
Standing Variations Modeling Captures Inter-Individual Heterogeneity in a Deterministic Model of Prostate Cancer Response to Combination Therapy

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
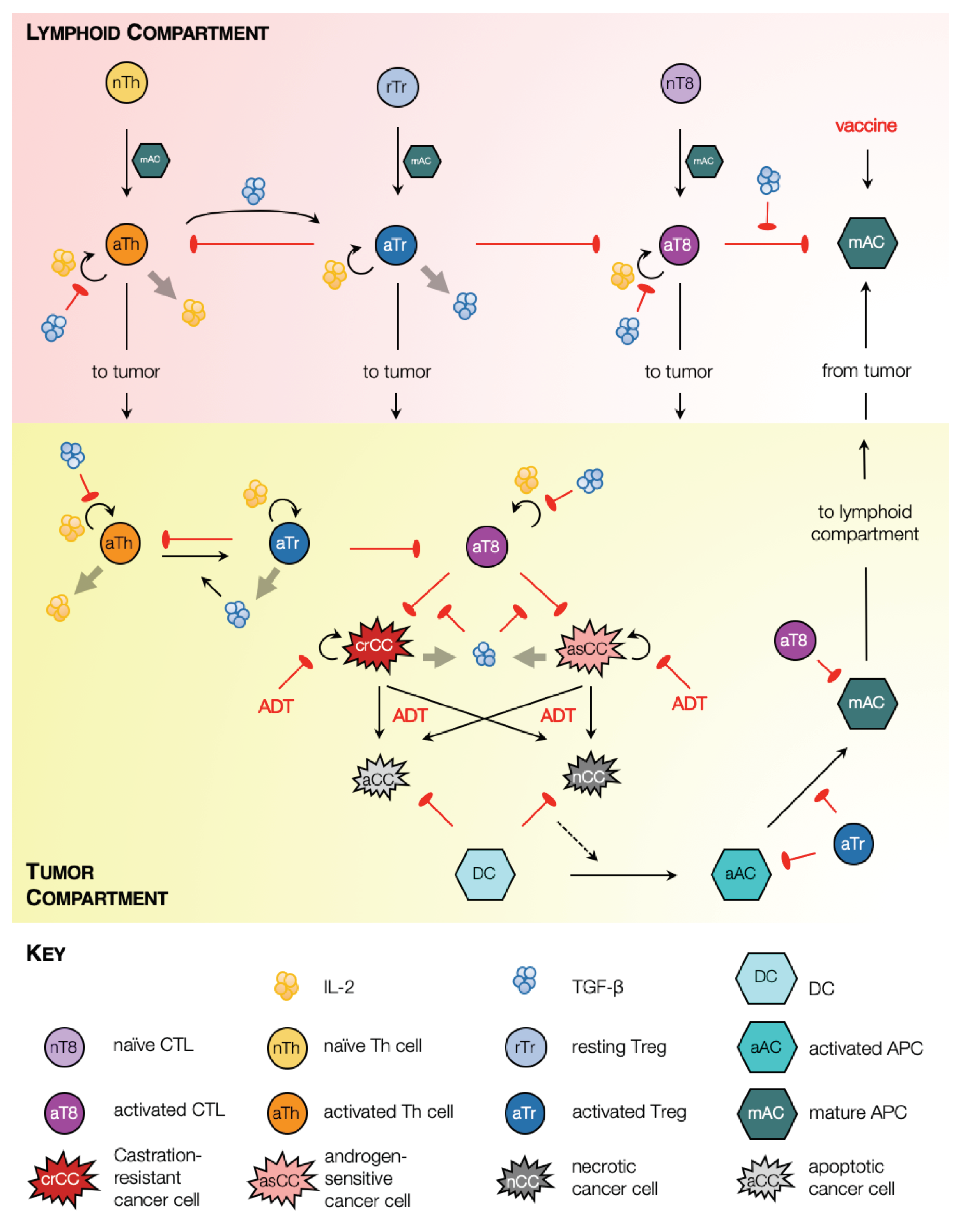
2.1. Model Schematic
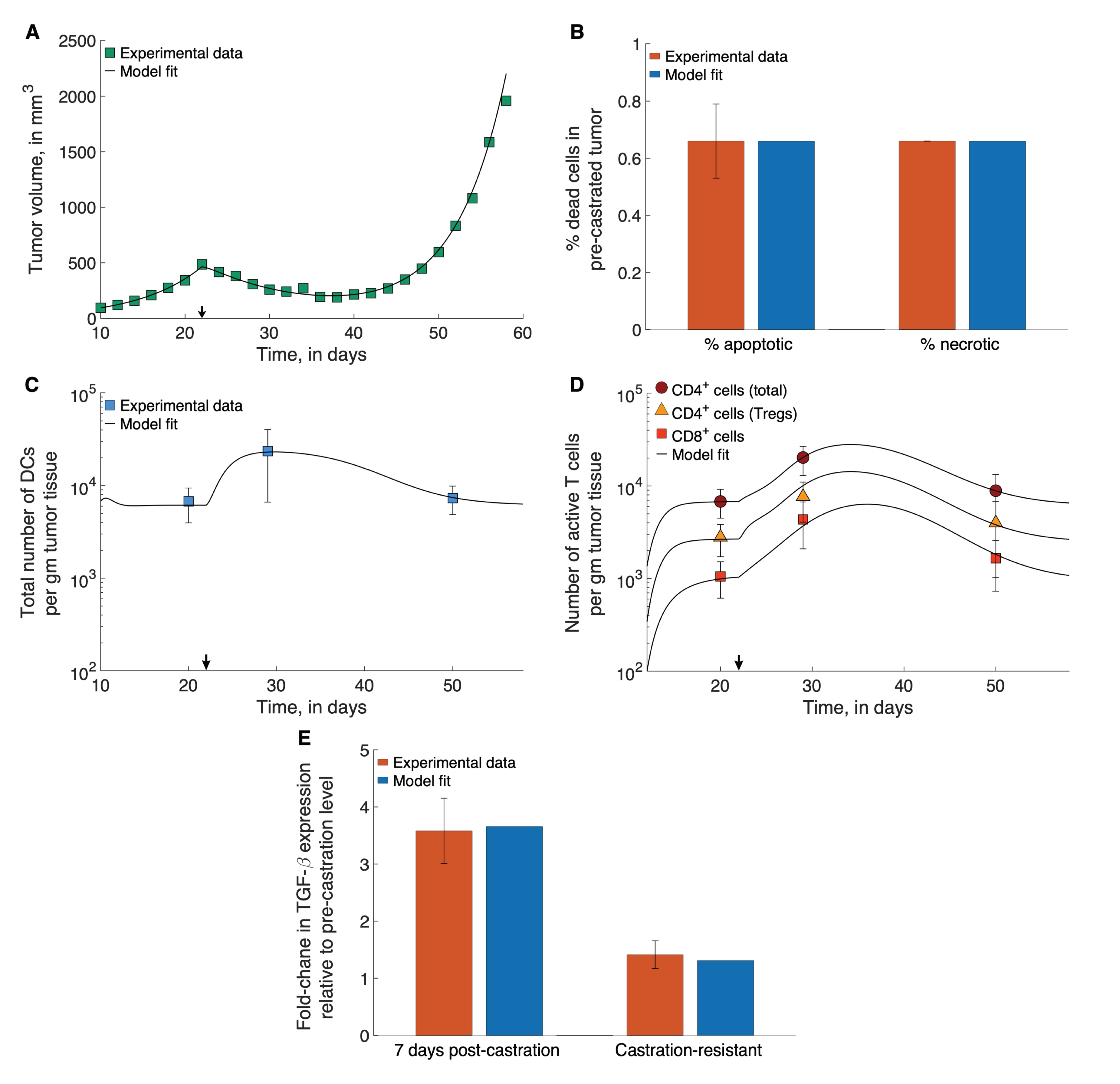
2.2. Parameter Estimation
2.3. The Standing Variations Method
2.4. In Silico Preclinical Trial
2.5. Approximation of Sensitivity Analysis
2.6. Treatment Optimization
3. Results
3.1. Sipuleucel-T Alone Does Not Significantly Improve Overall Survival of Mice
3.2. Optimization of Vaccine Scheduling
3.3. Characteristics of Response to Vaccine
3.4. ADT and Vaccination Combination Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Androgen deprivation therapy |
| APC | Antigen-presenting cell |
| CTL | CD8 Cytotoxic T lymphocyte |
| DC | Dendritic cell |
| mCRPC | Metastatic castration-resistant prostate cancer |
| ODE | Ordinary differential Equation |
| PAP | Prostatic acid phosphatase |
| PBMC | Peripheral blood mononuclear cell |
| PCa | Prostate cancer |
| Th cell | CD4 T helper cell |
| Treg | CD4 regulatory T cell |
References
- Printz, C. Prostate cancer mortality projections reach a new high: With prostate cancer deaths projected to rise to their highest level in 20 years, some experts worry that changes to screening guidelines made in 2012 could be a factor. Cancer 2021, 126, 3893–3894. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 70, 7–33. [Google Scholar] [CrossRef]
- Lewis, D.D.; Cropp, C.D. The impact of African ancestry on prostate cancer disparities in the era of precision medicine. Genes 2020, 11, 1471. [Google Scholar] [CrossRef]
- Perlmutter, M.A.; Lepor, H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev. Urol. 2007, 9, S3–S8. [Google Scholar]
- Tucci, M.; Zichi, C.; Buttigliero, C.; Vignani, F.; Scagliotti, G.V.; Di Maio, M. Enzalutamide-resistant castration-resistant prostate cancer: Challenges and solutions. OncoTargets Ther. 2018, 11, 7353–7368. [Google Scholar] [CrossRef]
- Bilusic, M.; Madan, R.A.; Gulley, J.L. Immunotherapy of prostate cancer: Facts and hopes. Clin. Cancer Res. 2017, 23, 6764–6770. [Google Scholar] [CrossRef]
- Sumanasuriya, S.; De Bono, J. Treatment of advanced prostate cancer—A review of current therapies and future promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.K.; Cutting, E.; Martin, J.; Oh, W.K. The role of cabazitaxel in the treatment of metastatic castration-resistant prostate cancer. Ther. Adv. Urol. 2014, 6, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yang, F.; Chen, D.; Zhao, Q.; Chen, D.; Ping, H.; Xing, N. Quercetin reverses docetaxel resistance in prostate cancer via androgen receptor and PI3K/Akt signaling pathways. Int. J. Biol. Sci. 2020, 16, 1121. