

Dendritic Cell Subsets in Melanoma: Pathophysiology, Clinical Prognosis and Therapeutic Exploitation

Abstract
Simple Summary
Abstract
1. Introduction
2. Different DC Subsets at Basal State and during Inflammation
2.1. Main Peripheral DC Subsets
2.2. Other DC Subsets
2.2.1. At the Basal State
2.2.2. During Inflammation
2.3. Heterogeneity of Intratumoral DC Subsets
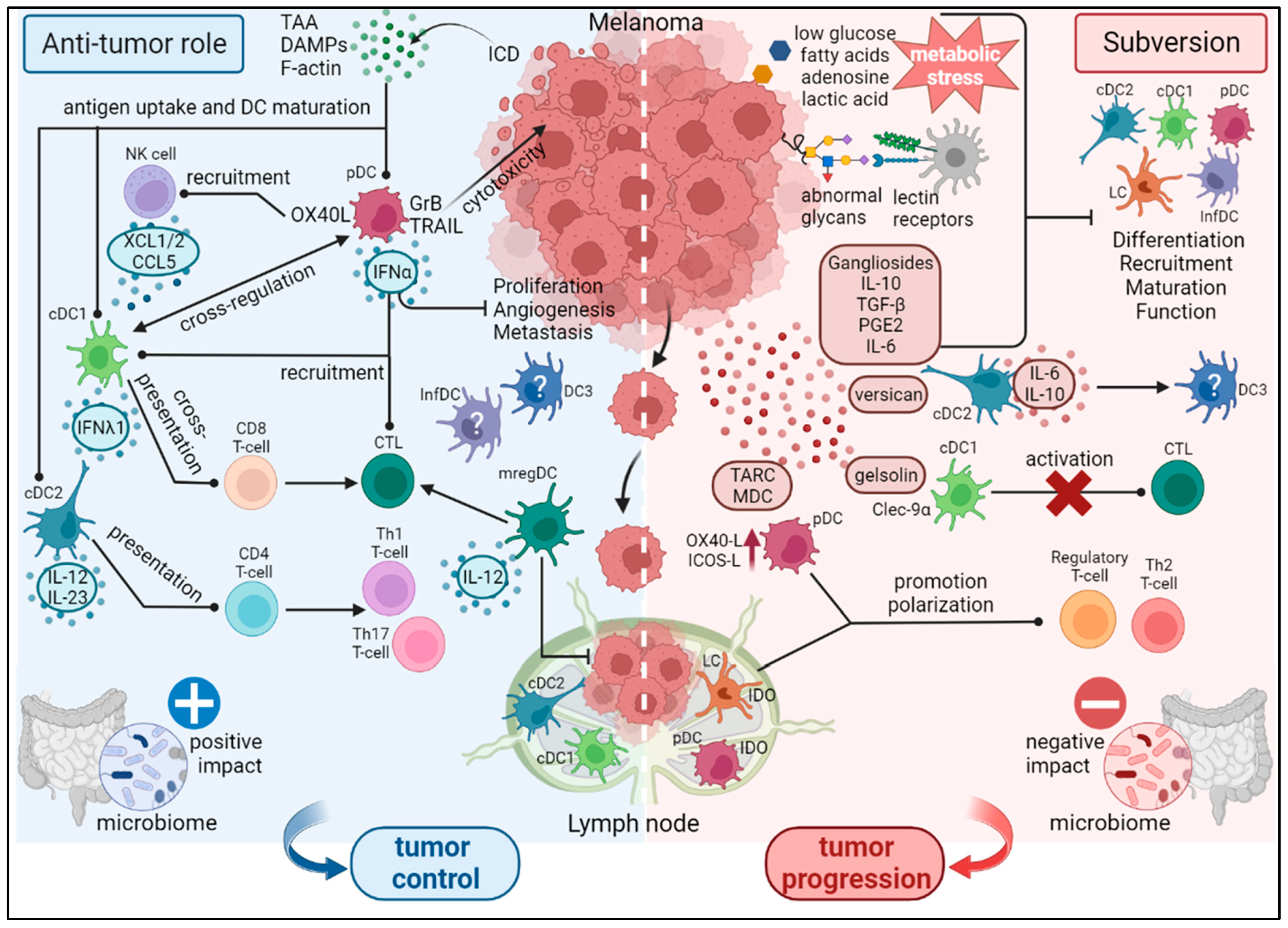
3. DC Subsets in Melanoma: Heterogeneity, Function, Subversion and Prognosis
3.1. Anti-Tumor Activity of DCs
3.2. Subversion of DCs within the TME
3.3. Factors Influencing DC Function in Melanoma
3.3.1. Melanoma-Derived Factors
3.3.2. Tumor Glycocode
3.3.3. Impact of the Metabolism on DCs’ Function
3.3.4. Role of the Microbiome in Shaping DC Immunogenicity
3.4. Prognostic Impact of DC Subsets in Melanoma
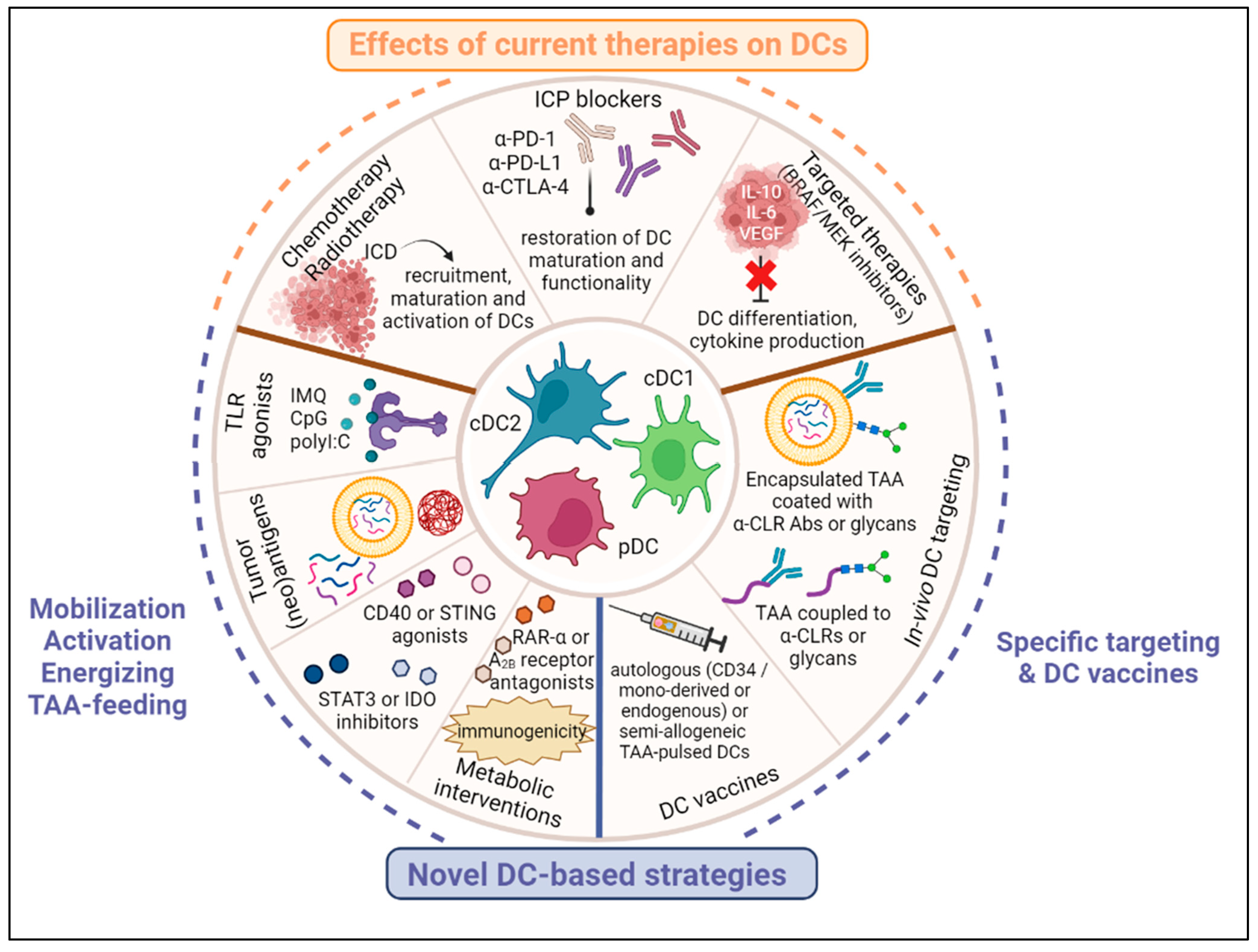
4. Effects of Current Melanoma Therapies on DC Subsets’ Features
4.1. Chemotherapy and Radiotherapy
4.2. Targeted Therapies
4.3. Immunotherapies
5. Harnessing DC Subsets for Therapeutic Strategies in Melanoma (Figure 2)
5.1. In Vivo Activation, Mobilization and TAA-Feeding of DCs
5.2. Targeting DC Subsets In Vivo
5.3. DC Vaccines
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, S.J.; den Dunnen, J.; Gringhuis, S.I.; Geijtenbeek, T.B.; van Kooyk, Y. Innate signaling and regulation of Dendritic cell immunity. Curr. Opin. Immunol. 2007, 19, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Kvedaraite, E.; Ginhoux, F. Human dendritic cells in cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Bachem, A.; Guttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- van der Aa, E.; van Montfoort, N.; Woltman, A.M. BDCA3+CLEC9A+ human dendritic cell function and development. Semin. Cell Dev. Biol. 2015, 41, 39–48. [Google Scholar] [CrossRef]
- Poulin, L.F.; Reyal, Y.; Uronen-Hansson, H.; Schraml, B.U.; Sancho, D.; Murphy, K.M.; Hakansson, U.K.; Moita, L.F.; Agace, W.W.; Bonnet, D.; et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood 2012, 119, 6052–6062. [Google Scholar] [CrossRef]
- Schreibelt, G.; Klinkenberg, L.J.; Cruz, L.J.; Tacken, P.J.; Tel, J.; Kreutz, M.; Adema, G.J.; Brown, G.D.; Figdor, C.G.; de Vries, I.J. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012, 119, 2284–2292. [Google Scholar] [CrossRef]
- Nizzoli, G.; Krietsch, J.; Weick, A.; Steinfelder, S.; Facciotti, F.; Gruarin, P.; Bianco, A.; Steckel, B.; Moro, M.; Crosti, M.; et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood 2013, 122, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef]
- Colonna, M.; Trinchieri, G.; Liu, Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef]
- Alcantara-Hernandez, M.; Leylek, R.; Wagar, L.E.; Engleman, E.G.; Keler, T.; Marinkovich, M.P.; Davis, M.M.; Nolan, G.P.; Idoyaga, J. High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 2017, 47, 1037–1050.e6. [Google Scholar] [CrossRef] [PubMed]
- See, P.; Dutertre, C.A.; Chen, J.; Gunther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Handler, K.; Duan, K.; et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science 2017, 356, eaag3009. [Google Scholar] [CrossRef]
- Valladeau, J.; Ravel, O.; Dezutter-Dambuyant, C.; Moore, K.; Kleijmeer, M.; Liu, Y.; Duvert-Frances, V.; Vincent, C.; Schmitt, D.; Davoust, J.; et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 2000, 12, 71–81. [Google Scholar] [CrossRef]
- Klechevsky, E.; Morita, R.; Liu, M.; Cao, Y.; Coquery, S.; Thompson-Snipes, L.; Briere, F.; Chaussabel, D.; Zurawski, G.; Palucka, A.K.; et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 2008, 29, 497–510. [Google Scholar] [CrossRef]
- Fehres, C.M.; Bruijns, S.C.; Sotthewes, B.N.; Kalay, H.; Schaffer, L.; Head, S.R.; de Gruijl, T.D.; Garcia-Vallejo, J.J.; van Kooyk, Y. Phenotypic and Functional Properties of Human Steady State CD14+ and CD1a+ Antigen Presenting Cells and Epidermal Langerhans Cells. PLoS ONE 2015, 10, e0143519. [Google Scholar] [CrossRef]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a pDC: Recent Advances in Understanding Plasmacytoid DC Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef] [PubMed]
- Heger, L.; Hofer, T.P.; Bigley, V.; de Vries, I.J.M.; Dalod, M.; Dudziak, D.; Ziegler-Heitbrock, L. Subsets of CD1c+ DCs: Dendritic Cell Versus Monocyte Lineage. Front. Immunol. 2020, 11, 559166. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Yu, H.; Jin, X.; Li, J.; Guo, H.; Shi, Q.; Yin, Z.; Xu, Y.; Wang, X.; Liu, R.; et al. Human Blood CD1c+ Dendritic Cells Encompass CD5high and CD5low Subsets That Differ Significantly in Phenotype, Gene Expression, and Functions. J. Immunol. 2017, 198, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Bourdely, P.; Anselmi, G.; Vaivode, K.; Ramos, R.N.; Missolo-Koussou, Y.; Hidalgo, S.; Tosselo, J.; Nunez, N.; Richer, W.; Vincent-Salomon, A.; et al. Transcriptional and Functional Analysis of CD1c+ Human Dendritic Cells Identifies a CD163+ Subset Priming CD8+ CD103+ T Cells. Immunity 2020, 53, 335–352.e8. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallee, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e24. [Google Scholar] [CrossRef] [PubMed]
