


The Secondary Myelodysplastic Neoplasms (MDS) Jigsaw



Simple Summary
Abstract
1. Introduction
2. The Pieces of the Jigsaw
2.1. MDS Post-Cytotoxic Therapy
2.2. Germline Predisposition
2.3. Clonal Hematopoiesis
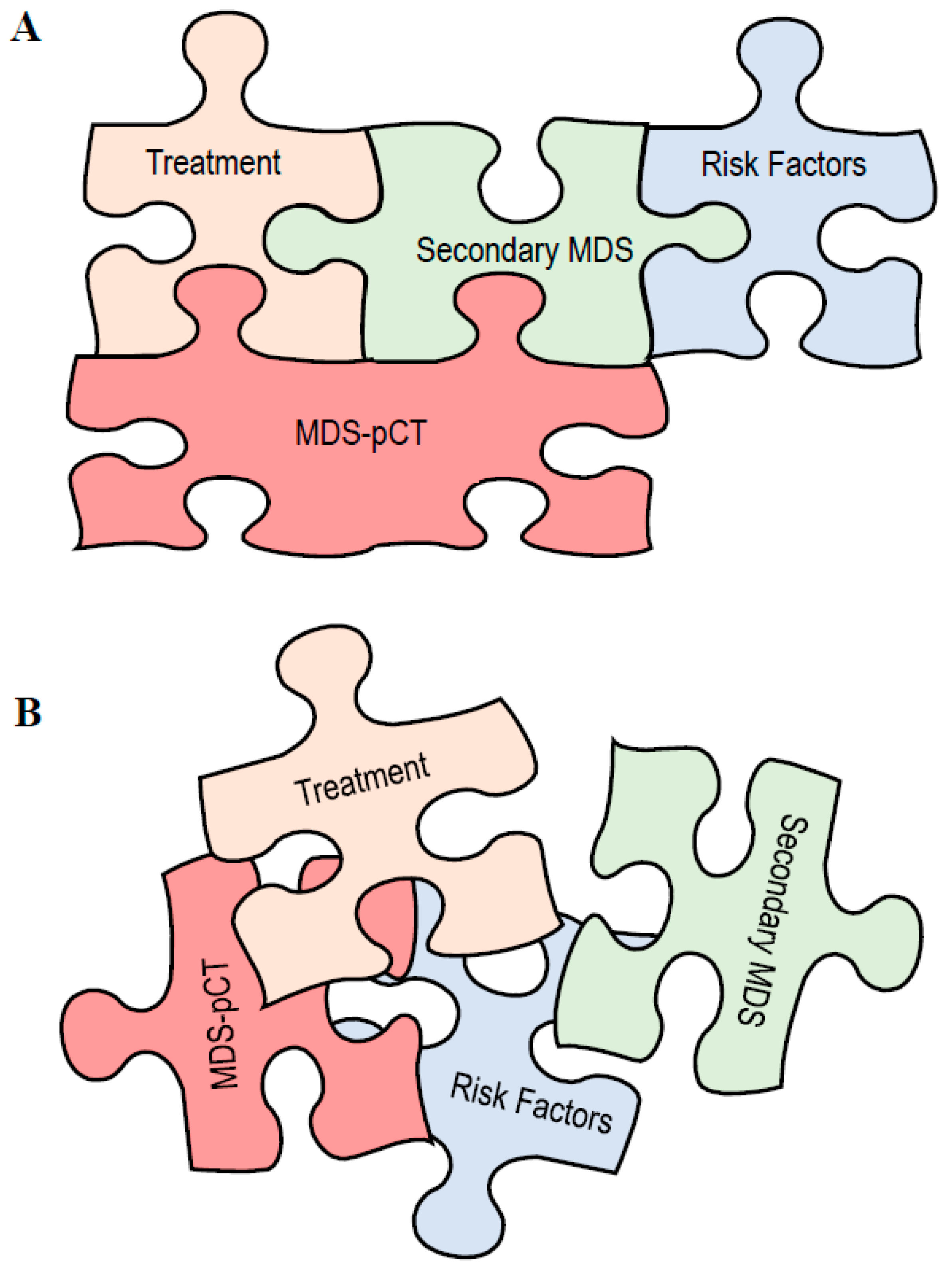
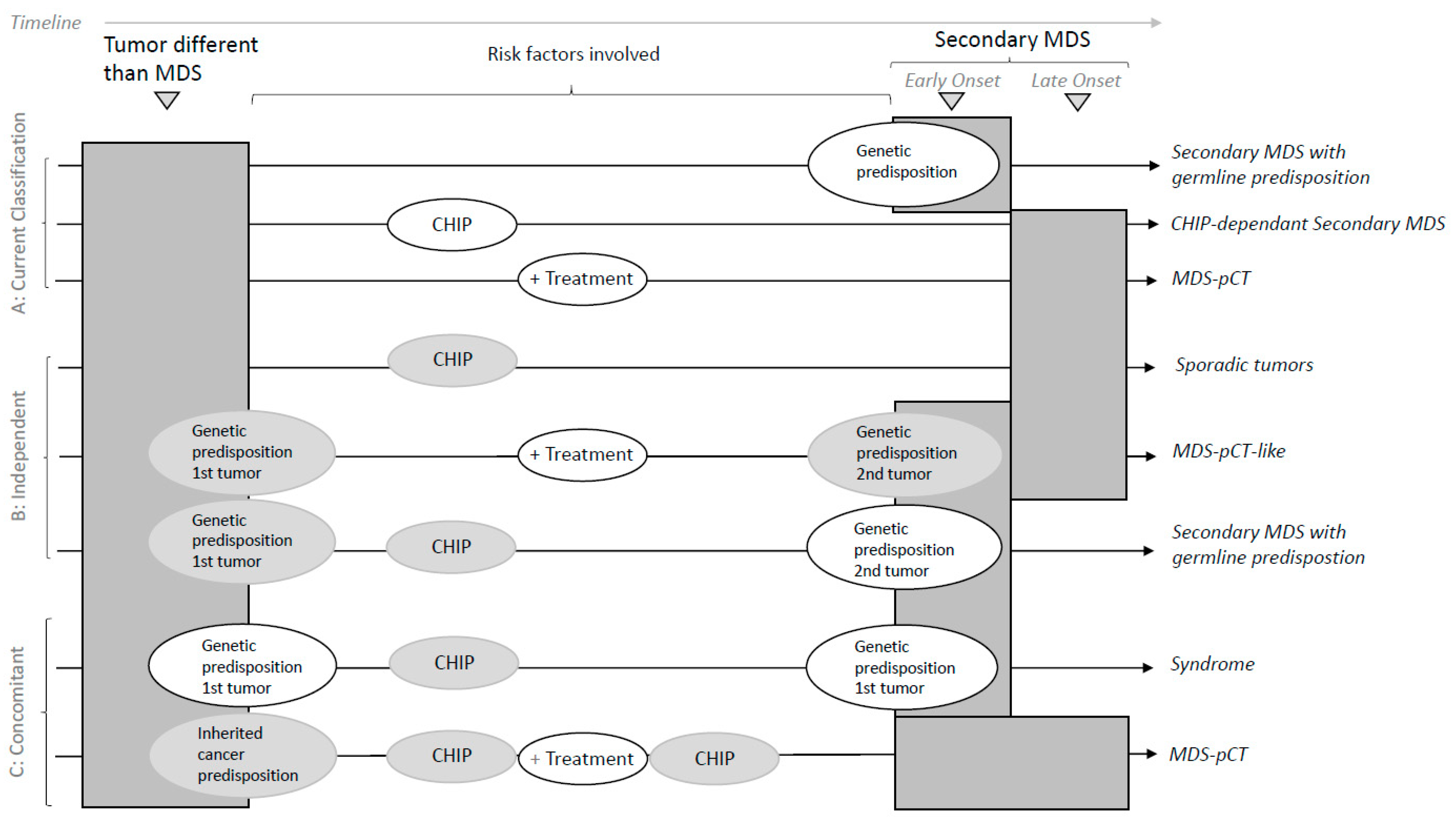
2.4. Current Classifications and Secondary MDS
3. Discussion: How to Put the Jigsaw Pieces Together
- solely contribute to MDS without other known risk factors;
- trigger CHIP or drive an increase in CHIP;
- contribute depending on germline predisposition or susceptibility;
- contribute together with CHIP and the germline landscape.