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zieren, R.C.; Xue, W.; de Reijke, T.M.; Pienta, K.J. Metastatic prostate cancer remains incurable, why? Asian J. Urol. 2019, 6, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Swami, U.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N. Advanced prostate cancer: Treatment advances and future directions. Trends Cancer 2020, 6, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef]
- Han, P.; Hanlon, D.; Sobolev, O.; Chaudhury, R.; Edelson, R.L. Ex vivo dendritic cell generation—A critical comparison of current approaches. In International Review of Cell and Molecular Biology; Lhuillier, C., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 349, pp. 251–307. [Google Scholar]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Provenge—FDA. Available online: https://www.fda.gov/media/78511/download (accessed on 5 January 2020).
- Shen, Y.C.; Ghasemzadeh, A.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Lopez-Bujanda, Z.A.; Haro, M.A.C.; Tam, A.; Anders, R.A.; Selby, M.J.; et al. Combining intratumoral Treg depletion with androgen deprivation therapy (ADT): Preclinical activity in the Myc-CaP model. Prostate Cancer Prostatic Dis. 2018, 21, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Birdsell, J.B. Evolution, genetics, and man. By Theodosius Dobzhansky, 1955. John Wiley and Sons, New York, IX, 398 pp., $5.50. Am. J. Phys. Anthropol. 1956, 14, 665–668. [Google Scholar] [CrossRef]
- Nei, M. Genetic polymorphism and neomutationism. In Evolutionary Dynamics of Genetic Diversity; Mani, G.S., Ed.; Springer: Berlin/Heidelberg, Germany, 1984; pp. 214–241. [Google Scholar]
- Ideta, A.M.; Tanaka, G.; Takeuchi, T.; Aihara, K. A mathematical model of intermittent androgen suppression for prostate cancer. J. Nonlinear Sci. 2008, 18, 593–614. [Google Scholar] [CrossRef]
- Jackson, T.L. A mathematical model of prostate tumor growth and androgen-independent relapse. Discret. Contin. Dyn. Syst. Ser. B 2004, 4, 187–201. [Google Scholar] [CrossRef]
- Jackson, T.L. A mathematical investigation of the multiple pathways to recurrent prostate cancer: Comparison with experimental data. Neoplasia 2004, 6, 697–704. [Google Scholar] [CrossRef]
- Kronik, N.; Kogan, Y.; Elishmereni, M.; Halevi-Tobias, K.; Vuk-Pavlović, S.; Agur, Z. Predicting outcomes of prostate cancer immunotherapy by personalized mathematical models. PLoS ONE 2010, 5, e15482. [Google Scholar] [CrossRef]
- Tanaka, G.; Hirata, Y.; Goldenberg, S.; Bruchovsky, N.; Aihara, K. Mathematical modelling of prostate cancer growth and its application to hormone therapy. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2010, 368, 5029–5044. [Google Scholar] [CrossRef]