- Alculumbre, S.G.; Saint-Andre, V.; Di Domizio, J.; Vargas, P.; Sirven, P.; Bost, P.; Maurin, M.; Maiuri, P.; Wery, M.; Roman, M.S.; et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat. Immunol. 2018, 19, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Onodi, F.; Bonnet-Madin, L.; Meertens, L.; Karpf, L.; Poirot, J.; Zhang, S.Y.; Picard, C.; Puel, A.; Jouanguy, E.; Zhang, Q.; et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J. Exp. Med. 2021, 218, e20201387. [Google Scholar] [CrossRef] [PubMed]
- Sosa Cuevas, E.; Bendriss-Vermare, N.; Mouret, S.; De Fraipont, F.; Charles, J.; Valladeau-Guilemond, J.; Chaperot, L.; Aspord, C. Diversification of circulating and tumor-infiltrating plasmacytoid DCs towards the P3 (CD80+ PDL1−)-pDC subset negatively correlated with clinical outcomes in melanoma patients. Clin. Transl. Immunol. 2022, 11, e1382. [Google Scholar] [CrossRef]
- Cheng, S.; Li, Z.; Gao, R.; Xing, B.; Gao, Y.; Yang, Y.; Qin, S.; Zhang, L.; Ouyang, H.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809.e23. [Google Scholar] [CrossRef] [PubMed]
- Hubert, M.; Gobbini, E.; Bendriss-Vermare, N.; Caux, C.; Valladeau-Guilemond, J. Human Tumor-Infiltrating Dendritic Cells: From In Situ Visualization to High-Dimensional Analyses. Cancers 2019, 11, 1082. [Google Scholar] [CrossRef] [PubMed]
- Marmonti, E.; Oliva-Ramirez, J.; Haymaker, C. Dendritic Cells: The Long and Evolving Road towards Successful Targetability in Cancer. Cells 2022, 11, 3028. [Google Scholar] [CrossRef]
- Gupta, Y.H.; Khanom, A.; Acton, S.E. Control of Dendritic Cell Function within the Tumour Microenvironment. Front. Immunol. 2022, 13, 733800. [Google Scholar] [CrossRef]
- Del Prete, A.; Sozio, F.; Barbazza, I.; Salvi, V.; Tiberio, L.; Laffranchi, M.; Gismondi, A.; Bosisio, D.; Schioppa, T.; Sozzani, S. Functional Role of Dendritic Cell Subsets in Cancer Progression and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 3930. [Google Scholar] [CrossRef]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond cDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Bonecchi, R.; Facchetti, F.; Bianchi, D.; Sozzani, S.; Festa, S.; Berenzi, A.; Cella, M.; Colonna, M. Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. J. Pathol. 2003, 200, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Leccia, M.T.; Charles, J.; Plumas, J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer Immunol. Res. 2013, 1, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 938. [Google Scholar] [CrossRef]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef]
- Sosa Cuevas, E.; Ouaguia, L.; Mouret, S.; Charles, J.; De Fraipont, F.; Manches, O.; Valladeau-Guilemond, J.; Bendriss-Vermare, N.; Chaperot, L.; Aspord, C. BDCA1+ cDC2s, BDCA2+ pDCs and BDCA3+ cDC1s reveal distinct pathophysiologic features and impact on clinical outcomes in melanoma patients. Clin. Transl. Immunol. 2020, 9, e1190. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Reis, E.S.C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Cancel, J.C.; Crozat, K.; Dalod, M.; Mattiuz, R. Are Conventional Type 1 Dendritic Cells Critical for Protective Antitumor Immunity and How? Front. Immunol. 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Hubert, M.; Gobbini, E.; Couillault, C.; Manh, T.V.; Doffin, A.C.; Berthet, J.; Rodriguez, C.; Ollion, V.; Kielbassa, J.; Sajous, C.; et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci. Immunol. 2020, 5, eaav3942. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Deauvieau, F.; Ollion, V.; Doffin, A.C.; Achard, C.; Fonteneau, J.F.; Verronese, E.; Durand, I.; Ghittoni, R.; Marvel, J.; Dezutter-Dambuyant, C.; et al. Human natural killer cells promote cross-presentation of tumor cell-derived antigens by dendritic cells. Int. J. Cancer 2015, 136, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Komori, S.; Kotani, T.; Murata, Y.; Matozaki, T. The Role of Type-2 Conventional Dendritic Cells in the Regulation of Tumor Immunity. Cancers 2022, 14, 1976. [Google Scholar] [CrossRef] [PubMed]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef]
- van Beek, J.J.; Gorris, M.A.; Skold, A.E.; Hatipoglu, I.; Van Acker, H.H.; Smits, E.L.; de Vries, I.J.; Bakdash, G. Human blood myeloid and plasmacytoid dendritic cells cross activate each other and synergize in inducing NK cell cytotoxicity. Oncoimmunology 2016, 5, e1227902. [Google Scholar] [CrossRef]
