
Bone Marrow Fibrosis at Diagnosis and during the Course of Disease Is Associated with TP53 Mutations and Adverse Prognosis in Primary Myelodysplastic Syndrome

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients and Methods
2.1. Patient Samples
2.2. BM Fibrosis Assessment
2.3. Mutational Analysis
2.4. Statistical Methods
3. Results
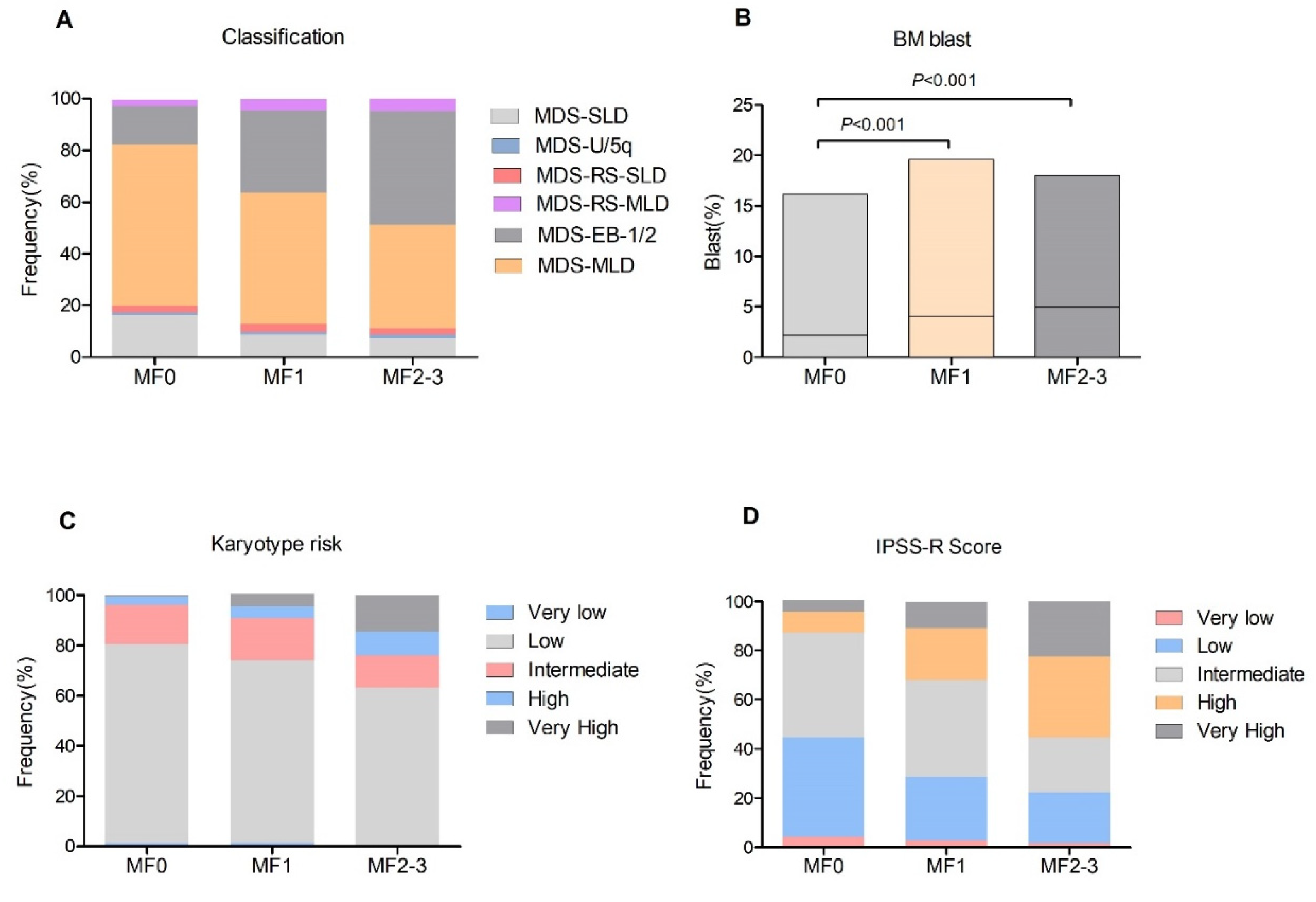
3.1. MDS Patients with BM Fibrosis Emerged Adverse Clinical Feature
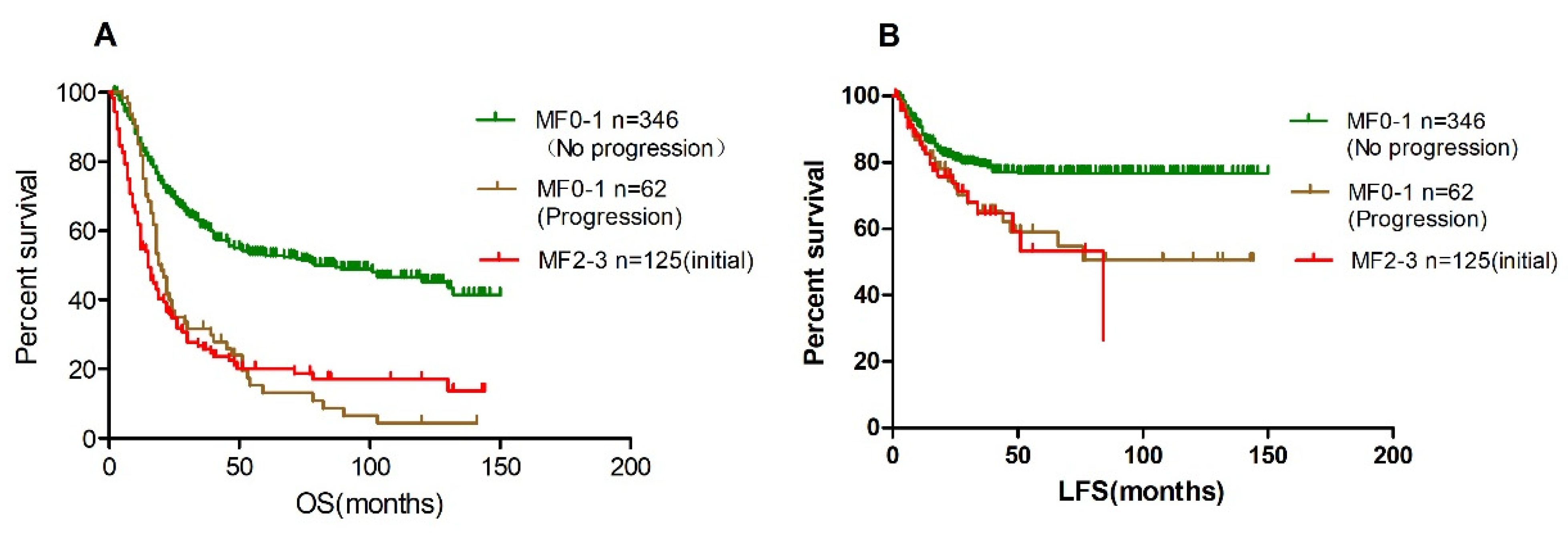
3.2. BM Fibrosis Independently Affect Overall Survival
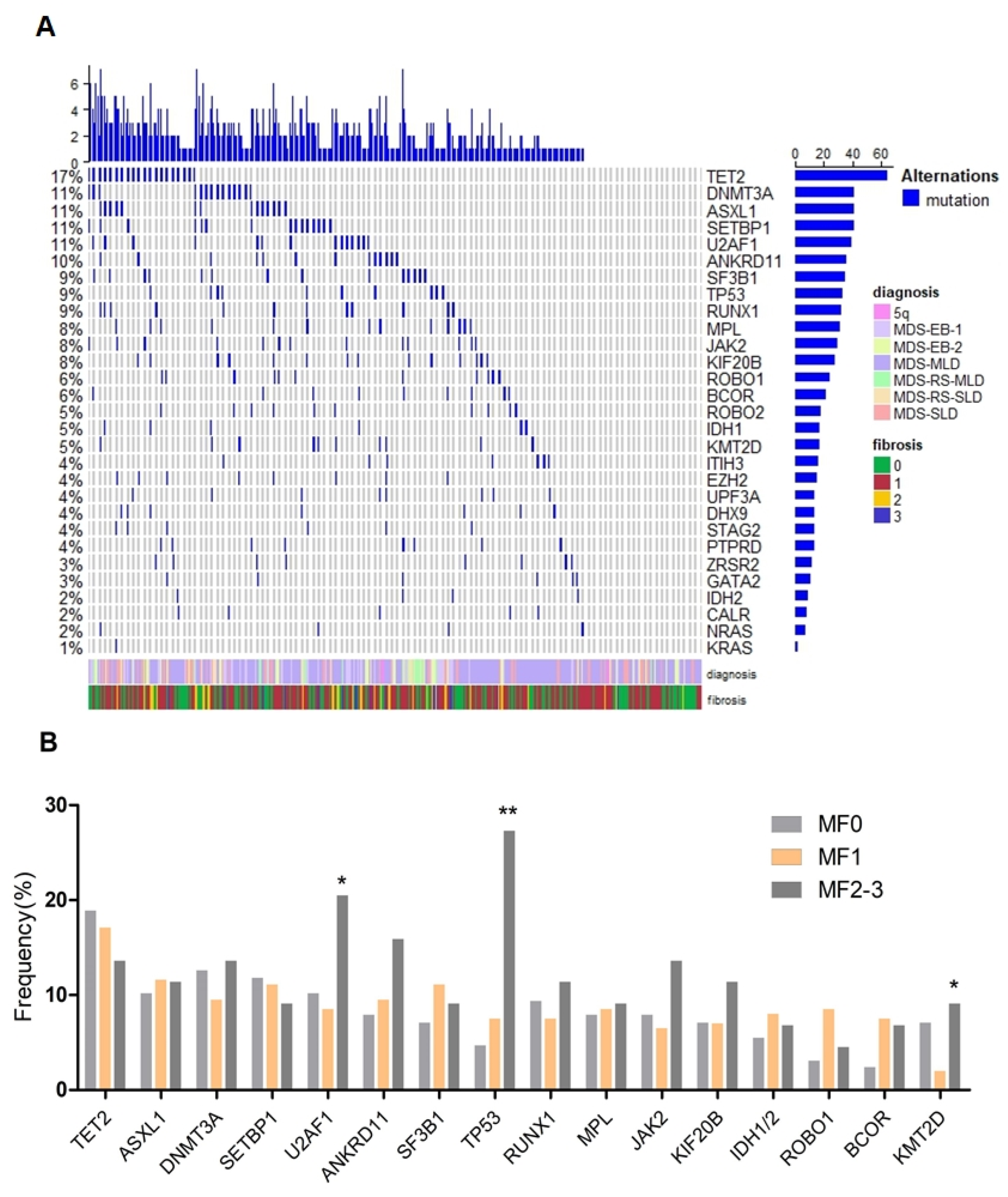
3.3. BM Fibrosis Associated with Gene Mutations of TP53, U2AF1 and KMT2D
3.4. Bone Marrow Fibrosis Progression during the Course of Myelodysplastic Syndromes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tefferi, A.; Vardiman, J.W. Myelodysplastic syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Malcovati, L.; Boveri, E.; Travaglino, E.; Pietra, D.; Pascutto, C.; Passamonti, F.; Invernizzi, R.; Castello, A.; Magrini, U.; et al. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J. Clin. Oncol. 2009, 27, 754–762. [Google Scholar] [CrossRef]
- Fu, B.; Ok, C.Y.; Goswami, M.; Xei, W.; Jaso, J.M.; Muzzafar, T.; Bueso-Ramos, C.; Verstovsek, S.; Garcia-Manero, G.; Medeiros, L.J.; et al. The clinical importance of moderate/severe bone marrow fibrosis in patients with therapy-related myelodysplastic syndromes. Ann. Hematol. 2013, 92, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Buesche, G.; Teoman, H.; Wilczak, W.; Ganser, A.; Hecker, H.; Wilkens, L.; Gohring, G.; Schlegelberger, B.; Bock, O.; Georgii, A.; et al. Marrow fibrosis predicts early fatal marrow failure in patients with myelodysplastic syndromes. Leukemia 2008, 22, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, J.; Kvasnicka, H.M.; Facchetti, F.; Franco, V.; van der Walt, J.; Orazi, A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005, 90, 1128–1132. [Google Scholar] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Montoro, J.; Robledo, C.; Zamora, L.; Valcárcel, D.; Ramos, F. Calreticulin mutations are exceedingly rare in patients with myelodysplastic syndromes with myelofibrosis. Ann. Hematol. 2017, 96, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.; Robledo, C.; Izquierdo-García, F.M.; Suárez-Vilela, D.; Benito, R.; Fuertes, M.; Insunza, A.; Barragán, E.; Del Rey, M.; De Morales, J.M.G.-R.; et al. Bone marrow fibrosis in myelodysplastic syndromes: A prospective evaluation including mutational analysis. Oncotarget 2016, 7, 30492–30503. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loghavi, S.; Al-Ibraheemi, A.; Zuo, Z.; Garcia-Manero, G.; Yabe, M.; Wang, S.A.; Kantarjian, H.M.; Yin, C.C.; Miranda, R.N.; Luthra, R.; et al. TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis. Br. J. Haematol. 2015, 171, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.P.; Martin, P.; Roboz, G.J. Practical Implications of the 2016 Revision of the World Health Organization Classification of Lymphoid and Myeloid Neoplasms and Acute Leukemia. J. Clin. Oncol. 2017, 35, 2708–2715. [Google Scholar] [CrossRef]
- Fang, Y.; Guo, J.; Wu, D.; Wu, L.Y.; Song, L.X.; Zhang, Z.; Zhao, Y.-S.; Chang, C.-K. Integration Analysis of JAK2 or RUNX1 Mutation with Bone Marrow Blast Can Improve Risk Stratification in the Patients With Lower Risk Myelodysplastic Syndrome. Front. Oncol. 2020, 10, 610525. [Google Scholar] [CrossRef]