- Sporadic, correlative tumors with no shared risk factors;
- MDS-pCT-like, in which cytotoxic therapy does not participate in the development of a secondary myeloid tumor.
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lim, U.; Park, Y.; Mayne, S.T.; Wang, R.; Hartge, P.; Hollenbeck, A.R.; Schatzkin, A. Obesity, Lifestyle Factors, and Risk of Myelodysplastic Syndromes in a Large US Cohort. Am. J. Epidemiol. 2009, 169, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Kuendgen, A.; Nomdedeu, M.; Tuechler, H.; Garcia-Manero, G.; Komrokji, R.S.; Sekeres, M.A.; della Porta, M.G.; Cazzola, M.; DeZern, A.E.; Roboz, G.J.; et al. Therapy-Related Myelodysplastic Syndromes Deserve Specific Diagnostic Sub-Classification and Risk-Stratification—An Approach to Classification of Patients with t-MDS. Leukemia 2021, 35, 835–849. [Google Scholar] [CrossRef]
- Furutani, E.; Shimamura, A. Germline Genetic Predisposition to Hematologic Malignancy. J. Clin. Oncol. 2017, 35, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Faltas, B.; Douglas Smith, B.; Gore, S. Myelodysplastic Syndromes: What Do Hospitalists Need to Know? J. Hosp. Med. 2013, 8, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Horny, H.-P.; Bennett, J.M.; Fonatsch, C.; Germing, U.; Greenberg, P.; Haferlach, T.; Haase, D.; Kolb, H.-J.; Krieger, O.; et al. Definitions and Standards in the Diagnosis and Treatment of the Myelodysplastic Syndromes: Consensus Statements and Report from a Working Conference. Leuk. Res. 2007, 31, 727–736. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Philip, P.; Mortensen, B.T.; Ersbøll, J.; Jensen, G.; Panduro, J.; Thomsen, M. Acute Nonlymphocytic Leukemia, Preleukemia, and Acute Myeloproliferative Syndrome Secondary to Treatment of Other Malignant Diseases. Clinical and Cytogenetic Characteristics and Results of in Vitro Culture of Bone Marrow and HLA Typing. Blood 1981, 57, 712–723. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the Classification of the Myelodysplastic Syndromes. Br. J. Haematol. 1982, 51, 189–199. [Google Scholar] [CrossRef]
- Streuli, R.A.; Testa, J.R.; Vardiman, J.W.; Mintz, U.; Golomb, H.M.; Rowley, J.D. Dysmyelopoietic Syndrome: Sequential Clinical and Cytogenetic Studies. Blood 1980, 55, 636–644. [Google Scholar] [CrossRef]
- McNerney, M.E.; Godley, L.A.; le Beau, M.M. Therapy-Related Myeloid Neoplasms: When Genetics and Environment Collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Hsu, W.-L.; Soda, M.; Takasaki, Y.; Tawara, M.; Joh, T.; Amenomori, T.; Yamamura, M.; Yoshida, Y.; Koba, T.; et al. Risk of Myelodysplastic Syndromes in People Exposed to Ionizing Radiation: A Retrospective Cohort Study of Nagasaki Atomic Bomb Survivors. J. Clin. Oncol. 2011, 29, 428–434. [Google Scholar] [CrossRef]
- Sun, L.-M.; Lin, C.-L.; Lin, M.-C.; Liang, J.-A.; Kao, C.-H. Radiotherapy- and Chemotherapy-Induced Myelodysplasia Syndrome. Medicine 2015, 94, e737. [Google Scholar] [CrossRef] [PubMed]