- He, M.; Roussak, K.; Ma, F.; Borcherding, N.; Garin, V.; White, M.; Schutt, C.; Jensen, T.I.; Zhao, Y.; Iberg, C.A.; et al. CD5 expression by dendritic cells directs T cell immunity and sustains immunotherapy responses. Science 2023, 379, eabg2752. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Stary, G.; Bangert, C.; Tauber, M.; Strohal, R.; Kopp, T.; Stingl, G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J. Exp. Med. 2007, 204, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Tramcourt, L.; Leloup, C.; Molens, J.P.; Leccia, M.T.; Charles, J.; Plumas, J. Imiquimod inhibits melanoma development by promoting pDC cytotoxic functions and impeding tumor vascularization. J. Investig. Dermatol. 2014, 134, 2551–2561. [Google Scholar] [CrossRef]
- Aspord, C.; Leloup, C.; Reche, S.; Plumas, J. pDCs efficiently process synthetic long peptides to induce functional virus- and tumour-specific T-cell responses. Eur. J. Immunol. 2014, 44, 2880–2892. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Tel, J.; van der Leun, A.M.; Figdor, C.G.; Torensma, R.; de Vries, I.J. Harnessing human plasmacytoid dendritic cells as professional APCs. Cancer Immunol. Immunother. CII 2012, 61, 1279–1288. [Google Scholar] [CrossRef]
- Liu, C.; Lou, Y.; Lizee, G.; Qin, H.; Liu, S.; Rabinovich, B.; Kim, G.J.; Wang, Y.H.; Ye, Y.; Sikora, A.G.; et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J. Clin. Investig. 2008, 118, 1165–1175. [Google Scholar] [CrossRef]
- Martinek, J.; Lin, J.; Kim, K.I.; Wang, V.G.; Wu, T.C.; Chiorazzi, M.; Boruchov, H.; Gulati, A.; Seeniraj, S.; Sun, L.; et al. Transcriptional profiling of macrophages in situ in metastatic melanoma reveals localization-dependent phenotypes and function. Cell Rep. Med. 2022, 3, 100621. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Rodriguez, E.; Kruijssen, L.J.W.; Crommentuijn, M.H.W.; Boon, L.; Van den Bossche, J.; Den Haan, J.M.M.; Van Kooyk, Y. Monocyte-derived APCs are central to the response of PD1 checkpoint blockade and provide a therapeutic target for combination therapy. J. Immunother. Cancer 2020, 8, e000588. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Duurland, C.L.; Jordanova, E.J.; van Ham, V.J.; Ehsan, I.; Loof, N.M.; Narang, V.; Dutertre, C.A.; Ginhoux, F.; van Egmond, S.L.; et al. CD163+ cytokine-producing cDC2 stimulate intratumoral type 1 T cell responses in HPV16-induced oropharyngeal cancer. J. Immunother. Cancer 2020, 8, e001053. [Google Scholar] [CrossRef]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Movassagh, M.; Spatz, A.; Davoust, J.; Lebecque, S.; Romero, P.; Pittet, M.; Rimoldi, D.; Lienard, D.; Gugerli, O.; Ferradini, L.; et al. Selective accumulation of mature DC-Lamp+ dendritic cells in tumor sites is associated with efficient T-cell-mediated antitumor response and control of metastatic dissemination in melanoma. Cancer Res. 2004, 64, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Smalley, I.; Chen, Z.; Phadke, M.; Li, J.; Yu, X.; Wyatt, C.; Evernden, B.; Messina, J.L.; Sarnaik, A.; Sondak, V.K.; et al. Single-Cell Characterization of the Immune Microenvironment of Melanoma Brain and Leptomeningeal Metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 4109–4125. [Google Scholar] [CrossRef] [PubMed]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef]
- Monti, M.; Consoli, F.; Vescovi, R.; Bugatti, M.; Vermi, W. Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma. Cells 2020, 9, 417. [Google Scholar] [CrossRef]
- Lee, Y.S.; O’Brien, L.J.; Walpole, C.M.; Pearson, F.E.; Leal-Rojas, I.M.; Masterman, K.A.; Atkinson, V.; Barbour, A.; Radford, K.J. Human CD141+ dendritic cells (cDC1) are impaired in patients with advanced melanoma but can be targeted to enhance anti-PD-1 in a humanized mouse model. J. Immunother. Cancer 2021, 9, e001963. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Giampazolias, E.; Schulz, O.; Lim, K.H.J.; Rogers, N.C.; Chakravarty, P.; Srinivasan, N.; Gordon, O.; Cardoso, A.; Buck, M.D.; Poirier, E.Z.; et al. Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell 2021, 184, 4016–4031.e22. [Google Scholar] [CrossRef]
- Gerner, M.Y.; Mescher, M.F. Antigen processing and MHC-II presentation by dermal and tumor-infiltrating dendritic cells. J. Immunol. 2009, 182, 2726–2737. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef]
- Santana-Magal, N.; Farhat-Younis, L.; Gutwillig, A.; Gleiberman, A.; Rasoulouniriana, D.; Tal, L.; Netanely, D.; Shamir, R.; Blau, R.; Feinmesser, M.; et al. Melanoma-Secreted Lysosomes Trigger Monocyte-Derived Dendritic Cell Apoptosis and Limit Cancer Immunotherapy. Cancer Res. 2020, 80, 1942–1956. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Diao, J.; Gu, H.; Khatri, I.; Zhao, J.; Cattral, M.S. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep. 2015, 13, 2851–2864. [Google Scholar] [CrossRef]