- Jain, H.V.; Clinton, S.K.; Bhinder, A.; Friedman, A. Mathematical modeling of prostate cancer progression in response to androgen ablation therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19701–19706. [Google Scholar] [CrossRef]
- Portz, T.; Kuang, Y.; Nagy, J.D. A clinical data validated mathematical model of prostate cancer growth under intermittent androgen suppression therapy. AIP Adv. 2012, 2, 011002. [Google Scholar] [CrossRef]
- Jain, H.V.; Friedman, A. Modeling prostate cancer response to continuous versus intermittent androgen ablation therapy. Discret. Contin. Dyn. Syst. Ser. B 2013, 18, 945–967. [Google Scholar]
- Morken, J.D.; Packer, A.; Everett, R.A.; Nagy, J.D.; Kuang, Y. Mechanisms of resistance to intermittent androgen deprivation in patients with prostate cancer identified by a novel computational method. Cancer Res. 2014, 74, 3673–3683. [Google Scholar] [CrossRef]
- Brady-Nicholls, R.; Nagy, J.D.; Gerke, T.A.; Zhang, T.; Wang, A.Z.; Zhang, J.; Gatenby, R.A.; Enderling, H. Prostate-specific antigen dynamics predict individual responses to intermittent androgen deprivation. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Mahlbacher, G.E.; Reihmer, K.C.; Frieboes, H.B. Mathematical modeling of tumor-immune cell interactions. J. Theor. Biol. 2019, 469, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, K.P. A Review of Mathematical Models of Cancer–Immune Interactions in the Context of Tumor Dormancy. In Systems Biology of Tumor Dormancy; Enderling, H., Almog, N., Hlatky, L., Eds.; Springer: New York, NY, USA, 2013; pp. 201–234. [Google Scholar]
- Bądziul, D.; Jakubczyk, P.; Chotorlishvili, L.; Toklikishvilie, Z.; Traciak, J.; Jakubowicz-Gil, J.; Chmiel-Szajner, S. Mathematical prostate cancer evolution: Effect of immunotherapy based on controlled vaccination strategy. Comput. Math. Methods Med. 2020, 2020, 7970265. [Google Scholar] [CrossRef]
- Coletti, R.; Leonardelli, L.; Parolo, S.; Marchetti, L. A QSP model of prostate cancer immunotherapy to identify effective combination therapies. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef]
- Rutter, E.M.; Kuang, Y. Global dynamics of a model of joint hormone treatment with dendritic cell vaccine for prostate cancer. Discret. Contin. Dyn. Syst. Ser. B 2017, 22, 1001–1021. [Google Scholar] [CrossRef]
- Radunskaya, A.; Kim, R.; Woods, T., II. Mathematical modeling of tumor immune interactions: A closer look at the role of a PD-L1 inhibitor in cancer immunotherapy. Spora J. Biomath. 2018, 4, 25–41. [Google Scholar] [CrossRef]
