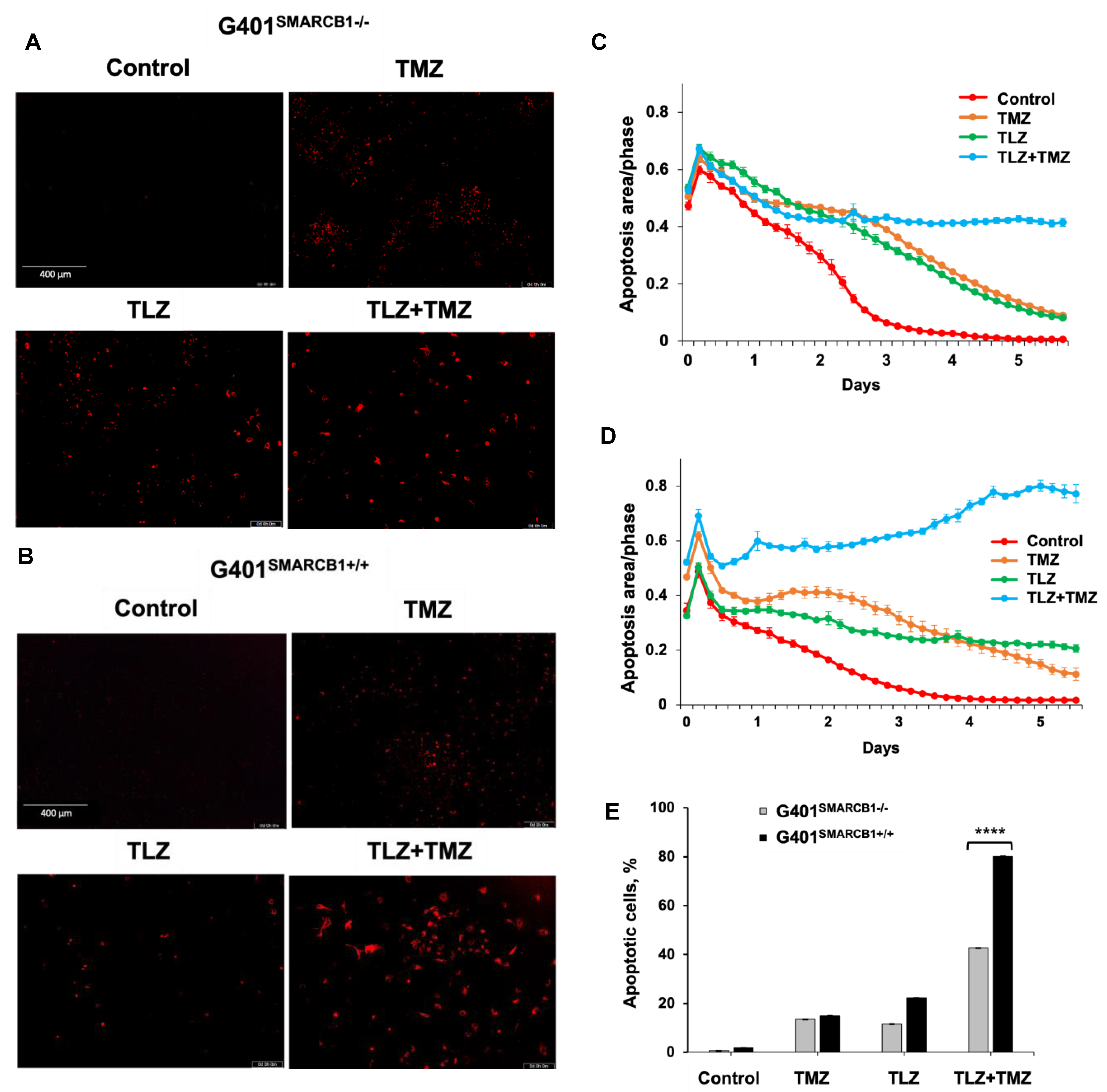
3.3. Effect of TLZ and TMZ on Cell Cycle Progression and Apoptosis in MRT Cells
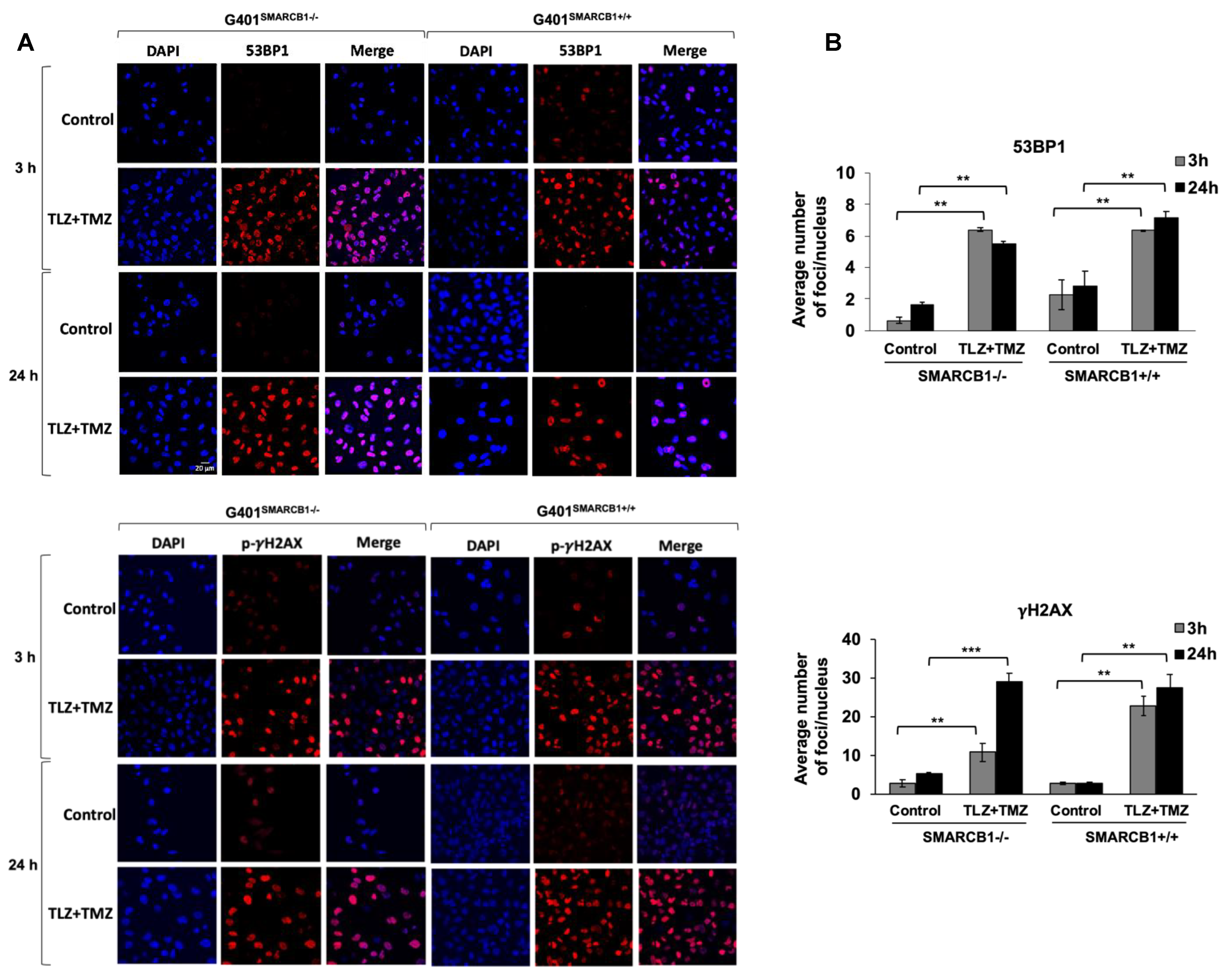
3.4. Assessment of DNA Damage Induced by TLZ and TMZ in MRT Cells
3.5. Evaluation of PEG~TLZ and TMZ Antitumor Activity in MRT Xenografts In Vivo
3.6. Dependency of PEG~TLZ+TMZ Antitumor Activity on DDR and Other Signaling Pathways in MRT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beckwith, J.B.; Palmer, N.F. Histopathology and prognosis of Wilms tumors: Results from the First National Wilms’ Tumor Study. Cancer 1978, 41, 1937–1948. [Google Scholar] [CrossRef]
- Judkins, A.R.; Mauger, J.; Ht, A.; Rorke, L.B.; Biegel, J.A. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am. J. Surg. Pathol. 2004, 28, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Nemes, K.; Johann, P.D.; Tuchert, S.; Melchior, P.; Vokuhl, C.; Siebert, R.; Furtwangler, R.; Fruhwald, M.C. Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors. Cancer Manag. Res. 2022, 14, 479–498. [Google Scholar] [CrossRef]
- Nakata, K.; Colombet, M.; Stiller, C.A.; Pritchard-Jones, K.; Steliarova-Foucher, E.; IICC-3 Contributors. Incidence of childhood renal tumours: An international population-based study. Int. J. Cancer 2020, 147, 3313–3327. [Google Scholar] [CrossRef]
- Blaney, S.M.; Kocak, M.; Gajjar, A.; Chintagumpala, M.; Merchant, T.; Kieran, M.; Pollack, I.F.; Gururangan, S.; Geyer, R.; Phillips, P.; et al. Pilot study of systemic and intrathecal mafosfamide followed by conformal radiation for infants with intracranial central nervous system tumors: A pediatric brain tumor consortium study (PBTC-001). J. Neurooncol. 2012, 109, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.N.; Zimmerman, M.A.; Yao, X.; Cohen, K.J.; Burger, P.; Biegel, J.A.; Rorke-Adams, L.B.; Fisher, M.J.; Janss, A.; Mazewski, C.; et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J. Clin. Oncol. 2009, 27, 385–389. [Google Scholar] [CrossRef]
- Dufour, C.; Beaugrand, A.; Le Deley, M.C.; Bourdeaut, F.; Andre, N.; Leblond, P.; Bertozzi, A.I.; Frappaz, D.; Rialland, X.; Fouyssac, F.; et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: A multicenter study. Cancer 2012, 118, 3812–3821. [Google Scholar] [CrossRef]
- Geyer, J.R.; Sposto, R.; Jennings, M.; Boyett, J.M.; Axtell, R.A.; Breiger, D.; Broxson, E.; Donahue, B.; Finlay, J.L.; Goldwein, J.W.; et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: A report from the Children’s Cancer Group. J. Clin. Oncol. 2005, 23, 7621–7631. [Google Scholar] [CrossRef]