- Gerlini, G.; Urso, C.; Mariotti, G.; Di Gennaro, P.; Palli, D.; Brandani, P.; Salvadori, A.; Pimpinelli, N.; Reali, U.M.; Borgognoni, L. Plasmacytoid dendritic cells represent a major dendritic cell subset in sentinel lymph nodes of melanoma patients and accumulate in metastatic nodes. Clin. Immunol. 2007, 125, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Charles, J.; Di Domizio, J.; Salameire, D.; Bendriss-Vermare, N.; Aspord, C.; Muhammad, R.; Lefebvre, C.; Plumas, J.; Leccia, M.T.; Chaperot, L. Characterization of circulating dendritic cells in melanoma: Role of CCR6 in plasmacytoid dendritic cell recruitment to the tumor. J. Investig. Dermatol. 2010, 130, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Machelon, V.; Coulomb-L’Hermin, A.; Borvak, J.; Nome, F.; Isaeva, T.; Wei, S.; Krzysiek, R.; Durand-Gasselin, I.; Gordon, A.; et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat. Med. 2001, 7, 1339–1346. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Moller, H.J.; Donskov, F.; Hoyer, M.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012, 118, 2476–2485. [Google Scholar] [CrossRef]
- Monti, M.; Vescovi, R.; Consoli, F.; Farina, D.; Moratto, D.; Berruti, A.; Specchia, C.; Vermi, W. Plasmacytoid Dendritic Cell Impairment in Metastatic Melanoma by Lactic Acidosis. Cancers 2020, 12, 2085. [Google Scholar] [CrossRef]
- Camisaschi, C.; De Filippo, A.; Beretta, V.; Vergani, B.; Villa, A.; Vergani, E.; Santinami, M.; Cabras, A.D.; Arienti, F.; Triebel, F.; et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: Involvement of LAG-3. J. Investig. Dermatol. 2014, 134, 1893–1902. [Google Scholar] [CrossRef]
- Hack, K.; Reilly, L.; Proby, C.; Fleming, C.; Leigh, I.; Foerster, J. Wnt5a inhibits the CpG oligodeoxynucleotide-triggered activation of human plasmacytoid dendritic cells. Clin. Exp. Dermatol. 2012, 37, 557–561. [Google Scholar] [CrossRef]
- Schnurr, M.; Toy, T.; Shin, A.; Hartmann, G.; Rothenfusser, S.; Soellner, J.; Davis, I.D.; Cebon, J.; Maraskovsky, E. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood 2004, 103, 1391–1397. [Google Scholar] [CrossRef]
- Girard, P.; Charles, J.; Cluzel, C.; Degeorges, E.; Manches, O.; Plumas, J.; De Fraipont, F.; Leccia, M.T.; Mouret, S.; Chaperot, L.; et al. The features of circulating and tumor-infiltrating gammadelta T cells in melanoma patients display critical perturbations with prognostic impact on clinical outcome. Oncoimmunology 2019, 8, 1601483. [Google Scholar] [CrossRef]
- Girard, P.; Sosa Cuevas, E.; Ponsard, B.; Mouret, S.; Gil, H.; Col, E.; De Fraipont, F.; Sturm, N.; Charles, J.; Manches, O.; et al. Dysfunctional BTN3A together with deregulated immune checkpoints and type I/II IFN dictate defective interplay between pDCs and gammadelta T cells in melanoma patients, which impacts clinical outcomes. Clin. Transl. Immunol. 2021, 10, e1329. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Gerlini, G.; Di Gennaro, P.; Mariotti, G.; Urso, C.; Chiarugi, A.; Pimpinelli, N.; Borgognoni, L. Indoleamine 2,3-dioxygenase+ cells correspond to the BDCA2+ plasmacytoid dendritic cells in human melanoma sentinel nodes. J. Investig. Dermatol. 2010, 130, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Gerlini, G.; Di Gennaro, P.; Mariotti, G.; Urso, C.; Chiarugi, A.; Caporale, R.; Pimpinelli, N.; Borgognoni, L. Human Langerhans cells are immature in melanoma sentinel lymph nodes. Blood 2012, 119, 4807–4808; author reply 4809–4810. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, R.; van den Hout, M.F.; Lindenberg, J.J.; Sluijter, B.J.; van Leeuwen, P.A.; Lougheed, S.M.; Meijer, S.; van den Tol, M.P.; Scheper, R.J.; de Gruijl, T.D. Characterization of four conventional dendritic cell subsets in human skin-draining lymph nodes in relation to T-cell activation. Blood 2011, 118, 2502–2510. [Google Scholar] [CrossRef]
- Romoli, M.R.; Di Gennaro, P.; Gerlini, G.; Sestini, S.; Brandani, P.; Ferrone, S.; Borgognoni, L. High Antigen Processing Machinery component expression in Langerhans cells from melanoma patients’ sentinel lymph nodes. Cell. Immunol. 2017, 320, 29–37. [Google Scholar] [CrossRef]
- Gerlini, G.; Di Gennaro, P.; Pimpinelli, N.; Sestini, S.; Borgognoni, L. Tolerogenic IDO1+CD83− Langerhans Cells in Sentinel Lymph Nodes of Patients with Melanoma. Int. J. Mol. Sci. 2022, 23, 3441. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, S.; van Wigcheren, G.F.; Becker, A.; van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nat. Commun. 2020, 11, 2749. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, G.; Buschow, S.I.; Gorris, M.A.; Halilovic, A.; Hato, S.V.; Skold, A.E.; Schreibelt, G.; Sittig, S.P.; Torensma, R.; Duiveman-de Boer, T.; et al. Expansion of a BDCA1+CD14+ Myeloid Cell Population in Melanoma Patients May Attenuate the Efficacy of Dendritic Cell Vaccines. Cancer Res. 2016, 76, 4332–4346. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M. Strategies to Improve the Efficacy of Dendritic Cell-Based Immunotherapy for Melanoma. Front. Immunol. 2017, 8, 1594. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Peguet-Navarro, J.; Sportouch, M.; Popa, I.; Berthier, O.; Schmitt, D.; Portoukalian, J. Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J. Immunol. 2003, 170, 3488–3494. [Google Scholar] [CrossRef]