- Robertson-Tessi, M.; El-Kareh, A.; Goriely, A. A mathematical model of tumor–immune interactions. J. Theor. Biol. 2012, 294, 56–73. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Green, D.; Oguin, T.; Martinez, J. The clearance of dying cells: Table for two. Cell Death Differ. 2016, 23, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Ravichandran, K.S. The dynamics of apoptotic cell clearance. Dev. Cell 2016, 38, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Mitra, S.; Leonard, W.J. Biology of IL-2 and its therapeutic modulation: Mechanisms and strategies. J. Leukoc. Biol. 2018, 103, 643–655. [Google Scholar] [CrossRef]
- Boissonnas, A.; Fetler, L.; Zeelenberg, I.S.; Hugues, S.; Amigorena, S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med. 2007, 204, 345–356. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Wing, J.B.; Tanaka, A.; Sakaguchi, S. Human FOXP3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity 2019, 50, 302–316. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef]
- Yamazaki, S.; Iyoda, T.; Tarbell, K.; Olson, K.; Velinzon, K.; Inaba, K.; Steinman, R.M. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 2003, 198, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Bluestone, J.A.; Stephan, S. Peripherally induced tregs–role in immune homeostasis and autoimmunity. Front. Immunol. 2013, 4, 232. [Google Scholar] [CrossRef]
- Alonso, R.; Flament, H.; Lemoine, S.; Sedlik, C.; Bottasso, E.; Péguillet, I.; Prémel, V.; Denizeau, J.; Salou, M.; Darbois, A.; et al. Induction of anergic or regulatory tumor-specific CD4+ T cells in the tumor-draining lymph node. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.C.; Wong, L.Y.; Jang, T.; Shah, A.H.; Park, I.; Yang, X.; Zhang, Q.; Lonning, S.; Teicher, B.A.; Lee, C. Tumor evasion of the immune system by converting CD4+ CD25- T cells into CD4+ CD25+ T regulatory cells: Role of tumor-derived TGF-β. J. Immunol. 2007, 178, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Flavell, R.A. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. USA 2005, 102, 5126–5131. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Flavell, R.A. ‘Yin–Yang’ functions of transforming growth factor-β and T regulatory cells in immune regulation. Immunol. Rev. 2007, 220, 199–213. [Google Scholar] [CrossRef]
- McKarns, S.C.; Schwartz, R.H. Distinct effects of TGF-β1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: A role for T cell intrinsic Smad3. J. Immunol. 2005, 174, 2071–2083. [Google Scholar] [CrossRef]
- Larmonier, N.; Marron, M.; Zeng, Y.; Cantrell, J.; Romanoski, A.; Sepassi, M.; Thompson, S.; Chen, X.; Andreansky, S.; Katsanis, E. Tumor-derived CD4+ CD25+ regulatory T cell suppression of dendritic cell function involves TGF-β and IL-10. Cancer Immunol. Immunother. 2007, 56, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Ye, F.; Jiang, Z.; Chu, Y.; Xiong, S.; Wang, Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4+ CD25+ regulatory T cells. Cell Death Differ. 2007, 14, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.X.; Firus, J.; Prieur, B.; Jia, W.; Rennie, P.S. To die or to survive, a fatal question for the destiny of prostate cancer cells after androgen deprivation therapy. Cancers 2011, 3, 1498–1512. [Google Scholar] [CrossRef]