- Machherndl-Spandl, S.; Sega, W.; Bösmüller, H.; Germing, U.; Gruber, C.; Nachtkamp, K.; Reinecke, P.; Sperr, W.R.; Wimazal, F.; Müllauer, L.; et al. Prognostic impact of blast cell counts in dysplastic bone marrow disorders (MDS and CMML I) with concomitant fibrosis. Ann. Hematol. 2014, 93, 57–64. [Google Scholar] [CrossRef]
- Maschek, H.; Georgii, A.; Kaloutsi, V.; Werner, M.; Bandecar, K.; Kressel, M.G.; Choritz, H.; Freund, M.; Hufnagl, D. Myelofibrosis in primary myelodysplastic syndromes: A retrospective study of 352 patients. Eur. J. Haematol. 1992, 48, 208–214. [Google Scholar] [CrossRef]
- Duarte, F.B.; Barbosa, M.C.; Jesus Dos Santos, T.E.; Lemes, R.P.G.; Vasconcelos, J.P.; de Vasconcelos, P.R.L.; Rocha, F.D.; Zalcberg, I.; Coutinho, D. Bone marrow fibrosis at diagnosis is associated with TP53 overexpression and adverse prognosis in low-risk myelodysplastic syndrome. Br. J. Haematol. 2018, 181, 547–549. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.G.; Zhang, L.; Bennett, J.M.; Komrokji, R. Myelodysplastic Syndromes with Bone Marrow Fibrosis: An Update. Ann. Lab. Med. 2022, 42, 299–305. [Google Scholar] [CrossRef]
- Melody, M.; Al Ali, N.; Zhang, L.; Ramadan, H.; Padron, E.; Sallman, D.; Sweet, K.; Lancet, J.; List, A.; Bennett, J.M.; et al. Decoding Bone Marrow Fibrosis in Myelodysplastic Syndromes. Clin. Lymphoma Myeloma Leuk. 2020, 20, 324–328. [Google Scholar] [CrossRef]
- Zhang, L.; McGraw, K.L.; Sallman, D.A.; List, A.F. The role of p53 in myelodysplastic syndromes and acute myeloid leukemia: Molecular aspects and clinical implications. Leuk. Lymphoma 2017, 58, 1777–1790. [Google Scholar] [CrossRef]