- Mao, Y.; Poschke, I.; Wennerberg, E.; Pico de Coana, Y.; Egyhazi Brage, S.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef]
- Lindenberg, J.J.; van de Ven, R.; Lougheed, S.M.; Zomer, A.; Santegoets, S.J.; Griffioen, A.W.; Hooijberg, E.; van den Eertwegh, A.J.; Thijssen, V.L.; Scheper, R.J.; et al. Functional characterization of a STAT3-dependent dendritic cell-derived CD14+ cell population arising upon IL-10-driven maturation. Oncoimmunology 2013, 2, e23837. [Google Scholar] [CrossRef]
- Agrawal, P.; Fontanals-Cirera, B.; Sokolova, E.; Jacob, S.; Vaiana, C.A.; Argibay, D.; Davalos, V.; McDermott, M.; Nayak, S.; Darvishian, F.; et al. A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer Cell 2017, 31, 804–819.e7. [Google Scholar] [CrossRef]
- De Vellis, C.; Pietrobono, S.; Stecca, B. The Role of Glycosylation in Melanoma Progression. Cells 2021, 10, 2136. [Google Scholar] [CrossRef]
- RodrIguez, E.; Schetters, S.T.T.; van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Reviews. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef]
- Sosa Cuevas, E.; Valladeau-Guilemond, J.; Mouret, S.; Roubinet, B.; de Fraipont, F.; Landemarre, L.; Charles, J.; Bendriss-Vermare, N.; Chaperot, L.; Aspord, C. Unique CLR expression patterns on circulating and tumor-infiltrating DC subsets correlated with clinical outcome in melanoma patients. Front. Immunol. 2022, 13, 1040600. [Google Scholar] [CrossRef] [PubMed]
- Sosa Cuevas, E.; Roubinet, B.; Mouret, S.; Thepaut, M.; de Fraipont, F.; Charles, J.; Fieschi, F.; Landemarre, L.; Chaperot, L.; Aspord, C. The melanoma tumor glyco-code impacts human dendritic cells’ functionality and dictates clinical outcomes. Front. Immunol. 2023, 14, 1120434. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Bullock, T.N. Metabolic influences that regulate dendritic cell function in tumors. Front. Immunol. 2014, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef]
- Xu, C.; Ruan, B.; Jiang, Y.; Xue, T.; Wang, Z.; Lu, H.; Wei, M.; Wang, S.; Ye, Z.; Zhai, D.; et al. Antibiotics-induced gut microbiota dysbiosis promotes tumor initiation via affecting APC-Th1 development in mice. Biochem. Biophys. Res. Commun. 2017, 488, 418–424. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Ladanyi, A.; Kiss, J.; Somlai, B.; Gilde, K.; Fejos, Z.; Mohos, A.; Gaudi, I.; Timar, J. Density of DC-LAMP+ mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol. Immunother. CII 2007, 56, 1459–1469. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef]
- Tucci, M.; Stucci, L.S.; Mannavola, F.; Passarelli, A.; D’Oronzo, S.; Lospalluti, L.; Giudice, G.; Silvestris, F. Defective levels of both circulating dendritic cells and T-regulatory cells correlate with risk of recurrence in cutaneous melanoma. Clin. Transl. Oncol. 2019, 21, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, R.; Monti, M.; Moratto, D.; Paolini, L.; Consoli, F.; Benerini, L.; Melocchi, L.; Calza, S.; Chiudinelli, M.; Rossi, G.; et al. Collapse of the Plasmacytoid Dendritic Cell Compartment in Advanced Cutaneous Melanomas by Components of the Tumor Cell Secretome. Cancer Immunol. Res. 2019, 7, 12–28. [Google Scholar] [CrossRef]
- Kim, C.W.; Kim, K.D.; Lee, H.K. The role of dendritic cells in tumor microenvironments and their uses as therapeutic targets. BMB Rep. 2021, 54, 31–43. [Google Scholar] [CrossRef]
- Reddy, S.M.; Reuben, A.; Wargo, J.A. Influences of BRAF Inhibitors on the Immune Microenvironment and the Rationale for Combined Molecular and Immune Targeted Therapy. Curr. Oncol. Rep. 2016, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Palermo, B.; Del Bello, D.; Sottini, A.; Serana, F.; Ghidini, C.; Gualtieri, N.; Ferraresi, V.; Catricala, C.; Belardelli, F.; Proietti, E.; et al. Dacarbazine treatment before peptide vaccination enlarges T-cell repertoire diversity of melan-a-specific, tumor-reactive CTL in melanoma patients. Cancer Res. 2010, 70, 7084–7092. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Henry, T.; Baranda, S.J.; Frleta, D.; Manches, O.; Bogunovic, D.; Bhardwaj, N. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol. Immunother. CII 2013, 62, 811–822. [Google Scholar] [CrossRef]
- Oba, T.; Long, M.D.; Keler, T.; Marsh, H.C.; Minderman, H.; Abrams, S.I.; Liu, S.; Ito, F. Overcoming primary and acquired resistance to anti-PD-L1 therapy by induction and activation of tumor-residing cDC1s. Nat. Commun. 2020, 11, 5415. [Google Scholar] [CrossRef]
