
Introduction of Mutant GNAQ into Endothelial Cells Induces a Vascular Malformation Phenotype with Therapeutic Response to Imatinib

 ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Development of MS1 GNAQ Q209L Cells
2.2. In Vivo Studies
2.3. Imatinib Treatment
2.4. RNA Extraction
2.5. Assay for Transposase-Accessible Chromatin Using Sequencing (ATAC-Seq)
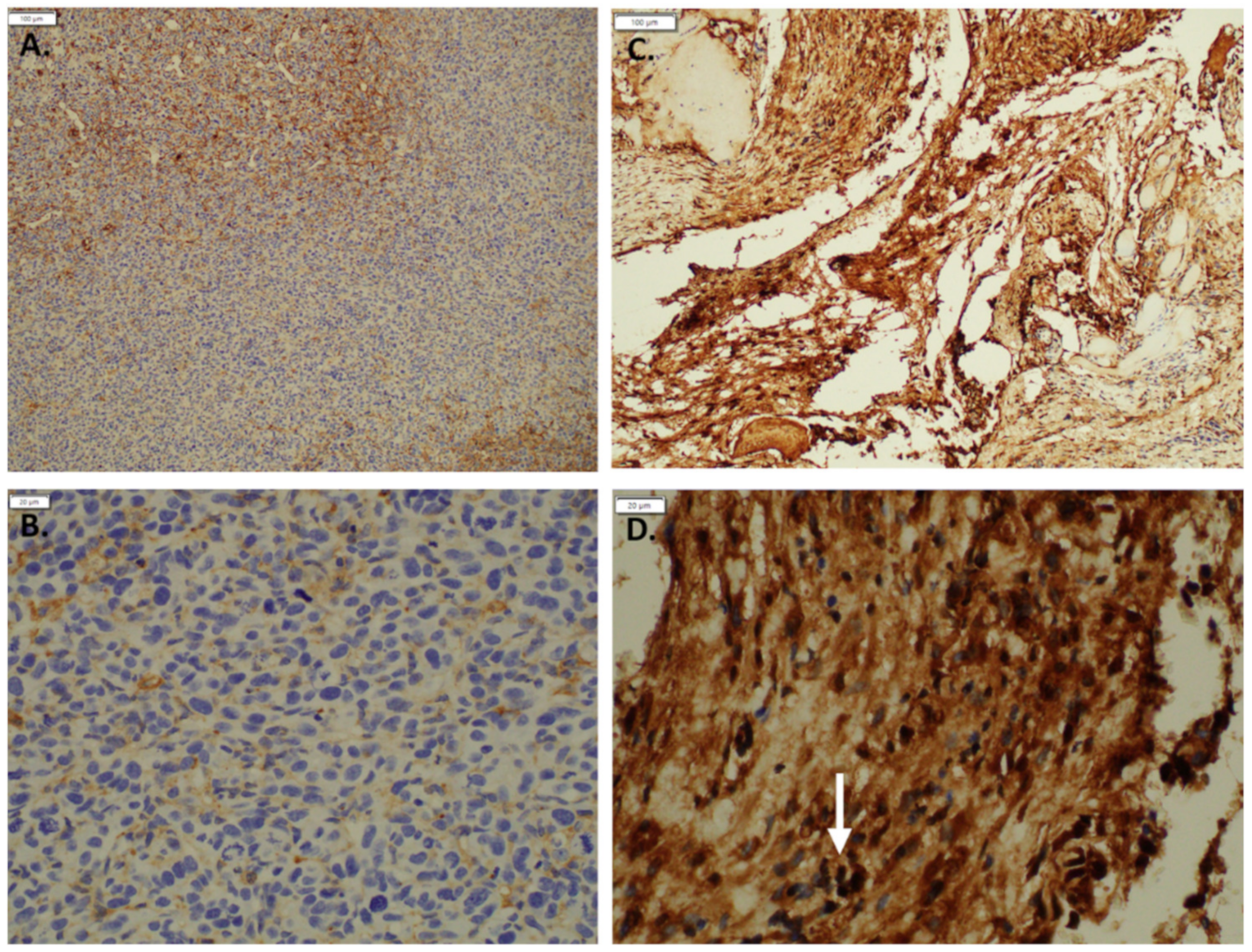
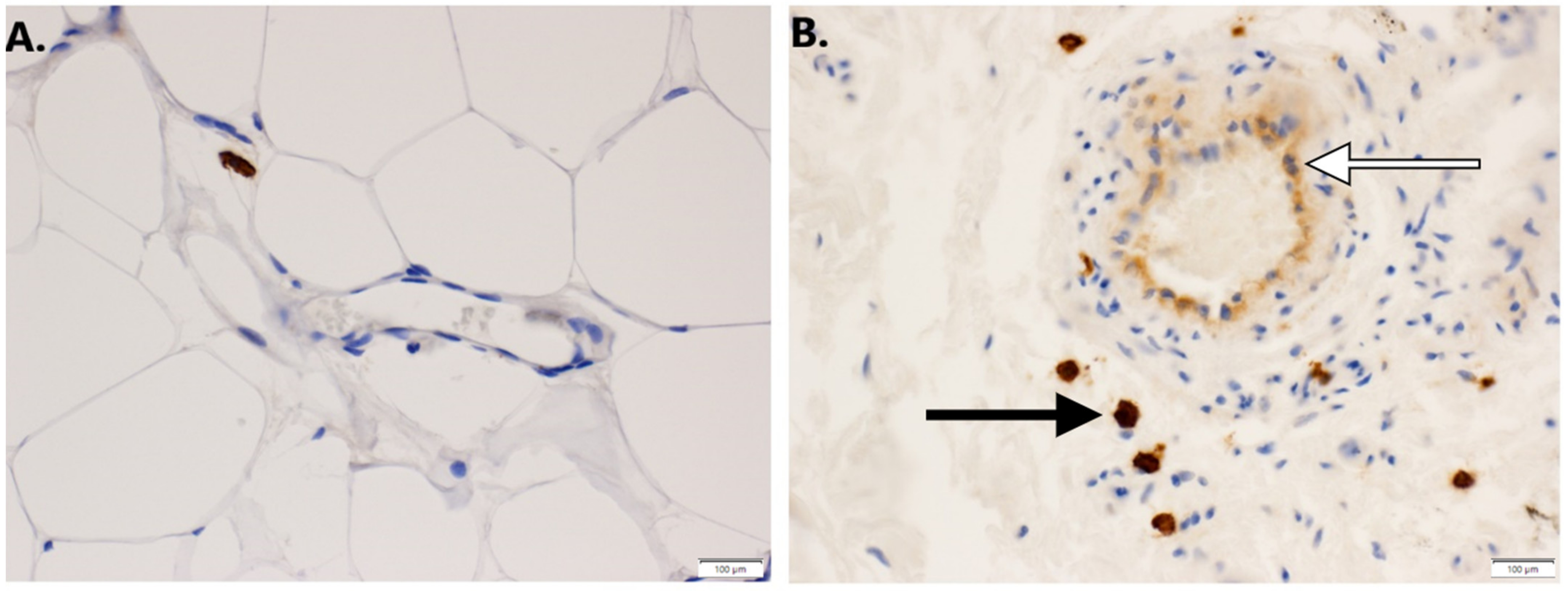
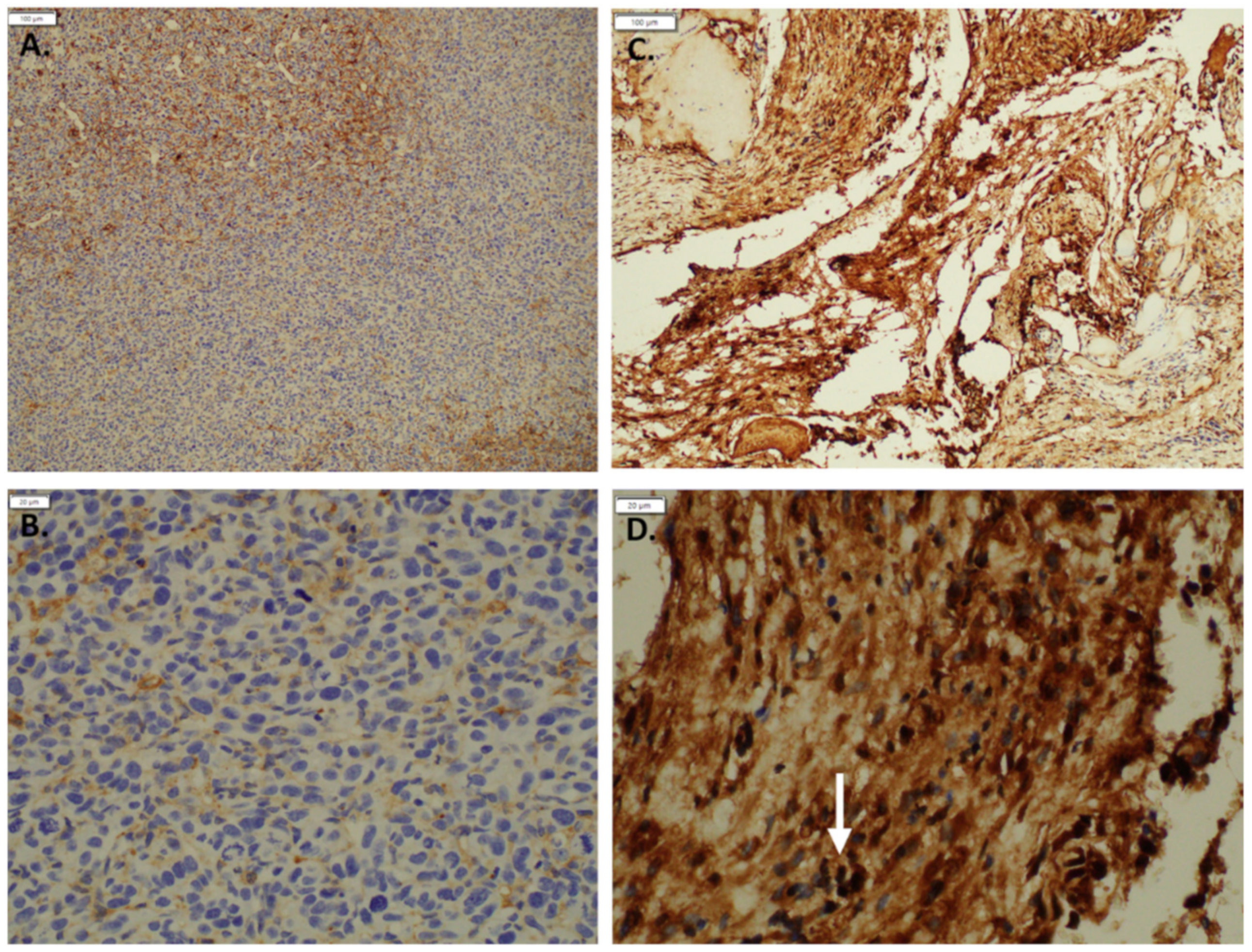
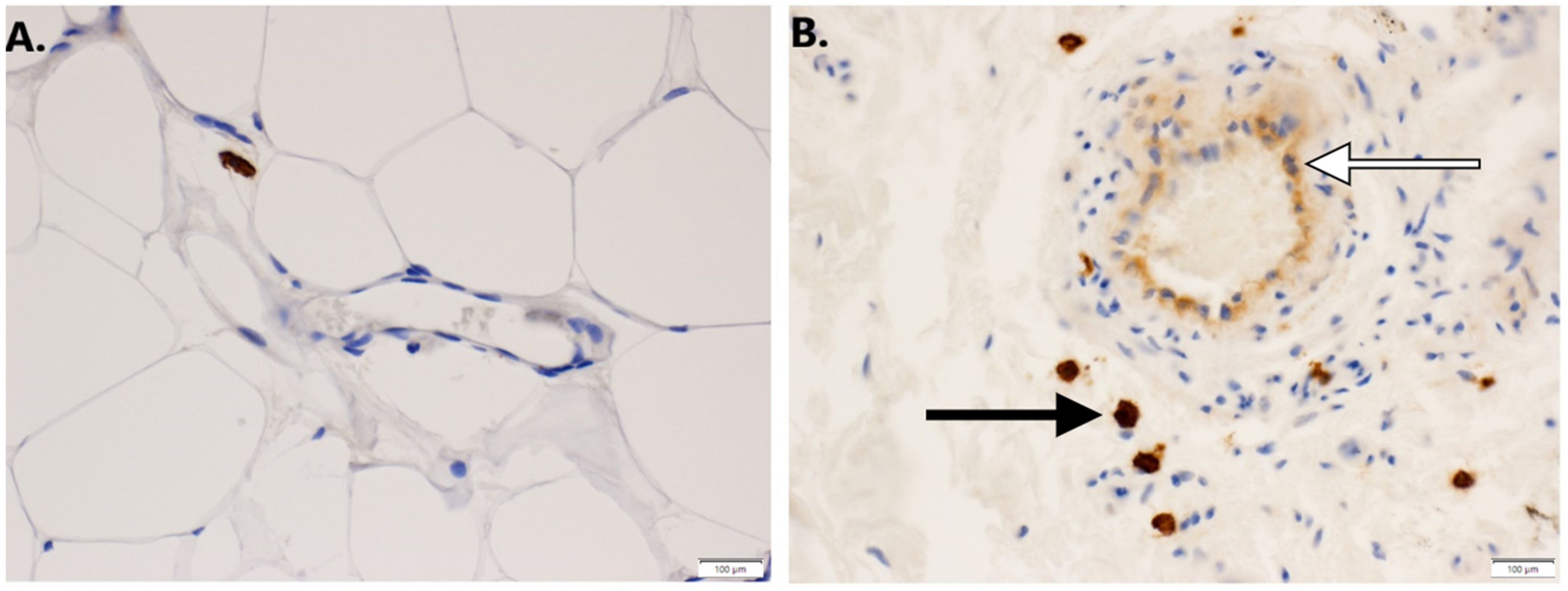
2.6. Immunohistochemistry
2.7. Statistical Analysis
3. Results
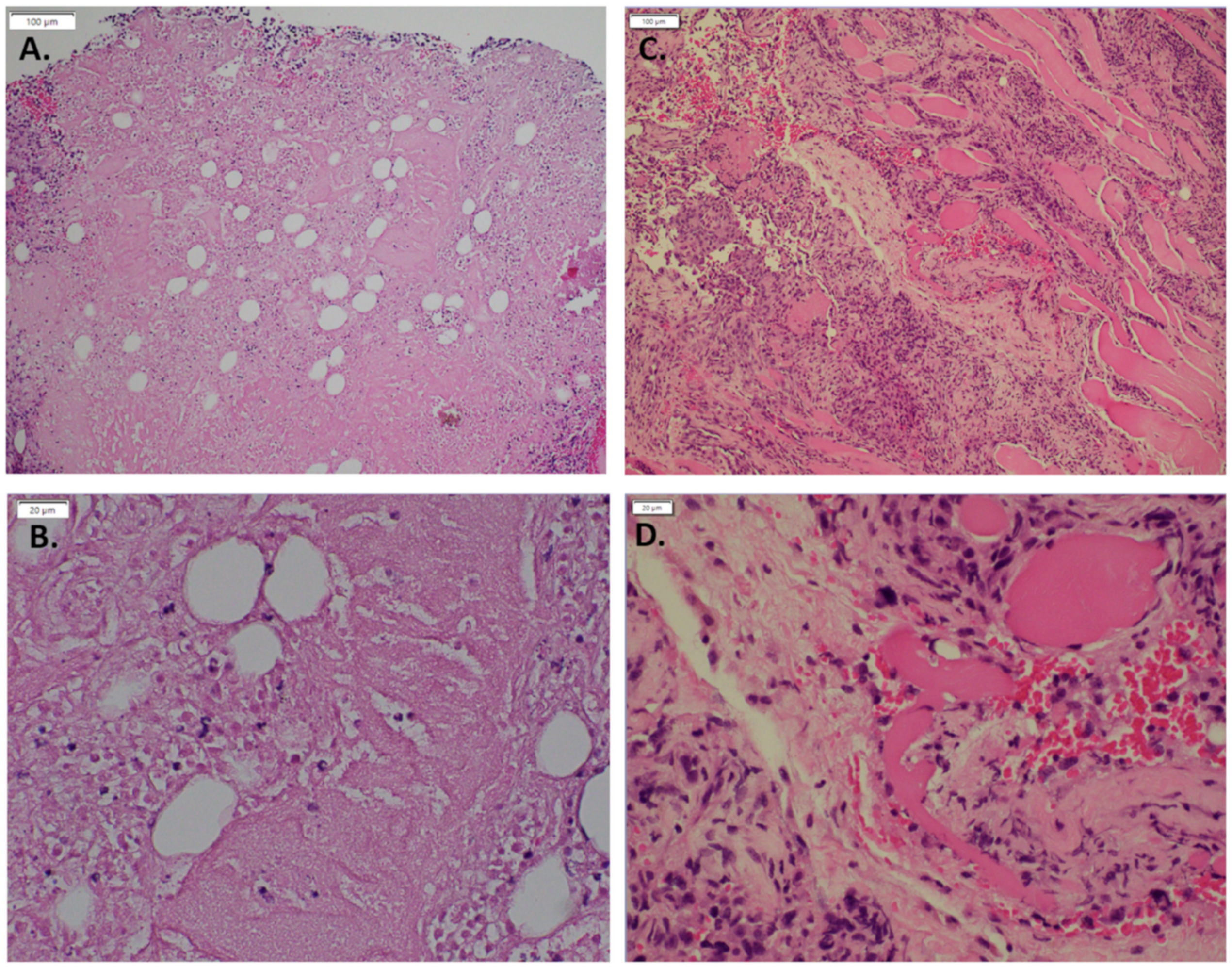
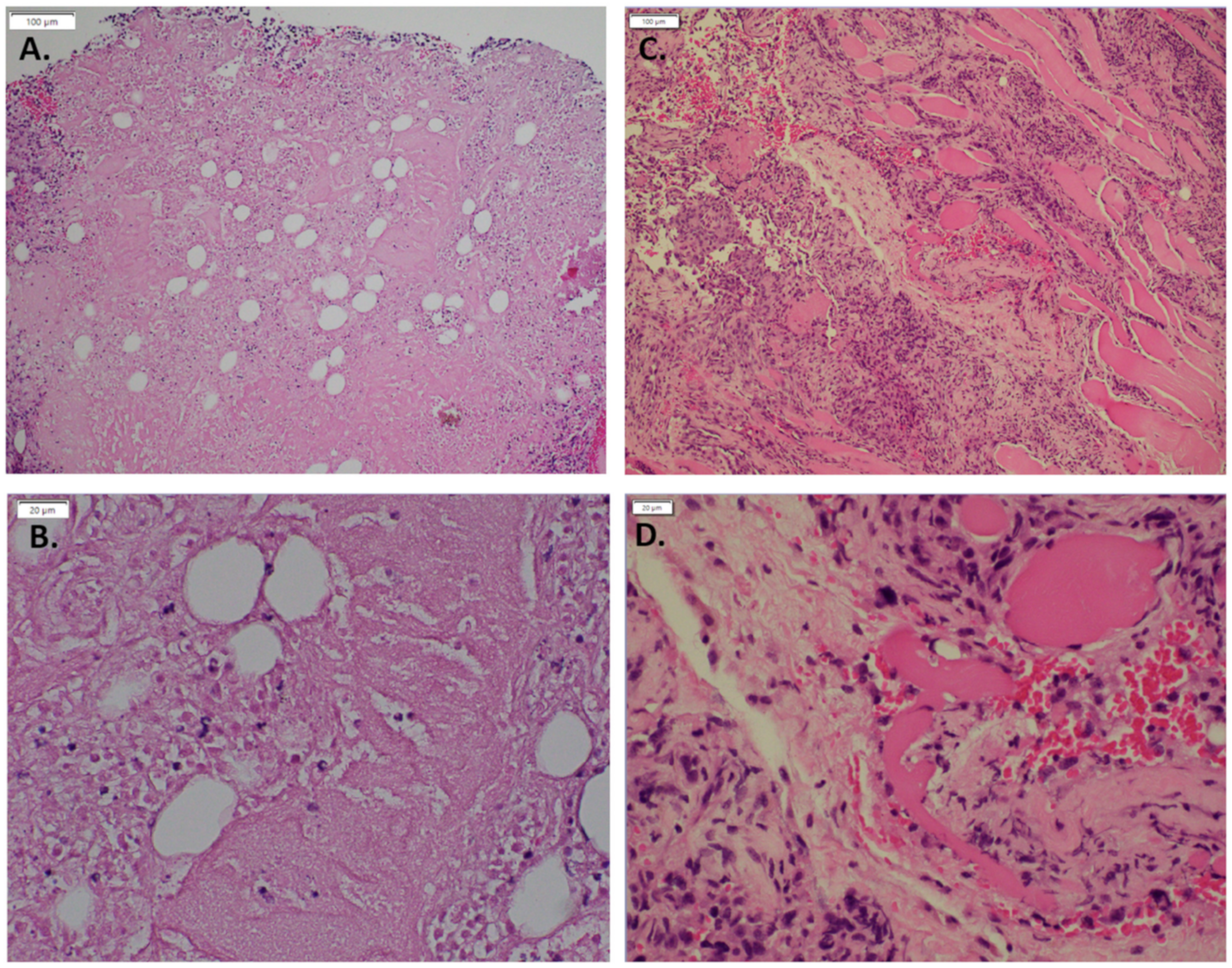
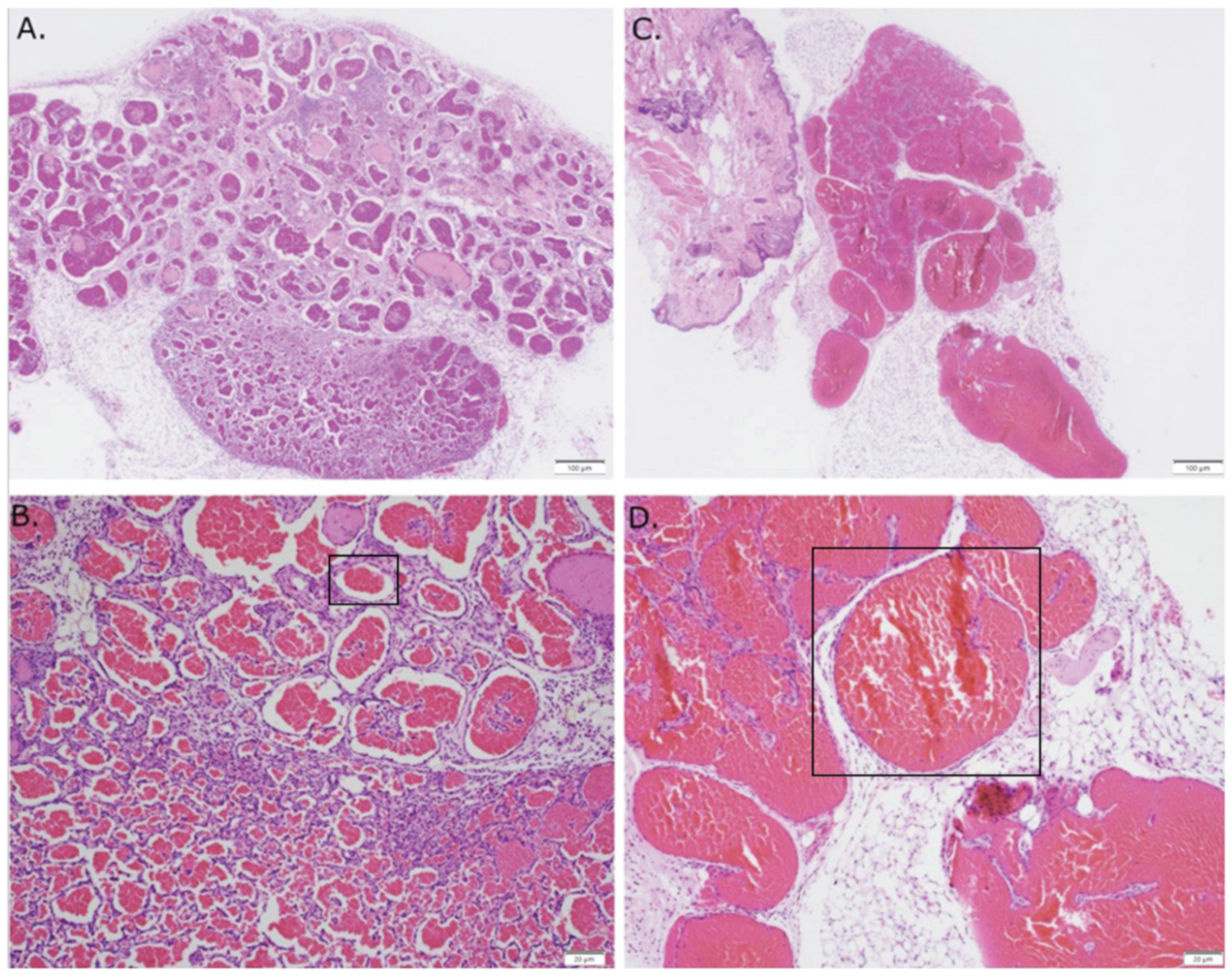
3.1. Expression of GNAQ Q209L in MS1 Cells Results in Vascular Malformations in Mice
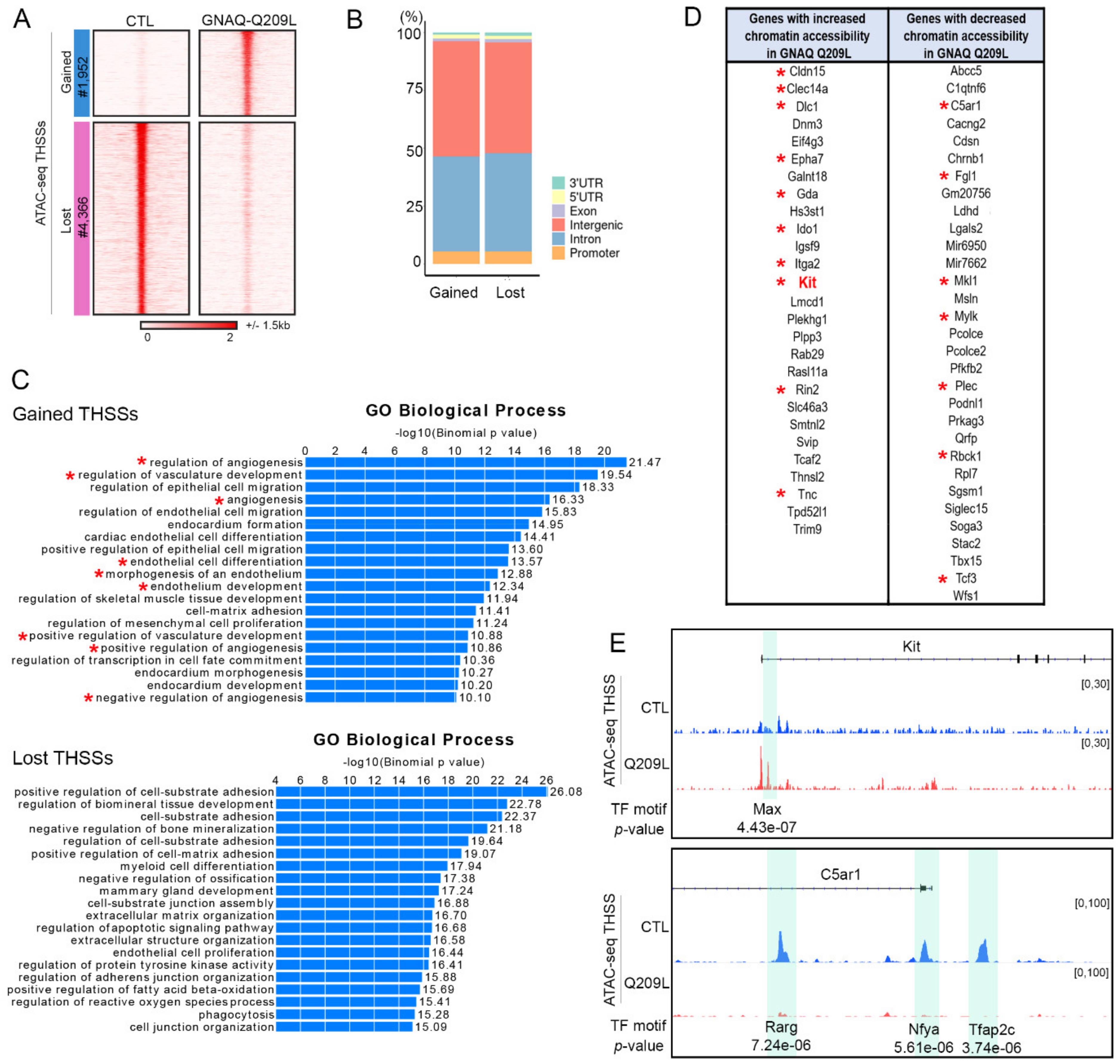
3.2. Expression of GNAQ Q209L Results in Alterations in Chromatin Accessibility
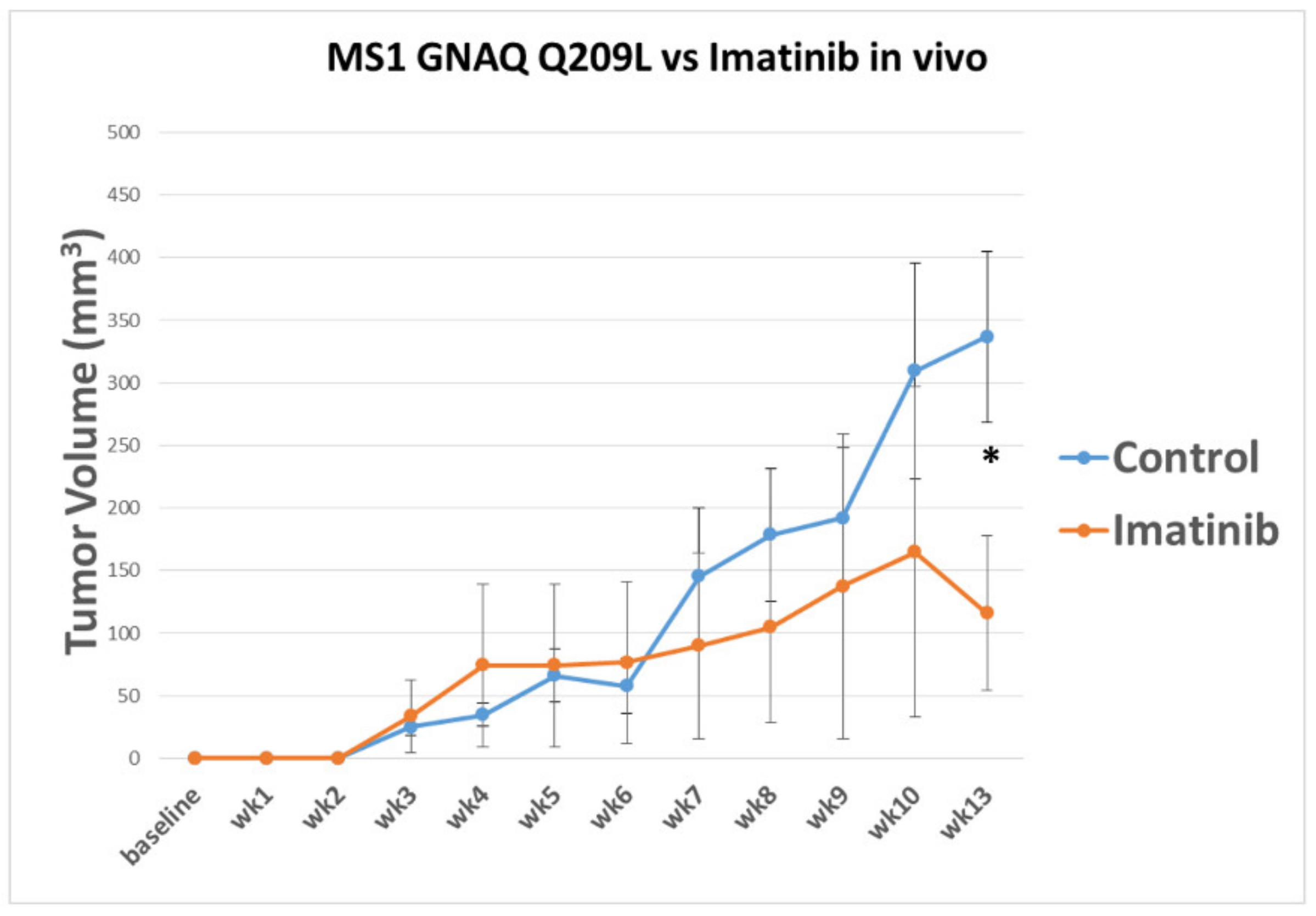
3.3. Imatinib Inhibits Growth of Sturge–Weber Model Vascular Malformations In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higueros, E.; Roe, E.; Granell, E.; Baselga, E. Síndrome de Sturge-Weber: Revisión. Actas Dermo-Sifiliográficas 2017, 108, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Shimakawa, S.; Fukui, M.; Tanabe, T.; Tamai, H. Lamotrigine for intractable migraine-like headaches in Sturge–Weber syndrome. Brain Dev. 2014, 36, 399–401. [Google Scholar] [CrossRef]