- Abou Zahr, A.; Kavi, A.M.; Mukherjee, S.; Zeidan, A.M. Therapy-Related Myelodysplastic Syndromes, or Are They? Blood Rev. 2017, 31, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O.; Carbone, P.P.; Connors, J.M.; Levine, A.; Bennett, J.M.; Kroll, S. Treatment-Related Myelodysplasia and Acute Leukemia in Non-Hodgkin’s Lymphoma Patients. J. Clin. Oncol. 2003, 21, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Neuberg, D.; Flinn, I.W.; Grever, M.R.; Lazarus, H.M.; Rowe, J.M.; Dewald, G.; Bennett, J.M.; Paietta, E.M.; Byrd, J.C.; et al. Incidence of Therapy-Related Myeloid Neoplasia after Initial Therapy for Chronic Lymphocytic Leukemia with Fludarabine-Cyclophosphamide versus Fludarabine: Long-Term Follow-up of US Intergroup Study E2997. Blood 2011, 118, 3525–3527. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Shah, D.; Kantarjian, H.; Orlowski, R.Z.; Nogueras González, G.M.; Baladandayuthapani, V.; Jain, N.; Wagner, V.; Garcia-Manero, G.; Shah, J.; et al. Characteristics and Outcomes of Patients with Multiple Myeloma Who Develop Therapy-Related Myelodysplastic Syndrome, Chronic Myelomonocytic Leukemia, or Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 110–114. [Google Scholar] [CrossRef]
- Bendari, M.; Khoubila, N. Cytogenetic and Genetic Advances in Myelodysplasia Syndromes. In Cytogenetics—Classical and Molecular Strategies for Analysing Heredity Material; IntechOpen: London, UK, 2021. [Google Scholar]
- Schanz, J.; Braulke, F.; Haase, D. Rare Cytogenetic Abnormalities in Myelodysplastic Syndromes. Mediterr. J. Hematol. Infect. Dis. 2015, 7, e2015034. [Google Scholar] [CrossRef]
- Churpek, J.E.; Larson, R.A. The Evolving Challenge of Therapy-Related Myeloid Neoplasms. Best Pract. Res. Clin. Haematol. 2013, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Shahjahani, M.; Hadad, E.H.; Azizidoost, S.; Nezhad, K.C.; Shahrabi, S. Complex Karyotype in Myelodysplastic Syndromes: Diagnostic Procedure and Prognostic Susceptibility. Oncol. Rev. 2019, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.F.; Malik, U.A.; Sohail, M.; Hassan, I.N.; Ali, S.; Shaukat, M.H.S. Cytogenetic Abnormalities in Myelodysplastic Syndromes: An Overview. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 231–239. [Google Scholar] [PubMed]
- Leone, G.; Fabiani, E.; Voso, M.T. De Novo and Therapy-Related Myelodysplastic Syndromes: Analogies and Differences. Mediterr. J. Hematol. Infect. Dis. 2022, 14, e2022030. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Wang, F.; Kantarjian, H.; Song, X.; Patel, K.; Neelapu, S.; Gumbs, C.; Little, L.; Tippen, S.; Thornton, R.; et al. Copy Number Alterations Detected as Clonal Hematopoiesis of Indeterminate Potential. Blood Adv. 2017, 1, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A. Therapy-Related Myeloid Neoplasms. Haematologica 2009, 94, 454–459. [Google Scholar] [CrossRef]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.N.; et al. New Insights into the Prognostic Impact of the Karyotype in MDS and Correlation with Subtypes: Evidence from a Core Dataset of 2124 Patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef]
- Heuser, M. Therapy-Related Myeloid Neoplasms: Does Knowing the Origin Help to Guide Treatment? Hematology 2016, 2016, 24–32. [Google Scholar] [CrossRef]
- Rowley, J.D.; Olney, H.J. International Workshop on the Relationship of Prior Therapy to Balanced Chromosome Aberrations in Therapy-Related Myelodysplastic Syndromes and Acute Leukemia: Overview Report. Genes Chromosomes Cancer 2002, 33, 331–345. [Google Scholar] [CrossRef]