- Klein, O.; Ebert, L.M.; Zanker, D.; Woods, K.; Tan, B.S.; Fucikova, J.; Behren, A.; Davis, I.D.; Maraskovsky, E.; Chen, W.; et al. Flt3 ligand expands CD4+ FoxP3+ regulatory T cells in human subjects. Eur. J. Immunol. 2013, 43, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.; Kandalaft, L.E. In vivo cancer vaccination: Which dendritic cells to target and how? Cancer Treat. Rev. 2018, 71, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Guinn, Z.P.; Petro, T.M. IFN-gamma synergism with poly I:C reduces growth of murine and human cancer cells with simultaneous changes in cell cycle and immune checkpoint proteins. Cancer Lett. 2018, 438, 1–9. [Google Scholar] [CrossRef]
- Martins, K.A.; Bavari, S.; Salazar, A.M. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev. Vaccines 2015, 14, 447–459. [Google Scholar] [CrossRef]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: TLR3 agonists in cancer therapy. Oncoimmunology 2020, 9, 1771143. [Google Scholar] [CrossRef]
- Kyi, C.; Roudko, V.; Sabado, R.; Saenger, Y.; Loging, W.; Mandeli, J.; Thin, T.H.; Lehrer, D.; Donovan, M.; Posner, M.; et al. Therapeutic Immune Modulation against Solid Cancers with Intratumoral Poly-ICLC: A Pilot Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4937–4948. [Google Scholar] [CrossRef]
- Pashenkov, M.; Goess, G.; Wagner, C.; Hormann, M.; Jandl, T.; Moser, A.; Britten, C.M.; Smolle, J.; Koller, S.; Mauch, C.; et al. Phase II trial of a toll-like receptor 9-activating oligonucleotide in patients with metastatic melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 5716–5724. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.J.; den Boer, A.T.; Medema, J.P.; van der Voort, E.I.; Fransen, M.F.; Offringa, R.; Melief, C.J.; Toes, R.E. CD40 stimulation leads to effective therapy of CD40− tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc. Natl. Acad. Sci. USA 2002, 99, 5561–5566. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Young, A.; Blake, S.J.; Hill, G.R.; Yagita, H.; Teng, M.W.; Korman, A.J.; Smyth, M.J. Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res. 2016, 76, 6266–6277. [Google Scholar] [CrossRef] [PubMed]
- Oosterhoff, D.; Lougheed, S.; van de Ven, R.; Lindenberg, J.; van Cruijsen, H.; Hiddingh, L.; Kroon, J.; van den Eertwegh, A.J.; Hangalapura, B.; Scheper, R.J.; et al. Tumor-mediated inhibition of human dendritic cell differentiation and function is consistently counteracted by combined p38 MAPK and STAT3 inhibition. Oncoimmunology 2012, 1, 649–658. [Google Scholar] [CrossRef]
- O’Sullivan, D.; Sanin, D.E.; Pearce, E.J.; Pearce, E.L. Metabolic interventions in the immune response to cancer. Nat. Rev. Immunol. 2019, 19, 324–335. [Google Scholar] [CrossRef]
- Galvin, K.C.; Dyck, L.; Marshall, N.A.; Stefanska, A.M.; Walsh, K.P.; Moran, B.; Higgins, S.C.; Dungan, L.S.; Mills, K.H. Blocking retinoic acid receptor-alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer Immunol. Immunother. CII 2013, 62, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.W.L.; et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- van Dinther, D.; Stolk, D.A.; van de Ven, R.; van Kooyk, Y.; de Gruijl, T.D.; den Haan, J.M.M. Targeting C-type lectin receptors: A high-carbohydrate diet for dendritic cells to improve cancer vaccines. J. Leukoc. Biol. 2017, 102, 1017–1034. [Google Scholar] [CrossRef]
- Birkholz, K.; Schwenkert, M.; Kellner, C.; Gross, S.; Fey, G.; Schuler-Thurner, B.; Schuler, G.; Schaft, N.; Dorrie, J. Targeting of DEC-205 on human dendritic cells results in efficient MHC class II-restricted antigen presentation. Blood 2010, 116, 2277–2285. [Google Scholar] [CrossRef]
- Tsuji, T.; Matsuzaki, J.; Kelly, M.P.; Ramakrishna, V.; Vitale, L.; He, L.Z.; Keler, T.; Odunsi, K.; Old, L.J.; Ritter, G.; et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J. Immunol. 2011, 186, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Friedlander, P.A.; Pavlick, A.C.; Ernstoff, M.S.; Gastman, B.R.; Hanks, B.A.; Curti, B.D.; Albertini, M.R.; Luke, J.J.; Blazquez, A.B.; et al. Flt3 ligand augments immune responses to anti-DEC-205-NY-ESO-1 vaccine through expansion of dendritic cell subsets. Nat. Cancer 2020, 1, 1204–1217. [Google Scholar] [CrossRef]
- Joffre, O.P.; Sancho, D.; Zelenay, S.; Keller, A.M.; Reis e Sousa, C. Efficient and versatile manipulation of the peripheral CD4+ T-cell compartment by antigen targeting to DNGR-1/CLEC9A. Eur. J. Immunol. 2010, 40, 1255–1265. [Google Scholar] [CrossRef]
- Sancho, D.; Mourao-Sa, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef]