- Hilden, J.M.; Meerbaum, S.; Burger, P.; Finlay, J.; Janss, A.; Scheithauer, B.W.; Walter, A.W.; Rorke, L.B.; Biegel, J.A. Central nervous system atypical teratoid/rhabdoid tumor: Results of therapy in children enrolled in a registry. J. Clin. Oncol. 2004, 22, 2877–2884. [Google Scholar] [CrossRef]
- Zaky, W.; Dhall, G.; Ji, L.; Haley, K.; Allen, J.; Atlas, M.; Bertolone, S.; Cornelius, A.; Gardner, S.; Patel, R.; et al. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: The Head Start III experience. Pediatr. Blood Cancer 2014, 61, 95–101. [Google Scholar] [CrossRef]
- Tomlinson, G.E.; Breslow, N.E.; Dome, J.; Guthrie, K.A.; Norkool, P.; Li, S.; Thomas, P.R.; Perlman, E.; Beckwith, J.B.; D’Angio, G.J.; et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: Age at diagnosis as a prognostic factor. J. Clin. Oncol. 2005, 23, 7641–7645. [Google Scholar] [CrossRef] [PubMed]
- Weeks, D.A.; Beckwith, J.B.; Mierau, G.W.; Luckey, D.W. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am. J. Surg. Pathol. 1989, 13, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar] [PubMed]
- Hasselblatt, M.; Nagel, I.; Oyen, F.; Bartelheim, K.; Russell, R.B.; Schuller, U.; Junckerstorff, R.; Rosenblum, M.; Alassiri, A.H.; Rossi, S.; et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014, 128, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Holsten, T.; Bens, S.; Oyen, F.; Nemes, K.; Hasselblatt, M.; Kordes, U.; Siebert, R.; Fruhwald, M.C.; Schneppenheim, R.; Schuller, U. Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: A meta-analysis. Eur. J. Hum. Genet. 2018, 26, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.W.; Biegel, J.A. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol. Ther. 2009, 8, 412–416. [Google Scholar] [CrossRef]
- Geller, J.I.; Roth, J.J.; Biegel, J.A. Biology and Treatment of Rhabdoid Tumor. Crit. Rev. Oncog. 2015, 20, 199–216. [Google Scholar] [CrossRef]
- Wang, X.; Lee, R.S.; Alver, B.H.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.M.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef]