- Seifert, H.; Mohr, B.; Thiede, C.; Oelschlägel, U.; Schäkel, U.; Illmer, T.; Soucek, S.; Ehninger, G.; Schaich, M.; Study Alliance Leukemia (SAL). The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009, 23, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.K.; Zhao, Y.S.; Xu, F.; Guo, J.; Zhang, Z.; He, Q.; Wu, D.; Wu, L.-Y.; Su, J.-Y.; Song, L.-X.; et al. TP53 mutations predict decitabine-induced complete responses in patients with myelodysplastic syndromes. Br. J. Haematol. 2017, 176, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Kantarjian, H.M.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Cortes, J.E.; Pemmaraju, N.; Pierce, S.R.; Newberry, K.J.; Daver, N.; et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk. Res. 2015, 39, 950–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gale, R.P.; Xu, Z.; Qin, T.; Song, Z.; Zhang, P.; Bai, J.; Zhang, L.; Zhang, Y.; Liu, J.; et al. Non-driver mutations in myeloproliferative neoplasm-associated myelofibrosis. J. Hematol. Oncol. 2017, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Q.; Zhang, H.; He, Y.; Huang, Y.; Zhang, R.; Wei, J.; Ma, Q.; Pang, A.; Yang, D.; et al. Moderate to Severe Marrow Fibrosis as a More Advanced Risk Factor for MDS and MDS-AML Patients With Excess of Blasts Receiving Allogeneic Hematopoietic Stem Cell Transplantation. Transplant. Cell. Ther. 2021, 27, 666.e1–666.e9. [Google Scholar] [CrossRef]
- Fu, B.; Jaso, J.M.; Sargent, R.L.; Goswami, M.; Verstovsek, S.; Medeiros, L.J.; Wang, S.A. Bone marrow fibrosis in patients with primary myelodysplastic syndromes has prognostic value using current therapies and new risk stratification systems. Mod. Pathol. 2014, 27, 681–689. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Total N = 814 | MF0 N = 211 | MF1 N = 478 | MF(2, 3) N = 125 | p Value |
|---|---|---|---|---|---|
| Median age | 59 | 58 | 59 | 63 | 0.039 |
| Sex | |||||