- Gou, S.; Wang, S.; Liu, W.; Chen, G.; Zhang, D.; Du, J.; Yan, Z.; Wang, H.; Zhai, W.; Sui, X.; et al. Adjuvant-free peptide vaccine targeting Clec9a on dendritic cells can induce robust antitumor immune response through Syk/IL-21 axis. Theranostics 2021, 11, 7308–7321. [Google Scholar] [CrossRef] [PubMed]
- Ghinnagow, R.; De Meester, J.; Cruz, L.J.; Aspord, C.; Corgnac, S.; Macho-Fernandez, E.; Soulard, D.; Fontaine, J.; Chaperot, L.; Charles, J.; et al. Co-delivery of the NKT agonist alpha-galactosylceramide and tumor antigens to cross-priming dendritic cells breaks tolerance to self-antigens and promotes antitumor responses. Oncoimmunology 2017, 6, e1339855. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Pots, J.M.; Torensma, R.; Buschow, S.I.; Figdor, C.G. Comparison of antibodies and carbohydrates to target vaccines to human dendritic cells via DC-SIGN. Biomaterials 2012, 33, 4229–4239. [Google Scholar] [CrossRef]
- Unger, W.W.; Mayer, C.T.; Engels, S.; Hesse, C.; Perdicchio, M.; Puttur, F.; Streng-Ouwehand, I.; Litjens, M.; Kalay, H.; Berod, L.; et al. Antigen targeting to dendritic cells combined with transient regulatory T cell inhibition results in long-term tumor regression. Oncoimmunology 2015, 4, e970462. [Google Scholar] [CrossRef]
- Horrevorts, S.K.; Stolk, D.A.; van de Ven, R.; Hulst, M.; van Het Hof, B.; Duinkerken, S.; Heineke, M.H.; Ma, W.; Dusoswa, S.A.; Nieuwland, R.; et al. Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination. Cancers 2019, 11, 1266. [Google Scholar] [CrossRef]
- Li, R.E.; Hogervorst, T.P.; Achilli, S.; Bruijns, S.C.; Arnoldus, T.; Vives, C.; Wong, C.C.; Thepaut, M.; Meeuwenoord, N.J.; van den Elst, H.; et al. Systematic Dual Targeting of Dendritic Cell C-Type Lectin Receptor DC-SIGN and TLR7 Using a Trifunctional Mannosylated Antigen. Front. Chem. 2019, 7, 650. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, A.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Schwarze, J.K.; Geeraerts, X.; Tuyaerts, S.; Neyns, B. Current “state of the art” on dendritic cell-based cancer vaccines in melanoma. Curr. Opin. Oncol. 2023, 35, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Alvarez-Dominguez, C.; Calderon-Gonzalez, R.; Teran-Navarro, H.; Salcines-Cuevas, D.; Garcia-Castano, A.; Freire, J.; Gomez-Roman, J.; Rivera, F. Dendritic cell therapy in melanoma. Ann. Transl. Med. 2017, 5, 386. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, M.; Jaafari, M.R.; Verdi, J.; Alani, B.; Noureddini, M.; Badiee, A. Ex vivo-generated dendritic cell-based vaccines in melanoma: The role of nanoparticulate delivery systems. Immunotherapy 2020, 12, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Yewdall, A.W.; Drutman, S.B.; Jinwala, F.; Bahjat, K.S.; Bhardwaj, N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS ONE 2010, 5, e11144. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.S.; Jiang, A. DC-Based Vaccines for Cancer Immunotherapy. Vaccines 2020, 8, 706. [Google Scholar] [CrossRef]
- Thordardottir, S.; Schaap, N.; Louer, E.; Kester, M.G.; Falkenburg, J.H.; Jansen, J.; Radstake, T.R.; Hobo, W.; Dolstra, H. Hematopoietic stem cell-derived myeloid and plasmacytoid DC-based vaccines are highly potent inducers of tumor-reactive T cell and NK cell responses ex vivo. Oncoimmunology 2017, 6, e1285991. [Google Scholar] [CrossRef]
- van Eck van der Sluijs, J.; van Ens, D.; Thordardottir, S.; Vodegel, D.; Hermens, I.; van der Waart, A.B.; Falkenburg, J.H.F.; Kester, M.G.D.; de Rink, I.; Heemskerk, M.H.M.; et al. Clinically applicable CD34+-derived blood dendritic cell subsets exhibit key subset-specific features and potently boost anti-tumor T and NK cell responses. Cancer Immunol. Immunother. CII 2021, 70, 3167–3181. [Google Scholar] [CrossRef]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.; Duiveman-de Boer, T.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef]
- van Beek, J.J.P.; Florez-Grau, G.; Gorris, M.A.J.; Mathan, T.S.M.; Schreibelt, G.; Bol, K.F.; Textor, J.; de Vries, I.J.M. Human pDCs Are Superior to cDC2s in Attracting Cytolytic Lymphocytes in Melanoma Patients Receiving DC Vaccination. Cell Rep. 2020, 30, 1027–1038.e4. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, C.; Kim, G.J.; Liu, Y.J.; Hwu, P.; Wang, G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J. Immunol. 2007, 178, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Charles, J.; Leccia, M.T.; Laurin, D.; Richard, M.J.; Chaperot, L.; Plumas, J. A novel cancer vaccine strategy based on HLA-A*0201 matched allogeneic plasmacytoid dendritic cells. PLoS ONE 2010, 5, e10458. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.T.; Salameire, D.; Laurin, D.; Chaperot, L.; Charles, J.; Plumas, J. HLA-A*0201+ plasmacytoid dendritic cells provide a cell-based immunotherapy for melanoma patients. J. Investig. Dermatol. 2012, 132, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Charles, J.; Chaperot, L.; Hannani, D.; Bruder Costa, J.; Templier, I.; Trabelsi, S.; Gil, H.; Moisan, A.; Persoons, V.; Hegelhofer, H.; et al. An innovative plasmacytoid dendritic cell line-based cancer vaccine primes and expands antitumor T-cells in melanoma patients in a first-in-human trial. Oncoimmunology 2020, 9, 1738812. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Silva, Z.; Marques, G.; Ferro, T.; Goncalves, M.; Monteiro, M.; van Vliet, S.J.; Mohr, E.; Lino, A.C.; Fernandes, A.R.; et al. Sialic acid removal from dendritic cells improves antigen cross-presentation and boosts anti-tumor immune responses. Oncotarget 2016, 7, 41053–41066. [Google Scholar] [CrossRef]