- Armand, P.; Kim, H.T.; DeAngelo, D.J.; Ho, V.T.; Cutler, C.S.; Stone, R.M.; Ritz, J.; Alyea, E.P.; Antin, J.H.; Soiffer, R.J. Impact of Cytogenetics on Outcome of De Novo and Therapy-Related AML and MDS after Allogeneic Transplantation. Biol. Blood Marrow Transplant. 2007, 13, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. [Google Scholar] [CrossRef]
- Todisco, E.; Gigli, F.; Ronchini, C.; Amato, V.; Sammassimo, S.; Pastano, R.; Parma, G.; Lapresa, M.T.; Bertolini, F.; Corsini, C.; et al. Hematological Disorders after Salvage PARPi Treatment for Ovarian Cancer: Cytogenetic and Molecular Defects and Clinical Outcomes. Int. J. Cancer 2022, 151, 1791–1803. [Google Scholar] [CrossRef]
- Martin, J.E.; Khalife-Hachem, S.; Grinda, T.; Kfoury, M.; Garciaz, S.; Pasquier, F.; Vargaftig, J.; Uzunov, M.; Belhabri, A.; Bertoli, S.; et al. Therapy-Related Myeloid Neoplasms Following Treatment with PARP Inhibitors: New Molecular Insights. Ann. Oncol. 2021, 32, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Mailankody, S. Update on Second Primary Malignancies in Multiple Myeloma: A Focused Review. Leukemia 2014, 28, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Guerra, V.A.; Kennedy, J.A.; Yan, Y.; Hsu, J.I.; Wang, F.; Nguyen, A.T.; Miller, P.G.; McConkey, M.E.; Quevedo Barrios, V.A.; et al. Lenalidomide Promotes the Development of TP53 -Mutated Therapy-Related Myeloid Neoplasms. Blood 2022, 140, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Saleem, K.; Franz, J.; Klem, M.L.; Yabes, J.G.; Boyiadzis, M.; Jones, J.R.; Shaikh, N.; Lontos, K. Second Primary Malignancies in Patients with Haematological Cancers Treated with Lenalidomide: A Systematic Review and Meta-Analysis. Lancet Haematol. 2022, 9, e906–e918. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Daver, N.; Kim, H.; Dinardo, C.; Jabbour, E.; Kadia, T.; Borthakur, G.; Pierce, S.; Shan, J.; Cardenas-Turanzas, M.; et al. A Prognostic Model of Therapy-Related Myelodysplastic Syndrome for Predicting Survival and Transformation to Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2014, 14, 401–410. [Google Scholar] [CrossRef]
- Singhal, D.; Wee, L.Y.A.; Kutyna, M.M.; Chhetri, R.; Geoghegan, J.; Schreiber, A.W.; Feng, J.; Wang, P.P.-S.; Babic, M.; Parker, W.T.; et al. The Mutational Burden of Therapy-Related Myeloid Neoplasms Is Similar to Primary Myelodysplastic Syndrome but Has a Distinctive Distribution. Leukemia 2019, 33, 2842–2853. [Google Scholar] [CrossRef]
- Khalife-Hachem, S.; Saleh, K.; Pasquier, F.; Willekens, C.; Tarabay, A.; Antoun, L.; Grinda, T.; Castilla-Llorente, C.; Duchmann, M.; Quivoron, C.; et al. Molecular Landscape of Therapy-Related Myeloid Neoplasms in Patients Previously Treated for Gynecologic and Breast Cancers. Hemasphere 2021, 5, e632. [Google Scholar] [CrossRef] [PubMed]
- DeZern, A.E.; Malcovati, L.; Ebert, B.L. CHIP, CCUS, and Other Acronyms: Definition, Implications, and Impact on Practice. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 400–410. [Google Scholar] [CrossRef]
- Huang, K.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.-L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic Analysis of Germ Line and Somatic Variants in Familial Myelodysplasia/Acute Myeloid Leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef]