| Male | 471 | 117 (24.8) | 272 (57.8) | 82 (17.4) | |
| Female | 343 | 94 (27.4) | 206 (60.1) | 43 (12.5) | |
| MDS classification | <0.001 | ||||
| MDS-SLD | 85 | 34 (16.1) | 42 (8.8) | 9 (7.2) | |
| MDS-MLD | 424 | 131 (62.1) | 243 (50.8) | 50 (40.0) | |
| MDS-RS-SLD | 24 | 6 (2.8) | 15 (3.1) | 3 (2.4) | |
| MDS-RS-MLD | 33 | 5 (2.4) | 22 (4.6) | 6 (4.8) | |
| RA-EB-1/2 | 238 | 32 (15.2) | 151 (31.6) | 55 (44.0) | |
| MDS-U/5q | 10 | 3 (1.0) | 5 (1.0) | 2 (1.6) | |
| Cytopenia | |||||
| Hemoglobin (g/L) | 78.9 | 82.1 | 79.2 | 72.6 | 0.001 |
| WBC counts | |||||
| (×109/L) | 3.8 | 4.0 | 3.6 | 4.2 | 0.204 |
| Platelet counts | |||||
| (×109/L) | 86.8 | 83.3 | 86.2 | 94.8 | 0.592 |
| BM blast | <0.001 | ||||
| <5% | 581 | 180 (85.3) | 329 (68.8) | 72 (57.6) | |
| ≥5% | 233 | 31 (14.7) | 149 (31.2) | 53 (42.4) | |
| Karyotype | <0.001 | ||||
| Very good | 8 | 2 (1.0) | 6 (1.3) | 0 (0) | |
| Good | 595 | 168 (79.6) | 348 (72.8) | 79 (63.2) | |
| Intermediate | 130 | 33 (15.6) | 81 (16.9) | 16 (12.8) | |
| Poor | 41 | 7 (3.3) | 22 (4.6) | 12 (9.6) | |
| Very poor | 40 | 1 (0.5) | 21 (4.4) | 18 (14.4) | |
| IPSS-R | <0.001 | ||||
| Very low | 24 | 9 (4.3) | 13 (2.7) | 2 (1.6) | |
| Low | 235 | 85 (40.3) | 124 (25.9) | 26 (20.8) | |
| Intermediate | 307 | 90 (42.7) | 189 (39.5) | 28 (22.4) | |
| High | 159 | 18 (8.5) | 100 (20.9) | 41 (32.8) | |
| Very high | 89 | 9 (4.3) | 52 (10.9) | 28 (22.4) |
| Mutation | Total N = 370 | MF0 (n/%) N = 127 | MF1 (n/%) N = 199 | MF2-3 (n/%) N = 44 | p Value |
|---|---|---|---|---|---|
| TET2 | 64 (17.3) | 24 (18.9) | 34 (17.1) | 6 (13.6) | 0.755 |
| ASXL1 | 41 (11.1) | 13 (10.2) | 23 (11.6) | 5 (11.4) | 0.944 |
| DNMT3A | 41 (11.1) | 16 (12.6) | 19 (9.5) | 6 (13.6) | 0.542 |