- Ludewig, B.; Krebs, P.; Junt, T.; Metters, H.; Ford, N.J.; Anderson, R.M.; Bocharov, G. Determining control parameters for dendritic cell-cytotoxic T lymphocyte interaction. Eur. J. Immunol. 2004, 34, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Bladou, F.; Vessella, R.L.; Buhler, K.R.; Ellis, W.J.; True, L.D.; Lange, P.H. Cell proliferation and apoptosis during prostatic tumor xenograft involution and regrowth after castration. Int. J. Cancer 1996, 67, 785–790. [Google Scholar] [CrossRef]
- Søgaard, C.K.; Moestue, S.A.; Rye, M.B.; Kim, J.; Nepal, A.; Liabakk, N.B.; Bachke, S.; Bathen, T.F.; Otterlei, M.; Hill, D.K. APIM-peptide targeting PCNA improves the efficacy of docetaxel treatment in the TRAMP mouse model of prostate cancer. Oncotarget 2018, 9, 11752–11766. [Google Scholar] [CrossRef]
- Eisenberg, M.C.; Jain, H.V. A confidence building exercise in data and identifiability: Modeling cancer chemotherapy as a case study. J. Theor. Biol. 2017, 431, 63–78. [Google Scholar] [CrossRef]
- Rubin, D.B. Using the SIR algorithm to simulate posterior distributions. Bayesian Stat. 1988, 3, 395–402. [Google Scholar]
- Dosne, A.G.; Bergstrand, M.; Harling, K.; Karlsson, M.O. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J. Pharmacokinet. Pharmacodyn. 2016, 43, 583–596. [Google Scholar] [CrossRef]
- Raftery, A.E.; Bao, L. Estimating and projecting trends in HIV/AIDS generalized epidemics using incremental mixture importance sampling. Biometrics 2010, 66, 1162–1173. [Google Scholar] [CrossRef]
- Vanlier, J.; Tiemann, C.A.; Hilbers, P.A.; van Riel, N.A. A Bayesian approach to targeted experiment design. Bioinformatics 2012, 28, 1136–1142. [Google Scholar] [CrossRef]
- Sobol, I.; Levitan, Y.L. A pseudo-random number generator for personal computers. Comput. Math. Appl. 1999, 37, 33–40. [Google Scholar] [CrossRef]
- Saltelli, A.; Annoni, P.; Azzini, I.; Campolongo, F.; Ratto, M.; Tarantola, S. Variance based sensitivity analysis of model output. Design and estimator for the total sensitivity index. Comput. Phys. Commun. 2010, 181, 259–270. [Google Scholar] [CrossRef]
- Holland, J.H. Adaptation in Natural and Artificial Systems: An Introductory Analysis with Applications to Biology, Control and Artificial Intelligence; MIT Press: Cambridge, MA, USA, 1992. [Google Scholar]
- Wei, Y.; Kehm, R.D.; Goldberg, M.; Terry, M.B. Applications for quantile regression in epidemiology. Curr. Epidemiol. Rep. 2019, 6, 191–199. [Google Scholar] [CrossRef]