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating Germline Variant Assessment into Routine Clinical Practice for Myelodysplastic Syndrome and Acute Myeloid Leukaemia: Current Strategies and Challenges. Br. J. Haematol. 2022, 196, 1293–1310. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Tesi, B.; Wartiovaara-Kautto, U.; Stray-Pedersen, A.; Friis, L.S.; Dybedal, I.; Hovland, R.; Jahnukainen, K.; Raaschou-Jensen, K.; Ljungman, P.; et al. Nordic Guidelines for Germline Predisposition to Myeloid Neoplasms in Adults: Recommendations for Genetic Diagnosis, Clinical Management and Follow-Up. Hemasphere 2019, 3, e321. [Google Scholar] [CrossRef] [PubMed]
- Schlegelberger, B.; Mecucci, C.; Wlodarski, M. Review of Guidelines for the Identification and Clinical Care of Patients with Genetic Predisposition for Hematological Malignancies. Fam. Cancer 2021, 20, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Churpek, J.E. Familial Myelodysplastic Syndrome/Acute Myeloid Leukemia. Best Pract. Res. Clin. Haematol. 2017, 30, 287–289. [Google Scholar] [CrossRef]
- Brown, A.L.; Churpek, J.E.; Malcovati, L.; Döhner, H.; Godley, L.A. Recognition of Familial Myeloid Neoplasia in Adults. Semin. Hematol. 2017, 54, 60–68. [Google Scholar] [CrossRef]
- Babushok, D.V.; Bessler, M.; Olson, T.S. Genetic Predisposition to Myelodysplastic Syndrome and Acute Myeloid Leukemia in Children and Young Adults. Leuk. Lymphoma 2016, 57, 520–536. [Google Scholar] [CrossRef]
- Chai-Ho, W.; Schiller, G.J. Myelodysplastic Syndromes: An Update on Pathophysiology and Management. In Recent Developments in Myelodysplastic Syndromes; IntechOpen: London, UK, 2019. [Google Scholar]
- Kennedy, A.L.; Shimamura, A. Genetic Predisposition to MDS: Clinical Features and Clonal Evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef]
- Palomino-Echeverría, S.; Vázquez, I.; Alfonso-Piérola, A.; José Larrayoz, M.; Aguilera-Díaz, A.; Ariceta, B.; Daniela Urribarri, A.; Mañú, A.; Blasco-Iturri, Z.; Prósper, F.; et al. Predisposición a Neoplasias Mieloides: El Nuevo Desafío En La Consulta de Hematología; Genética medica y genómica: Valencia, Spain, 2020; Volume 4. [Google Scholar]
- Crysandt, M.; Brings, K.; Beier, F.; Thiede, C.; Brümmendorf, T.H.; Jost, E. Germ Line Predisposition to Myeloid Malignancies Appearing in Adulthood. Expert Rev. Hematol. 2018, 11, 625–636. [Google Scholar] [CrossRef]
- Babushok, D.V.; Bessler, M. Genetic Predisposition Syndromes: When Should They Be Considered in the Work-up of MDS? Best Pract. Res. Clin. Haematol. 2015, 28, 55–68. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Pich, O.; Reyes-Salazar, I.; Gonzalez-Perez, A.; Lopez-Bigas, N. Discovering the Drivers of Clonal Hematopoiesis. Nat. Commun. 2022, 13, 4267. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. ICUS, IDUS, CHIP and CCUS: Diagnostic Criteria, Separation from MDS and Clinical Implications. Pathobiology 2019, 86, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Senguttuvan, N.B.; Subramanian, V.; Venkatesan, V.; Muralidharan, T.R.; Sankaranarayanan, K. Clonal Hematopoiesis of Indeterminate Potential (CHIP) and Cardiovascular Diseases—An Updated Systematic Review. J. Genet. Eng. Biotechnol. 2021, 19, 105. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, 7. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Schulz, E.; Valentin, A.; Ulz, P.; Beham-Schmid, C.; Lind, K.; Rupp, V.; Lackner, H.; Wölfler, A.; Zebisch, A.; Olipitz, W.; et al. Germline Mutations in the DNA Damage Response Genes BRCA1, BRCA2, BARD1 and TP53 in Patients with Therapy Related Myeloid Neoplasms. J. Med. Genet. 2012, 49, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Churpek, J.E.; Marquez, R.; Neistadt, B.; Claussen, K.; Lee, M.K.; Churpek, M.M.; Huo, D.; Weiner, H.; Bannerjee, M.; Godley, L.A.; et al. Inherited Mutations in Cancer Susceptibility Genes Are Common among Survivors of Breast Cancer Who Develop Therapy-Related Leukemia. Cancer 2016, 122, 304–311. [Google Scholar] [CrossRef]
- Voso, M.T.; Fabiani, E.; Zang, Z.; Fianchi, L.; Falconi, G.; Padella, A.; Martini, M.; Li Zhang, S.; Santangelo, R.; Larocca, L.M.; et al. Fanconi Anemia Gene Variants in Therapy-Related Myeloid Neoplasms. Blood Cancer J. 2015, 5, e323. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer Therapy Shapes the Fitness Landscape of Clonal Hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.; Yoder, S.J.; Balasis, M.E.; Mesa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal Haemopoiesis and Therapy-Related Myeloid Malignancies in Elderly Patients: A Proof-of-Concept, Case-Control Study. Lancet Oncol. 2017, 18, 112–121. [Google Scholar] [CrossRef]
- Desai, P.; Roboz, G.J. Clonal Hematopoiesis and Therapy Related MDS/AML. Best Pract. Res. Clin. Haematol. 2019, 32, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Baer, C.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C. Mutational Patterns and Their Correlation to CHIP-Related Mutations and Age in Hematological Malignancies. Blood Adv. 2021, 5, 4426–4434. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Karyotype | Genetic Risk Factors | CHIP | De Novo MDS | MDS-pCT |
|---|---|---|---|---|
| Altered karyotypes | Not described | 21% [25] | 40–60% [20] | 70–90% [19,20] |
| Complex karyotypes * | Not described | Not described | 30% [22] | 46–70% [12,21] |
| Most frequent unique alterations | dup(1q), 3q+, −7/del(7q), i(17)(q10), +8, +21, del(20q), del(11q) [24] | del(20q), del(13q), del(11q), +8, del(5q), del(17p) [6,12] | del(5q), −7/del(7q), +8, −Y [20,23] | Post-alkylating agents: −7, del(7q), del(5q), −5 [22,26] Topoisomerase II inhibidors: t(11;21)(q23;q22), t(15;17), inv(16)(p13q22), t(17;19)(q22;q12) [24] |
| Most frequent complex karyotypes | Not described | Not described | del(5q), −7/del(7q), −18/−18q (7%), +8, −20q. Other: +1/+1q, −5, +11, −13/13q−, −17/17p−, −21, +mar [27] | −5/del(5q), −7/del(7q) Other: der(21q), +8, der(12q), t(1;7), −12, der(17q), der(3q), der(3q), and −18 [22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvete, O.; Mestre, J.; Jerez, A.; Solé, F. The Secondary Myelodysplastic Neoplasms (MDS) Jigsaw. Cancers 2023, 15, 1483. https://doi.org/10.3390/cancers15051483
Calvete O, Mestre J, Jerez A, Solé F. The Secondary Myelodysplastic Neoplasms (MDS) Jigsaw. Cancers. 2023; 15(5):1483. https://doi.org/10.3390/cancers15051483
Chicago/Turabian StyleCalvete, Oriol, Julia Mestre, Andrés Jerez, and Francesc Solé. 2023. "The Secondary Myelodysplastic Neoplasms (MDS) Jigsaw" Cancers 15, no. 5: 1483. https://doi.org/10.3390/cancers15051483
APA StyleCalvete, O., Mestre, J., Jerez, A., & Solé, F. (2023). The Secondary Myelodysplastic Neoplasms (MDS) Jigsaw. Cancers, 15(5), 1483. https://doi.org/10.3390/cancers15051483