| SETBP1 | 41 (11.1) | 15 (11.8) | 22 (11.1) | 4 (9.1) | 0.917 |
| U2AF1 | 39 (10.5) | 13 (10.2) | 17 (8.5) | 9 (20.5) | 0.079 |
| ANKRD11 | 36 (9.7) | 10 (7.9) | 19 (9.5) | 7 (15.9) | 0.294 |
| SF3B1 | 35 (9.5) | 9 (7.1) | 22 (11.1) | 4 (9.1) | 0.498 |
| TP53 | 33 (8.9) | 6 (4.7) | 15 (7.5) | 12 (27.3) | <0.001 |
| RUNX1 | 32 (8.6) | 12 (9.4) | 15 (7.5) | 5 (11.4) | 0.581 |
| MPL | 31 (8.4) | 10 (7.9) | 17 (8.5) | 4 (9.1) | 0.964 |
| JAKA2 | 29 (7.8) | 10 (7.9) | 13 (6.5) | 6 (13.6) | 0.269 |
| KIF20B | 28 (7.6) | 9 (7.1) | 14 (7.0) | 5 (11.4) | 0.564 |
| IDH1/2 | 26 (7.0) | 7 (5.5) | 16 (8.0) | 3 (6.8) | 0.705 |
| ROBO1 | 23 (6.2) | 4 (3.1) | 17 (8.5) | 2 (4.5) | 0.142 |
| BCOR | 21 (5.7) | 3 (2.4) | 15 (7.5) | 3 (6.8) | 0.102 |
| ROBO2 | 18 (4.9) | 4 (3.1) | 12 (6.0) | 2 (4.5) | 0.502 |
| KMT2D | 17 (4.6) | 9 (7.1) | 4 (2.0) | 4 (9.1) | 0.022 |
| ITIH3 | 16 (4.3) | 6 (4.7) | 8 (4.0) | 2 (4.5) | 0.936 |
| EZH2 | 15 (4.1) | 4 (3.1) | 7 (3.5) | 4 (9.1) | 0.221 |
| UPF3A | 13 (3.5) | 5 (3.9) | 7 (3.5) | 1 (2.3) | 1.000 |
| PTPRD | 13 (3.5) | 4 (3.1) | 6 (3.0) | 3 (6.8) | 0.401 |
| STAG2 | 13 (3.5) | 2 (1.6) | 8 (4.0) | 3 (6.8) | 0.183 |
| DHX9 | 13 (3.5) | 4 (3.1) | 6 (3.0) | 3 (6.8) | 0.401 |
| ZRSR2 | 12 (3.2) | 5 (3.9) | 7 (3.5) | 0 (0) | 0.538 |
| GATA2 | 11 (3.0) | 5 (3.9) | 5 (2.5) | 1 (2.3) | 0.821 |
| NRAS/KRAS | 9 (2.4) | 1 (0.8) | 7 (3.5) | 1 (2.3) | / |
| CALR | 8 (2.2) | 2 (1.6) | 5 (2.5) | 1 (2.3) | / |
| Variable | Total N = 408 | MF0-1 (Stable) N = 346 | MF0-1 (Progression) N = 62 | p Value |
|---|---|---|---|---|
| Median age | 58 | 56 | 60 | 0.038 |
| MDS classification | 0.068 | |||
| MDS-SLD/5q/MDS-U | 35 | 29 (8.4) | 6 (9.7) | |
| MDS-RS-SLD/MLD | 26 | 21 (6.1) | 5 (8.1) | |
| MDS-MLD | 222 | 198 (57.2) | 24 (38.7) | |