- Roberts, C.W.; Orkin, S.H. The SWI/SNF complex--chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Brennan, B.; Stiller, C.; Bourdeaut, F. Extracranial rhabdoid tumours: What we have learned so far and future directions. Lancet Oncol. 2013, 14, e329–e336. [Google Scholar] [CrossRef]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Erickson, S.W.; Earley, E.; Smith, M.A.; Houghton, P.J. In vivo evaluation of the EZH2 inhibitor (EPZ011989) alone or in combination with standard of care cytotoxic agents against pediatric malignant rhabdoid tumor preclinical models-A report from the Pediatric Preclinical Testing Consortium. Pediatr. Blood Cancer 2021, 68, e28772. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Poirier, G.G.; Lindahl, T. Dual function for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry 1994, 33, 7099–7106. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Rouleau, M.; Aubin, R.A.; Poirier, G.G. Poly(ADP-ribosyl)ated chromatin domains: Access granted. J. Cell Sci. 2004, 117, 815–825. [Google Scholar] [CrossRef]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef]
- Sasaki, M.; Ogiwara, H. Synthetic lethal therapy based on targeting the vulnerability of SWI/SNF chromatin remodeling complex-deficient cancers. Cancer Sci. 2020, 111, 774–782. [Google Scholar] [CrossRef]
- Alimova, I.; Murdock, G.; Pierce, A.; Wang, D.; Madhavan, K.; Brunt, B.; Venkataraman, S.; Vibhakar, R. The PARP inhibitor Rucaparib synergizes with radiation to attenuate atypical teratoid rhabdoid tumor growth. Neurooncol. Adv. 2023, 5, vdad010. [Google Scholar] [CrossRef]
- Schafer, E.S.; Rau, R.E.; Berg, S.L.; Liu, X.; Minard, C.G.; Bishop, A.J.R.; Romero, J.C.; Hicks, M.J.; Nelson, M.D., Jr.; Voss, S.; et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatr. Blood Cancer 2020, 67, e28073. [Google Scholar] [CrossRef]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef]
- Del Pozo, V.; Robles, A.J.; Fontaine, S.D.; Liu, Q.; Michalek, J.E.; Houghton, P.J.; Kurmasheva, R.T. PEGylated talazoparib enhances therapeutic window of its combination with temozolomide in Ewing sarcoma. iScience 2022, 25, 103725. [Google Scholar] [CrossRef]
- Fontaine, S.D.; Ashley, G.W.; Houghton, P.J.; Kurmasheva, R.T.; Diolaiti, M.; Ashworth, A.; Peer, C.J.; Nguyen, R.; Figg, W.D., Sr.; Beckford-Vera, D.R.; et al. A Very Long-Acting PARP Inhibitor Suppresses Cancer Cell Growth in DNA Repair-Deficient Tumor Models. Cancer Res. 2021, 81, 1076–1086. [Google Scholar] [CrossRef]
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog. Nucleic Acid. Res. Mol. Biol. 1995, 51, 167–223. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef]