- Balkuv, E.; Isik, N.; Aydin, I.; Işik, N.; Basaran, R.; Canturk, I.A. Sturge-weber syndrome: A case report with persistent headache. Pan Afr. Med. J. 2014, 18, 87. [Google Scholar] [CrossRef]
- Hatfield, L.A.; Kossoff, E.H. Comorbidity of Epilepsy and Headache in Patients With Sturge-Weber Syndrome. J. Child Neurol. 2005, 20, 678–682. [Google Scholar] [CrossRef]
- Shirley, M.; Tang, H.; Gallione, C.J.; Baugher, J.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.; Comi, A.M.; Pevsner, J. Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Arbiser, J.L.; Moses, M.A.; Fernandez, C.A.; Ghiso, N.; Cao, Y.; Klauber, N.; Frank, D.; Brownlee, M.; Flynn, E.; Parangi, S.; et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. USA 1997, 94, 861–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, G.R.; Joseph, N.M.; Gill, R.M.; Folpe, A.L.; Horvai, A.E.; Umetsu, S.E. Recurrent GNAQ mutations in anastomosing hemangiomas. Mod. Pathol. 2017, 30, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Ayturk, U.M.; Couto, J.A.; Hann, S.; Mulliken, J.B.; Williams, K.L.; Huang, A.Y.; Fishman, S.J.; Boyd, T.K.; Kozakewich, H.P.; Bischoff, J.; et al. Somatic Activating Mutations in GNAQ and GNA11 Are Associated with Congenital Hemangioma. Am. J. Hum. Genet. 2016, 98, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; Von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.H.; Bacchiocchi, A.; Qiu, J.; Straub, R.; Bruckner, A.; Bercovitch, L.; Narayan, D.; McNiff, J.; Ko, C.; Robinson-Bostom, L.; et al. GNA14 Somatic Mutation Causes Congenital and Sporadic Vascular Tumors by MAPK Activation. Am. J. Hum. Genet. 2016, 99, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Reber, L.L.; Starkl, P.; Balbino, B.; Sibilano, R.; Gaudenzio, N.; Rogalla, S.; Sensarn, S.; Kang, D.; Raghu, H.; Sokolove, J.; et al. The tyrosine kinase inhibitor imatinib mesylate suppresses uric acid crystal-induced acute gouty arthritis in mice. PLoS ONE 2017, 12, e0185704. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Morita, R.; Suzuki, M.; Kasahara, H.; Shimizu, N.; Shichita, T.; Sekiya, T.; Kimura, A.; Sasaki, K.-I.; Yasukawa, H.; Yoshimura, A. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, M.; James, D.; Ding, B.