| MDS-EB-1/2 | 125 | 98 (28.3) | 27 (43.5) | |
| Cytopenia | ||||
| Hemoglobin (mean g/L) | 57.5 | 58.5 | 51.8 | 0.073 |
| WBC counts | ||||
| (mean × 10 9/L) | 3.6 | 3.4 | 4.4 | 0.061 |
| Platelet counts | ||||
| (mean × 10 9/L) | 75.8 | 72.2 | 96.0 | 0.03 |
| Blast | 3.9 | 3.6 | 5.1 | <0.001 |
| Karyotype | 0.002 | |||
| Very good/Good | 303 | 263 (76.0) | 40 (64.5) | |
| Intermediate | 71 | 62 (17.9) | 9 (14.5) | |
| Poor | 19 | 13 (3.8) | 6 (9.7) | |
| Very poor | 15 | 8 (2.3) | 7 (11.3) | |
| IPSS-R | 0.04 | |||
| Very low/Low | 124 | 110 (31.8) | 14 (22.6) | |
| Intermediate | 161 | 139 (40.2) | 22 (35.5) | |
| High | 82 | 68 (19.7) | 14 (22.6) | |
| Very high | 41 | 29 (8.4) | 12 (19.4) |
| Mutation | Total N = 170 | MF0-1 (Stable) N = 142 | MF0-1 (Progression) N = 28 | p Value |
|---|---|---|---|---|
| TET2 | 28 (16.5) | 21 (14.8) | 7 (25.0) | 0.262 |
| DNMT3A | 22 (12.9) | 20 (14.1) | 2 (7.1) | 0.537 |
| SETBP1 | 18 (10.6) | 15 (10.6) | 3 (10.7) | 1.000 |
| ASXL1 | 17 (10.0) | 13 (9.2) | 4 (14.3) | 0.487 |
| ROBO1 | 16 (9.4) | 16 (11.3) | 0 (0) | 0.077 |
| SF3B1 | 14 (8.2) | 10 (7.0) | 4 (14.3) | 0.252 |
| RUNX1 | 14 (8.2) | 9 (5.3) | 5 (17.9) | 0.058 |
| JAKA2 | 14 (8.2) | 10 (7.0) | 4 (14.3) | 0.252 |
| KIF20B | 14 (8.2) | 12 (8.5) | 2 (7.1) | 1.000 |
| U2AF1 | 13 (7.6) | 10 (7.0) | 3 (10.7) | 0.452 |
| ANKRD11 | 13 (7.6) | 10 (7.0) | 3 (10.7) | 0.452 |
| MPL | 13 (7.6) | 11 (7.7) | 2 (7.1) | 1.000 |
| IDH1/2 | 13 (7.6) | 11 (7.7) | 2 (7.1) | 1.000 |
| BCOR | 13 (7.6) | 12 (8.5) | 1 (3.6) | 0.696 |
| ITIH3 | 9 (5.3) | 8 (5.6) | 1 (3.6) | 1.000 |
| TP53 | 8 (4.7) | 3 (2.1) | 5 (17.9) | 0.003 |
| ROBO2 | 8 (4.7) | 8 (5.6) | 0 (0) | 0.356 |
| ZRSR2 | 8 (4.7) | 6 (4.2) | 2 (7.1) | 0.620 |
| PTPRD | 7 (4.1) | 7 (4.9) | 0 (0) | 0.601 |