- Koob, L.; Friskes, A.; van Bergen, L.; Feringa, F.M.; van den Broek, B.; Koeleman, E.S.; van Beek, E.; Schubert, M.; Blomen, V.A.; Brummelkamp, T.R.; et al. MND1 enables homologous recombination in somatic cells primarily outside the context of replication. Mol. Oncol. 2023, 17, 1192–1211. [Google Scholar] [CrossRef]
- Houghton, P.J.; Morton, C.L.; Tucker, C.; Payne, D.; Favours, E.; Cole, C.; Gorlick, R.; Kolb, E.A.; Zhang, W.; Lock, R.; et al. The pediatric preclinical testing program: Description of models and early testing results. Pediatr. Blood Cancer 2007, 49, 928–940. [Google Scholar] [CrossRef]
- Peterson, J.K.; Houghton, P.J. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur. J. Cancer 2004, 40, 837–844. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef]
- Turinetto, V.; Giachino, C. Multiple facets of histone variant H2AX: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef]
- Groesser, T.; Chang, H.; Fontenay, G.; Chen, J.; Costes, S.V.; Helen Barcellos-Hoff, M.; Parvin, B.; Rydberg, B. Persistence of gamma-H2AX and 53BP1 foci in proliferating and non-proliferating human mammary epithelial cells after exposure to gamma-rays or iron ions. Int. J. Radiat. Biol. 2011, 87, 696–710. [Google Scholar] [CrossRef]
- Cropper, J.D.; Alimbetov, D.S.; Brown, K.T.G.; Likhotvorik, R.I.; Robles, A.J.; Guerra, J.T.; He, B.; Chen, Y.; Kwon, Y.; Kurmasheva, R.T. PARP1-MGMT complex underpins pathway crosstalk in O(6)-methylguanine repair. J. Hematol. Oncol. 2022, 15, 146. [Google Scholar] [CrossRef]
- Hindle, A.; Koneru, B.; Makena, M.R.; Lopez-Barcons, L.; Chen, W.H.; Nguyen, T.H.; Reynolds, C.P. The O6-methyguanine-DNA methyltransferase inhibitor O6-benzylguanine enhanced activity of temozolomide + irinotecan against models of high-risk neuroblastoma. Anticancer Drugs 2021, 32, 233–247. [Google Scholar] [CrossRef]
- Darr, J.; Klochendler, A.; Isaac, S.; Geiger, T.; Eden, A. Phosphoproteomic analysis reveals Smarcb1 dependent EGFR signaling in Malignant Rhabdoid tumor cells. Mol. Cancer 2015, 14, 167. [Google Scholar] [CrossRef]
- Holdhof, D.; Johann, P.D.; Spohn, M.; Bockmayr, M.; Safaei, S.; Joshi, P.; Masliah-Planchon, J.; Ho, B.; Andrianteranagna, M.; Bourdeaut, F.; et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021, 141, 291–301. [Google Scholar] [CrossRef]