-S.; Nolan, D.; Geng, F.; Butler, J.M.; Schachterle, W.; Pulijaal, V.R.; Mathew, S.; Chasen, S.T.; et al. Efficient Direct Reprogramming of Mature Amniotic Cells into Endothelial Cells by ETS Factors and TGFβ Suppression. Cell 2012, 151, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Rafail, S.; Kourtzelis, I.; Foukas, P.G.; Markiewski, M.M.; DeAngelis, R.A.; Guariento, M.; Ricklin, D.; Grice, E.; Lambris, J.D. Complement Deficiency Promotes Cutaneous Wound Healing in Mice. J. Immunol. 2015, 194, 1285–1291. [Google Scholar] [CrossRef] [Green Version]
- Arbiser, J.L.; Govindarajan, B.; Bai, X.; Onda, H.; Kazlauskas, A.; Lim, S.D.; Amin, M.B.; Claesson-Welsh, L. Functional tyrosine kinase inhibitor profiling: A generally applicable method points to a novel role of platelet-derived growth factor receptor-beta in tuberous sclerosis. Am. J. Pathol. 2002, 161, 781–786. [Google Scholar] [CrossRef]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef]
- Revencu, N.; Boon, L.M.; Mendola, A.; Cordisco, M.R.; Dubois, J.; Clapuyt, P.; Hammer, F.; Amor, D.; Irvine, A.; Baselga, E.; et al. RASA1Mutations and Associated Phenotypes in 68 Families with Capillary Malformation-Arteriovenous Malformation. Hum. Mutat. 2013, 34, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, E.; Coma, S.; Luks, V.L.; Greene, A.K.; Klagsbrun, M.; Warman, M.L.; Bischoff, J. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis 2015, 18, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Limaye, N.; Kangas, J.; Mendola, A.; Godfraind, C.; Schlögel, M.J.; Helaers, R.; Eklund, L.; Boon, L.M.; Vikkula, M. Somatic Activating PIK3CA Mutations Cause Venous Malformation. Am. J. Hum. Genet. 2015, 97, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Revencu, N.; Boon, L.M.; Mulliken, J.B.; Enjolras, O.; Cordisco, M.R.; Burrows, P.E.; Clapuyt, P.; Hammer, F.; Dubois, J.; Baselga, E.; et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused byRASA1 mutations. Hum. Mutat. 2008, 29, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, F.; Cohen, C.; Fried, L.; Arbiser, J.L. Mammalian Target of Rapamycin (mTOR) is Activated in Cutaneous Vascular Malformations in Vivo. Lymphat. Res. Biol. 2007, 5, 233–236. [Google Scholar] [CrossRef]
- Adams, D.M.; Trenor, C.C.; Hammill, A.M.; Vinks, A.A.; Patel, M.N.; Chaudry, G.; Wentzel, M.S.; Mobberley-Schuman, P.S.; Campbell, L.M.; Brookbank, C.; et al. Efficacy and Safety of Sirolimus in the Treatment of Complicated Vascular Anomalies. Pediatrics 2016, 137, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, K.W.; Hammill, A.M.; Mobberley-Schuman, P.; Nelson, S.C.; Blatt, J.; Bender, J.L.G.; McCuaig, C.C.; Synakiewicz, A.; Frieden, I.J.; Adams, D.M. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham–Stout disease. Pediatr. Blood Cancer 2019, 66, e27614. [Google Scholar] [CrossRef] [PubMed]