- Drake, C.G. Update on prostate cancer vaccines. Cancer J. 2011, 17, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Maini, P.K. Mathematical oncology: Cancer summed up. Nature 2003, 421, 321. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A phase 2 study of galunisertib (TGF-β1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef] [PubMed]
- Manogue, C.; Cotogno, P.; Ledet, E.; Lewis, B.; Wyatt, A.W.; Sartor, O. Biomarkers for programmed death-1 inhibition in prostate cancer. Oncologist 2019, 24, 444–448. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimensions | Effect Size | SE | Z | p-Value | |
|---|---|---|---|---|---|
| mm | 52.4147 | <0.0001 | |||
| mm | 105.2884 | 33.4320 | 3.1493 | 0.0016 | |
| 1/day | 109.8302 | <0.0001 | |||
| 1/(day) | 201.5437 | 45.3648 | 4.4427 | <0.0001 | |
| 1/day | 1800.5782 | 156.7306 | 11.4884 | <0.0001 | |
| 1/(day) | 77.1509 | <0.0001 | |||
| 1/(day) | 152.1312 | 47.9072 | 3.1755 | 0.0015 | |
| 1/day | 544.2696 | 54.5942 | 9.9694 | <0.0001 | |
| 1/(day) | 37.7388 | <0.0001 | |||
| 11.6798 | <0.0001 | ||||
| 149.0218 | <0.0001 | ||||
| 277.4565 | 73.9001 | 3.7545 | 0.0002 | ||
| 45.4733 | <0.0001 | ||||
| 1/day | 1196.2169 | 100.5631 | 11.8953 | <0.0001 | |
| 1/(day) | 49.1828 | <0.0001 | |||
| cells/mm | 58.8709 | <0.0001 | |||
| (cells)/mm | 220.5233 | 37.2676 | 5.9173 | <0.0001 | |
| dimensionless | 721.4055 | <0.0001 | |||
| dimensionless | 827.6841 | 0.0008 | |||
| dimensionless | 426.4977 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, H.V.; Sorribes, I.C.; Handelman, S.K.; Barnaby, J.; Jackson, T.L. Standing Variations Modeling Captures Inter-Individual Heterogeneity in a Deterministic Model of Prostate Cancer Response to Combination Therapy. Cancers 2021, 13, 1872. https://doi.org/10.3390/cancers13081872
Jain HV, Sorribes IC, Handelman SK, Barnaby J, Jackson TL. Standing Variations Modeling Captures Inter-Individual Heterogeneity in a Deterministic Model of Prostate Cancer Response to Combination Therapy. Cancers. 2021; 13(8):1872. https://doi.org/10.3390/cancers13081872
Chicago/Turabian StyleJain, Harsh Vardhan, Inmaculada C. Sorribes, Samuel K. Handelman, Johnna Barnaby, and Trachette L. Jackson. 2021. "Standing Variations Modeling Captures Inter-Individual Heterogeneity in a Deterministic Model of Prostate Cancer Response to Combination Therapy" Cancers 13, no. 8: 1872. https://doi.org/10.3390/cancers13081872
APA StyleJain, H. V., Sorribes, I. C., Handelman, S. K., Barnaby, J., & Jackson, T. L. (2021). Standing Variations Modeling Captures Inter-Individual Heterogeneity in a Deterministic Model of Prostate Cancer Response to Combination Therapy. Cancers, 13(8), 1872. https://doi.org/10.3390/cancers13081872