- Jeibmann, A.; Buerger, H.; Fruhwald, M.; Hasselblatt, M. No evidence for epidermal growth factor receptor amplification and overexpression in atypical teratoid/rhabdoid tumors. Acta Neuropathol. 2006, 112, 513–514. [Google Scholar] [CrossRef]
- Terashima, M.; Kobayashi, M.; Motomiya, M.; Inoue, N.; Yoshida, T.; Okano, H.; Iwasaki, N.; Minami, A.; Matsuoka, I. Analysis of the expression and function of BRINP family genes during neuronal differentiation in mouse embryonic stem cell-derived neural stem cells. J. Neurosci. Res. 2010, 88, 1387–1393. [Google Scholar] [CrossRef]
- Chun, H.E.; Lim, E.L.; Heravi-Moussavi, A.; Saberi, S.; Mungall, K.L.; Bilenky, M.; Carles, A.; Tse, K.; Shlafman, I.; Zhu, K.; et al. Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 2016, 29, 394–406. [Google Scholar] [CrossRef]
- van der Weyden, L.; Arends, M.J.; Rust, A.G.; Poulogiannis, G.; McIntyre, R.E.; Adams, D.J. Increased tumorigenesis associated with loss of the tumor suppressor gene Cadm1. Mol. Cancer 2012, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Alfaro-Cervello, C.; Andrade-Gamarra, V.; Nieto, G.; Navarro, L.; Martin-Vano, S.; Garcia de la Torre, J.P.; Bengoa Caamano, M.; Garcia Maurino, M.L.; Noguera, R.; Navarro, S. Congenital undifferentiated sarcoma associated to BCOR-CCNB3 gene fusion. Pathol. Res. Pract. 2017, 213, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, S.; Li, S.; Wang, J.; Lin, H.; Fu, W. High expression of oncogene cadherin-6 correlates with tumor progression and a poor prognosis in gastric cancer. Cancer Cell Int. 2021, 21, 493. [Google Scholar] [CrossRef] [PubMed]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef]
- Wohrle, S.; Weiss, A.; Ito, M.; Kauffmann, A.; Murakami, M.; Jagani, Z.; Thuery, A.; Bauer-Probst, B.; Reimann, F.; Stamm, C.; et al. Fibroblast growth factor receptors as novel therapeutic targets in SNF5-deleted malignant rhabdoid tumors. PLoS ONE 2013, 8, e77652. [Google Scholar] [CrossRef]
- Maeda, T.; Kanzaki, H.; Chiba, T.; Ao, J.; Kanayama, K.; Maruta, S.; Kusakabe, Y.; Saito, T.; Kobayashi, K.; Kiyono, S.; et al. Serum fibroblast growth factor 19 serves as a potential novel biomarker for hepatocellular carcinoma. BMC Cancer 2019, 19, 1088. [Google Scholar] [CrossRef]
- Zhu, F.; Dai, S.N.; Xu, D.L.; Hou, C.Q.; Liu, T.T.; Chen, Q.Y.; Wu, J.L.; Miao, Y. EFNB2 facilitates cell proliferation, migration, and invasion in pancreatic ductal adenocarcinoma via the p53/p21 pathway and EMT. Biomed. Pharmacother. 2020, 125, 109972. [Google Scholar] [CrossRef]