- Léauté-Labrèze, C.; De La Roque, E.D.; Hubiche, T.; Boralevi, F.; Thambo, J.-B.; Taïeb, A. Propranolol for Severe Hemangiomas of Infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.R.; Fuh, B.; Blatt, J.; Gupta, A.; Merrow, A.C.; Hammill, A.; Adams, D. Malignant transformation of infantile hemangioma to angiosarcoma: Response to chemotherapy with bevacizumab. Pediatr. Blood Cancer 2014, 61, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.; Bonner, M.Y.; Rao, S.; Gilbert, L.; Sasaki, M.; Elsey, J.; MacKelfresh, J.; Arbiser, J.L. Establishment of a Temperature-Sensitive Model of Oncogene-Induced Senescence in Angiosarcoma Cells. Cancers 2020, 12, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogler, C.; Beck, C.; Kulozik, A.; Penzel, R.; Schirmacher, P.; Breuhahn, K. Elevated expression of c-kit in small venous malformations of blue rubber bleb nevus syndrome. Rare Tumors 2010, 30, e36. [Google Scholar]
- Wu, J.K.; Kitajewski, C.; Reiley, M.; Keung, C.H.; Monteagudo, J.; Andrews, J.P.; Liou, P.; Thirumoorthi, A.; Wong, A.; Kandel, J.J.; et al. Aberrant lymphatic endothelial progenitors in lymphatic malformation development. PLoS ONE 2015, 10, e0117352. [Google Scholar]