| UPF3A | 6 (3.5) | 5 (3.5) | 1 (3.6) | 1.000 |
| DHX9 | 6 (3.5) | 5 (3.5) | 1 (3.6) | 1.000 |
| GATA2 | 6 (3.5) | 4 (2.8) | 2 (7.1) | / |
| EZH2 | 5 (2.9) | 3 (2.1) | 2 (7.1) | / |
| STAG2 | 4 (2.4) | 4 (2.8) | 0 (0) | / |
| KMT2D | 2 (1.2) | 2 (1.4) | 0 (0) | / |
| NRAS/KRAS | 2 (1.2) | 1 (0.7) | 1 (3.6) | / |
| CALR | 2 (1.2) | 2 (1.4) | 0 (0) | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Guo, J.; Zhao, S.; Wang, R.; Shi, L.; Fang, Y.; Zhang, Z.; Song, L.; Wu, D.; Chang, C. Bone Marrow Fibrosis at Diagnosis and during the Course of Disease Is Associated with TP53 Mutations and Adverse Prognosis in Primary Myelodysplastic Syndrome. Cancers 2022, 14, 2984. https://doi.org/10.3390/cancers14122984
Zhao Y, Guo J, Zhao S, Wang R, Shi L, Fang Y, Zhang Z, Song L, Wu D, Chang C. Bone Marrow Fibrosis at Diagnosis and during the Course of Disease Is Associated with TP53 Mutations and Adverse Prognosis in Primary Myelodysplastic Syndrome. Cancers. 2022; 14(12):2984. https://doi.org/10.3390/cancers14122984
Chicago/Turabian StyleZhao, Youshan, Juan Guo, Sida Zhao, Roujia Wang, Lei Shi, Ying Fang, Zheng Zhang, Luxi Song, Dong Wu, and Chunkang Chang. 2022. "Bone Marrow Fibrosis at Diagnosis and during the Course of Disease Is Associated with TP53 Mutations and Adverse Prognosis in Primary Myelodysplastic Syndrome" Cancers 14, no. 12: 2984. https://doi.org/10.3390/cancers14122984
APA StyleZhao, Y., Guo, J., Zhao, S., Wang, R., Shi, L., Fang, Y., Zhang, Z., Song, L., Wu, D., & Chang, C. (2022). Bone Marrow Fibrosis at Diagnosis and during the Course of Disease Is Associated with TP53 Mutations and Adverse Prognosis in Primary Myelodysplastic Syndrome. Cancers, 14(12), 2984. https://doi.org/10.3390/cancers14122984