- Banham, A.H.; Beasley, N.; Campo, E.; Fernandez, P.L.; Fidler, C.; Gatter, K.; Jones, M.; Mason, D.Y.; Prime, J.E.; Trougouboff, P.; et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001, 61, 8820–8829. [Google Scholar] [PubMed]
- Fox, S.B.; Brown, P.; Han, C.; Ashe, S.; Leek, R.D.; Harris, A.L.; Banham, A.H. Expression of the forkhead transcription factor FOXP1 is associated with estrogen receptor alpha and improved survival in primary human breast carcinomas. Clin. Cancer Res. 2004, 10, 3521–3527. [Google Scholar] [CrossRef]
- Toma, M.I.; Grosser, M.; Herr, A.; Aust, D.E.; Meye, A.; Hoefling, C.; Fuessel, S.; Wuttig, D.; Wirth, M.P.; Baretton, G.B. Loss of heterozygosity and copy number abnormality in clear cell renal cell carcinoma discovered by high-density affymetrix 10K single nucleotide polymorphism mapping array. Neoplasia 2008, 10, 634–642. [Google Scholar] [CrossRef]
- van Keimpema, M.; Gruneberg, L.J.; Mokry, M.; van Boxtel, R.; Koster, J.; Coffer, P.J.; Pals, S.T.; Spaargaren, M. FOXP1 directly represses transcription of proapoptotic genes and cooperates with NF-kappaB to promote survival of human B cells. Blood 2014, 124, 3431–3440. [Google Scholar] [CrossRef]
- Yamashita, S.I.; Masuda, Y.; Yoshida, N.; Matsuzaki, H.; Kurizaki, T.; Haga, Y.; Ikei, S.; Miyawaki, M.; Kawano, Y.; Chujyo, M.; et al. p53AIP1 expression can be a prognostic marker in non-small cell lung cancer. Clin. Oncol. (R Coll. Radiol.) 2008, 20, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Betz, B.L.; Strobeck, M.W.; Reisman, D.N.; Knudsen, E.S.; Weissman, B.E. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 2002, 21, 5193–5203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Hampton, O.A.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr. Blood Cancer 2015, 62, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef]
- Kim, J.H.; Penson, A.V.; Taylor, B.S.; Petrini, J.H.J. Nbn-Mre11 interaction is required for tumor suppression and genomic integrity. Proc. Natl. Acad. Sci. USA 2019, 116, 15178–15183. [Google Scholar] [CrossRef]
- Ali, D.M.; Ansari, S.S.; Zepp, M.; Knapp-Mohammady, M.; Berger, M.R. Optineurin downregulation induces endoplasmic reticulum stress, chaperone-mediated autophagy, and apoptosis in pancreatic cancer cells. Cell Death Discov. 2019, 5, 128. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, P.; Gao, H.; Gu, Y.; Yang, J.; Peng, H.; Xu, X.; Wang, H.; Yang, M.; Liu, X.; et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014, 26, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Nevanlinna, H.; Bartek, J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006, 25, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Sodha, N.; Houlston, R.S.; Bullock, S.; Yuille, M.A.; Chu, C.; Turner, G.; Eeles, R.A. Increasing evidence that germline mutations in CHEK2 do not cause Li-Fraumeni syndrome. Hum. Mutat. 2002, 20, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Sodha, N.; Williams, R.; Mangion, J.; Bullock, S.L.; Yuille, M.R.; Eeles, R.A. Screening hCHK2 for mutations. Science 2000, 289, 359. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. Ku regulates signaling to DNA damage response pathways through the Ku70 von Willebrand A domain. Mol. Cell Biol. 2012, 32, 76–87. [Google Scholar] [CrossRef]
- Tsang, T.; He, Q.; Cohen, E.B.; Stottrup, C.; Lien, E.C.; Zhang, H.; Lau, C.G.; Chin, Y.R. Upregulation of Receptor Tyrosine Kinase Activity and Stemness as Resistance Mechanisms to Akt Inhibitors in Breast Cancer. Cancers 2022, 14, 5006. [Google Scholar] [CrossRef]
- Bagrodia, S.; Smeal, T.; Abraham, R.T. Mechanisms of intrinsic and acquired resistance to kinase-targeted therapies. Pigment. Cell Melanoma Res. 2012, 25, 819–831. [Google Scholar] [CrossRef]


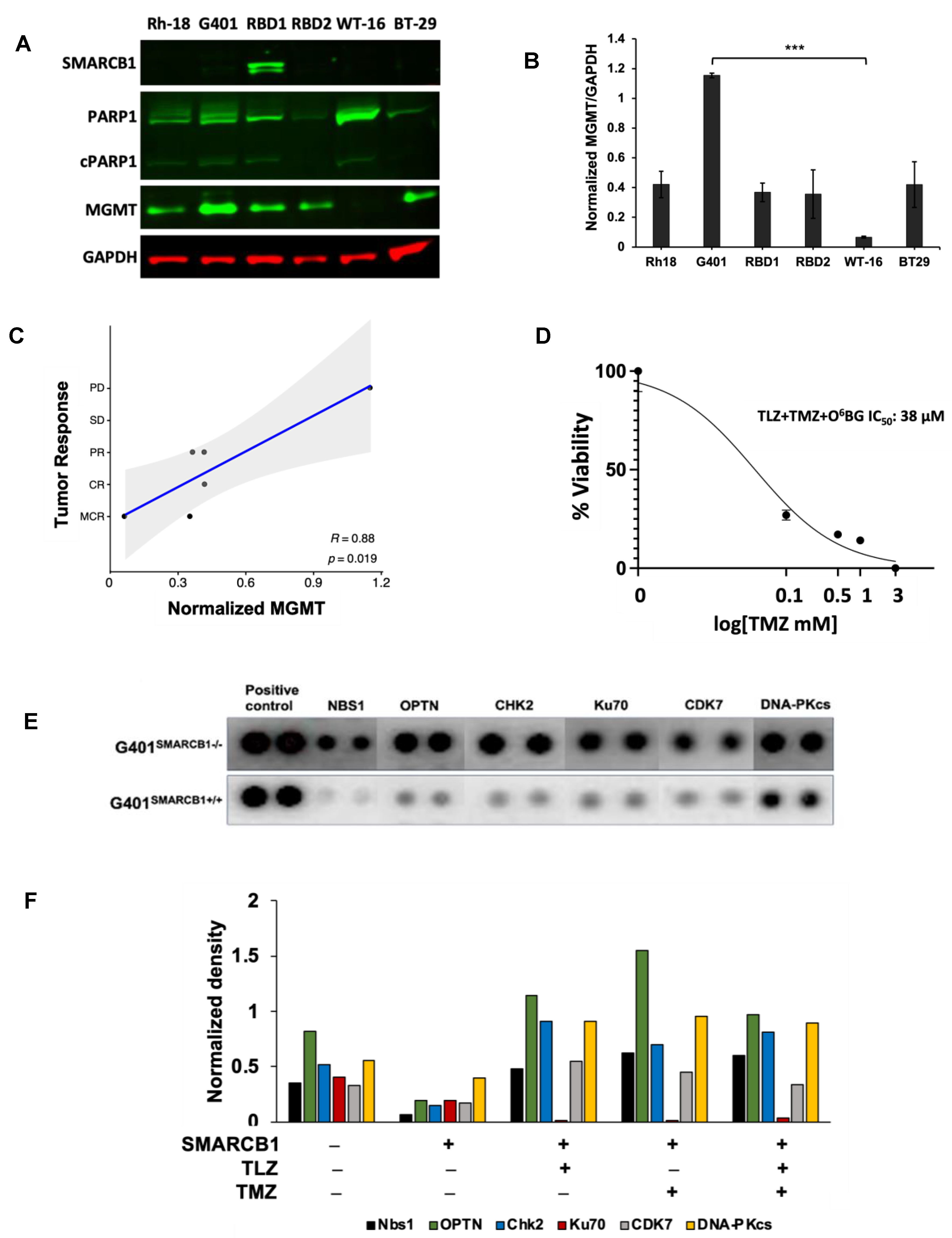

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model ID | Passage | Subtype | Age, Years | Sex | Site of Origin | Diagnosis or Relapse |
|---|---|---|---|---|---|---|
| BT-29 | p18 | AT/RT | 2 | M | Frontal lobe | Diagnosis |
| G401 | p10/10 | RTK | 3 m | M | Kidney | Diagnosis |
| WT-16 | p29 | RTK | 10 m | F | Kidney | Relapse |
| NCH-RBD-1 | p25 | Extrarenal | 1 | F | Lung | Diagnosis |
| NCH-RBD-2 | p13 | Extrarenal | 9 | F | Liver | Diagnosis |
| Rh18 | p45 | Extrarenal | 2 | F | Perineum | Diagnosis |
| Tumor | Histology | Median Time to Event | Antitumor Activity | |
|---|---|---|---|---|
| Control (Days) | Treated (Days) | |||
| NCH-RBD2 | Extrarenal | 15.2 | 49.5 | MCR |
| Rh-18 | Extrarenal | 17.3 | 83.8 | CR |
| WT-16 | RTK | 36.3 | >161 | MCR |
| NCH-RBD1 | Extrarenal | 126.6 | 31.7 | PR |
| BT-29 | AT/RT | 10.2 | 34.9 | PR |
| G401 | RTK | 13.3 | 17.8 | PD |
| Gene Symbol | Gene Name | Fold Change (FPKM) | Raw p-Value |
|---|---|---|---|
| BRINP3 [58,59] | BMP/retinoic acid inducible neural specific 3 | 52.6 | 3.8 × 10−6 * |