- Tian, L.; Goldstein, A.; Wang, H.; Lo, H.C.; Kim, I.S.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Log2(FC) | p-Value |
|---|---|---|
| Cldn15 | 2.9 | 2.10 × 10−11 |
| Clec14a | 4.53 | 2.85 × 10−06 |
| Dlc1 | 4.05 | 3.80 × 10−07 |
| Dnm3 | 4.43 | 8.97 × 10−09 |
| Eif4g3 | 3.86 | 0.0000381 |
| Epha7 | 4.54 | 2.74 × 10−09 |
| Galnt18 | 4.53 | 1.00 × 10−05 |
| Gda | 2.52 | 5.69 × 10−07 |
| Hs3st1 | 3.83 | 7.23 × 10−09 |
| Ido1 | 2.68 | 4.58 × 10−05 |
| Igsf9 | 2.89 | 6.50 × 10−09 |
| Itga2 | 2.54 | 2.48 × 10−06 |
| Kit | 3.95 | 1.36 × 10−07 |
| Lmcd1 | 5.64 | 2.48 × 10−09 |
| Plekhg1 | 5.24 | 1.42 × 10−07 |
| Plpp3 | 3.53 | 2.41 × 10−10 |
| Rab29 | 2.53 | 1.37 × 10−08 |
| Rasl11a | 4.03 | 7.50 × 10−17 |
| Rin2 | 5.73 | 0.0000106 |
| Slc46a3 | 1.79 | 5.86 × 10−08 |
| Smtnl2 | 4.31 | 1.42 × 10−06 |
| Svip | 2.89 | 2.01 × 10−08 |
| Tcaf2 | 2.8 | 3.17 × 10−10 |
| Thnsl2 | 4.4 | 1.29 × 10−08 |
| Tnc | 6.12 | 5.03 × 10−14 |
| Tpd52l1 | 2.88 | 3.94 × 10−06 |
| Trim9 | 4.17 | 4.42 × 10−05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasaki, M.; Jung, Y.; North, P.; Elsey, J.; Choate, K.; Toussaint, M.A.; Huang, C.; Radi, R.; Perricone, A.J.; Corces, V.G.; et