- Shadbad, M.A.; Hajiasgharzadeh, K.; Derakhshani, A.; Silvestris, N.; Baghbanzadeh, A.; Racanelli, V.; Baradaran, B. From Melanoma Development to RNA-Modified Dendritic Cell Vaccines: Highlighting the Lessons from the Past. Front. Immunol. 2021, 12, 623639. [Google Scholar] [CrossRef]
- Van Nuffel, A.M.; Benteyn, D.; Wilgenhof, S.; Pierret, L.; Corthals, J.; Heirman, C.; van der Bruggen, P.; Coulie, P.G.; Neyns, B.; Thielemans, K.; et al. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1063–1074. [Google Scholar] [CrossRef]
- Hobo, W.; Novobrantseva, T.I.; Fredrix, H.; Wong, J.; Milstein, S.; Epstein-Barash, H.; Liu, J.; Schaap, N.; van der Voort, R.; Dolstra, H. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol. Immunother. CII 2013, 62, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, K.; Hosoi, A.; Iino, T.; Morishita, Y.; Matsushita, H.; Kakimi, K. Dendritic cell vaccine induces antigen-specific CD8+ T cells that are metabolically distinct from those of peptide vaccine and is well-combined with PD-1 checkpoint blockade. Oncoimmunology 2018, 7, e1395124. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, J.K.; Awada, G.; Cras, L.; Tijtgat, J.; Forsyth, R.; Dufait, I.; Tuyaerts, S.; Van Riet, I.; Neyns, B. Intratumoral Combinatorial Administration of CD1c (BDCA-1)+ Myeloid Dendritic Cells Plus Ipilimumab and Avelumab in Combination with Intravenous Low-Dose Nivolumab in Patients with Advanced Solid Tumors: A Phase IB Clinical Trial. Vaccines 2020, 8, 670. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, J.; Tazzari, M.; Granato, A.M.; Ridolfi, L.; Maiocchi, S.; de Rosa, F.; Petrini, M.; Pancisi, E.; Gentili, G.; Vergani, B.; et al. Dendritic Cell Vaccination in Metastatic Melanoma Turns “Non-T Cell Inflamed” Into “T-Cell Inflamed” Tumors. Front. Immunol. 2019, 10, 2353. [Google Scholar] [CrossRef]
- Wang, C.; Barnoud, C.; Cenerenti, M.; Sun, M.; Caffa, I.; Kizil, B.; Bill, R.; Liu, Y.; Pick, R.; Garnier, L.; et al. Dendritic cells direct circadian anti-tumour immune responses. Nature 2023, 614, 136–143. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| DC Subset | Transcription Factors or Inducers | Markers | Main Functions | Ref. |
|---|---|---|---|---|
| cDC1s | ID2, IRF8, BATF3 | BDCA3/CD141, CD11cint, DNGR1/CLEC9A, XCR1, CADM1, BTLA, CD26 | CD8 T-cell activation Type III IFN secretion | [5,9,10,11] |
| cDC2s | ID2, ZEB2, IRF4, Notch2 | BDCA1/CD1c, CD11chi, CD11b, CD5, FCεR1, SIRPA/CD172a, CCR2, BTLA CLEC10A+ (cDC2B 1) or CLEC10A− (cDC2A) | CD4 T-cell activation IL-12 secretion | [5,12,25] |
| pDCs | E2-2, ZEB2, IRF8, IRF4, IRF7 | BDCA2/CD303, CD123, BDCA4/CD304, FCεR1, ILT3, ILT7 | Antiviral immunity Type I IFN secretion | [14] |
| LCs | ID2, RUNX3 | Langerin, CD1a, EpCAM, TROP2, E-cadherin | Induction of Th2 T-cells IL-15 secretion | [18,19] |
| DC3s | GM-CSF | BDCA1, CD163, CD11c, CD14lo to hi, MGL/CLEC10A, CD36, FCεR1 | T-cell activation IL-12 & IL-23 secretion | [24] |
| moDCs/InfDCs | MAFB, KLF4 IRF8 | BDCA1, CD14, CD11c, MR/CD206, CD1a, DC-SIGN, SIRPA | Inflammation (IL-1β, IL-6, IL-23, TNF-α) | [3] |
| mregDCs | C/EBPα | LAMP3/DC-LAMP, CD11b, XCR1, CD11c, CD103, CCR7hi | Tumor control (IL-12) Immune regulation (PD-L1, PD-L2) | [5,29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosa Cuevas, E.; Saas, P.; Aspord, C. Dendritic Cell Subsets in Melanoma: Pathophysiology, Clinical Prognosis and Therapeutic Exploitation. Cancers 2023, 15, 2206. https://doi.org/10.3390/cancers15082206
Sosa Cuevas E, Saas P, Aspord C. Dendritic Cell Subsets in Melanoma: Pathophysiology, Clinical Prognosis and Therapeutic Exploitation. Cancers. 2023; 15(8):2206. https://doi.org/10.3390/cancers15082206
Chicago/Turabian StyleSosa Cuevas, Eleonora, Philippe Saas, and Caroline Aspord. 2023. "Dendritic Cell Subsets in Melanoma: Pathophysiology, Clinical Prognosis and Therapeutic Exploitation" Cancers 15, no. 8: 2206. https://doi.org/10.3390/cancers15082206
APA StyleSosa Cuevas, E., Saas, P., & Aspord, C. (2023). Dendritic Cell Subsets in Melanoma: Pathophysiology, Clinical Prognosis and Therapeutic Exploitation. Cancers, 15(8), 2206. https://doi.org/10.3390/cancers15082206