| EGFR [55,57] | Epithelial growth factor receptor | 41.8 | 1.4 × 10−3 |
| EPHA5 [56] | Ephrin type-A receptor 5 | 26.9 | 1.9 × 10−4 |
| FOXP1 [67,68,69,70] | Forkhead box P1 | 0.005 | 9.0 × 10−7 ** |
| MEIS3P1 | Meis homeobox 3 pseudogene 1 | 106.8 | 3.6 × 10−4 |
| VEGFC | Vascular endothelial growth factor C | 412.3 | 5.2 × 10−4 |
| Gene Symbol | Gene Name | Fold Change (FPKM) | Raw p-Value |
|---|---|---|---|
| CADM1 [60] | Cell adhesion molecule 1 | 31.59 | 2.2 × 10−3 ** |
| CCNB3 [61] | Cyclin B3 | 18.06 | 1.1 × 10−2 |
| CDH6 [62] | Cadherin 6 | 173.19 | 8.2 × 10−11 **** |
| CDH9 | Cadherin 9 | 591.03 | 1.2 × 10−6 *** |
| CLDN10 [63] | Claudin 10 | 94.47 | 2.2 × 10−3 |
| EFNB1 | Ephrin B1 | 12.42 | 1.6 × 10−3 |
| EFNB2 [66] | Ephrin B2 | 7.41 | 6.7 × 10−3 |
| FGF3 [64] | Fibroblast growth factor 3 | 54.00 | 1.1 × 10−2 |
| FGF4 | Fibroblast growth factor 4 | 72.56 | 5.4 × 10−3 |
| FGF19 [65] | Fibroblast growth factor 19 | 2892.42 | 2.4 × 10−20 **** |
| KIT | Proto-oncogene, receptor tyrosine kinase | 28.85 | 1.9 × 10−2 |
| PIK3AP1 [71] | Phosphoinositide-3-kinase adaptor protein 1 | 64.85 | 4.2 × 10−5 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mironova, E.; Molinas, S.; Pozo, V.D.; Bandyopadhyay, A.M.; Lai, Z.; Kurmashev, D.; Schneider, E.L.; Santi, D.V.; Chen, Y.; Kurmasheva, R.T. Synergistic Antitumor Activity of Talazoparib and Temozolomide in Malignant Rhabdoid Tumors. Cancers 2024, 16, 2041. https://doi.org/10.3390/cancers16112041
Mironova E, Molinas S, Pozo VD, Bandyopadhyay AM, Lai Z, Kurmashev D, Schneider EL, Santi DV, Chen Y, Kurmasheva RT. Synergistic Antitumor Activity of Talazoparib and Temozolomide in Malignant Rhabdoid Tumors. Cancers. 2024; 16(11):2041. https://doi.org/10.3390/cancers16112041
Chicago/Turabian StyleMironova, Elena, Sebastian Molinas, Vanessa Del Pozo, Abhik M. Bandyopadhyay, Zhao Lai, Dias Kurmashev, Eric L. Schneider, Daniel V. Santi, Yidong Chen, and Raushan T. Kurmasheva. 2024. "Synergistic Antitumor Activity of Talazoparib and Temozolomide in Malignant Rhabdoid Tumors" Cancers 16, no. 11: 2041. https://doi.org/10.3390/cancers16112041
APA StyleMironova, E., Molinas, S., Pozo, V. D., Bandyopadhyay, A. M., Lai, Z., Kurmashev, D., Schneider, E. L., Santi, D. V., Chen, Y., & Kurmasheva, R. T. (2024). Synergistic Antitumor Activity of Talazoparib and Temozolomide in Malignant Rhabdoid Tumors. Cancers, 16(11), 2041. https://doi.org/10.3390/cancers16112041