al. Introduction of Mutant GNAQ into Endothelial Cells Induces a Vascular Malformation Phenotype with Therapeutic Response to Imatinib. Cancers 2022, 14, 413. https://doi.org/10.3390/cancers14020413
Sasaki M, Jung Y, North P, Elsey J, Choate K, Toussaint MA, Huang C, Radi R, Perricone AJ, Corces VG, et al. Introduction of Mutant GNAQ into Endothelial Cells Induces a Vascular Malformation Phenotype with Therapeutic Response to Imatinib. Cancers. 2022; 14(2):413. https://doi.org/10.3390/cancers14020413
Chicago/Turabian StyleSasaki, Maiko, Yoonhee Jung, Paula North, Justin Elsey, Keith Choate, Michael Andrew Toussaint, Christina Huang, Rakan Radi, Adam J. Perricone, Victor G. Corces, and et al. 2022. "Introduction of Mutant GNAQ into Endothelial Cells Induces a Vascular Malformation Phenotype with Therapeutic Response to Imatinib" Cancers 14, no. 2: 413. https://doi.org/10.3390/cancers14020413
APA StyleSasaki, M., Jung, Y., North, P., Elsey, J., Choate, K., Toussaint, M. A., Huang, C., Radi, R., Perricone, A. J., Corces, V. G., & Arbiser, J. L. (2022). Introduction of Mutant GNAQ into Endothelial Cells Induces a Vascular Malformation Phenotype with Therapeutic Response to Imatinib. Cancers, 14(2), 413. https://doi.org/10.3390/cancers14020413